

Sir,
Rhabdomyolysis refers to the acute muscle fiber necrosis, the breakdown of striated muscle.[1] Clinically, it is manifested as acute muscle weakness associated with myalgia and dark urine, which can lead to acute renal insufficiency.[1] The major causes of rhabdomyolysis are acquired etiology, namely, physical exertion, crush injury, tissue ischemia, drugs, and toxins.[1] However, underlying inherited conditions should be considered in the following situations: (1) recurrent rhabdomyolysis evoked by minimal exercise, fever, heat or cold exposure, or fasting, (2) previous history of malignant hyperthermia, (3) family history of rhabdomyolysis, malignant hyperthermia or myopathy, and (4) patients with hyperCKemia between attacks.[2]
Repeated exertional rhabdomyolysis is not only a remarkable feature of inherited metabolic myopathies but also one of the uncommon symptoms in muscular dystrophies.[2] Here, we reported a patient with a novel mutation in the DMD gene who presented as recurrent exertional rhabdomyolysis.
A 17-year-old boy was referred to the department of neurology with recurrent myalgia following exercise. He had suffered an easy fatigability and myalgia several minutes after exercise such as running or trekking the mountain every time since childhood. These symptoms had recovered spontaneously within a day. The second wind phenomenon was not seen. Incidentally, he was found to have an elevated aspartate aminotransferase (73 U/L, normal range <40 U/L, AST) and alanine aminotransferase (65 U/L, normal range <41 U/L) at age of 13 years [Figure 1a]. Spontaneous recovery had left him undiagnosed. At 14 years of age, he experienced dark urine for a day after 500 m running [Figure 1a]. At 17 years, he presented with myalgia on the bilateral thigh with dark urine after 10-minute walking. The laboratory performed on a same day in referring hospital showed an elevated level of serum creatine kinase (73,529 U/L, normal range 39-308 U/L, CK), AST (88 U/L), myoglobin (>1000 ng/mL, normal range 0-110 ng/mL), and myoglobinuria [Figure 1a]. These laboratory derangements brought the patient to our hospital 4 months after the latest attack.
Figure 1.

The timeline of clinical information, results of the ischemic forearm exercise test, IHC using dystrophin C-terminal antibody, and genetic analysis. (a) Clinical complaints and the results of the blood test corresponding to symptoms are depicted depending on the age. (b) Exercise-associated lactate and ammonia production is identified. (c) Mildly increased fiber size variation and increased number of fibers with internal nuclei are seen in hematoxylin and eosin stain. Absence of immune activity against dystrophin C-terminal antibody. Scale bar, 100 μm. (d) The mutation c.119T > A (p.Leu40His, NM_004006.2) in the DMD gene is confirmed by Sanger sequence in both patient and mother. AST, aspartate transaminase, U/L; ALT, alanine transaminase, U/L; CK, creatine kinase; U/L; myoglobin, ng/mL
On neurological examination, the muscle strengths of the extremities were normal. The deep tendon reflexes were mildly reduced. The atrophy, calf hypertrophy or arthrogryposis was not identified. The routine laboratory showed an elevated level of serum CK (1,484 U/L), AST (45 U/L), myoglobin (430 ng/mL), and aldolase (185 U/L, normal range <7.6 U/L, Figure 1a). Complete blood count, thyroid stimulating hormone, alanine aminotransferase, and urine myoglobin were normal. The ischemic forearm exercise test was normal, showing an elevated serum level of lactic acid and ammonia after exercise [Figure 1b]. The electrocardiogram showed normal sinus rhythm. The findings of nerve conduction studies were normal. The needle electromyography demonstrated positive sharp waves with small amplitude and short duration motor unit action potentials, indicating active myopathic changes.
The muscle pathology from left biceps brachii biopsy showed a mildly increased fiber size variation and increased number of fibers with internal nuclei on hematoxylin and eosin stain in paraffin block [Figure 1c]. Necrotic or regenerating fibers were not seen. However, the immunohistochemistry (IHC) revealed an absence of dystrophin expression using the antibody against the C-terminal region (Thermofisher scientific, PA5-16734, USA, 1:200 dilution, Figure 1c). During the hospitalization, we suspected the metabolic myopathies based on the normal neurological examination and episodic attack and performed the whole exome sequencing before getting the histologic results. WES revealed a novel hemizygotic missense mutation c. 119T > A (p. Leu40His, exon 3, NM_004006.2, NG_012232.1, Figure 1c) in DMD gene by investigating variants which affect protein function, show a depth of more than 30, and filtered by allele frequency of PopFreqMax less than 0.0001 consisting of the Genome Aggregation Database (gnomAD), the Exome Aggregation Consortium (ExAC), and 1000 Genome (1000 genome). This variant is not present in the Human Gene Mutation Database (HGMD) or Leiden Open Variation Database (https://databases.lovd.nl/shared/genes/DMD) and predicted to be pathogenic by using SIFT/PROVEAN and Mutation taster system. Structure of mutated dystrophin protein was predicted to be destabilizing using SDM web server (ΔΔG = 0.88 kcal/mol) and FoldX (ΔΔG = 19.5414 kcal/mol).[3,4] Both wild type (Leu40) and mutated (His40) residues were expected not to be part of aggregation-prone regions by an Aggrescan3D server,[5] although the mutated residue (His40) can become solvent exposed by JPred4 server.[6] Moreover, this variant was also identified in the asymptomatic mother as heterozygote by Sanger sequence [Figure 1d].
Initially, we had suspected metabolic myopathies for the cause of recurrent exertional rhabdomyolysis. However, we could diagnose Becker muscular dystrophy (BMD) based on the absent immunoreactivity to dystrophin C-terminal, elevated resting CK levels, and a novel pathogenic mutation DMD c. 119T > A (p. Leu40His) in genetic analysis.
Patients with muscular dystrophies have been reported with exercise intolerance, namely, anoctaminopathy, caveolinopathy, dysferlinopathy, dystrophinopathy, fukutin-related proteinopathy, and sarcoglycanopathy.[2,7] The extremely high level of serum CK and mild-to-moderate myoglobinuria are common features in muscular dystrophies presenting rhabdomyolysis.[7] Muscle ache usually occurs in BMD. In fukutin-related proteinopathy, muscle weakness and muscle biopsy can be normal, therefore, the IHC for alpha-dystroglycan is necessary.[7] Dysferlinopathy often shows a marked inflammatory both on muscle biopsy and magnetic resonance imaging.[7] Therefore, multidisciplinary tools are necessary for the differential diagnosis of myopathies associated with rhabdomyolysis.
In cases of BMD with exertional rhabdomyolysis or myalgia,[2,8,9] genetic analysis has identified the in-frame deletion in the most cases[2,8] and rarely the missense mutations of the rod domain.[9] The mutant p. Leu40His found in our case is located on N-terminal actin-binding domain (N-ABD) of dystrophin, which has not been reported previously to have pathogenicity for exertional rhabdomyolysis or myalgia. Recently, reported case harboring p. Asn76Ile in dystrophin also supports the mutation in N-ABD can be pathogenic for BMD.[10]
Concerning the immunoblotting, IHC of dystrophin rod-domain was absent or markedly reduced in patients with in-frame deletions,[2,8] whereas IHC of dystrophin C-terminal and rod domain was normal in patients with missense mutations.[9] However, absent immunostaining of dystrophin C-terminal has been reported in a patient with a missense mutation in N-ABD,[10] which is compatible with our patient.
In terms of pathomechanism, the loss of hydrophobicity in the actin-binding domain has been suggested in the previous study.[11] An amino acid substitution from leucine to arginine at position 54 is well formulated, which is associated with Duchenne muscular dystrophy.[11] Similarly, in our case, the leucine at position 40 is surrounded by the residue with a hydrophobic side chain at the position Phe21, Val25, Phe41, and Ile111 [Figure 2a]. The amino acid change from leucine to histidine at position 40 is expected to disrupt the interaction in the vicinity by losing hydrophobicity [Figure 2b]. Stability analysis and secondary structure prediction also showed destabilization and solvent exposed nature of p. Leu40His mutation, respectively, although the predicted value of energy transfer differs among prediction systems. However, the mutant was predicted not to be an aggregation-prone region.
Figure 2.
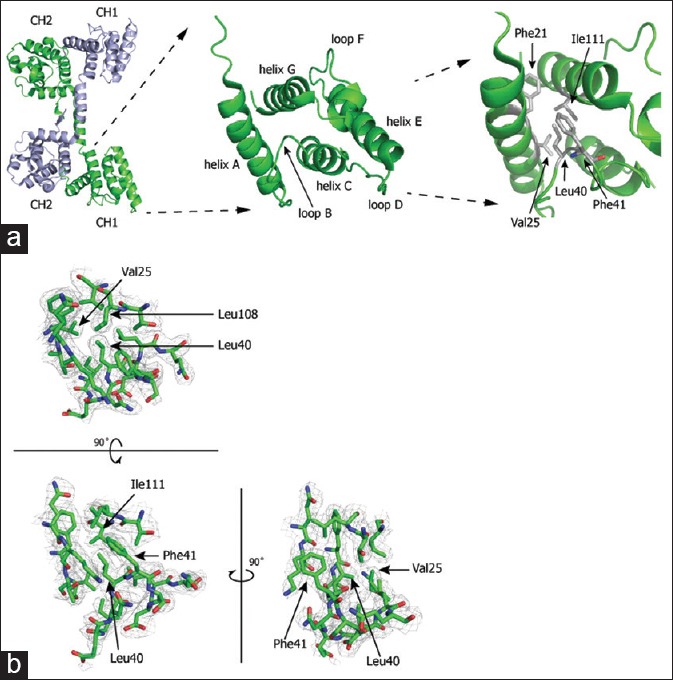
Structure of human dystrophin and a pathogenic missense mutation in the dystrophin N-ABD. (a) Left panel: dimer of human dystrophin N-ABDs, Middle panel: structure of the CH1 subdomain, Right panel: Leucine residue at the position 40 and its vicinities. (b) Stereoview of the 2Fo-Fc electron-density map in the vicinity of leucine at residue 40. The electron-density map is defined at 1.0σ
Therefore, maintaining a high degree of clinical suspicion of BMD is advised when patients present with recurrent exertional rhabdomyolysis.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Warren JD, Blumbergs PC, Thompson PD. Rhabdomyolysis: A review. Muscle Nerve. 2002;25:332–47. doi: 10.1002/mus.10053. [DOI] [PubMed] [Google Scholar]
- 2.Liewluck T, Tian X, Wong LJ, Pestronk A. Dystrophinopathy mimicking metabolic myopathies. Neuromuscul Disord. 2015;25:653–7. doi: 10.1016/j.nmd.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Pandurangan AP, Ochoa-Montano B, Ascher DB, Blundell TL. Sdm: A server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017;45:229–35. doi: 10.1093/nar/gkx439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The foldx web server: An online force field. Nucleic Acids Res. 2005;33:382–8. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zambrano R, Jamroz M, Szczasiuk A, Pujols J, Kmiecik S, Ventura S. Aggrescan3d (a3d): Server for prediction of aggregation properties of protein structures. Nucleic Acids Res. 2015;43:306–13. doi: 10.1093/nar/gkv359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drozdetskiy A, Cole C, Procter J, Barton GJ. Jpred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015;43:389–94. doi: 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quinlivan R, Jungbluth H. Myopathic causes of exercise intolerance with rhabdomyolysis. Dev Med Child Neurol. 2012;54:886–91. doi: 10.1111/j.1469-8749.2012.04320.x. [DOI] [PubMed] [Google Scholar]
- 8.Minetti C, Tanji K, Chang HW, Medori R, Cordone G, DiMauro S, et al. Dystrophinopathy in two young boys with exercise-induced cramps and myoglobinuria. Eur J Pediatr. 1993;152:848–51. doi: 10.1007/BF02073385. [DOI] [PubMed] [Google Scholar]
- 9.Veerapandiyan A, Shashi V, Jiang YH, Gallentine WB, Schoch K, Smith EC. Pseudometabolic presentation of dystrophinopathy due to a missense mutation. Muscle Nerve. 2010;42:975–9. doi: 10.1002/mus.21823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koczok K, Mero G, Szabo GP, Madar L, Gombos E, Ajzner E, et al. Anovel point mutation affecting asn76 of dystrophin protein leads to dystrophinopathy. Neuromuscul Disord. 2018;28:129–36. doi: 10.1016/j.nmd.2017.12.003. [DOI] [PubMed] [Google Scholar]
- 11.Norwood FL, Sutherland-Smith AJ, Keep NH, Kendrick-Jones J. The structure of the n-terminal actin-binding domain of human dystrophin and how mutations in this domain may cause duchenne or becker muscular dystrophy. Structure. 2000;8:481–91. doi: 10.1016/s0969-2126(00)00132-5. [DOI] [PubMed] [Google Scholar]