

SUMMARY
Telomeres use shelterin to protect chromosome ends from activating the DNA damage sensor MRE11-RAD50-NBS1 (MRN), repressing ataxia-telangiectasia, mutated (ATM) and ATM and Rad3-related (ATR) dependent DNA damage checkpoint responses. The MRE11 nuclease is thought to be essential for the resection of the 5′ C-strand to generate the microhomologies necessary for alternative non-homologous end joining (A-NHEJ) repair. In the present study, we uncover DNA damage signaling and repair pathways engaged by components of the replisome complex to repair dysfunctional telomeres. In cells lacking MRN, single-stranded telomeric overhangs devoid of POT1-TPP1 do not recruit replication protein A (RPA), ATR-interacting protein (ATRIP), and RAD 51. Rather, components of the replisome complex, including Claspin, Proliferating cell nuclear antigen (PCNA), and Downstream neighbor of SON (DONSON), initiate DNA-PKcs-mediated p-CHK1 activation and A-NHEJ repair. In addition, Claspin directly interacts with TRF2 and recruits EXO1 to newly replicated telomeres to promote 5′ end resection. Our data indicate that MRN is dispensable for the repair of dysfunctional telomeres lacking POT1-TPP1 and highlight the contributions of the replisome in telomere repair.
Graphical Abstract
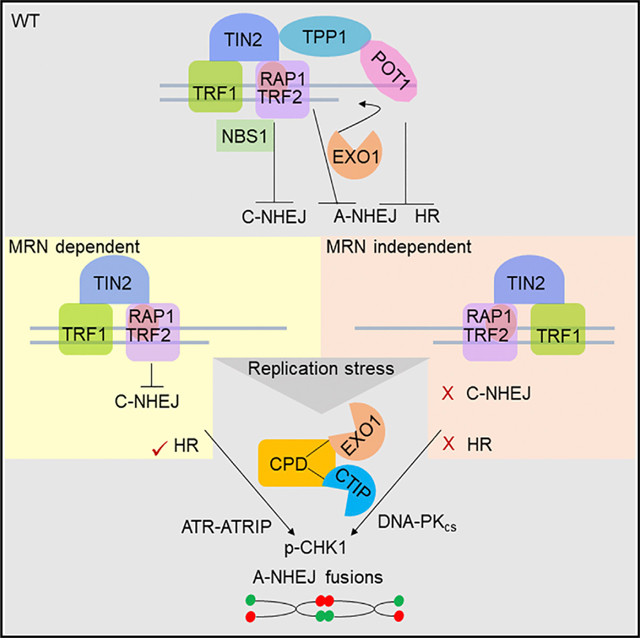
In Brief
Rai et al. define roles for the DNA replisome components Claspin, PCNA, and DONSON in the sensing and repair of telomeres lacking POT1-TPP1. In cells lacking MRN, CPD initiates DNA-PKcs-mediated p-CHK1 activation and A-NHEJ repair. Claspin directly interacts with TRF2 and recruits EXO1 to promote 5′ C-strand end resection.
INTRODUCTION
DNA double-strand breaks (DSBs) are genotoxic lesions that threaten genomic integrity. The failure to repair DSBs has deleterious consequences, leading to chromosomal translocations and genomic instability that can progress to cell death or neoplastic transformation (Aguilera and Gómez-González, 2008; Jackson and Bartek, 2009). In mammalian cells, the DNA damage response (DDR) pathway senses, signals, and repairs the damage by activating multiple DNA checkpoint and repair pathways (Ciccia and Elledge, 2010; MacDougall et al., 2007). In mammalian cells, DSBs are repaired primarily by classical non-homologous end joining (C-NHEJ), homologous recombination (HR), or alternative non-homologous end joining (A-NHEJ) repair pathways. C-NHEJ repairs DSBs through direct ligation of the broken DNA ends, with little or no end processing, and thus is error prone (Lieber, 2010). In contrast, HR uses homologous sister chromatids as templates to repair the break in an error-free manner and is initiated by extensive nucleolytic processing of the 5′ end of a DSB by DNA end resection (Huertas, 2010; Kass and Jasin, 2010; Symington, 2016). A-NHEJ repair is initiated by limited end resection and involves some of the same factors that comprise the HR end resection machinery (Sfeir and Symington, 2015; Truong et al., 2013). DNA end resection generates 3′ single-stranded DNA (ssDNA), which, if not removed by endonucleases, mitigates the activation of the ataxia-telangiectasia mutated-checkpoint kinase 2 (ATM-CHK2) checkpoint pathway that inhibits C-NHEJ repair (Huertas, 2010; Lieber, 2010). ssDNA overhangs are further sensed and bound by replication protein A (RPA) to recruit ATR interacting protein (ATRIP) and ATR to damage sites (Cortez et al., 2001; Zou and Elledge, 2003). RAD17 loads the RAD9-RAD1-HUS1 (9-1-1) complex to ssDNA to activate ATR-mediated CHK1 phosphorylation, which initiates cell-cycle arrest and DNA repair (Cimprich and Cortez, 2008; Jazayeri et al., 2006; Lee and Dunphy, 2010; Zou et al., 2002). Similar to resected ssDNA, stalled DNA replication forks possess regions of ssDNA that potently activate ATR-CHK1 by coordinating components of the replisome complex, including Claspin, AND-1, Timeless, and Tipin. These factors recruit CHK1 to ssDNA to enable CHK1 activation by ATR so as to maintain genome stability (Chini and Chen, 2003; Hao et al., 2015; Kemp et al., 2010; Kumagai et al., 2004; Lindsey-Boltz et al., 2009).
Another mediator of genome stability is telomeres, repetitive DNA-protein complexes that are protected from inappropriately activating DNA DDR checkpoints by a complex of six core telomere-specific-binding proteins called shelterin (de Lange, 2018). The duplex telomere-binding proteins TRF1 and TRF2-RAP1 and the single-stranded telomere DNA-binding protein POT1 (POT1a/b in mice) are integral members of this complex. POT1 forms a heterodimer with TPP1, and TIN2 tethers POT1-TPP1 to TRF1 and TRF2 (Wu et al., 2006). The targeted removal of specific shelterin components leads to uncapped chromosome ends that are recognized as DSBs, revealing that unique members of this complex evolved to protect telomeres from engaging in specific DNA repair pathways. In eukaryotes, the MRE11-RAD50-NBS1 (MRN) complex is the primary sensor of DSBs. Deletion of TRF2 in the G1 phase of the cell cycle activates MRN-ATM-CHK2-dependent C-NHEJ-mediated repair (Attwooll et al., 2009; Celli and de Lange, 2005; Deng et al., 2009; Dimitrova and de Lange, 2009). Removal of TRF2 and POT1a/b-TPP1 activates ATR-CHK1-dependent A-NHEJ-mediated repair (Badie et al., 2015; Denchi and de Lange, 2007; Guo et al., 2007; Kibe et al., 2016; Kratz and de Lange, 2018; Rai et al., 2010; Sfeir and de Lange, 2012). In addition, the removal of RAP1 together with the basic domain of TRF2 leads to rapid telomere attrition and formation of end-to-end chromosome fusions due to the activation of HR-mediated repair (Chen et al., 2011; Rai et al., 2016). We recently showed that the interaction of NBS1 with TRF2 dictates the telomere repair pathway choice (Rai et al., 2017). The C-NHEJ-mediated repair of telomeres lacking TRF2 requires phosphorylated NBS1 at serine 432 (NBS1S432) to activate ATM, while the interaction of de-phosphorylated NBS1S432 with TRF2 promotes the A-NHEJ repair of telomeres lacking POT1a/b-TPP1. In addition, immediately after DNA replication, TRF2 recruits the Apollo/SNM1B nuclease to resect the leading C-strand telomere to generate a short 3′ overhang (Chen et al., 2008; Lam et al., 2010; Rai et al., 2017; Wu et al., 2010). Further resection by EXO1 generates longer overhangs that are inhibitory to NHEJ repair but favor HR (Kibe et al., 2016; Wu et al., 2010, 2012).
Due to their highly repetitive nature and their propensity to adopt aberrant secondary structures, including G-quadru-plexes, telomeres represent significant challenges for the replication machinery (Verdun and Karlseder, 2007). TRF1 helps to address this problem by recruiting Timeless, a component of the replisome fork protection complex, to telomeres to prevent replication fork stalling (Leman et al., 2012). However, how the replisome contributes to the repair of damaged DNA is not known. In the present study, we uncover telomere sensing and repair roles engaged by components of the replisome Claspin, Proliferating cell nuclear antigen (PCNA), and Downstream neighbor of SON (DONSON) (CPD). CPD promotes A-NHEJ-mediated repair of dysfunctional telomeres lacking POT1-TPP1 or TRF2-POT1-TPP1 in an MRN- and ATR-ATRIP-independent manner, demonstrating that MRN is not absolutely required for A-NHEJ repair. In the absence of MRN, DNA-PKcs is required to promote CHK1 phosphorylation and A-NHEJ repair. In addition, we show that TRF2 directly interacts with Claspin and, in conjunction with DONSON, promotes the recruitment of EXO1 and CTIP to newly replicated telomeres to mediate C-strand resection and telomere end protection. Our work highlights the contribution of the replisome in mediating the repair of dysfunctional telomeres.
RESULTS
The A-NHEJ Pathway Repairs Telomeres Lacking TRF2 and POT1-TPP1 in an MRN-Independent Manner
We and others have previously shown that C-NHEJ-mediated repair of telomeres devoid of TRF2 requires functional MRN (Attwooll et al., 2009; Deng et al., 2009; Dimitrova and de Lange, 2009). To explore the requirement of the MRN complex in the repair of telomeres lacking shelterin components, we removed POT1a/b-TPP1, TRF2, and POT1a/b-TPP1 or TRF2-TIN2 in WT or Nbs1 null cell lines. More than 60% of WT mouse embryonic fibroblasts (MEFs) exhibited γ-H2AX or 53BP1+ dysfunctional telomere-induced foci (TIFs) after the removal of POT1a/b-TPP1 with the dominant negative TPP1ΔRD allele (Figures S1A and S1B) (Deng et al., 2009; Guo et al., 2007; Hu et al., 2017; Rai et al., 2010, 2011, 2017; Xin et al., 2007). Similar treatment of Nbs1−/− MEFs or human NBS-ILB1 cells lacking functional NBS1 (Falck et al., 2012; Matsuura et al., 2004) resulted in ~30% of cells displaying TIFs. A similar number of TIFs were observed in both WT and Nbs1−/− MEFs lacking both TRF2 and POT1a/b-TPP1 (Figures S1A and S1B). These results indicate that the DDR is not abrogated at telomeres lacking POT1a/b-TPP1 in the absence of functional MRN.
We next examined the number of telomere fusions in Nbs1−/− MEFs to distinguish between C-NHEJ chromosome fusions stemming from the depletion of TRF2 from A-NHEJ or HR-mediated chromosome fusions stemming from the loss of POT1-TPP1. As expected, only ~6% of telomeres lacking TRF2 were fused in MEFs lacking Nbs1. In contrast, the removal of POT1-TPP1 from WT or Nbs1−/− MEFs or NBS-ILB1 cells generated fusions involving ~15% of all chromosomes, accompanied by robust CHK1 phosphorylation (Figures 1A–1C). In WT MEFs lacking both TRF2 and POT1a/b-TPP1, 80% of all chromosomes were fused, while in Nbs1−/− MEFs or NBS-ILB1 cells, these fusions decreased to involve ~40% of chromosomes (Figures 1A–1C). Comparable numbers of chromosome fusions (30%–40%) were observed in Nbs1−/− MEFs lacking TRF2-TIN2 (Hu et al., 2017). Similar phenotypes were observed in Nbs1−/− MEFs expressing TRF2ΔBΔM and small hairpin Tpp1 (shTpp1) (Figures S1C and S1D). Chromosome orientation-fluorescence in situ hybridization (CO-FISH) (Bailey et al., 2001) revealed that chromosomal fusions lacking both TRF2 and POT1a/b-TPP1 took place in G1 phase of the cell cycle, with a small percentage of G2 chromosome and sister chromatid fusions that occurred post-replicatively (Lam et al., 2010) (Figure S1E). These observations indicate that telomeric ends lacking both TRF2 and POT1-TPP1 are efficiently repaired in an MRN-independent manner and that this MRN-independent pathway also repairs ~50% of all dysfunctional telomeres lacking both TRF2 and POT1-TPP1 in WT cells. In agreement with previously published results (Yang et al., 2006), treatment of Nbs1−/− MEFs with 2 Gy ionizing radiation (IR) also promoted the accumulation of 53BP1 and γ-H2AX to genomic DSBs, accompanied by an increase in the number of genomic chromosomal fusions (Figures S1F–S1H). These results are indicative of a role for MRN-independent repair of both telomeres lacking POT1-TPP1, TRF2, and POT1-TPP1, as well as IR-induced damaged DNA.
Figure 1. Telomeres Devoid of TRF2 and POT1-TPP1 Are Repaired in an MRN-Independent Manner.

(A) Nbs1+/+ and Nbs1−/− MEFs expressing the indicated DNA constructs for 120 h and TelG-FAM (green, leading-strand), TelC-Cy3 (red, lagging-strand), and DAPI (blue) were used to visualize fused chromosomes (arrowheads). Scale bar for all panels: 5 μm.
(B) Telomere fusion frequencies in NBS-ILB1 cells and in (A). Data are the average of 2 independent experiments ± SD from a minimum of 80 metaphases. ***p = 0.0002, ****p < 0.0001 by 1-way ANOVA. ns, non-significant.
(C) Immunoblot to detect total (T)-CHK1, T-CHK2, phosphorylated (p)-CHK1, and p-CHK2 in Nbs1+/+ and Nbs1−/− cells expressing indicated proteins. γ-Tubulin, loading control. NS, nonspecific band.
(D) Nbs1−/− MEFs and NBS-ILB1 cells expressed the indicated DNA constructs for 120 h. TelG-FAM (green), TelC-Cy3 (red), and DAPI (blue) were used to visualize fused chromosomes (arrowheads). Scale bar for all panels: 5 μm.
(E) Telomere fusion frequencies in (D). Data are the average of 3 independent experiments ± SD from a minimum of 120 metaphases. **p = 0.002, ***p = 0.0002, ****p < 0.0001 by 1-way ANOVA. Ns, non-significant.
We next determined which DNA repair pathways are involved in the MRN-independent repair of telomeres lacking both TRF2 and POT1a/b-TPP1. shRNA-mediated depletion of factors involved in A-NHEJ, including ligase III (Audebert et al., 2004; Rai et al., 2010; Sfeir and de Lange, 2012; Wang et al., 2005), polymerase q encoded by Polq (Ceccaldi et al., 2015; Mateos-Gomez et al., 2015, 2017; Sfeir and de Lange, 2012), and poly (ADP-ribose) polymerase-1 (PARP1) (Audebert et al., 2004; Sfeir and de Lange, 2012), reduced chromosomal fusions by 3- to 5-fold (Figures 1D and 1E). In contrast, the depletion of Rad51 and Rad52, which are known to play an important role in HR and break-induced repair (BIR), respectively (Sotiriou et al., 2016; Verma and Greenberg, 2016), did not appreciably reduce the number of chromosomal fusions (Figures 1D and 1E). We therefore conclude that the repair of dysfunctional telomeres that are devoid of TRF2 and POT1a/b-TPP1 is mediated by MRN-independent A-NHEJ and does not involve C-NHEJ, HR, or BIR pathways.
Replisome Components Claspin, DONSON, and PCNA Localize to Dysfunctional Telomeres Lacking Both TRF2 and POT1-TPP1
We found that the accumulation of ssDNA damage sensor protein RPA at IR-induced DNA damaged foci was reduced by ~80% in the absence of NBS1 (Figure S1F), which supports previously published reports demonstrating that in the absence of functional MRN, RPA localized poorly to damaged ssDNA, preventing the activation of ATR and downstream damage signaling (Jazayeri et al., 2006; Manthey et al., 2007; Stiff et al., 2005; Yuan and Chen, 2010). Similarly, in Nbs1−/− MEFs lacking POT1a/b-TPP1 or TRF2 and POT1a/b-TPP1, neither phosphorylated RPA32 (p-RPA32), ATRIP, nor the HR repair protein RAD51 were found to localize to dysfunctional telomeres (Figures 2A, 2B, and S2A). p-RPA32 and ATRIP+ TIFs were detected on dysfunctional telomeric ends only after the reconstitution of NBS-ILB1 cells with WT NBS1 (Figures 2A, 2B, S2B, and S2C). These results further support the observation that in the absence of functional MRN, telomeres lacking both TRF2 and POT1a/b-TPP1 are not repaired by HR. In addition, failure to detect p-RPA32 and ATRIP at these telomeres suggest that ATR is unlikely to be the kinase that phosphorylates CHK1 at these dysfunctional telomeres.
Figure 2. Recruitment of CPD to Telomeres Lacking TRF2 and POT1a/b-TPP1.

(A) Co-localization of p-RPA32, RAD51, and Flag-ATRIP with telomeres (arrowheads) in Nbs1+/+, Nbs1−/− MEFs, or NBS-ILB1 cells. Telomeres were visualized by peptide nucleic acid-fluorescent in situ hybridization (PNA-FISH) (red), indicated proteins (green), and nuclei (blue). Scale bar for all panels: 5 μm.
(B) Percentage of cells containing ≥5 TIFs in (A). Data are the mean of 2 independent experiments ± SD; n > 200 nuclei analyzed per experiment. ****p < 0.0001 by 1-way ANOVA. ns, non-significant.
(C) Co-IP of HA-Claspin with GFP-DONSON or Myc-PCNA in 293T cells. Input, 5% of the total cell lysate. γ-Tubulin, loading control.
(D–F) Localization of Claspin (D), PCNA (E), and DONSON (F) in Nbs1−/− MEFs expressing indicated DNAs. Endogenous DONSON TIFs were detectable only in HA-claspin-expressing cells. Telomeres were visualized by PNA-FISH (red), and endogenous CPD was visualized with the indicated antibodies (green). Scale bar for all panels: 5 μm.
(G) Percentage of cells containing ≥5 CPD+ foci co-localizing with telomeres, detected in (D)–(F).
(H) Co-expression of HA-Claspin with GFP-DONSON (top panel) or HA-Claspin, Myc-PCNA, and GFP-DONSON (bottom panel) in U2OS cells. Top panel: GFP-DONSON (green) and HA-Claspin(red). Bottom panel: Myc-PCNA (red) and GFP-DONSON (green). For bothpanels, anti-TRF2 antibody visualized telomeres (white).
(I) HA-Claspin and GFP-DONSON co-localized with BrdU+ foci in U2OS cells. GFP-DONSON (green), BrdU (red), and DAPI (blue). Scale bar for all panels: 5 μm.
We therefore searched for proteins that mediate both CHK1 activation and DNA repair and identified Claspin, an integral component of the replisome complex (Dungrawala et al., 2015) and an adaptor protein previously shown to mediate CHK1 phosphorylation after DNA damage (Chini and Chen, 2003; Kumagai et al., 2004; Lindsey-Boltz et al., 2009). Using the Fucci system, which uses the fluorescent G1 reporter CDT1 and the S/G2 reporter Geminin (Sakaue-Sawano et al., 2008), we found that Claspin preferentially localizes to the nuclei in S/G2 phases of the cell cycle (Figure S2D) and interacts with a large number of proteins involved in DNA repair and replication (Smits et al., 2019). Co-immunoprecipitation (Co-IP) experiments revealed that Claspin directly interacts with several replisome components, including PCNA and DONSON (Figure 2C) (Reynolds et al., 2017; Yang et al., 2008, 2016). Claspin has been shown to recognize and bind to branched/forked DNA structures containing both double-stranded and ssDNA (Lee et al., 2003; Sar et al., 2004). In vitro DNA-binding assays reveal that Claspin binds to a variety of ssDNAs, including telomeric G-rich oligos, telomeric C-rich oligos, and oligo-dT (Figures S2E and S2F). While individual endogenous or epitope-tagged Claspin, PCNA, and DONSON (abbreviated CPD) components localized poorly to dysfunctional telomeres lacking POT1a/b-TPP1 or TRF2 and POT1a/b-TPP1 in WT or Nbs1−/− MEFs (Figures 2D–2G, S2G, and S2H), co-expression of any 2 replisome components stabilized complex formation and resulted in a 4-fold increase in CPD localization at both functional and dysfunctional telomeres in U2OS cells (Figures 2H and S2I). shRNA-mediated depletion of any individual CPD component resulted in the instability of the other 2 proteins, suggesting that complex formation promotes CPD stability (Figure S2J). Finally, bromodeoxyuridine (BrdU)-labeling experiments in U2OS cells revealed that CPD readily co-localized with BrdU to newly replicated telomeres (Figures 2I and S2K).
CPD Promotes A-NHEJ-Mediated Repair of Telomeres Lacking Both TRF2 and POT1a/b-TPP1
shRNA-mediated depletion of any of the CPD components individually in WT MEFs lacking both TRF2 and POT1a/b-TPP1 resulted in a ~50% reduction in the number of γ-H2AX+ TIFs and end-to-end chromosome fusions observed, revealing that CPD participates in the A-NHEJ-mediated repair of dysfunctional telomeres (Figures 3A and 3B). In both Nbs1−/− MEFs and NBS-ILB1 cells lacking POT1a/b-TPP1, depleting CPD components individually reduced the number of γ-H2AX or 53BP1+ TIFs as well as end-to-end telomere fusions by ~3- to 5-fold without adversely affecting cell-cycle profiles, indicating that CPD plays critical roles in the sensing and repair of these dysfunctional telomeric ends independent of MRN function (Figures 3A–3C and S3A–S3F). CPD also plays a role in the HR-mediated repair of telomeres lacking POT1a/b-TPP1, since Rad51 localization and telomere sister chromatid exchanges (T-SCEs), both hallmarks of HR-mediated repair, were reduced by 3- to 4-fold in the absence of CPD (Figures S3G and S3H). In contrast, CPD is not involved in the C-NHEJ repair of telomeres lacking TRF2 (Figures 3B, 3C, and S3B–S3D).
Figure 3. CPD Promotes A-NHEJ-Mediated Chromosome Fusions in the Absence of NBS1.

(A) Percentage of cells containing ≥5 γ-H2AX+ TIFs in Nbs1+/+ and Nbs1−/− MEFs expressing the indicated DNAs. Data are the mean of 3 independent experiments ± SD; n > 300 nuclei analyzed per experiment. ***p = 0.0002, ****p < 0.0001 by 1-way ANOVA.
(B) Telomere fusion frequencies in Nbs1+/+ cells treated with shTrf2, TPP1ΔRD, or both. Data are the average of 3 independent experiments ± SD from a minimum of 140 metaphases. *p = 0.01, **p = 0.001, ****p < 0.0001 by 1-way ANOVA. ns, non-significant.
(C) Telomere fusion frequencies in Nbs1−/− treated with shTrf2, TPP1ΔRD, or both. Data are the average of 3 independent experiments ± SD from a minimum of 140 metaphases. **p = 0.001, ***p = 0.0001, ****p < 0.0001 by 1-way ANOVA. ns, non-significant.
(D) Domain organization of Claspin. Replication fork-interacting domain, basic patch I, basic patch II, PCNA-interacting domain, CHK1-binding domain, and C-terminal acidic patch. The red dashed line separates ClaspinN-Ter (amino acids [aa] 1–679) and ClaspinC-Ter (aa 679–1,332).
(E) Nbs1−/− MEFs expressed the indicated DNA constructs for 120 h. TelG-FAM, TelC-Cy3, and DAPI-labeled metaphase spreads to visualize fused chromosomes (arrowheads). Scale bar for all panels: 5 μm.
(F) Percentage of cells containing ≥5 γ-H2AX TIFs observed in (E). Data are the mean of 2 independent experiments ± SD; n > 200 nuclei analyzed per experiment. **p = 0.01, ***p = 0.001, by 1-way ANOVA. ns, non-significant.
(G) Telomere fusion frequencies in (E). Data are the average of 2 independent experiments ± SD from a minimum of 70 metaphases. **p = 0.001, ***p = 0.0001, ****p < 0.0001 by 1-way ANOVA. ns, non-significant.
(H) Co-IP of HA-ClaspinWT, HA-ClaspinN-Ter, or HA-ClaspinC-Ter with GFP-DONSON in 293T cells. Input, 5% of the total cell lysate. γ-Tubulin, loading control.
(I) U2OS cells transiently transfected with HA-ClaspinWT, HA-Claspin3A, HA-ClaspinN-Ter, or HA-ClaspinC-Ter with GFP-DONSON for 48 h. GFP-DONSON (green), HA-Claspin (red), and DAPI to visualize nuclei (blue). Scale bar for all panels: 5 μm.
(J) Percentage of cells containing ≥5 γ-H2AX+ TIFs in Nbs1−/− MEFs infected with indicated DNA constructs. Data are the mean of 2 independent experiments ± SD; n > 200 nuclei analyzed per experiment. **p = 0.001, ***p = 0.0003 by 1-way ANOVA. ns, non-significant.
(K) Telomere fusion frequencies. Data are the average of 3 independent experiments ± SD from a minimum of 120 metaphases. ***p = 0.001, ****p < 0.0001 by 1-way ANOVA. ns, non-significant.
Claspin contains several functional domains, including a PCNA-interacting protein motif (PIP) near its N terminus and a CHK1-binding domain (CKBD) along with an acidic patch (AP) in its C terminus (Figure 3D) (Smits et al., 2019; Yang et al., 2016). Phosphorylation of the AP by CDC7 prevents the intramolecular interaction of Claspin, promoting its binding to PCNA and DNA substrates (Yang et al., 2016). Three phosphorylation sites in the CKBD domain of Claspin and the mutation of these to al-anines (the Claspin3A mutant) abolished the ability of Claspin to promote TIF formation, end-to-end chromosome fusions, and CHK1 phosphorylation (Figures 3E–3G, S3I, S3J, S4A, and S4B) (Lindsey-Boltz et al., 2009). The number of TIFs, chromosome fusions, and CHK1 phosphorylations was almost fully restored when shRNA-resistant WT Claspin but not the Claspin3A mutant was reconstituted into Nbs1−/− MEFs lacking TRF2-POT1a/b-TPP1 (Figures 3E–3G, S3I, S3J, S4A, and S4B), revealing that Claspin-mediated CHK1 phosphorylation is essential for the A-NHEJ repair of telomeres lacking POT1a/b-TPP1. We also found that the Claspin N terminus (ClaspinN-Ter) interacts with PCNA, while the C terminus (ClaspinC-Ter) interacts with DONSON (Figures 3H and S4C). Claspin3A and ClaspinC-Ter but not ClaspinN-Ter are able to co-localize with GFP-DONSON on telomeres, but only ClaspinC-Ter promotes CHK1 phosphorylation (Figures 3I, S4B, and S4D). Depletion of CPD individually abolished TIF formation, chromosome fusion, and CHK1 phosphorylation in Nbs1−/− MEFs lacking both TRF2 and POT1a/b/TPP1, and only the expression of WT CPD rescued these phenotypes (Figures 3E–3G, 3J, 3K, S4B, and S4D–S4I). These results suggest that CPD is required for A-NHEJ repair at dysfunctional telomeres lacking both TRF2 and POT1a/b-TPP1 by promoting CHK1 phosphorylation in an MRN-independent manner.
DNA-PKcs Is Required for MRN-Independent CHK1 Phosphorylation
In the absence of functional MRN, telomeres lacking both TRF2 and POT1a/b-TPP1 fail to recruit RPA, ATRIP, and RAD51, suggesting that ATR-ATRIP cannot phosphorylate CHK1 at these telomeres (Figures 2A and 2B). Since expression of the Claspin3A mutant reduced both CHK1 phosphorylation and the A-NHEJ repair of dysfunctional telomeres in Nbs1−/− MEFs (Figures 3E–3G and S4B), our data suggest that Claspin-mediated phosphorylation of CHK1 is required for A-NHEJ repair of dysfunctional telomeres in the absence of functional MRN. In support of this observation, Claspin has been shown to participate in ATR-independent phosphorylation of CHK1 (Kumagai et al., 2004; Rodríguez-Bravo et al., 2006). Previous studies also reveal that DNA-dependent protein kinase (DNA-PKcs), a key regulator of C-NHEJ repair, is required to maintain Claspin-CHK1 complex stability and is activated by DSBs in the absence of functional MRN (Hartlerode et al., 2015; Lin et al., 2014). We postulated that DNA-PKcs may participate in the phosphorylation of CHK1 at dysfunctional telomeres when MRN function is abrogated. To test this hypothesis, we treated Nbs1−/− MEFs bearing dysfunctional telomeres devoid of TRF2 and POT1a/b-TPP1 with the specific DNA-PKcs inhibitor NU7441. The presence of 10 μM of NU7441 decreased both γ-H2AX and 53BP1 TIF formation by 3-fold and resulted in a 4-fold reduction in the number of chromosomal fusions repaired by A-NHEJ (Figures 4A–4D). These levels of TIF and fusion reductions are similar to those observed in cells expressing the Claspin3A mutant. A 4-fold reduction in the level of CHK1 phosphorylation was also observed in Nbs1−/− MEFs expressing TIN2A110R or TPP1ΔRD to generate dysfunctional telomeres lacking either TRF2-TIN2 or POT1a/b-TPP1 (Figures 4E and 4F). These data support a critical role for DNA-PKcs to promote efficient CHK1 phosphorylation in MRN-deficient cells bearing dysfunctional telomeres.
Figure 4. Inhibition of DNA-PKcs Abolishes DDR and A-NHEJ-Mediated Chromosome Fusions in Nbs1−/− Cells.

(A) Nbs1−/− cells expressing the indicated DNA constructs were treated with DMSO or 10 μM DNA-PKcs inhibitor NU7441 and γ-H2AX, and 53BP1+ TIFs were detected. Telomeres were visualized by PNA-FISH (red), proteins (green), and nuclei (blue). Scale bar for all panels: 5 μm.
(B) Percentage of cells containing positive γ-H2AX and 53BP1 TIFs in (A). Data are the mean of 2 independent experiments ± SD; n > 200 nuclei analyzed per experiment. ***p = 0.0002, ****p < 0.0001 by 1-way ANOVA.
(C) Nbs1−/− MEFs expressing indicated DNA constructs were treated with DMSO or 10 μM NU7441 for 120 h. TelG-FAM, TelC-Cy3, and DAPI were used to visualize fused chromosomes (arrowheads).
(D) Telomere fusion frequencies in (C). Data are the average of 2 independent experiments ± SD from a minimum of 80 metaphases. **p = 0.001, ***p =0.0005 by 1-way ANOVA. Scale bar for all panels: 5 μm.
(E and F) Immunoblot for p-CHK1 levels in Nbs1−/− cells expressing either TIN2A110R (E) or TPP1ΔRD (F) treated with DMSO or NU7441. γ-tubulin, loading control. NS, nonspecific band.
CPD Promote DNA End Resection at Telomeres Devoid of TRF2 and POT1a/b-TPP1
Telomeres lacking TRF2-POT1a/b-TPP1 recruit BARD1, CTIP, and BRCA1 in a CPD-dependent manner (Figures 5A and 5B). The shRNA-mediated depletion of CPD decreased the telomeric localization of BARD1 and BRCA1 by ~3-fold (Figures 5C, 5D, S5A, and S5B). These results suggest that BRCA1 and BARD1 play important roles in the repair of telomeres lacking TRF2-POT1a/b-TPP1 in the absence of functional MRN, a notion that is further supported by the observation that the depletion of BRCA1 in NBS1-ILB1 cells devoid of TRF2-POT1-TPP1 reduced telomere fusions by 3-fold (Figures 5E, 5F, and S5C). We also found that the shRNA-mediated depletion of exonucleases EXO1 and CTIP in Nbs1−/− MEFs devoid of TRF2 and POT1a/b-TPP1 reduced end-to-end chromosome fusions by ~4-fold, suggesting that EXO1-mediated nucleolytic processing of the telomeric C-strand is required for A-NHEJ-mediated, MRN-independent repair of dysfunctional telomeres (Figures 5E, 5F, and S5D). To test this notion, we performed telomere restriction fragment (TRF) southern to monitor the length of the G-overhang, which also provides an indication of the amount of C-strand processing. For example, increased C-strand processing would manifest as an increase in the amount of ss telomeric G-strand overhang observed (Gu et al., 2018; Lam et al., 2010). TRF southern of Nbs1−/− MEFs devoid of TRF2 and POT1a/b-TPP1 and treated with 2 independent shRNAs targeting either EXO1, CTIP, or CPD individually revealed a reduction in the amount of G-overhang detected, suggesting a defect in C-strand processing (Figures 5G and 5H). To rule out the possibility that this increase in G-overhang elongation is due to telomerase activity, we examined G-overhang formation in telomerase null generation 1 (G1) mTR−/− MEFs after removing TRF2 and POT1a/b-TPP1. The lack of telomerase did not abolish the formation of γ-H2AX TIFs, A-NHEJ-mediated end-to-end chromosome fusion, or increased G-overhang formation on these dysfunctional telomeres, ruling out a role for telomerase in G-overhang elongation (Figures S5E–S5J). The depletion of Exo1 in G1 mTR−/− MEFs resulted in decreased G-overhang formation, further supporting the notion that increased nucleolytic C-strand processing by EXO1 mediates the length of the G-strand at telomeres lacking POT1a/b-TPP1 (Figures S5I and S5J). The shRNA-mediated depletion of CPD, EXO1, and CTIP also resulted in a corresponding decrease in CHK1 phosphorylation in NBS-ILB1, Nbs1−/−, and G1 mTR−/− MEFs (Figures 5I, S5D, and S5K). Finally, we addressed the possibility that compromised C-strand fill-in machinery resulted in increased G-overhang. The CTC1-STN1-TEN1 (CST) complex promotes C-strand fill-in with DNA Pol-α, and CST localization to telomeres requires POT1b (Figures S5L and S5M) (Gu et al., 2018; Wu et al., 2012). The expression of TPP1ΔRD prevents STN1 localization to the 5′ telomeric ends by removing POT1b from telomeres (Figure S5M), compromising the C-strand fill-in machinery. However, even when C-strand fill-in synthesis is inhibited by TPP1ΔRD, the 3′ overhang is still elongated (Figures 5G, 5H, S5I, and S5J). These results suggest that CPD, EXO1, and CTIP are required for the processing of the telomeric C-strand, generating the ss 3′ telomeric overhangs necessary for A-NHEJ-mediated repair.
Figure 5. CPD Complex Promotes DNA End Resection at Telomeres Devoid of TRF2 and POT1-TPP1.

(A) BARD1, CTIP, and BRCA1 co-localized with telomeres (arrowheads) in Nbs1−/− MEFs and NBS1-ILB1 cells treated with shTrf2 and TPP1ΔRD. Telomeres were visualized by PNA-FISH (red), proteins (green), and nuclei (blue). Scale bar for all panels: 5 μm.
(B) Percentage of cells containing ≥5 TIFs in (A). Data represent the mean of 3 independent experiments ± SD; n > 250 nuclei analyzed per experiment. ***p = 0.0001, ****p < 0.0001 by 1-way ANOVA.
(C) Percentage of cells with ≥5 BARD1+ TIFs in Nbs1+/+ or Nbs1−/− MEFs treated with the indicated shRNAs and shTrf2 + TPP1ΔRD. Data represent the average of 3 independent experiments ± SD; n > 250 nuclei analyzed per experiment. *p = 0.02, ****p < 0.0001 by 1-way ANOVA.
(D) Percentage of cells with ≥5 BRCA1+ TIFs in NBS1-ILB1 cells treated with shClaspin 1 or 2 and TRF2ΔBΔM + TPP1ΔRD. Data represent the average of 2 independent experiments ± SD from a minimum of 200 nuclei analyzed per experiment. **p = 0.002, ***p = 0.0008 by 1-way ANOVA. ns, non-significant.
(E) Nbs1−/− or NBS1-ILB1 cells were treated with the indicated DNA constructs for 120 h. TelG-FAM, TelC-Cy3, and DAPI were used to visualize fused chromosomes (arrowheads). Scale bar for all panels: 5 μm.
(F) Telomere fusion frequencies in (E). Data represent the average of 2 independent experiments ± SD from a minimum of 90 metaphases. ***p = 0.0002, ****p < 0.0001 by 1-way ANOVA. ns, non-significant.
(G) Effect of indicated shRNAs on telomere end resection in Nbs1−/− MEFs using in-gel hybridization with radiolabeled (CCCTAA)4 probes to detect 3′ ss overhang under native conditions (left) and denaturing conditions to detect total TTAGGG repeats (right).
(H) Quantification of telomeric overhang signals in (G). Single-stranded TTAGGG signals were normalized to the total TTAGGG signal in the same lane and set to 100, and all of the other values are displayed relative to this value.
(I) Immunoblots were examined for the level of indicated proteins in Nbs1−/− MEFs expressing indicated DNA constructs and shRNAs targeting CPD. γ-tubulin, loading control. NS, nonspecific band.
TRF2 Interacts with Claspin to Recruit CPD and EXO1 to Dysfunctional Telomeres That Resemble Stalled Replication Forks
Telomeric DNA are difficult to replicate regions due to their repetitive nature and propensity to form G4 quadruplexes that can impede the progression of replicative DNA polymerases. POT1 represses the formation of these structures, and its depletion results in replication defects at telomeres (Pinzaru et al., 2016; Rizzo et al., 2009; Zaug et al., 2005). The localization of CPD to telomeres lacking POT1a/b-TPP1 suggests that these dysfunctional telomeres likely adopt difficult-to-repair secondary structures resembling stalled replication forks. Claspin has been shown to recognize and bind to branched/forked DNA structures containing both double-stranded and ssDNA to facilitate phosphorylation of CHK1 by ATR (Lee et al., 2003; Sar et al., 2004). We postulate that CPD is required to help resolve aberrant structures at ss telomeres lacking POT1-TPP1 to promote A-NHEJ repair. To test this hypothesis, we examined the localization of SMARCAL1 (SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A-like 1), an ATP-dependent annealing helicase that localizes to stalled replication forks to promote replication restart (Bansbach et al., 2009; Cox et al., 2016; Poole et al., 2015; Yusufzai and Kadonaga, 2008). In addition, SMARCAL1 localizes to telomeres undergoing replication stress (Cox et al., 2016). While cells lacking TRF2 displayed only background levels of SMARCAL1+ TIFs, 15% of U2OS cells lacking POT1-TPP1 displayed >5 SMARCAL1+ TIFs (Figures 6A and S6A). Removal of TRF2 and POT1-TPP1 from telomeres increased the number of cells bearing >5 SMARCAL1+ TIFs to 25%. These results suggest that telomeres devoid of POT1-TPP1 adopt secondary structures that recruit CPD and SMARCAL1 to maintain telomere stability. In support of this observation, WT GFP-DONSON readily co-localized with HA-Claspin to telomeres lacking POT1-TPP1 and repressed the telomeric localization of SMARCAL1 to background levels (Figures S6B–S6D).
Figure 6. CPD and EXO1 Are Recruited by TRF2 to Dysfunctional Telomeres That Resemble Stalled Replication Forks.

(A) SMARCAL1 co-localizes with telomeres in U2OS cells expressing the indicated cDNAs. Telomere PNA-FISH (red), antibody staining (green), and DAPI (blue). Scale bar for all panels: 5 μm.
(B) Co-localization of GFP-DONSON and HA-Claspin with endogenous EXO1 at telomeres. Data represent the mean of 2 independent experiments ± SD; n > 150 nuclei analyzed per experiment. *p = 0.01, **p = 0.001 by 1-way ANOVA. ns, non-significant.
(C) WT GFP-DONSON or mutant DONSON interactions with V5-EXO1. Inputs represent 5% of the total cell lysate used for the IP. γ-tubulin, loading control.
(D) (Top) diagram illustrating that the TRF2TRFH domain interacts with the ClaspinTBM domain in its C terminus. (Bottom) representative proteins that contain the TBM (F/Y/H-X-L-X-P) are indicated.
(E) ITC measurements of the interaction between TRF2TRFH and ClaspinTBM peptides.
(F) Chromosome and chromatid fusions in Apollo/SNM1B−/− MEFs treated with indicated shRNAs for 120 h. TelG-FAM, TelC-Cy3, and DAPI were used to visualize fused chromosomes (arrowheads). Representative G1/G2 chromosome fusions (white arrows), G2 chromosome fusions (orange arrows), leading-leading strand (green arrows), sister-sister telomere fusion (pink arrows), and interstitial telomeres (red arrows) are indicated. Scale bar for all panels: 5 μm.
(G) Quantification of G1/G2 and G2 chromosome-type fusions in (F). Data represent the mean of 2 independent experiments ± SD from a minimum of 50 metaphases analyzed per experiment.
(H) Quantification of the chromatid-type and sister fusions in (F). Data represent the mean of 2 independent experiments ± SD from a minimum of 50 metaphases analyzed per experiment.
A recent report identified hypomorphic mutations in DONSON patients that increased the formation of stalled replication forks (Reynolds et al., 2017). DONSON mutants failed to co-localize with HA-Claspin to telomeres lacking POT1-TPP1 and are unable to suppress the telomeric localization of SMARCAL1 (Figures S6B–S6D). These results suggest that telomeres lacking POT1-TPP1 adopted aberrant secondary structures resembling stalled replication forks, leading to the recruitment of SMARCAL1 and CPD. Using immunofluorescence and fluorescence in situ hybridization (IF-FISH), we also found that endogenous EXO1 co-localized to dysfunctional telomeres with GFP-DONSON and HA-Claspin (Figures 6B and S6E). Co-immunoprecipitation (Co-IP) experiments revealed that DONSON but not Claspin or PCNA interacts directly with EXO1 and CTIP (Figures 6C and S6F). With the exception of DONSONK489T, all other DONSON mutants examined showed decreased interaction with EXO1 (Tomimatsu et al., 2014), CTIP, and Claspin (Figures 6C, S6G, and S6H).
Following DNA replication, telomeres synthesized from the leading-strand template are blunt ended, while lagging-strand telomeres possess short ssDNA overhangs due to the removal of the terminal Okazaki fragment. Nucleolytic processing of the C-strand by MRE11 and Apollo/SNM1B, followed by more extensive processing by EXO1, generates the 3′ overhang at leading-strand telomeres inhibitory to both leading- and lagging-strand chromatid fusions (Lam et al., 2010; Wu et al., 2012). It is unclear how EXO1 is recruited to newly replicated telomeres. We postulate that the interaction of EXO1 with DONSON (and by extension, CPD) is required for its recruitment to newly replicated telomeres. In support of this notion, sequencing analysis revealed that the C terminus of Claspin possesses a highly evolutionarily conserved TRF2-binding motif (TBM), 1286Y/F/H-X-L-X-P1290 (where X is any amino acid), found only in proteins that interact with the TRF2 homology (TRFH) domain (Figures 6D and S6I) (Chen et al., 2008). Isothermal titration calorimetry revealed that the ClaspinTBM peptide binds to TRF2TRFH peptide with a Kd of 80 μM (Figure 6E). To further validate TRF2 and Claspin interaction in vivo, we performed Co-IP experiments with endogenous proteins in both 293T and Nbs1−/− MEFs. Co-IP results revealed that endogenous Claspin interacts with endogenous TRF2, and this interaction was completely abolished by the TRF2F120A mutation, which has been shown to disrupt the ability of the TRF2TRFH domain to interact with TBM-containing proteins (Figures S7A–S7C). These results suggest that the recruitment of Claspin to telomeres occurs via its specific interaction with TRF2.
To characterize the ability of CPD to recruit EXO1 to protect newly replicated telomeres, we used CO-FISH to interrogate Apollo/SNM1B−/− MEFs (Lam et al., 2010; Wu et al., 2010). We found that CPD foci readily co-localized to Apollo/SNM1B−/− telomeres (Figure S7D). Using CO-FISH to distinguish leading-strand (green signal) and lagging-strand (red signal) telomeres, we found that the deletion of Apollo/SNM1B results in an increase (~5%) in the number of leading-leading-strand chromatid fusions characteristic of post-replicative repair, as well as increased G1/G2 and G2 chromosome fusions following progression through the cell cycle (Figures 6F–6H and S7E) (Lam et al., 2010; Wu et al., 2010). The shRNA-mediated depletion of CPD and EXO1 individually in Apollo/SNM1B−/− MEFs resulted in a 2- to 3-fold increase in the number of both leading-strand and lagging-strand chromatid fusions and subsequent G1/G2 and G2 chromosome fusions, suggesting that CPD and EXO1 participate in the C-strand processing of both the leading-ends and the lagging-ends without the need for the enzymatic activity of Apollo/SNM1B to initiate nucleolytic cleavage (Figures 6F–6H and S7F). Expression of the DONSON mutants but not WT DONSON in Apollo/SNM1B−/− MEFs increased the number of chromatid and chromosome aberrations, further supporting the notion that these cytogenetic aberrations arise due to defects in telomere replication (Figures S7G and S7H). We also detected a 10-fold increase in the number of chromosomes possessing interstitial telomeric signals (ITSs) of leading-strand and lagging-strand telomeres when CPD or EXO1 was individually depleted in Apollo/SNM1B−/− MEFs (Figures 6F and S7I). In addition, the hallmarks of DNA damage, including chromosome breaks, chromosome fragments, radial chromosomes, and fragile telomeres (indicative of telomere replication defects), were also observed when CPD or EXO1 was individually depleted (Figures S7I and S7J). These results suggest that TRF2-mediated recruitment of Apollo/SNM1B, CPD, DONSON, EXO1, and CTIP to newly replicated telomeres is required for nucleolytic processing of the C-strand at both leading and lagging chromosomes. EXO1 and CTIP generates the 3′ overhangs, enabling the loading of POT1-TPP1 to prevent the initiation of A-NHEJ-mediated chromosome and chromatid fusions. Depletion of CPD in an Apollo/SNM1B−/− background further compromised G-overhang formation, resulting in the increased cytogenetic aberrations observed.
Mouse Tumors without Pot1a Experience Increased Replication Stress
To determine whether the deletion of Pot1a promotes replication stress in solid tumors, we examined tumors derived from our MMTV-Cre+; Pot1aF/F; p53F/F mouse tumor model (Gu et al., 2017). We found that 17/24 of mammary, sarcoma, and salivary tumors generated were genotyped as Pot1aF/Δ; p53Δ/Δ, while 7/24 were Pot1aΔ/Δ; p53Δ/Δ (Figures 7A and S8A; data not shown). Examination of 5 MMTV-Cre+; Pot1aΔ/Δ; p53Δ/Δ tumors by western analysis revealed that they were segregated into 2 groups: a Claspin-low group (tumors 4 and 5) and a Claspin-high group (tumors 1–3) (Figure 7A). In the Claspin-low group, p-RPA and p-CHK1 levels were almost undetectable, and γ-H2AX and p-RPA32+ TIFs were detected in only 20% of nuclei (Figures 7A, 7D–7F, S8B, and S8C). SMARCAL1+ TIFs were also low in these tumors (Figures 7G and S8D), and end-to-end telomere fusions and interstitial telomeres involved only ~1.8% of all chromosome ends (Figures 7H and 7I). In contrast, Claspin-high tumors expressed high levels of p-RPA32, p-CHK1, and SMARCAL1 by western analysis with increase Claspin and PCNA TIFs (Figures 7A–7C). We also detected >5 γ-H2AX+ TIFs in 35% of cells and ≥3 p-RPA32+ TIFs in 12% of cells, along with increased co-localization of SMARCAL1 on telomeres (Figures 7D–7G and S8B–S8D). Amplification of ITS was found in ~12% of all chromosomes, with tumor number 3 showing such massive ITS amplifications that they can be detected in interphase nuclei (Figures 7H, 7I, and S8B–S8D). Only a small region of the large ITSs contain DSBs that co-localize with γ-H2AX (Figure S8B). These results suggest that the deletion of Pot1a results in increased replication stress at telomeres in certain Pot1a null tumors, necessitating the increased expression of Claspin and SMARCAL1 to maintain telomere stability. In support of this observation, telomeric replication stress was not detected in Pot1aΔ/Δ; p53Δ/Δ tumors, since γ-H2AX/p-RPA32+ TIFs, interstitial telomeres, and chromosomal aberrations were found at barely detectable levels (Figures 7D–7I and S8B–S8C). Correspondingly, Claspin and SMARCAL1 were undetectable by western analysis (Figure 7A). To further examine the correlation of POT1 and Claspin levels in human cancer, we analyzed the status of Claspin and POT1 gene expression in 308 human colon carcinomas in The Cancer Genome Atlas (TCGA) database. We found a significant negative correlation between POT1 and Claspin gene expression in these cancers (Figure 7J).
Figure 7. Genomic Instability in Tumors Derived from MMTV-Cre; p53F/F; Pot1aF/F Mice.

(A) CPD, SMARCAL1, p-CHK1, and p-RPA32 expression in MMTV-Cre; p53Δ/Δ; Pot1aΔ/Δ breast tumors (#1, 3, and 4), sarcoma (#2), and salivary gland tumor (#5). Tumor #6 is MMTV-Cre; p53Δ/Δ; Pot1aF/ΔΔ. γ-Tubulin, loading control. Bottom panel: PCR genotyping.
(B) Detection of Claspin and PCNA (green) and telomeres (red) in tumor #1.
(C) Percentage of cells containing ≥3 Claspin/PCNA (CP+) TIFs in (B). Scale bar for all panels: 5 μm.
(D) γ-H2AX TIFs in tumor cell lines. Telomere PNA-FISH (red), antibody staining (green), and nuclei (blue). Scale bar for all panels: 5 μm.
(E) Percentage of cells containing ≥5 γ-H2AX TIFs in (D). Data represent the mean of 2 independent experiments ± SD; n > 200 nuclei analyzed per experiment. *p = 0.01, **p = 0.001, ***p = 0.0001 by 1-way ANOVA.
(F) Percentage of cells containing ≥3 p-RPA32 TIFs in tumor cell lines.
(G) SMARCAL1 TIFs in tumor cell lines.
(H) Interstitial telomeres (red arrow), chromosome and sister chromatid fusions (orange arrow) in Pot1a deleted mouse tumor cell lines. TelG-FAM, TelC-Cy3, and DAPI were used to visualize fused chromosomes. Scale bar for all panels: 5 μm.
(I) Quantification of the number of interstitial telomeres and chromosome fusions in (H). Data represent the average of 2 independent experiments ± SD from a minimum of 70 metaphases.
(J) Spearman’s correlation between claspin and POT1 gene expression in colon adenocarcinoma obtained from TCGA data.
(K) Schematic depicting CPD functions. See text for details.
DISCUSSION
In this study, we describe unexpected roles for the replisome components Claspin, PCNA, and DONSON at telomeres. CPD is recruited to telomeres through the interaction of Claspin with TRF2-RAP1, while DONSON interacts with EXO1 and CTIP. Together, these proteins promote telomeric C-strand resection and the generation of 3′ G-overhangs, which serve as a platform for POT1-TPP1 binding to protect telomeres from initiating A-NHEJ repair (Figure 7Ka). Dysfunctional telomeres lacking POT1-TPP1 also generate aberrant secondary structures that are inhibitory to A-NHEJ repair, requiring resolution by CPD and SMARCAL (Figure 7Kb). In the absence of functional MRN, Claspin-mediated CHK1 phosphorylation and A-NHEJ repair requires DNA-PKcs (Figure 7Kc). Finally, mouse tumors lackingPot1a display increased telomere replication stress (RS) and elevated Claspin expression. Our results highlight the importance of CPD in promoting the A-NHEJ repair of dysfunctional telomeres lacking POT1-TPP1.
Role of Replisome Components in the Repair of Telomeres Lacking POT1-TPP1 by A-NHEJ
Telomere DNA is difficult to replicate due to its highly repetitive nature and ability to form aberrant secondary structures that impede polymerase progression (Crabbe et al., 2004; Gilson and Géli, 2007; Rizzo et al., 2009; Zaug et al., 2005). TRF1 promotes normal fork progression through telomeres by recruiting the helicases RTEL1 and BLM to facilitate lagging-strand telomere synthesis by resolving G4 quadruplexes (Sfeir et al., 2009; Vannier et al., 2012, 2013). The interaction of RTEL1 with PCNA is essential for proper telomere replication. Our findings also highlight a role for the replisome in mediating the repair of dysfunctional telomeres. Telomeres devoid of POT1-TPP1 activate ATR-dependent hyper-resection of the 5′ C-strand, resulting in the generation of long 3′ ss overhangs (Kibe et al., 2016). Since POT1-TPP1 functions to repress RPA from loading onto 3′ G-overhangs (Flynn et al., 2011; Gong and de Lange, 2010), we postulate that in the absence of POT1-TPP1, telomeres adopt aberrant secondary DNA structures similar to those found in stalled replication forks. Claspin specifically interacts with TRF2 to help resolve these structures and facilitate A-NHEJ repair independent of MRN status. In support of this notion, shRNA-mediated depletion of individual CPD components reduced A-NHEJ-mediated chromosome fusions to levels similar to those observed when Pol-q, PARP1, or ligase III are removed, indicative of their importance to promote the A-NHEJ repair of ss telomeric ends. Our data also suggest that CTIP, BRCA1, and EXO1 play roles in mediating the extensive 5′−3′ C-strand resection necessary to generate the long tracks of microhomologies necessary for A-NHEJ repair in the absence of TRF2-POT1-TPP1. Furthermore, localization of RPA to ssDNA represses Pol-q-mediated A-NHEJ repair and instead channels the ssDNA substrates for HR-mediated repair (Mateos-Gomez et al., 2017). Since telomeres lacking POT1-TPP1 cannot recruit RPA in the absence of MRN, they must be repaired through A-NHEJ.
While ITC revealed that the interaction between ClaspinTBM and TRF2TRFH peptides is relatively weak, it is important to note that the ability of Claspin to localize to telomeres increases when both PCNA and DONSON are present (Figures 2H and S2I), suggesting that the interaction of the CPD complex with TRF2 is likely substantially stronger than in vitro peptide interactions.
DNA-PKcs-Dependent, MRN-Independent Phosphorylation of CHK1 for A-NHEJ Repair
In the absence of the functional MRN complex, RPA localized poorly to damaged ssDNA, preventing the activation of ATR-ATRIP and downstream damage signaling through CHK1 (Jazayeri et al., 2006; Manthey et al., 2007; Stiff et al., 2005; Yuan and Chen, 2010). However, NBS1 has been shown to directly activate ATR independently of MRE11 and TOPBP1 function (Kobayashi et al., 2013). The Ustilago maydis Claspin homolog Mrc1 but not MRN is required to initiate the CHK1-dependent DNA damage response necessary for cell-cycle arrest (Tenorio-Gómez et al., 2015). This result suggests that MRN activation of the ATR/CHK1 pathway is dispensable under certain DNA damage conditions, and that Claspin could substitute for MRN function. In the absence of functional MRN, RPA and ATRIP fail to localize to telomeres lacking POT1-TPP1, suggesting that the MRN-ATRIP-ATR cascade required for CHK1 phosphorylation is dispensable for A-NHEJ repair. We now show that DNA-PKcs can substitute for MRN function by promoting CHK1 phosphorylation and A-NHEJ-mediated repair at telomeres lacking POT1-TPP1, an observation that is in line with a previous report suggesting that DNA-PKcs is required to maintain Claspin-CHK1 complex stability (Lin et al., 2014).
CTIP Functions Independently of MRN for A-NHEJ Repair of Dysfunctional Telomeres Lacking POT1a/b-TPP1
In mammalian cells, MRE11-CTIP nucleases are required to resect DSBs to generate the resected 3′ overhangs necessary for annealing and A-NHEJ repair (Buis et al., 2012; Dinkelmann et al., 2009; Rass et al., 2009; Xie et al., 2009). While MRN-CTIP is essential for C-NHEJ-mediated repair of telomeres lacking TRF2 (Attwooll et al., 2009; Deng et al., 2009; Dinkelmann et al., 2009), our data indicate that MRN but not CTIP is dispensable for A-NHEJ-mediated repair of telomeres lacking POT1a/b-TPP1. While CTIP has been shown to enhance the nuclease activity of MRE11 to promote A-NHEJ repair (Badie et al., 2015; Bennardo et al., 2008; Kibe et al., 2016; Lee-Theilen et al., 2011; Rass et al., 2009; Sfeir and de Lange, 2012; Xie et al., 2009; Zhang and Jasin, 2011), CTIP nuclease activity independent of MRN function has also been reported (Lee-Theilen et al., 2011). Our data strongly suggest that at least at telomeres lacking POT1a/b-TPP1, CTIP is required to promote A-NHEJ mediated repair in the absence of the functional MRN complex, a notion that is consistent with the MRN-independent nuclease activity of CTIP/Sae2 (Lengsfeld et al., 2007; Makharashvili et al., 2014; Przetocka et al., 2018; Wang et al., 2014; Yu et al., 2018).
POT1 Protects Tumors from Replication Stress
At stalled replication forks, RPA serves as a platform to recruit fork repair proteins to promote fork restart and restore replication (Bhat and Cortez, 2018). After fork restart, RPA must be displaced for DNA replication to proceed normally. POT1 has been shown to participate in RPA exclusion at telomeres (Flynn et al., 2011). We postulate that POT1 may serve an analogous function at telomere replication forks. The deletion of POT1 is thus expected to result in increased replicative stress at telomeres. A subset of MMTV-Cre; Pot1aΔ/Δ; p53Δ/Δ tumors show high levels of Claspin expression, elevated p-CHK1 levels, and increased localization of γ-H2AX and SMARCAL1 to telomeres, all indicative of increased telomere replication stress. Our data suggest that the upregulation of Claspin is required to maintain genome stability in tumors with high levels of RS. In support of this notion, a recent report demonstrates that the increased expression of Claspin, Timeless, and CHK1 is observed in a diverse array of primary human tumors to enable tolerance to increased RS and maintain the integrity of the replication fork (Bianco et al., 2019).
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Sandy Chang (s.chang@yale.edu). All plasmids and cell lines generated in this study are available on request without restrictions.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Human NBS-ILB1 cells, Nbs1+/+, Nbs1−/−, G1 mTR−/− and Apollo/SNM1B−/− MEFs and 293T were cultured in DMEM supplemented with 10% FBS and maintained in 5% CO2 at 37° C. U2OS cells were cultured in Macoy’s medium. Source of U2OS, 293T and MEFs used in this study are listed in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti phospho-CHKI | Cell Signaling Technology | 2348; RRID: AB_331212 |
| Mouse anti phospho-CHK2 | BD Biosciences | 611570; RRID: AB_399016 |
| Mouse anti γ-H2AX | Millipore | 05–636; RRID: AB_309864 |
| Mouse anti-TRF2 | Millipore | 05–521; RRID: AB_2303145 |
| Mouse anti-Myc | Millipore | 05–724, RRID: AB_309938 |
| Rabbit anti-RAD51 | Santacruz | sc-8349, RRID: AB_2253533 |
| Mouse anti-BRCA1 | Santacruz | sc-6954, RRID: AB_626761 |
| Rabbit anti-BARD1 | Santacruz | sc-11438; RRID: AB_2061240 |
| Rabbit anti-CTIP | Santacruz | sc-22838, RRID: AB_2175257 |
| Mouse anti-SMARCAL1 | Santacruz | sc-376377, RRID: AB_10987841 |
| Rabbit anti-PCNA | Santacruz | sc-7907, RRID: AB_2160375 |
| Mouse anti-GFP | Santacruz | sc-9996; RRID: AB_627695 |
| Rabbit anti-53BP1 | Santacruz | sc-22760; RRID: AB_2256326 |
| Rabbit anti-EXO1 | Santacruz | sc-33194, RRID: AB_2101433 |
| Mouse RAD52 | Santacruz | sc-365341, RRID: AB_10851346 |
| Mouse anti-Flag | Sigma-Aldrich | F3165; RRID: AB_259529 |
| Mouse anti-HA | Sigma-Aldrich | H3663; RRID: AB_262051 |
| Rabbit anti-V5 | Sigma-Aldrich | V8137, RRID: AB_261889 |
| Mouse anti-γ-Tubulin (clone GTU-488) | Sigma-Aldrich | T6557; RRID: AB_477584 |
| Mouse anti-Myc agarose beads | Sigma-Aldrich | A7470; RRID: AB_10109522 |
| Mouse anti-Flag M2 affinity Gel | Sigma-Aldrich | A2220; RRID: AB_10063035 |
| Mouse anti-HA agarose beads | Sigma-Aldrich | A2095, RRID: AB_257974 |
| Rabbit anti-DONSON | Sigma-Aldrich | AV45862, RRID: AB_1847828 |
| Rabbit anti-DONSON | Invitrogen | PA5–61865, RRID: AB_2640751 |
| Rabbit anti-Claspin | Abcam | ab3720, RRID: AB_2245123 |
| Rabbit anti-Claspin | Novus | NB100–248, RRID: AB_2082897 |
| Rabbit anti-Claspin | Bethyl Laboratories | A300–266A, RRID: AB_155895 |
| Rabbit anti phospho-RPA32 (S4/S8) | Bethyl Laboratories | A300–245A, RRID: AB_210547 |
| Mouse anti PCNA | Bio-Rad | VMA00018 |
| Rabbit anti-mTRF2 | Karlsedar Lab | N/A |
| Streptavidin-Agarose resin | Thermo Scientific | 20359 |
| Bacterial and Virus Strains | ||
| XL1-Blue Supercompetent cells | Agilent Technologies | 200518 |
| XL10-Gold Ultracompetent Cells | Agilent Technologies | 200315 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DNA-PKcs inhibitor NU7441 | Selleckchem | S2638 |
| Critical Commercial Assays | ||
| Site-directed mutagenesis | Stratagene | 200521 |
| TRAPeze Telomerase Detection Kit | Fisher Scientific | S7700 |
| Experimental Models: Cell Lines | ||
| Human NBS-ILB1 cells | Falck et al., 2012, EMBO Reports. | N/A |
| U2OS | ATCC | N/A |
| 293T | ATCC | CRL-3216, RRID: CVCL_0063 |
| Nbs1−/− MEFs | Matsuura et al., 2004, Adv Biophys. | N/A |
| Apollo/SNM1B−/− MEFs | Lam et al., 2010, EMBO J. | N/A |
| G1 mTR−/− MEFs | This paper | N/A |
| MMTV-Cre+; Pot1aF/Δ; p53Δ/Δ MEFs | Gu et al., 2017 | N/A |
| MMTV-Cre+; Pot1aΔ/Δ; p53Δ/Δ MEFs | ||
| Experimental Models: Organisms/Strains | ||
| MMTV-Cre+; Pot1aF/F; p53F/F mouse tumor model | Gu et al., 2017 | N/A |
| Oligonucleotides | ||
| Biotin-Tel-G (IIAGGG)6 | This paper | N/A |
| Biotin-Tel-C (CCCTAA)6 | This paper | N/A |
| Biotin Oligo (dT)6 | This paper | N/A |
| TelC-Cy3 (CCCTAA)3 | PNABio | F1002 |
| TelG-FAM (TTAGGG)3 | PNABio | F1005 |
| Recombinant DNA | ||
| pBabe puro Myc-hTRF2ΔBΔM | Rai et al., 2017, Molecular Cell | N/A |
| pQCXIP puro Flag-hTPP1ΔRD | Rai et al., 2017, Molecular Cell | N/A |
| pQCXIP puro HA-hTPP1ΔRD | Rai et al., 2017, Molecular Cell | N/A |
| pQCXIP puro HA-mTPP1ΔRD | Rai et al., 2017, Molecular Cell | N/A |
| pQCXIP puro Flag-mTPP1ΔRD | Rai et al., 2017, Molecular Cell | NA |
| pQCXIP puro Flag-mNBS1WT | Rai et al., 2017, Molecular Cell | N/A |
| pQCXIP Flag-TIN2A110R | Hu et al., 2017, Cell Res. | N/A |
| pQCXIP Myc-mPOT1a and OB fold mutant mPOT1aF62A | Wu et al., 2006, Cell | N/A |
| pCDNA3.1 Myc-TRF2 | Chen et al., 2011, NSMB | N/A |
| pEGFP-EXO1 | Tomimatsu et al., 2014, Nature Comm. | N/A |
| pLenti V5-EXO1 | Tomimatsu et al., 2014, Nature Comm. | N/A |
| Fucci mKO-CTD1 | Sakaue-Sawano et al., 2008, Cell | N/A |
| Fucci mAG-Geminin | Sakaue-Sawano et al., 2008, Cell | N/A |
| pGEX4T1 Human N-Terminal, C-Terminal, WT and Claspin 3A mutant | Lindsey-Boltz et al., 2009, JBC | N/A |
| pQCXIP HA-hClaspin WT and mutants | This paper | N/A |
| pQCXIP HA-mClaspin WT and mutants | This paper | N/A |
| pQCXIP HA-mPCNA | This paper | N/A |
| pCMV6 Myc-mPCNA | Origene | RC209379 |
| pQCXIP Myc hDonson | This paper | N/A |
| pEGFP-DONSON and disease mutants | Reynolds et al., 2017 | N/A |
| Flag-ATRIP | Lee Zou (Harvard Medical School) | N/A |
| pRetroSuper shTrf2 | Deng et al., 2009 | N/A |
| pMKO.1 shBRCA1 | Xiaohua Wu (Scrips Research Institute) | N/A |
| pRetroSuper shParp1 | Madalena Tarsounas (University of Oxford) | N/A |
| pRetroSuper shLigase 3 | Madalena Tarsounas | N/A |
| pRetroSuper shRad51 | Madalena Tarsounas | N/A |
| pGIPZ shClaspin | Zhenkun Lou (Mayo Clinic) | N/A |
| pGIPZ shRad52 | Ryan Jensen (Yale University) | V2LHS_171206 |
| V3LHS_376616 | ||
| PlK.01 Lenti shPolq | Agnel Sfeir (NYU) | N/A |
| PlK.01 Lenti shCtip | Sigma-Aldrich | TRCN0000305376 |
| PlK.01 Lenti shExo1 | Sigma-Aldrich | TRCN0000218614 |
| TRCN0000238466 | ||
| TRCN0000238468 | ||
| PlK.01 Lenti shClaspin | Sigma-Aldrich | TRCN0000193573 |
| TRCN0000175992 | ||
| TRCN0000193398 | ||
| PlK.01 Lenti shPcna | Sigma-Aldrich | TRCN0000294872 |
| TRCN0000294805 | ||
| TRCN0000287377 | ||
| PlK.01 Lenti shDonson | Sigma-Aldrich | TRCN0000377075 |
| TRCN0000249773 | ||
| TRCN0000201175 | ||
| PlK.01 Lenti shPCNA | Sigma-Aldrich | TRCN0000003862 |
| TRCN0000003864 | ||
| PlK.01 Lenti shDONSON | Sigma-Aldrich | TRCN0000275364 |
| TRCN0000275365 | ||
| TRCN0000275367 | ||
| Software and Algorithms | ||
| GraphPad Prism Software (Version 7) | San Diego, CA | RRID:SCR_002798 |
| NIS-Elements BR (Verson3.2) | Nikon | RRID:SCR_002776 |
| Origin 7 software | OriginLab | N/A |
| ImageQuantTL | GE Healthcare | RRID:SCR_014246 |
| FlowJo | FlowJo LLC | RRID:SCR_008520 |
Mouse tumor model for generation of MMTV-Cre; mPot1aF/F; p53F/F tumor cell lines
To generate MMTV-Cre; mPot1aF/F; p53F/F mice, MMTV-Cre mice (The Jackson Laboratory, Tg (MMTV-cre) 4Mam/J; stock number: 003553) were first crossed with mPot1aF/F mice (Wu et al., 2006) to generate MMTV-Cre; mPot1aF/F mice. MMTV-Cre; mPot1aF/F mice were then crossed with p53F/F (The Jackson Laboratory, B6.129P2-Trp53tm1Brn/J; Stock No:008462) mice to generate MMTV-Cre; mPot1aF/F; p53F/+ and MMTV-Cre; mPot1aF/F; p53F/F mice. mPot1aF/F and mPot1aF/F; p53F/F mice were generated as controls. All mice were maintained under basic care conditions according to the IACUC-approved protocols of Yale University. Sick mice (age 7–12 months old; Table S1) were sacrificed and tumors were harvested from breast glands or other organs. Chopped tumors were digested with 0.25% trypsin at 37° C for 15min followed by collagenase D treatment at 37° C for 30 min. The digested suspension was filtered through 40μm cell strainer and isolated tumor cells were pelleted by centrifugation and expanded by passaging in the DMEM with 10% FBS culture media.
METHOD DETAILS
Retroviral infection
DNA constructs were transfected into 293T cells using Fugene 6 and packaged into retro or lentiviral particles. Viral supernatant was collected 48–72 h after transfection, filtered and directly used to infect immortalized MEFs or human cells.
Western blot analysis
For immunoblotting, trypsinized cells were lysed in urea lysis buffer (8 M urea, 50 mM Tris-HCl, pH 7.4, and 150 mM β-mercaptoethanol). The lysates were denatured and then resolved on SDS–PAGE gel. The separated proteins were then blotted on a nitrocellulose plus membrane (Amersham), blocked with blocking solution (5% non-fat dry milk in PBS/0.1% Tween-20) for at least 1 h and incubated with appropriate primary antibody in blocking solution at least 2 h at room temperature or overnight at 4 °C. The membranes were washed 3 × 5 min with PBS/0.1% Tween-20 and incubated with appropriate secondary antibody in blocking solution for 1 h at room temperature. Chemiluminescence detection was performed using an ECL Western Blotting Detection kit from GE Healthcare.
Immunofluorescence and fluorescent in situ hybridization
Cells grown on coverslips were fixed for 10 min in 2% (w/v) sucrose and 2% (v/v) paraformaldehyde at room temperature followed by PBS washes. Coverslips were blocked in 0.2% (w/v) fish gelatin and 0.5% (w/v) BSA in PBS. Cells were incubated with primary antibodies and after PBS washes, cells were incubated with appropriate Alexa fluor secondary antibodies followed by washes in PBS + 0.1% Triton. IF-FISH was carried out using a TelC-Cy3 PNA telomere probe (PNA Bio). DNA was stained with DAPI, and digital images captured using NIS-Elements BR (Nikon) with a Nikon Eclipse 80i microscope and an Andor CCD camera.
Coimmunoprecipitation
293T cells grown in 10 cm plates were co-transfected with epitope tag cDNAs or vector control. 48 h after transfection, cells were harvested and lysed in buffer (20 mM HEPES, pH 7.5, 10% glycerol, 1mM EDTA, 0.5% (v/v) NP-40). Supernatants were immunopre-cipitated with appropriate endogenous or protein tagged antibody conjugated agarose beads (Sigma). Beads were washed thrice and eluted proteins analyzed by SDS-PAGE.
Isothermal titration calorimetry
The equilibrium dissociation constants between the WT and mutant TRF2TRFH and ClaspinTBM peptides were determined using an iTC200 calorimeter (MicroCal). The enthalpies of binding between the TRF2TRFH domain and the ClaspinTBM were measured at 20° C in 25 mM Tris-HCl (pH8.0) and 150 mM NaCl. ITC data were subsequently analyzed and fitted using Origin 7 software (OriginLab) with blank injections of peptides into buffer subtracted from the experimental titrations prior to data analysis.
DNA binding assay
To examine whether CPD complex bind to ss telomeric DNA in vitro, 293T cells expressing GFP DONSON or HA-Claspin lysed in TEB150 buffer (50 mM HEPES pH 7.3, 150 mM NaCl, 2 mM MgCl2, 5 mM EGTA, 0.5% Triton X-100, 10% Glycerol, proteinase inhibitors) were incubated with streptavidin–agarose beads (Thermo-Fisher) coated with biotin-Tel-G (TTAGGG)6 or TEL-C (CCCTAA)6 or Oligo (dT)6 DNA oligos overnight at 4 ° C. POT1aWT and POT1aF62 protein lysates used as positive control (Wu et al., 2006). Bound complexes were washed three times with the same buffer and eluted proteins analyzed by SDS-PAGE.
Cell Cycle analysis
For FACS analysis, cells washed in PBS fixed with 70% ice-cold ethanol by drop-wise mixing, and incubated for at least 24 h in-20° C. Cells washed twice with PBS and then resuspended in 0.5 mL PBS containing 50 μg/ml of propidium iodide and 100 μg/ml of RNase A. The samples were analyzed on a BD LSR Fortessa cytometer. Data analysis was performed using FlowJo software. Cell cycle dependent expression of Claspin in U2OS cells was determined using Fucci system based on fluorescent proteins with fragments of CDT1 and Geminin, for the G1 and S/G2 reporters, respectively (Sakaue-Sawano et al., 2008).
Chromosome analysis by telomere PNA-FISH and CO-FISH
Cells were treated with 0.5 μg/ml of Colcemid before harvest. Chromosomes were fixed and telomere PNA-FISH performed with aTelC-Cy3 probe (PNA Bio) as described (Rai et al., 2016; Wu et al., 2006). For CO-FISH, metaphase spreads were incubated sequentially with TelG-FAM and TelC-Cy3 probes. Images were captured as described above. The percent of chromosome fusions observed is defined as: total number of chromosome fusions in 30–50 metaphase spreads analyzed divided by the total number of chromosomes examined X 100%.
Telomere length analysis and G-Strand overhang assays
For in-gel detection of telomere length and G-stand overhang, a total of 1–2 × 106 cells were suspended in PBS, mixed 1:1 with 1.8% agarose in 1 × PBS and cast into plugs. The plugs were then digested overnight at 50 ° C with 20 mg ml−1 Proteinase K (Roche) in 10 mM sodium phosphate (pH 7.2) and 0.5 mM EDTA and 1% sodium lauryl sarcosine. DNA in plugs were subsequently digested by Hinf1/Rsa1 overnight at 37 ° C. The next morning, plugs were washed once with 1 × TE and equilibrated with 0.5 × TBE. The plugs were loaded onto a 0.8% agarose gel in 0.5 ×TBE and run on a CHEF-DRII pulse field electrophoresis apparatus (Bio-Rad). The electrophoresis parameters were as follows: Initial pulse: 0.3 s, final pulse: 16 s, voltage: 6 V cm−1, run time: 14 h. Dried gel pre-hybridized with Church mix for 2 h at 55 ° C and hybridized overnight at 55 ° C in Church mix with 32P-labeled T2AG3 oligonucleotides. After hybridization, the gel was washed three times for 30 min with 4 × SSC/0.1% SDS at 37 ° C, thrice with 4 ×SSC/0.1% SDS at 55 °C and exposed to a phosphoimager screen overnight. After exposure, the screen was scanned on a Typhoon Trio imager system and the gel was subsequently denatured and hybridized using the same probe.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics for TIFs and chromosome fusions were determined in term of p value from one-way Anova using GraphPad Prism software (Version 7.0).
DATA AND CODE AVAILABILITY
This study did not generate/analyze datasets/code.
Supplementary Material
Highlights.
A-NHEJ repair of telomeres lacking POT1-TPP1 occurs independently of MRN
Without MRN, Claspin and DNA-PKcs promote CHK1 phosphorylation and A-NHEJ repair
Claspin interacts with TRF2 and helps recruit EXO1 for telomere C-strand resection
Increased replication stress in tumors without POT1 stabilized by claspin
ACKNOWLEDGMENTS
We thank the members of the Chang lab for helpful suggestions. We thank Dr. Lee Zou and Dr. Kenshi Komatsu for kindly providing Nbs1−/− MEFs, Dr. Steve Jackson for human NBS-ILB1 cells, Dr. Grant S. Stewart for DONSON constructs, Dr. Sandeep Verma for EXO1 cDNA constructs, Dr. Lesley Devine for performing flow cytometry, and Yuping Zhang for analyzing Claspin and POT1 gene expression. This work was supported by the National Cancer Institute, United States (RO1 CA129037 and RO1 CA202816 to S.C.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.11.012.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aguilera A, and Gómez-González B (2008). Genome instability: a mechanistic view of its causes and consequences. Nat. Rev. Genet 9, 204–217. [DOI] [PubMed] [Google Scholar]
- Attwooll CL, Akpinar M, and Petrini JH (2009). The mre11 complex and the response to dysfunctional telomeres. Mol. Cell. Biol 29, 5540–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audebert M, Salles B, and Calsou P (2004). Involvement of poly(ADPribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem 279, 55117–55126. [DOI] [PubMed] [Google Scholar]
- Badie S, Carlos AR, Folio C, Okamoto K, Bouwman P, Jonkers J, and Tarsounas M (2015). BRCA1 and CtIP promote alternative non-homologous end-joining at uncapped telomeres. EMBO J. 34, 828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey SM, Cornforth MN, Kurimasa A, Chen DJ, and Goodwin EH (2001). Strand-specific postreplicative processing of mammalian telomeres. Science 293, 2462–2465. [DOI] [PubMed] [Google Scholar]
- Bansbach CE, Bétous R, Lovejoy CA, Glick GG, and Cortez D (2009). The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 23, 2405–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennardo N, Cheng A, Huang N, and Stark JM (2008). Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 4, e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KP, and Cortez D (2018). RPA and RAD51: fork reversal, fork protection, and genome stability. Nat. Struct. Mol. Biol 25, 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco JN, Bergoglio V, Lin YL, Pillaire MJ, Schmitz AL, Gilhodes J, Lusque A, Mazières J, Lacroix-Triki M, Roumeliotis TI, et al. (2019). Overexpression of Claspin and Timeless protects cancer cells from replication stress in a checkpoint-independent manner. Nat. Commun 10, 910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buis J, Stoneham T, Spehalski E, and Ferguson DO (2012). Mre11 regulates CtIP-dependent double-strand break repair by interaction with CDK2. Nat. Struct. Mol. Biol 19, 246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, O’Connor KW, Konstantinopoulos PA, Elledge SJ, Boulton SJ, et al. (2015). Homologous-recombination-deficient tumours are dependent on Polq-mediated repair. Nature 518, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli GB, and de Lange T (2005). DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol 7, 712–718. [DOI] [PubMed] [Google Scholar]
- Chen Y, Yang Y, van Overbeek M, Donigian JR, Baciu P, de Lange T, and Lei M (2008). A shared docking motif in TRF1 and TRF2 used for differential recruitment of telomeric proteins. Science 319, 1092–1096. [DOI] [PubMed] [Google Scholar]
- Chen Y, Rai R, Zhou ZR, Kanoh J, Ribeyre C, Yang Y, Zheng H, Damay P, Wang F, Tsujii H, et al. (2011). A conserved motif within RAP1 has diversified roles in telomere protection and regulation in different organisms. Nat. Struct. Mol. Biol 18, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini CC, and Chen J (2003). Human claspin is required for replication checkpoint control. J. Biol. Chem 278, 30057–30062. [DOI] [PubMed] [Google Scholar]
- Ciccia A, and Elledge SJ (2010). The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimprich KA, and Cortez D (2008). ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol 9, 616–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, and Elledge SJ (2001). ATR and ATRIP: partners in checkpoint signaling. Science 294, 1713–1716. [DOI] [PubMed] [Google Scholar]
- Cox KE, Maréchal A, and Flynn RL (2016). SMARCAL1 Resolves Replication Stress at ALT Telomeres. Cell Rep. 14, 1032–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe L, Verdun RE, Haggblom CI, and Karlseder J (2004). Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science 306, 1951–1953. [DOI] [PubMed] [Google Scholar]
- de Lange T (2018). Shelterin-Mediated Telomere Protection. Annu. Rev. Genet 52, 223–247. [DOI] [PubMed] [Google Scholar]
- Denchi EL, and de Lange T (2007). Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448, 1068–1071. [DOI] [PubMed] [Google Scholar]
- Deng Y, Guo X, Ferguson DO, and Chang S (2009). Multiple roles for MRE11 at uncapped telomeres. Nature 460, 914–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N, and de Lange T (2009). Cell cycle-dependent role of MRN at dysfunctional telomeres: ATM signaling-dependent induction of nonhomologous end joining (NHEJ) in G1 and resection-mediated inhibition of NHEJ in G2. Mol. Cell. Biol 29, 5552–5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkelmann M, Spehalski E, Stoneham T, Buis J, Wu Y, Sekiguchi JM, and Ferguson DO (2009). Multiple functions of MRN in end-joining pathways during isotype class switching. Nat. Struct. Mol. Biol 16, 808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, and Cortez D (2015). The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol. Cell 59, 998–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Forment JV, Coates J, Mistrik M, Lukas J, Bartek J, and Jackson SP (2012). CDK targeting of NBS1 promotes DNA-end resection, replication restart and homologous recombination. EMBO Rep. 13, 561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RL, Centore RC, O’Sullivan RJ, Rai R, Tse A, Songyang Z, Chang S, Karlseder J, and Zou L (2011). TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature 471, 532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson E, and Géli V (2007). How telomeres are replicated. Nat. Rev. Mol. Cell Biol 8, 825–838. [DOI] [PubMed] [Google Scholar]
- Gong Y, and de Lange T (2010). A Shld1-controlled POT1a provides support for repression of ATR signaling at telomeres through RPA exclusion. Mol. Cell 40, 377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu P, Wang Y, Bisht KK, Wu L, Kukova L, Smith EM, Xiao Y, Bailey SM, Lei M, Nandakumar J, and Chang S (2017). Pot1 OB-fold mutations unleash telomere instability to initiate tumorigenesis. Oncogene 36, 1939–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu P, Jia S, Takasugi T, Smith E, Nandakumar J, Hendrickson E, and Chang S (2018). CTC1-STN1 coordinates G- and C-strand synthesis to regulate telomere length. Aging Cell 17, e12783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Deng Y, Lin Y, Cosme-Blanco W, Chan S, He H, Yuan G, Brown EJ, and Chang S (2007). Dysfunctional telomeres activate an ATM-ATR-dependent DNA damage response to suppress tumorigenesis. EMBO J. 26, 4709–4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao J, de Renty C, Li Y, Xiao H, Kemp MG, Han Z, DePamphilis ML, and Zhu W (2015). And-1 coordinates with Claspin for efficient Chk1 activation in response to replication stress. EMBO J. 34, 2096–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlerode AJ, Morgan MJ, Wu Y, Buis J, and Ferguson DO (2015). Recruitment and activation of the ATM kinase in the absence of DNA-damage sensors. Nat. Struct. Mol. Biol 22, 736–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Rai R, Huang C, Broton C, Long J, Xu Y, Xue J, Lei M, Chang S, and Chen Y (2017). Structural and functional analyses of the mammalian TIN2-TPP1-TRF2 telomeric complex. Cell Res. 27, 1485–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P (2010). DNA resection in eukaryotes: deciding how to fix the break. Nat. Struct. Mol. Biol 17, 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, and Bartek J (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, and Jackson SP (2006). ATM and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol 8, 37–45. [DOI] [PubMed] [Google Scholar]
- Kass EM, and Jasin M (2010). Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett. 584, 3703–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp MG, Akan Z, Yilmaz S, Grillo M, Smith-Roe SL, Kang TH, Cordeiro-Stone M, Kaufmann WK, Abraham RT, Sancar A, and UnsalKaçmaz K (2010). Tipin-replication protein A interaction mediates Chk1 phosphorylation by ATR in response to genotoxic stress. J. Biol. Chem 285, 16562–16571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibe T, Zimmermann M, and de Lange T (2016). TPP1 Blocks an ATR-Mediated Resection Mechanism at Telomeres. Mol. Cell 61, 236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Hayashi N, Takata M, and Yamamoto K (2013). NBS1 directly activates ATR independently of MRE11 and TOPBP1. Genes Cells 18, 238–246. [DOI] [PubMed] [Google Scholar]
- Kratz K, and de Lange T (2018). Protection of telomeres 1 proteins POT1a and POT1b can repress ATR signaling by RPA exclusion, but binding to CST limits ATR repression by POT1b. J. Biol. Chem 293, 14384–14392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Kim SM, and Dunphy WG (2004). Claspin and the activated form of ATR-ATRIP collaborate in the activation of Chk1. J. Biol. Chem 279, 49599–49608. [DOI] [PubMed] [Google Scholar]
- Lam YC, Akhter S, Gu P, Ye J, Poulet A, Giraud-Panis MJ, Bailey SM, Gilson E, Legerski RJ, and Chang S (2010). SNMIB/Apollo protects leading-strand telomeres against NHEJ-mediated repair. EMBO J. 29, 2230–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, and Dunphy WG (2010). Rad17 plays a central role in establishment of the interaction between TopBP1 and the Rad9-Hus1-Rad1 complex at stalled replication forks. Mol. Biol. Cell 21, 926–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kumagai A, and Dunphy WG (2003). Claspin, a Chk1-regulatory protein, monitors DNA replication on chromatin independently of RPA, ATR, and Rad17. Mol. Cell 11, 329–340. [DOI] [PubMed] [Google Scholar]
- Lee-Theilen M, Matthews AJ, Kelly D, Zheng S, and Chaudhuri J (2011). CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nat. Struct. Mol. Biol 18, 75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leman AR, Dheekollu J, Deng Z, Lee SW, Das MM, Lieberman PM, and Noguchi E (2012). Timeless preserves telomere length by promoting efficient DNA replication through human telomeres. Cell Cycle 11, 2337–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengsfeld BM, Rattray AJ, Bhaskara V, Ghirlando R, and Paull TT (2007). Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol. Cell 28, 638–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR (2010). The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem 79, 181–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YF, Shih HY, Shang Z, Matsunaga S, and Chen BP (2014). DNA-PKcs is required to maintain stability of Chk1 and Claspin for optimal replication stress response. Nucleic Acids Res. 42, 4463–4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey-Boltz LA, Serçin O, Choi JH, and Sancar A (2009). Reconstitution of human claspin-mediated phosphorylation of Chk1 by the ATR (ataxia telangiectasia-mutated and rad3-related) checkpoint kinase. J. Biol. Chem 284, 33107–33114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDougall CA, Byun TS, Van C, Yee MC, and Cimprich KA (2007). The structural determinants of checkpoint activation. Genes Dev. 21, 898–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makharashvili N, Tubbs AT, Yang SH, Wang H, Barton O, Zhou Y, Deshpande RA, Lee JH, Lobrich M, Sleckman BP, et al. (2014). Catalytic and noncatalytic roles of the CtIP endonuclease in double-strand break end resection. Mol. Cell 54, 1022–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manthey KC, Opiyo S, Glanzer JG, Dimitrova D, Elliott J, and Oakley GG (2007). NBS1 mediates ATR-dependent RPA hyperphosphorylation following replication-fork stall and collapse. J. Cell Sci 120, 4221–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos-Gomez PA, Gong F, Nair N, Miller KM, Lazzerini-Denchi E, and Sfeir A (2015). Mammalian polymerase q promotes alternative NHEJ and suppresses recombination. Nature 518, 254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos-Gomez PA, Kent T, Deng SK, McDevitt S, Kashkina E, Hoang TM, Pomerantz RT, and Sfeir A (2017). The helicase domain of Polθ counteracts RPA to promote alt-NHEJ. Nat. Struct. Mol. Biol 24, 1116–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura S, Kobayashi J, Tauchi H, and Komatsu K (2004). Nijmegen breakage syndrome and DNA double strand break repair by NBS1 complex. Adv. Biophys 38, 65–80. [PubMed] [Google Scholar]
- Pinzaru AM, Hom RA, Beal A, Phillips AF, Ni E, Cardozo T, Nair N, Choi J, Wuttke DS, Sfeir A, and Denchi EL (2016). Telomere Replication Stress Induced by POT1 Inactivation Accelerates Tumorigenesis. Cell Rep. 15, 2170–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole LA, Zhao R, Glick GG, Lovejoy CA, Eischen CM, and Cortez D (2015). SMARCAL1 maintains telomere integrity during DNA replication. Proc. Natl. Acad. Sci. USA 112, 14864–14869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przetocka S, Porro A, Bolck HA, Walker C, Lezaja A, Trenner A, von Aesch C, Himmels SF, D’Andrea AD, Ceccaldi R, et al. (2018). CtIP-Mediated Fork Protection Synergizes with BRCA1 to Suppress Genomic Instability upon DNA Replication Stress. Mol. Cell 72, 568–582.e6. [DOI] [PubMed] [Google Scholar]
- Rai R, Zheng H, He H, Luo Y, Multani A, Carpenter PB, and Chang S (2010). The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. EMBO J. 29, 2598–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai R, Li JM, Zheng H, Lok GT, Deng Y, Huen MS, Chen J, Jin J, and Chang S (2011). The E3 ubiquitin ligase Rnf8 stabilizes Tpp1 to promote telomere end protection. Nat. Struct. Mol. Biol 18, 1400–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai R, Chen Y, Lei M, and Chang S (2016). TRF2-RAP1 is required to protect telomeres from engaging in homologous recombination-mediated deletions and fusions. Nat. Commun 7, 10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai R, Hu C, Broton C, Chen Y, Lei M, and Chang S (2017). NBS1 Phosphorylation Status Dictates Repair Choice of Dysfunctional Telomeres. Mol. Cell 65, 801–817.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, and Lopez BS (2009). Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat. Struct. Mol. Biol 16, 819–824. [DOI] [PubMed] [Google Scholar]
- Reynolds JJ, Bicknell LS, Carroll P, Higgs MR, Shaheen R, Murray JE, Papadopoulos DK, Leitch A, Murina O, Tarnauskaitė Z, et al. (2017). Mutations in DONSON disrupt replication fork stability and cause microcephalic dwarfism. Nat. Genet 49, 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo A, Salvati E, Porru M, D’Angelo C, Stevens MF, D’Incalci M, Leonetti C, Gilson E, Zupi G, and Biroccio A (2009). Stabilization of quadruplex DNA perturbs telomere replication leading to the activation of an ATR-dependent ATM signaling pathway. Nucleic Acids Res. 37, 5353–5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Bravo V, Guaita-Esteruelas S, Florensa R, Bachs O, and Agell N (2006). Chk1- and claspin-dependent but ATR/ATM- and Rad17-independent DNA replication checkpoint response in HeLa cells. Cancer Res. 66, 8672–8679. [DOI] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498. [DOI] [PubMed] [Google Scholar]
- Sar F, Lindsey-Boltz LA, Subramanian D, Croteau DL, Hutsell SQ, Griffith JD, and Sancar A (2004). Human claspin is a ring-shaped DNA-binding protein with high affinity to branched DNA structures. J. Biol. Chem 279, 39289–39295. [DOI] [PubMed] [Google Scholar]
- Sfeir A, and de Lange T (2012). Removal of shelterin reveals the telomere end-protection problem. Science 336, 593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, and Symington LS (2015). Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci 40, 701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, and de Lange T (2009). Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138, 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits VAJ, Cabrera E, Freire R, and Gillespie DA (2019). Claspin checkpoint adaptor and DNA replication factor. FEBS J. 286, 441–455. [DOI] [PubMed] [Google Scholar]
- Sotiriou SK, Kamileri I, Lugli N, Evangelou K, Da-Ré C, Huber F, Pa-dayachy L, Tardy S, Nicati NL, Barriot S, et al. (2016). Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Mol. Cell 64, 1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiff T, Reis C, Alderton GK, Woodbine L, O’Driscoll M, and Jeggo PA (2005). Nbs1 is required for ATR-dependent phosphorylation events. EMBO J. 24, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS (2016). Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol 51, 195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenorio-Gómez M, de Sena-Tomás C, and Pérez-Martín J (2015). MRNand 9-1-1-Independent Activation of the ATR-Chk1 Pathway during the Induction of the Virulence Program in the Phytopathogen Ustilago maydis. PLoS One 10, e0137192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomimatsu N, Mukherjee B, Catherine Hardebeck M, Ilcheva M, Vanessa Camacho C, Louise Harris J, Porteus M, Llorente B, Khanna KK, and Burma S (2014). Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nat. Commun 5, 3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H, Razavian N, Berns MW, and Wu X (2013). Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. USA 110, 7720–7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, and Boulton SJ (2012). RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 149, 795–806. [DOI] [PubMed] [Google Scholar]
- Vannier JB, Sandhu S, Petalcorin MI, Wu X, Nabi Z, Ding H, and Boulton SJ (2013). RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science 342, 239–242. [DOI] [PubMed] [Google Scholar]
- Verdun RE, and Karlseder J (2007). Replication and protection of telomeres. Nature 447, 924–931. [DOI] [PubMed] [Google Scholar]
- Verma P, and Greenberg RA (2016). Noncanonical views of homology-directed DNA repair. Genes Dev. 30, 1138–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Rosidi B, Perrault R, Wang M, Zhang L, Windhofer F, and Ilia-kis G (2005). DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res. 65, 4020–4030. [DOI] [PubMed] [Google Scholar]
- Wang H, Li Y, Truong LN, Shi LZ, Hwang PY, He J, Do J, Cho MJ, Li H, Negrete A, et al. (2014). CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol. Cell 54, 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Multani AS, He H, Cosme-Blanco W, Deng Y, Deng JM, Bachilo O, Pathak S, Tahara H, Bailey SM, et al. (2006). Pot1 deficiency initiates DNA damage checkpoint activation and aberrant homologous recombination at telomeres. Cell 126, 49–62. [DOI] [PubMed] [Google Scholar]
- Wu P, van Overbeek M, Rooney S, and de Lange T (2010). Apollo contributes to G overhang maintenance and protects leading-end telomeres. Mol. Cell 39, 606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Takai H, and de Lange T (2012). Telomeric 3′ overhangs derive from resection by Exo1 and Apollo and fill-in by POT1b-associated CST. Cell 150, 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie A, Kwok A, and Scully R (2009). Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat. Struct. Mol. Biol 16, 814–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin H, Liu D, Wan M, Safari A, Kim H, Sun W, O’Connor MS, and Songyang Z (2007). TPP1 is a homologue of ciliate TEBP-beta and interacts with POT1 to recruit telomerase. Nature 445, 559–562. [DOI] [PubMed] [Google Scholar]
- Yang YG, Saidi A, Frappart PO, Min W, Barrucand C, Dumon-Jones V, Michelon J, Herceg Z, and Wang ZQ (2006). Conditional deletion of Nbs1 in murine cells reveals its role in branching repair pathways of DNA double-strand breaks. EMBO J. 25, 5527–5538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XH, Shiotani B, Classon M, and Zou L (2008). Chk1 and Claspin potentiate PCNA ubiquitination. Genes Dev. 22, 1147–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CC, Suzuki M, Yamakawa S, Uno S, Ishii A, Yamazaki S, Fukatsu R, Fujisawa R, Sakimura K, Tsurimoto T, and Masai H (2016). Claspin recruits Cdc7 kinase for initiation of DNA replication in human cells. Nat. Commun 7, 12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu TY, Kimble MT, and Symington LS (2018). Sae2 antagonizes Rad9 accumulation at DNA double-strand breaks to attenuate checkpoint signaling and facilitate end resection. Proc. Natl. Acad. Sci. USA 115, E11961–E11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, and Chen J (2010). MRE11-RAD50-NBS1 complex dictates DNA repair independent of H2AX. J. Biol. Chem 285, 1097–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusufzai T, and Kadonaga JT (2008). HARP is an ATP-driven annealing helicase. Science 322, 748–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaug AJ, Podell ER, and Cech TR (2005). Human POT1 disrupts telomeric G-quadruplexes allowing telomerase extension in vitro. Proc. Natl. Acad. Sci. USA 102, 10864–10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, and Jasin M (2011). An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat. Struct. Mol. Biol 18, 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, and Elledge SJ (2003). Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548. [DOI] [PubMed] [Google Scholar]
- Zou L, Cortez D, and Elledge SJ (2002). Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 16, 198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets/code.