

Abstract
Histone lysine demethylase 1 (LSD1), the first identified histone demethylase, is overexpressed in multiple tumor types, including breast cancer. However, the mechanisms that cause LSD1 dysregulation in breast cancer remain largely unclear. Here, we report that protein arginine methyltransferase 4 (PRMT4 or CARM1) dimethylates LSD1 at R838, which promotes the binding of the deubiquitinase USP7, resulting in the deubiquitination and stabilization of LSD1. Moreover, CARM1‐ and USP7‐dependent LSD1 stabilization plays a key role in repressing E‐cadherin and activating vimentin transcription through promoter H3K4me2 and H3K9me2 demethylation, respectively, which promotes invasion and metastasis of breast cancer cells. Consistently, LSD1 arginine methylation levels correlate with tumor grade in human malignant breast carcinoma samples. Our findings unveil a unique mechanism controlling LSD1 stability by arginine methylation, also highlighting the role of the CARM1‐USP7‐LSD1 axis in breast cancer progression.
Keywords: arginine methylation, breast cancer, CARM1, LSD1, metastasis
Subject Categories: Cancer; Chromatin, Epigenetics, Genomics & Functional Genomics; Post-translational Modifications, Proteolysis & Proteomics
The protein arginine methyltransferase CARM1 targets the histone lysine demethylase LSD1 and promotes its stabilization via USP7‐dependent deubiqutination. Stabilised LSD1 enhances migration and invasion of breast cancer cells, further facilitating metastasis.
Introduction
Lysine‐specific demethylase 1A (LSD1/KDM1A/AOF2) is the first identified histone demethylase, which selectively removes the methyl group from mono‐ or dimethylated histone H3 at lysine 4 (H3K4me1/2) 1 and lysine 9 (H3K9me1/2) 2, 3, 4 to mediate gene repression and activation, respectively. In addition, nonhistone proteins such as p53 and Dnmt1 have also been found to be demethylated by LSD1 5, 6. Overexpression of LSD1 has been proved in numerous cancers, and high level of LSD1 causes tumor aggressiveness and poor prognosis in lung, prostate, colon and breast cancers 7, 8, 9, 10, 11, 12. As is known, protein abundance is regulated at both transcriptional and post‐transcriptional levels. Strikingly, multiple post‐transcriptional modifications have been reported regulating LSD1 stability 13, 14, 15, 16, 17, 18, which gained increasing appreciation. Our previous study also reports that high level of LSD1 contributes to breast cancer development 19, while phosphorylation of LSD1 was indispensable for breast cancer metastasis 20. To date, the mechanistic details of LSD1 dysregulation are largely unclear.
Previous studies have shown that LSD1 is an unstable protein and can be degraded by proteasome 21, 22. Ubiquitination plays important roles in the degradation of proteins 23. In contrast, deubiquitination, the reverse process that catalyzed by deubiquitylase, endows the proteins with the stabilization property 24, 25, and recent evidence reveals that LSD1 can be deubiquitinated and stabilized by deubiquitylases 13, 14, 15, 18. Notably, lysine methylation and phosphorylation have been learned to regulate LSD1 stability through ubiquitination modification. Lysine methyltransferase SUV39H2 trimethylates LSD1 at K322, which suppresses LSD1 ubiquitination, leading to the stabilization of the protein 17. Likewise, phosphorylation of LSD1 S683 mediated by GSK3β promotes the binding of deubiquitinase USP22 with LSD1, resulting in the deubiquitination and hence the stabilization of LSD1 13. However, whether the stability of LSD1 is regulated by arginine methylation is far from well understood.
As a prevalent post‐translational modification, protein arginine methylation is catalyzed by a family of enzymes termed protein arginine methyltransferases (PRMTs) 26. As a type I PRMTs, CARM1 catalyzes the formation of asymmetric dimethylarginine (me2a). Recent evidence shows that CARM1 is deregulated in numerous cancers 27, 28, 29. Strikingly, compelling evidence for oncogenic activities of CARM1 has recently been presented in breast cancer. In parallel to LSD1, CARM1 is highly expressed in breast cancer 30, 31, 32, 33, and elevated levels of CARM1 are associated with poor prognosis 32. In spite of the accumulating data for CARM1's roles in breast cancer progression, the mechanisms of its function remain elusive. Recent studies revealed that CARM1 methylates chromatin‐remodeling factor BAF155 at Arg1064; methylated BAF155 positively controls the expression of the c‐Myc pathway and enhances breast cancer progression and metastasis 34. Likewise, CARM1 methylates PKM2 at Arg445/447/455 and then promotes breast cancer metastasis by reprogramming oxidative phosphorylation to aerobic glycolysis 35. These studies provided clues that CARM1 promotes breast cancer progression through methylation of cancer‐relevant substrates. Among which, if LSD1 is a new substrate for CARM1 during breast cancer progression remains unclear.
The aim of this study was to explore the molecular mechanisms for the presence of high‐level LSD1 in breast cancer. We found that LSD1 is a novel substrate of CARM1, while CARM1 maintains LSD1 stability through promoting the binding between deubiquitinase USP7 and LSD1. We also demonstrated a significant correlation between LSD1, CARM1 and LSD1 R838me2a in human breast tumor samples. These findings may potentially provide a basis for the development of a novel therapy strategy against breast carcinogenesis.
Results
CARM1 dimethylates LSD1 at arginine 838
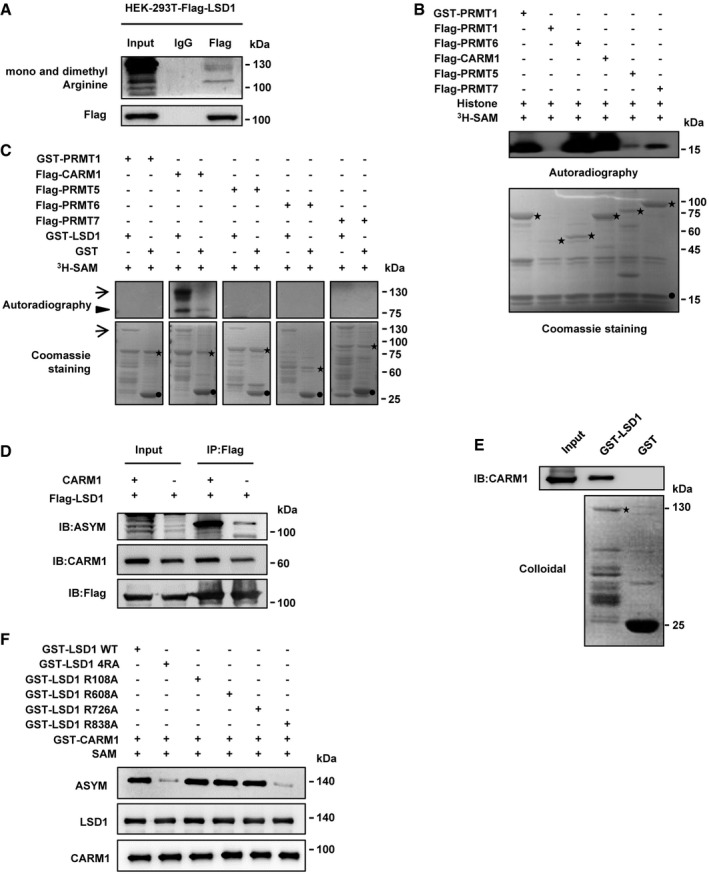
Numerous post‐translational modifications of LSD1 in human cancer cells have been found recently, such as lysine methylation, acetylation, and phosphorylation 13, 17, 36, 37, 38. Our previous work also found that phosphorylation of LSD1 was indispensable for breast cancer metastasis; however, whether LSD1 could be arginine‐methylated remains unknown. To explore whether LSD1 is arginine‐methylated, immunoprecipitation was conducted in HEK‐293T‐Flag‐LSD1 cells. The results revealed that LSD1 was indeed arginine‐methylated (Fig EV1A). To pinpoint the LSD1 methylation site(s), we performed liquid chromatography‐tandem mass spectrometry (LC‐MS/MS) analysis of LSD1 obtained from HEK‐293T‐Flag‐LSD1 cells. As the results, 4 methylated arginine residues (Arg108/608/726/838) were identified (Fig 1A, Appendix Fig S1A–C and Dataset EV1). To identify the PRMT(s) responsible for LSD1 methylation, we purified PRMT1, CARM1, PRMT5, PRMT6, and PRMT7 protein. In vitro methylation assays showed that all PRMTs displayed methyltransferase activity against histones (Fig EV1B); however, only CARM1 catalyzed LSD1 methylation (Fig EV1C). As a methyltransferase, CARM1 catalyzes the formation of asymmetric dimethylarginine; we did find that the asymmetric dimethylation status of LSD1 was markedly increased after CARM1 overexpression in HEK‐293T cells (Fig EV1D). The interaction of LSD1 and CARM1 was also confirmed in vivo (Fig 1B and C) and in vitro (Figs 1D and EV1E). These results suggest that LSD1 is methylated by CARM1.
Figure EV1. LSD1 is methylated by and interacts with CARM1.
-
AHEK‐293T cells were transfected with Flag‐tagged LSD1, and cell lysates were immunoprecipitated by anti‐Flag antibody and then subjected to immunoblotting.
-
B, CIn vitro methylation assays. Purified fusion proteins of GST‐tagged or Flag‐tagged PRMTs were incubated with core histones (B) and GST or GST‐LSD1 (C) in the presence of 3H‐SAM. Methylated proteins were detected by autoradiography, and the total amounts of proteins were visualized by Coomassie Blue staining (B, asterisks: PRMTs, filled circles: core histones; C, asterisks: PRMTs, arrows: LSD1, arrowheads: automethylation of CARM1, filled circles: GST).
-
DLSD1 asymmetric dimethylation status was examined by IP assay with anti‐Flag in HEK‐293T cells that transfected with the indicated plasmids.
-
EPurified GST‐tagged LSD1 was pulled down with the Flag‐CARM1 purified from HEK‐293T cells. The amounts of GST and GST‐tagged LSD1 were visualized by Coomassie Blue staining (asterisks: LSD1).
-
FIn vitro methylation assays. Purified fusion protein of GST‐tagged LSD1 WT or its mutants was incubated with GST‐CARM1 in the presence of SAM. Methylation of LSD1 protein was analyzed by Western blot.
Source data are available online for this figure.
Figure 1. CARM1 dimethylates LSD1 at arginine 838.
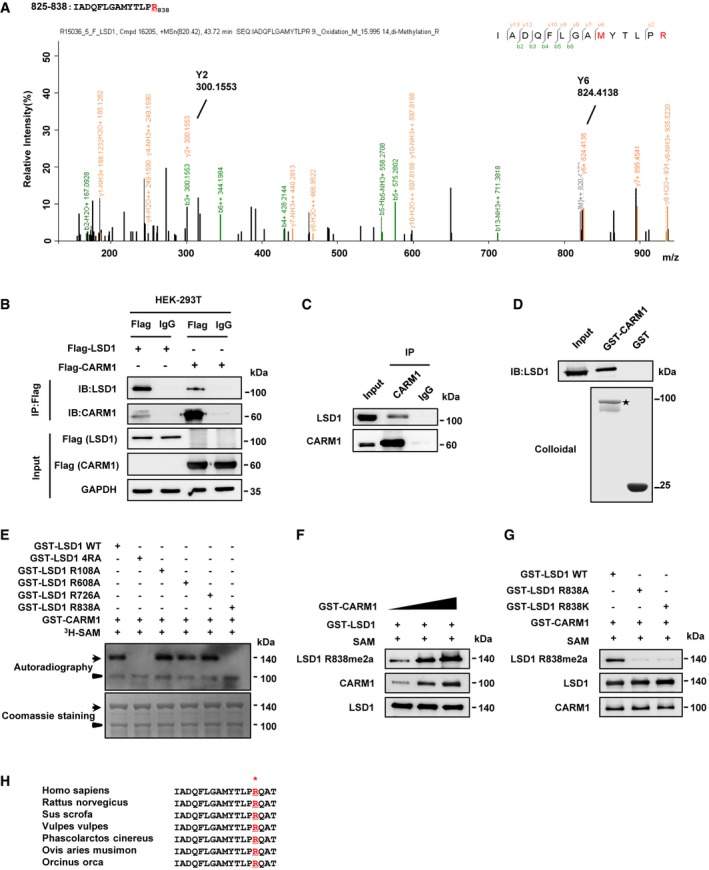
- Mass spectrometric analysis of LSD1 methylation. Immunoprecipitation (IP) was performed using anti‐Flag antibody in HEK‐293T cells overexpressing Flag‐tagged LSD1, followed by liquid chromatography coupled with tandem mass spectrometry (LC‐MS/MS) analysis. The fragmentation of the LSD1 peptide IADQFLGAMYTLPR identified a dimethylated residue at R838.
- Interactions between LSD1 and CARM1 were detected by co‐immunoprecipitation (Co‐IP) assay with anti‐Flag in HEK‐293T cells transfected with Flag‐LSD1 or Flag‐CARM1, respectively.
- Interaction of endogenous LSD1 with CARM1 was analyzed by Co‐IP assay using anti‐CARM1 antibody in MDA‐MB‐231 cells.
- Purified GST‐tagged CARM1 was pulled down with the Flag‐LSD1 purified from HEK‐293T cells. The amounts of GST and GST‐tagged CARM1 were visualized by Coomassie Blue staining (asterisk: GST‐CARM1).
- In vitro methylation assays. Purified fusion protein of GST‐tagged LSD1 WT or the four methylated arginine mutants (RA), or four methylated arginine mutants in combination (4RA), were incubated with GST‐CARM1 in the presence of 3H‐SAM. Methylated proteins were detected via autoradiography, and total amounts of proteins were visualized by Coomassie Blue staining (arrows: the position of LSD1; arrowheads: the position of CARM1).
- Increasing amounts of GST‐CARM1 incubated with GST‐LSD1 in the presence of SAM at 30°C for 1 h. And methylation of LSD1 was analyzed by immunoblotting with anti‐LSD1 R838me2a antibody.
- GST‐CARM1 incubated with GST‐LSD1 WT or GST‐LSD1 R838A/R838K in the presence of SAM at 30°C for 1 h, followed by immunoblotting with anti‐LSD1 R838me2a to detect LSD1 methylation.
- Sequence alignments of LSD1 among mammals. LSD1 residue at R838 is denoted in the protein sequences.
Source data are available online for this figure.
To determine the major methylation site(s) of LSD1, we substituted the 4 arginine residues with alanine, either individually or in combination, in GST‐LSD1. The in vitro methylation analysis showed that R838A, as well as mutations of R838 in combination with other sites, abolished CARM1‐mediated methylation of LSD1, whereas the other three sites (R108, R608, and R726) failed to do so (Figs 1E and EV1F). The LC‐MS/MS revealed that R838 was dimethylated (Fig 1A), indicating that CARM1 dimethylated LSD1 at R838. Since CARM1 catalyzes the formation of asymmetric dimethylarginine, we generated a polyclonal antibody specifically recognizing asymmetrically dimethylated R838 of LSD1 (anti‐LSD1 R838me2a), and the specificity was verified by ELISA, dot blotting, and immunoprecipitation assays (Appendix Fig S2A–C and Appendix Table S1).
To further confirm that CARM1 was responsible for LSD1 R838 methylation, increasing amounts of GST‐CARM1 were incubated with wild‐type LSD1 (LSD1 WT), and the resultant products were detected by immunoblotting with anti‐LSD1 R838me2a. As expected, methylation level of R838 was correlated with the increasing amounts of CARM1 (Fig 1F). Additionally, only LSD1 WT was methylated by CARM1, while R838K/A mutants (mimics of the unmethylated status) were not (Fig 1G). Consistently, we noticed that the methylation site (R838) of LSD1 is highly conserved among the mammals analyzed (Fig 1H). Collectively, these data indicate that LSD1 is asymmetrically dimethylated by CARM1 at R838.
LSD1 R838 methylation promotes the stabilization of LSD1
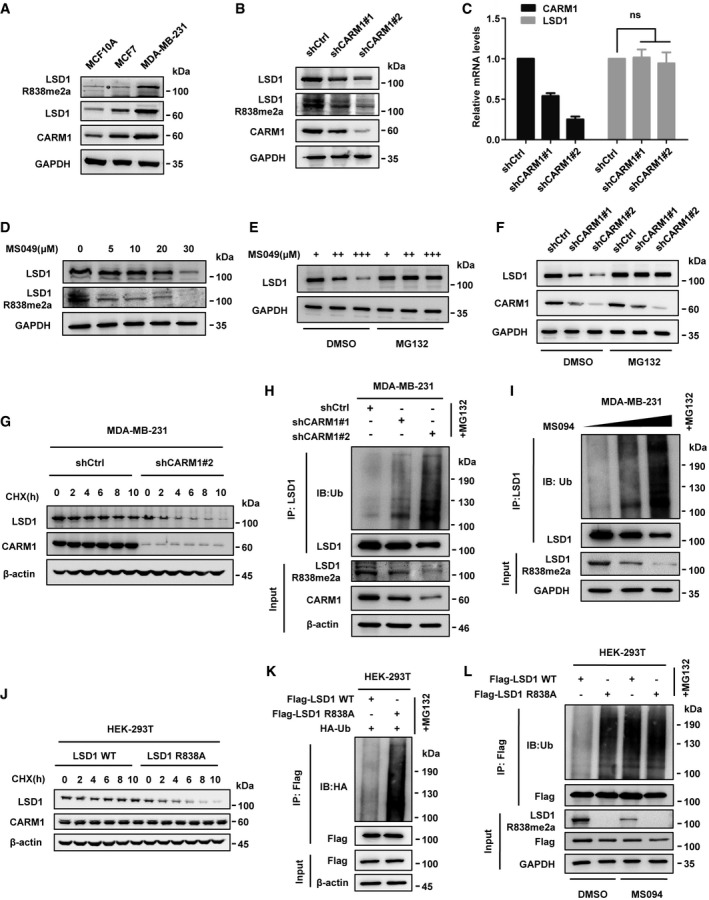
We then intended to clarify the function of LSD1 arginine methylation mediated by CARM1. We first examined the levels of CARM1, LSD1, and LSD1 R838me2a in MCF10A, MCF7, and MDA‐MB‐231 cell lines and found that they had the highest levels in MDA‐MB‐231 cells known to be highly metastatic (Fig 2A). In addition, both CARM1 and LSD1 were highly expressed in human breast cancerous tissues compared with the non‐cancerous tissues, and also with a high positive correlation (Fig EV2). These results implicate that CARM1 might play a role in maintaining high level of LSD1 in breast cancer. Actually, both LSD1 and LSD1 R838me2a levels were sharply decreased after knockdown of CARM1 in MDA‐MB‐231 cells (Fig 2B); however, the mRNA levels showed marginal change (Fig 2C). Consistently, LSD1 decreased in a dose‐dependent manner when MDA‐MB‐231 cells were treated with CARM1 inhibitor MS049 (Fig 2D). Results above implicate that CARM1‐mediated LSD1 arginine methylation might be responsible for its stability.
Figure 2. LSD1 R838 methylation promotes the stabilization of LSD1.
-
AMethylation of LSD1 was assessed in MCF10A, MCF7, and MDA‐MB‐231 cells by immunoblotting using anti‐LSD1 R838me2a.
-
B, CProtein and relative mRNA expression levels of LSD1 in MDA‐MB‐231 cells transfected with CARM1 shRNAs or control vector were assessed by Western blotting (B) and qPCR analysis (C) (Error bars represent the mean ± SD, n = 3 experimental replicates, ns = not significant, Student's t‐test).
-
DMDA‐MB‐231 cells were treated with the indicated amounts of MS049 for 48 h, LSD1 R838me2a and LSD1 levels were assessed by Western blotting using indicated antibodies.
-
EMDA‐MB‐231 cells were treated with increasing amounts of MS049 for 48 h in the presence of MG132 (10 μM) or DMSO, before LSD1 level was analyzed by immunoblotting.
-
FMDA‐MB‐231 cells were transfected with control or CARM1 shRNAs and then treated with MG132 or DMSO for 10 h, and the cell lysates were analyzed by Western blotting.
-
GMDA‐MB‐231 cells were transfected with CARM1 shRNA#2 or control vector, and then treated with CHX (100 μg/ml) for the indicated time; LSD1 protein level was examined by Western blotting.
-
HMDA‐MB‐231 cells were transfected with control or CARM1 shRNAs and then treated with MG132 for 10 h. Cell lysates were immunoprecipitated by anti‐LSD1 antibody and then analyzed by immunoblotting.
-
IMDA‐MB‐231 cells were treated with increasing amounts of MS049 for 48 h and MG132 for 10 h. The ubiquitination level of LSD1 was assessed by immunoblotting after IP with anti‐LSD1 antibody.
-
JHEK‐293T cells that transfected with LSD1 WT or LSD1 R838A was treated with CHX for the indicated time. LSD1 was examined by Western blotting analysis.
-
KHEK‐293T cells were transfected with the indicated plasmids and then treated with MG132 for 10 h. Cell lysates were immunoprecipitated using anti‐Flag antibody and then subjected to immunoblotting.
-
LHEK‐293T cells expressing LSD1 WT or LSD1 R838A were treated with or without MS049 for 48 h and MG132 for 10 h. Co‐IP assay was performed using anti‐Flag and then subjected to Western blotting.
Source data are available online for this figure.
Figure EV2. CARM1 and LSD1 are highly expressed in human breast cancerous tissues and positively correlated with each other.
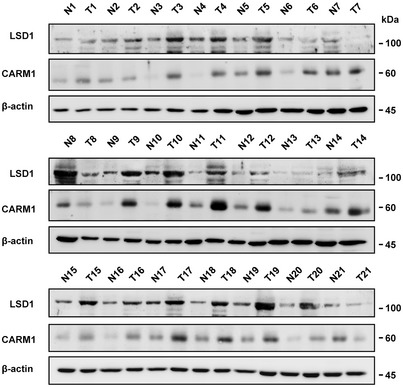
Immunoblotting analysis of LSD1 and CARM1 expression in human breast cancerous tissues and adjacent tissues (n = 21). N, normal; T, tumor.Source data are available online for this figure.
Since LSD1 has been reported degraded by proteasome 21, 22, we made a hypothesis that CARM1 affects the stability of LSD1 through ubiquitin‐proteasome pathway. To substantiate this assumption, we treated MDA‐MB‐231 cells with proteasome inhibitor MG132, and we observed that the inhibitory effect of MS049 and CARM1 knockdown on LSD1 was reversed (Fig 2E and F). Besides, CARM1 knockdown shortened the half‐life of LSD1 upon the addition of protein synthesis inhibitor cycloheximide (CHX) in MDA‐MB‐231 cells (Fig 2G). Furthermore, LSD1 ubiquitination was increased after CARM1 knockdown (Fig 2H), and it was increased in a dose‐dependent manner upon the addition of MS049 (Fig 2I). We further examined the ubiquitination and stabilization of LSD1 in MCF10A cells that barely express CARM1, and found that the half‐life of LSD1 in MCF10A was much shorter than that of MDA‐MB‐231 cells, at a similar level to that of CARM1‐knockdown MDA‐MB‐231 cells (Appendix Fig S3A and Fig 2G). Meanwhile, we observed a robust increase in LSD1 upon the addition of MG132 in MCF10A cells (Appendix Fig S3B), and the ubiquitination status of LSD1 in MCF10A was much stronger than that of MDA‐MB‐231 cells, at a similar level to that of MDA‐MB‐231‐shCARM1#2 cells in which CARM1 was efficiently knocked down (Appendix Fig S3C). These results support the notion that CARM1 stabilizes LSD1 by decreasing its ubiquitination, which intrigued us to determine whether the methylation of LSD1 R838 contributed to LSD1 stability. To verify this assumption, LSD1 WT or R838A mutant was overexpressed in HEK‐293T cells followed by CHX treatment, and we discovered that the half‐life of R838A mutant was much shorter than that of LSD1 WT (Fig 2J). Besides, R838A mutant exhibited higher ubiquitination levels than that of LSD1 WT (Fig 2K). In addition, the ubiquitination level of LSD1 WT was sharply elevated, whereas R838A mutant showed a negligible change upon the addition of MS049 in HEK‐293T cells overexpressing LSD1 WT or R838A mutant (Fig 2L). Taken together, these results indicate that CARM1‐mediated methylation of LSD1 R838 promotes LSD1 stabilization.
CARM1‐dependent methylation of LSD1 promotes deubiquitylation of LSD1 through USP7
Recent evidence suggests that LSD1 is stabilized by deubiquitylases 13, 14, 15, 18. As LSD1 R838 methylation decreases LSD1 ubiquitination, we hypothesized that R838 methylation may promote the binding of deubiquitylase to LSD1. To fulfill this hypothesis, we screened the deubiquitylases from our LC‐MS/MS data (Dataset EV2) and deubiquitylases which have been reported to interact with LSD1. In order to eliminate the influence of the background caused by high level of LSD1 in MDA‐MB‐231 cells, we first knockdown the endogenous LSD1 using lentivirus producing shRNAs targeting LSD1 mRNA 3′UTR. We then transfected the plasmids expressing Flag‐tagged LSD1 WT or LSD1 R838A/R838K/R838F (a mimic of constitutive methylated status) mutants in LSD1‐knockdown MDA‐MB‐231 cells, named MM‐231‐shLSD1‐WT/R838A/R838K/R838F. We found that the interaction between deubiquitylase USP7 and LSD1 was significantly attenuated in R838A mutant (Fig 3A), which indicates that LSD1 R838 methylation enhances the binding of USP7 to LSD1. To further dissect the interaction between LSD1 and USP7, we generated GST‐tagged or Flag‐tagged deletion mutants of LSD1 (Fig EV3A). The GST‐tagged mutants were purified and applied for GST pull‐down assays with Flag‐USP7. It was found that both the mutant with deletion of carboxyl terminus of LSD1 (◄CT), and the mutant with deletion of carboxyl terminal AOD (◄AOD‐C), dramatically weakened the pulldown of Flag‐USP7 (Fig EV3B). Furthermore, the Flag‐tagged mutants were overexpressed in HEK‐293T cells, followed by Co‐IP assays, and the same results as GST pulldown were obtained (Fig EV3C). These results indicate that the AOD‐C (520–852 aas) is required for the interaction between LSD1 and USP7.
Figure 3. CARM1‐dependent methylation of LSD1 promotes deubiquitylation of LSD1 through USP7.
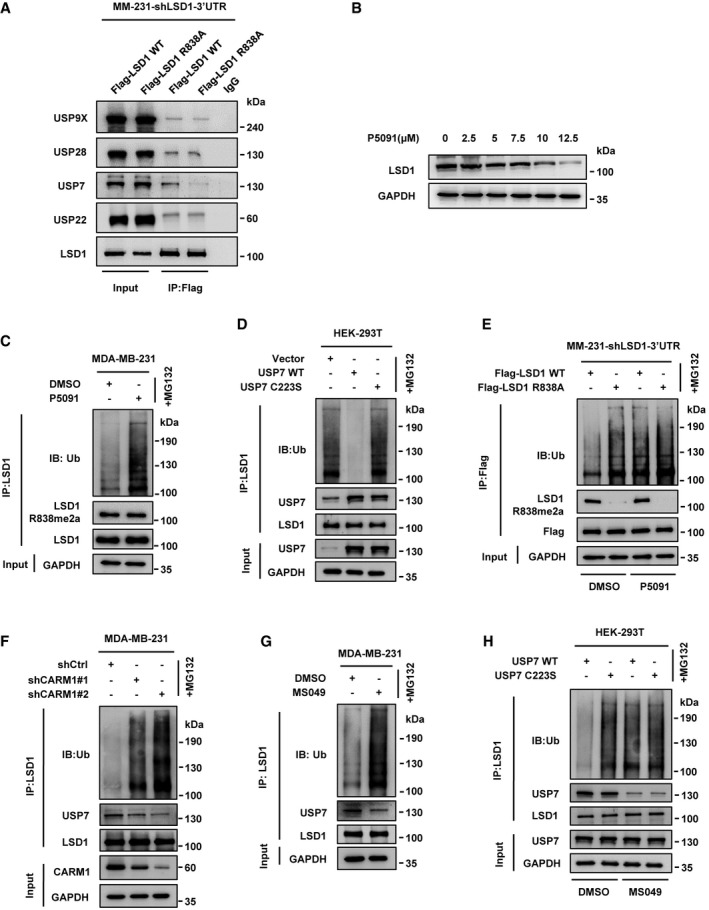
- Immunoprecipitation assay was performed using anti‐Flag antibody in MM‐231‐shLSD1‐WT/R838A cells. The interaction of LSD1 and DUBs was detected by immunoblotting with indicated antibodies.
- MDA‐MB‐231 cells were treated with the indicated amounts of P5091 for 48 h, LSD1 protein levels were assessed by immunoblotting.
- MDA‐MB‐231 cells were treated with P5091 (10 μM) for 48 h and MG132 for 10 h. The LSD1 ubiquitination level was measured by immunoblotting after immunoprecipitated with anti‐LSD1 antibody.
- HEK‐293T cells were transfected with USP7 WT or USP7 C223S, then treated with MG132 for 10 h. Cell lysates were immunoprecipitated using anti‐LSD1 antibody followed by immunoblotting analysis.
- MDA‐MB‐231 cells overexpressing Flag‐tagged LSD1 WT/R838A after knockdown of endogenous LSD1 and then treated with or without P5091 for 48 h and MG132 for 10 h. Cell lysates were immunoprecipitated by anti‐Flag antibody and then analyzed by immunoblotting.
- MDA‐MB‐231 cells were transfected with control or CARM1 shRNAs before treated with MG132 for 10 h. And cell lysates were immunoprecipitated with anti‐LSD1 then subjected to immunoblotting.
- MDA‐MB‐231 cells were treated with or without MS049 for 48 h and MG132 for 10 h, and cell lysates were immunoprecipitated using anti‐LSD1 antibody and then analyzed by immunoblotting.
- HEK‐293T cells were transfected with USP7 WT or USP7 C223S, then treated with or without MS049 for 48 h and MG132 for 10 h. Cell lysates were immunoprecipitated with anti‐LSD1 antibody followed by immunoblotting analysis.
Source data are available online for this figure.
Figure EV3. Carboxyl AOD domain (AOD‐C) of LSD1 is important for its interaction with USP7.
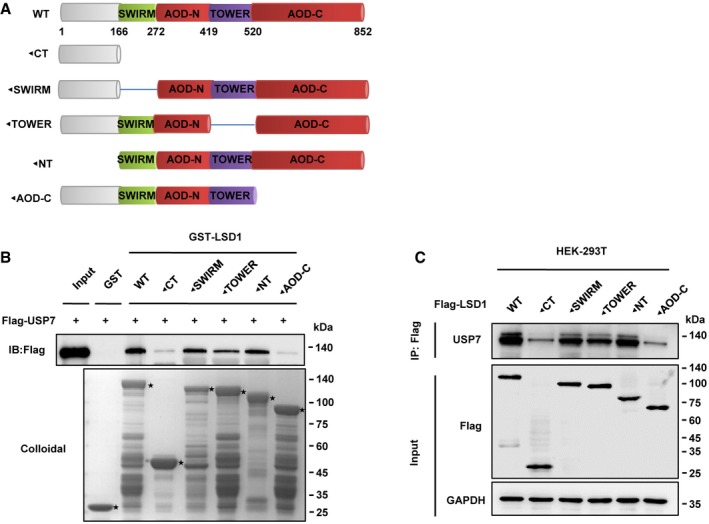
- Schematic diagram of LSD1. LSD1 encodes a polypeptide of 852 aas with a SWIRM domain, a TOWER domain in between the amino amine oxidase (AOD) domain (AOD‐N) and carboxyl AOD domain (AOD‐C). NT amino terminus; CT carboxyl terminus.
- The GST‐tagged LSD1 deletion mutants were applied for GST pull‐down assays with Flag‐USP7 purified from HEK‐293T cells. The amounts of GST or GST‐tagged mutants were visualized by Coomassie Blue staining (asterisk: GST or GST‐LSD1 mutants).
- Flag‐tagged LSD1 deletion mutants were overexpressed in HEK‐293T cells, followed by Co‐IP assays using anti‐Flag antibody, and their interaction with USP7 was assessed by Western blot.
Source data are available online for this figure.
We next assessed the effect of USP7 on LSD1 stability. We found that LSD1 protein levels were reduced when treated with USP7 inhibitor P5091 or knockdown USP7 in MDA‐MB‐231 cells (Figs 3B and EV4A). In contrast, USP7 WT overexpression increased LSD1 protein level in HEK‐293T cells, while USP7 C223S (a catalytic‐inactive mutant) failed to do so (Fig EV4B). As expected, knockdown or inhibition of USP7 dramatically increased LSD1 ubiquitination levels (Figs 3C and EV4C). Moreover, overexpression of USP7 WT, but not USP7 C223S, significantly weakened LSD1 ubiquitination in HEK‐293T cells (Fig 3D). These results demonstrate that USP7 deubiquitinates and stabilizes LSD1.
Figure EV4. Ubiquitin‐specific protease 7 increases LSD1 protein level.
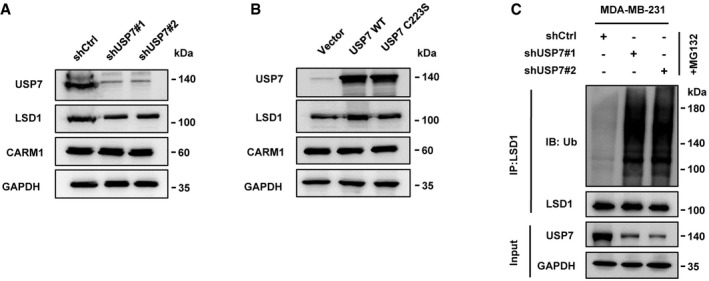
- MDA‐MB‐231 cells were transfected with USP7 shRNAs or control vector, and LSD1 protein level was assessed by Immunoblotting.
- HEK‐293T cells were transfected with USP7 WT or USP7 C223S, and LSD1 protein level was assessed by immunoblotting.
- MDA‐MB‐231 cells were transfected with USP7 shRNAs or control vector, and cell lysates were immunoprecipitated by anti‐LSD1 antibody and then subjected to immunoblotting.
Source data are available online for this figure.
We then attempted to determine whether the deubiquitination of LSD1 by USP7 depended on the methylation of LSD1 by CARM1. We demonstrated that P5091 could only increase the ubiquitination of LSD1 WT, whereas LSD1 R838A remained unchanged (Fig 3E). Moreover, CARM1 knockdown weakened the interaction between endogenous USP7 and LSD1 in MDA‐MB‐231 cells, which caused the enhancement of LSD1 ubiquitination (Fig 3F). MDA‐MB‐231 cells treated with MS049 yielded similar results (Fig 3G). Furthermore, inhibition of CARM1 attenuated the interaction between USP7 WT/C223S and LSD1 upon USP7 WT/C223S overexpression in HEK‐293T cells, whereas it only upregulated the ubiquitination of LSD1 in HEK‐293T‐USP7 WT cells (Fig 3H). These results implicate that CARM1‐dependent methylation of LSD1 is critical for LSD1 binding with USP7 that leads to the deubiquitination of the protein.
Methylation of LSD1 at R838 promotes breast cancer invasion and metastasis
We have reported that LSD1 induces the migration and invasion of breast cancer cells 19, 20. To further explore the role of LSD1 R838 methylation in breast cancer progression, we treated MM‐231‐shLSD1‐WT/R838A/R838K/R838F cells with CARM1 inhibitor MS049, and found that R838A/R838K mutants impaired migratory (Figs 4A and EV5A) and invasive (Fig 4B) capabilities of MDA‐MB‐231 cells. Besides, inhibition of CARM1 sharply decreased the migration (Figs 4A and EV5A) and invasion (Fig 4B) abilities of cells that expressed LSD1 WT, accompanied by decreased mesenchymal markers (Appendix Fig S4A), whereas had no effect on cells that expressed LSD1 R838A/R838K/R838F mutants (Figs 4A and B, and EV5A). Strikingly, we discovered that the cells expressed LSD1 R838F exhibited a high mobility no matter they were treated with or without MS049 (Figs 4A and B, and EV5A). Consistently, similar results can be seen in MCF7 cell lines that expressed LSD1‐WT/R838A/R838K/R838F under the same treatment (Figs 4C and D, and EV5B). Moreover, we co‐expressed CARM1 with LSD1 WT, or one of LSD1 mutants (R838A/R838K/R838F) in MCF10A cells. CARM1 overexpression remarkably increased migration (Figs EV5C and 5E) and invasion (Fig EV5D) abilities of cells that expressing LSD1 WT, together with a significant downregulation of epithelial marker E‐cadherin, and a strong up‐regulation of mesenchymal markers (Appendix Fig S4B). Collectively, these data clearly indicate that CARM1 enhances the LSD1‐induced cell migration and invasion depending on LSD1 R838 methylation.
Figure 4. LSD1 R838 methylation promotes breast cancer invasion and metastasis.
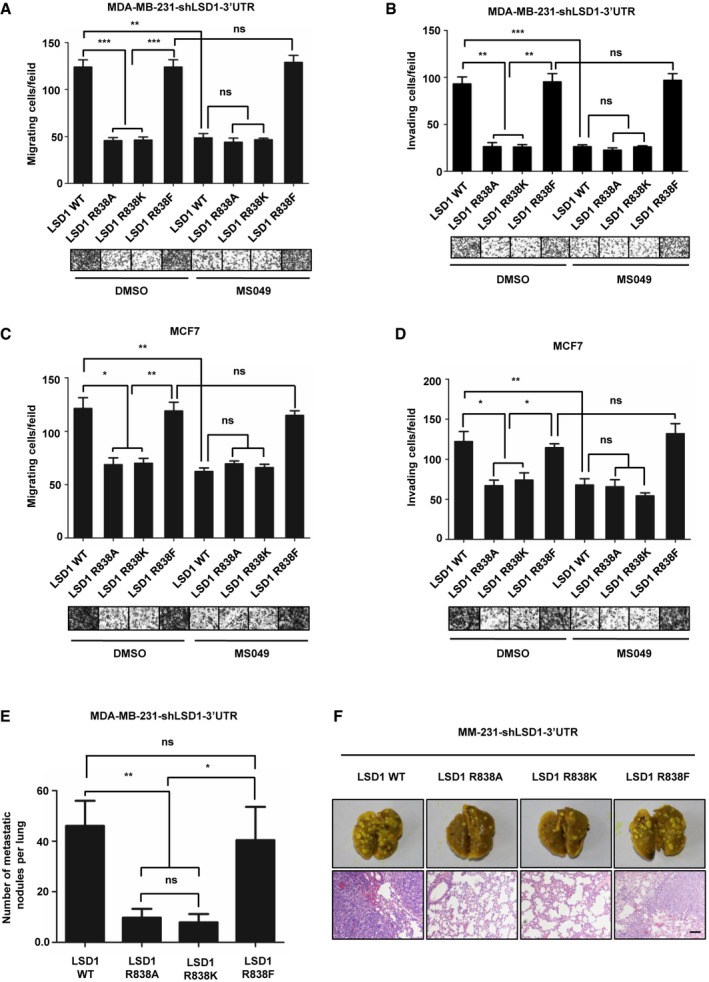
-
A, CMigration assays (24 h) of MM‐231‐shLSD1‐WT/R838A/R838K/R838F cells (A) and MCF7‐LSD1‐WT/R838A/R838K/R838F cells (C) with or without MS049 treatment. Representative micrographs of migrated cells are shown. Data represent the number of cells derived from mean cell counts of five fields (Error bars indicate the mean ± SD, n = 3 experimental replicates, *P < 0.05, **P < 0.01, ***P < 0.001, ns = not significant, Student's t‐test).
-
B, DInvasion assays (48 h) of MM‐231‐shLSD1‐WT/R838A/R838K/R838F cells (B) and MCF7‐LSD1‐WT/R838A/R838K/R838F cells (D) with or without MS049 treatment. Representative micrographs of invaded cells are shown. Data represent the number of cells derived from mean cell counts of five fields (Error bars represent mean ± SD, n = 3 experimental replicates, *P < 0.05, **P < 0.01, ***P < 0.001, ns = not significant, Student's t‐test).
-
EMM‐231‐shLSD1‐WT/R838A/R838K/R838F cells (1 × 106) were injected into the tail veins of 5‐week‐old female nude mice, and 7 weeks later, mice were sacrificed with euthanasia and lungs were fixed in paraformaldehyde solutions. The number of visible surface metastatic lesions in lungs was counted (mean of 7 mice) (Data are mean ± SD, n = 7 mice for each group, *P < 0.05, **P < 0.01, ns = not significant, Student's t‐test).
-
FLung metastatic nodules confirmed by hematoxylin and eosin staining. Scale bars, 100 μm.
Source data are available online for this figure.
Figure EV5. Methylation of LSD1 promotes migration and invasion of MDA‐MB‐231, MCF7, and MCF10A cells.
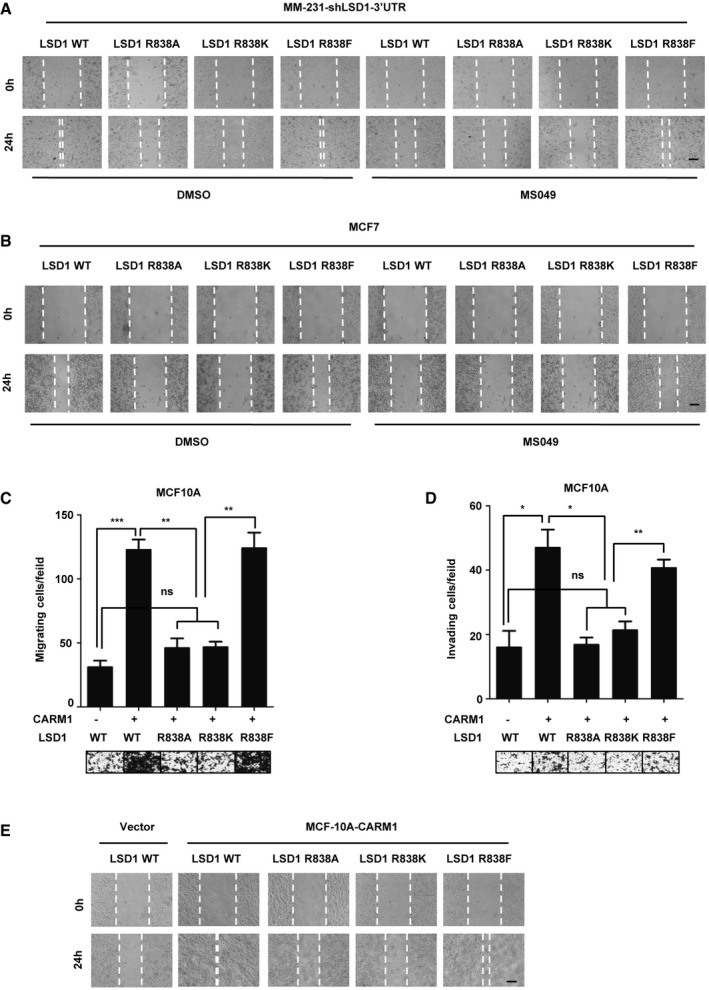
-
A, BWound‐healing assay of MM‐231‐shLSD1‐WT/R838A/R838K/R838F cells (A) and MCF7‐LSD1‐WT/R838A/R838K/R838F cells (B) with or without MS049 treatment. The wounds were created by scraping the cells with sterile pipette tip. Scale bars, 100 μm.
-
C, DMigration (C) and invasion assays (D) were conducted in MCF10A cells that co‐expressed CARM1 with LSD1 WT, or one of LSD1 mutants (R838A/R838K/R838F). Representative images of migrated and invaded cells are shown. Data represent the number of cells derived from mean cell counts of five fields (Error bars represent mean ± SD, n = 3 experimental replicates, *P < 0.05, **P < 0.01, ***P < 0.001, ns = not significant, Student's t‐test).
-
EWound‐healing assays of MCF10A cells that co‐expressed CARM1 with LSD1 WT, or one of LSD1 mutants (R838A/R838K/R838F). The wounds were created by scraping the cells with sterile pipette tip. Scale bars, 100 μm.
Source data are available online for this figure.
Figure 5. CARM1 and USP7 regulate the expression of E‐cadherin and vimentin depending on LSD1 R838 methylation.
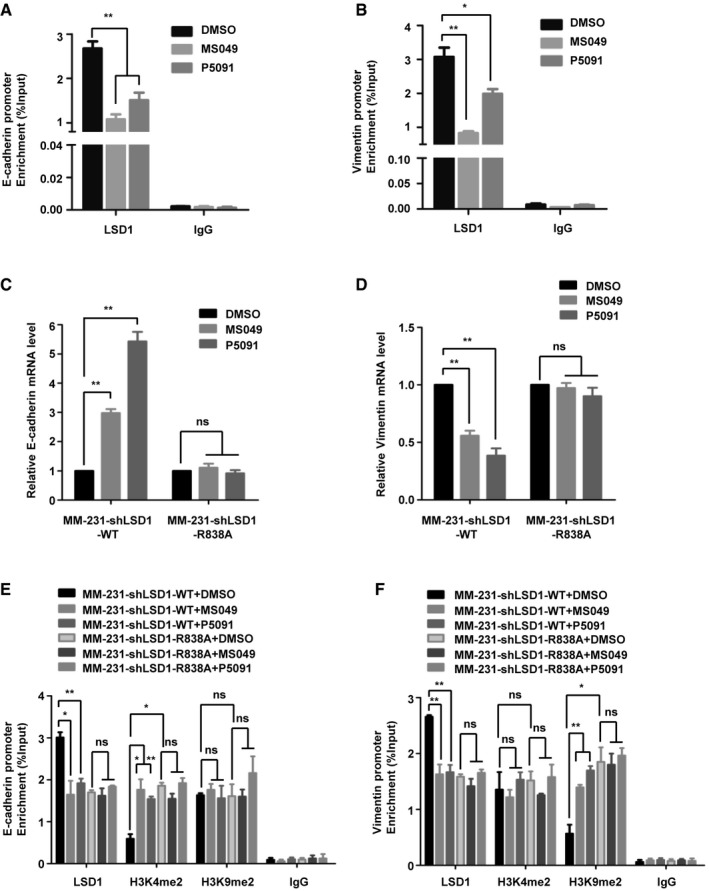
-
A, BMDA‐MB‐231 cells were treated with MS049 or P5091. ChIP assays were performed using anti‐LSD1 antibody, and the immunoprecipitated DNA was analyzed by qPCR using specific primers of E‐cadherin (A) and vimentin (B) promoters (Data are mean ± SD, n = 3 experimental replicates, *P < 0.05, **P < 0.01, Student's t‐test).
-
C, DMM‐231‐shLSD1‐WT/R838A cells were treated with MS049 or P5091, real‐time PCR was performed to analysis the E‐cadherin (C) and vimentin (D) mRNA levels (Data are presented as mean ± SD, n = 3 experimental replicates, **P < 0.01, ns = not significant, Student's t‐test).
-
E, FMM‐231‐shLSD1‐WT/R838A cells were treated with MS049 or P5091. ChIP assays were performed using anti‐LSD1 or anti‐H3K4me2 or anti‐H3K9me2 antibody, and the immunoprecipitated DNA was analyzed by qPCR using specific primers of E‐cadherin (E) and vimentin (F) promoters (Data are mean ± SD, n = 3 experimental replicates, *P < 0.05, **P < 0.01, ns = not significant, Student's t‐test).
Source data are available online for this figure.
We next evaluated the roles of LSD1 R838 methylation in mediating cell proliferation, and found that the growth rate of MDA‐MB‐231 cells expressing LSD1 WT was similar to that of LSD1 mutants (R838A/R838K/R838F) (Appendix Fig S5A). We then confirmed that inhibition of CARM1 had little influence on the growth velocity of MM‐231‐shLSD1‐WT/R838A cells (Appendix Fig S5B). Similar results can be seen in MCF7 cell lines under the same treatments (Appendix Fig S5C and D). These data suggest that methylation of LSD1 at R838 has no impact on cell proliferation.
To further evaluate the critical roles of LSD1 methylation in breast cancer metastasis in vivo, MM‐231‐shLSD1‐WT, MM‐231‐shLSD1‐LSD1 R838A, MM‐231‐shLSD1‐LSD1 R838K, and MM‐231‐shLSD1‐LSD1 R838F cells were injected into the tail veins of female nude mice. Strikingly, 7 weeks later, lung tissues were dissected and examined by macroscopically and H&E staining. We discovered that mice injected with MM‐231‐shLSD1 R838A/K cells formed fewer lung metastases foci than that injected with MM‐231‐shLSD1‐WT or MM‐231‐shLSD1‐LSD1 R838F cells (Fig 4E and F). These data are strongly indicative of the pivotal role of LSD1 methylation in promoting breast cancer metastasis in vivo.
CARM1 and USP7 regulate the expression of E‐cadherin and vimentin depending on LSD1 R838 methylation
We have shown that LSD1 decreases E‐cadherin while increases vimentin transcription by reducing H3K4me2 and H3K9me2 at their promoters, respectively, in breast cancer cells 19, 20. We intended to find out whether CARM1 and USP7 are involved in regulation of these genes by LSD1. Our ChIP results revealed that the presence of LSD1 at E‐cadherin and vimentin promoters was decreased under MS049 or P5091 treatment in MDA‐MB‐231 cells (Fig 5A and B). Moreover, the E‐cadherin mRNA level was increased while the vimentin was reduced after MS049 or P5091 treatment in MM‐231‐shLSD1‐WT cells, whereas MM‐231‐shLSD1 R838A cells displayed negligible changes (Fig 5C and D). These data revealed that CARM1 and USP7 regulate E‐cadherin and vimentin expression based on LSD1 R838 methylation.
We next investigated whether LSD1 methylation participates in the gene regulation through its function of histone demethylation modification. Our ChIP results revealed that R838A mutant led to a marked increase in H3K4me2 but unchanged H3K9me2 on E‐cadherin promoter (Fig 5E), meanwhile, an increased H3K9Me2 but unchanged H3K4me2 on vimentin promoter (Fig 5F). Besides, MS049 or P5091 treatment led to a decreased enrichment of LSD1 at E‐cadherin promoter, together with a notable increase in H3K4me2 but only a marginal change in H3K9me2 in MM‐231‐shLSD1‐WT cells (Fig 5E). Meanwhile, the enrichment of LSD1 at vimentin promoter was decreased accompanied by an increase in H3K9me2, but unchanged H3K4me2 (Fig 5F). In contrast, MS049 or P5091 treatment had no influence on the enrichment of LSD1 and histone modifications at both promoters in MM‐231‐shLSD1 R838A cells (Fig 5E and F). These results strongly suggest that CARM1 and USP7 epigenetically regulate the expression of E‐cadherin and vimentin via methylation of LSD1 at R838.
LSD1R838me2a/CARM1/LSD1 level is associated with grade of breast carcinoma
To investigate the clinical relevance of our findings, immunohistochemistry staining was performed with anti‐LSD1 R838me2a, anti‐LSD1, and anti‐CARM1 antibodies in human breast tumor samples. The results revealed that ~77.1% (54/70) of the samples were positive in LSD1 R838me2a immunoreactivity, while the proportion of LSD1 and CARM1 were ~78.6% (55/70) and ~75.6% (53/70), respectively. Among LSD1 R838me2a immunoreactivity positive samples, 12(22.2%) specimens showed weak staining, 9(16.7%) moderate staining, and 33 (61.1%) strong staining (Fig 6A). Likewise, among LSD1 and CARM1 immunoreactivity positive samples, 8 (14.5%) and 7 (13.2%) specimens displayed weak staining, 14 (25.5%) and 9 (17%) moderate staining, and 33 (60%) and 37 (70%) strong staining, respectively (Fig 6A). Furthermore, a high level of LSD1 R838me2a occurred in 66.7% of stage III, whereas only 34.9% in stage I/II breast cancer samples (Fig 6B and Appendix Table S2). Likewise, a high level of LSD1 and high CARM1 expression occurred in 70.4 and 75% of stage III, whereas only 32.6 and 38.1% in stage I/II breast cancer samples (Fig 6C and D, Appendix Table S2). To determine the clinical relevance of LSD1 R838me2a, LSD1, and CARM1, the immunohistochemistry staining was scored on a scale of 0–12. Consequently, data analysis showed that the correlation between LSD1, LSD1R838me2, and CARM1 was statistically significant (Fig 6E–G). These results suggest that high LSD1 R838me2a expression is driven by high‐level CARM1, which is correlated with malignant breast cancer.
Figure 6. LSD1R838me2a/CARM1/LSD1 level is associated with grade of breast carcinoma.
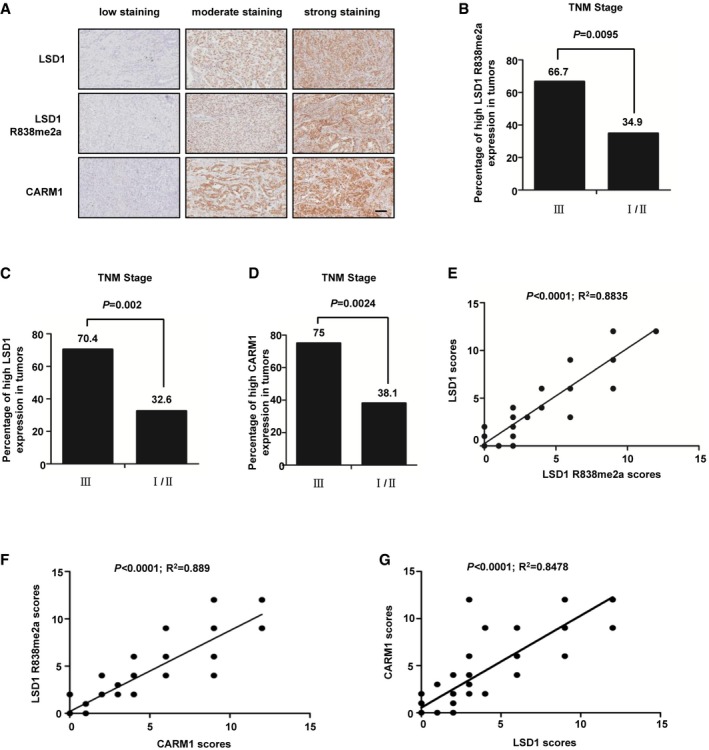
-
ATypical pictures of the immunohistochemical staining of LSD1 R838me2a, LSD1, and CARM1 in breast cancer samples. Scale bars, 100 μm.
-
B–DThe percentage of samples with high level of LSD1 R838me2a (B), LSD1 (C), and CARM1 (D) expression in different TNM stages of breast cancer (n = 70 breast tumor samples; B, P = 0.0095; C, P = 0.002; D, P = 0.0024; χ2 test).
-
E–GThe correlation of LSD1 R838me2a with LSD1 (E), CARM1 with LSD1 R838me2a (F), and CARM1 with LSD1 (G) was statistically significant among breast cancer specimens (n = 70 breast tumor samples; E, R 2 = 0.8835, P < 0.0001; F, R 2 = 0.889, P < 0.0001; G, R 2 = 0.8478, P < 0.0001; Pearson correlation test). Note that some samples have overlapping scores. IHC score was evaluated according to Feng et al 19.
Source data are available online for this figure.
Discussion
LSD1 has been frequently reported to be linked to tumorigenesis and is generally considered as an oncogene 9, 10, 11, 12, 19, 39, 40, with only a few exceptions 41. We have determined in this study that LSD1 is a tumor‐promoting gene for breast cancer. Significantly, we first describe in this report the critical role of LSD1 arginine methylation to its function in breast cancer, as well as a series molecular event involved in this process. Specifically, we have identified that CARM1 dimethylates LSD1 R838, which stabilizes LSD1 via enhancing the binding of LSD1 with USP7 leading to the deubiquitination of the protein, and this in turn promotes breast cancer tumorigenesis and metastasis. Thus, we have unraveled the novel critical function of the CARM1‐USP7‐LSD1 axis in breast carcinogenesis.
PRMTs are constantly found to be dysregulated in numerous cancers 27, 28; however, the effects of methylation by PRMTs on cancer metastasis remain poorly understood. Recently studies have shown that LSD1 and CARM1 co‐exist in the same complex 42. We confirmed in this study that LSD1 is directly interact with CARM1 (Figs 1D and EV1E) and LSD1 is a bona fide CARM1 substrate (Fig EV1C), and we identified that LSD1 R838 is the sole methylation site (Figs 1E and EV1F). In addition, the previously reported automethylation of CARM1 43, 44 was confirmed in our methylation assays (Fig 1E). Notably, recent studies indicate that the role of CARM1 is complex and context‐dependent in cancer development, which functions both as a tumor‐suppressor 45, 46, 47, 48, 49 and as a tumor‐promoting protein 28, 30, 34, 35, 50. Importantly, in vivo studies have shown the oncogenic activities of CARM1 in breast cancer 51; nevertheless, the molecular basis of this complexity in CARM1's action remained largely unexplored prior to this study. Our study demonstrates that CARM1 and LSD1 are highly expressed in human breast cancerous tissues compared with the non‐cancerous tissues (Fig EV2), and CARM1 expression is elevated in malignant breast cancer (Fig 6D) and is positively correlated with LSD1 R838me2a and LSD1 levels (Fig 6F and G), suggesting a tumor‐promoting role of this protein in breast carcinoma.
We find that the methylation of LSD1 R838 enhances its binding to E‐cadherin and vimentin promoters, to demethylate H3K4me2 and H3K9me2, respectively (Figs 5A and B, and 5E and F). However, it remains unclear how LSD1 mediate both H3K4me1/2 demethylation and H3K9me1/2 demethylation. One clue might be the interplay between different modifications, e.g., phosphorylation of LSD1 H3T6 or H3T11, switches its demethylation activity from H3K4me1/2 to H3K9me1/2 52, 53. Moreover, the interaction of LSD1 with different complexes may switch its substrate specificity between H3K4me1/2 and H3K9me1/2, indicating that the co‐repressor or co‐activator function of LSD1 might also be mediated at a target‐specific basis 54. It is known that association of LSD1 with CoREST or NuRD is involved in transcription repression 55, whereas when LSD1 interacts with androgen/estrogen receptor, it plays a role in transcriptional activation 3, 56, 57. Furthermore, LSD1+ 8a, an isoform of LSD1 in neurons, can only mediate H3K9me2 demethylation in neurons 4. In addition, as another neuron‐specific isoform of LSD1, LSD1n exhibits H4K20 demethylase activity and is able to remove H4K20me1/2 in vitro and in vivo 58. These data implicate that the alternative splicing of LSD1 can be a means by which it acquires selective substrate specificities. However, further studies are needed to determine whether the arginine methylation of LSD1 plays a role in the switch of substrate specificity.
Protein deubiquitination is a highly controlled process 59. In this report, we have provided evidence that arginine methylation of LSD1 suppresses its ubiquitination (Fig 2K and L), and at the molecular level, methylated LSD1 potentiates its association with deubiquitylase USP7, leading to deubiquitination of the protein (Fig 3F and G). In contrast, other deubiquitylases such as USP22 and USP28 that are reported to mediate LSD1 deubiquitination 13, 18 have no correlation with LSD1 R838me2a under our experimental system (Fig 3A). Moreover, previous studies have revealed that USP7 plays a key role in tumor progression by stabilizing a number of substrates, such as PHF8 and Ki‐67 60, 61. In line with these reports, our study demonstrates that inhibition of USP7 suppresses migration and invasion of breast cancer cells (Appendix Fig S6A–C).
Recently, Med12 was identified to be methylated by CARM1 62, and CARM1 inhibitor MS049 sharply decreases Med12‐Rme2a level in HEK‐293T cells 63. We prove that MS049 also reduces LSD1R838me2a level in MDA‐MB‐231 cells (Fig 2D), which leads to the downregulation of LSD1 by promoting LSD1 ubiquitination (Fig 2I and L). Strikingly, MS049 impairs the capabilities of migration and invasion of MDA‐MB‐231 and MCF7 cells, depending on LSD1 R838 methylation (Figs 4A–D and EV5A and B). Similar results can be seen when the cells are treated with USP7 inhibitor P5091 instead of MS049 (Appendix Fig S6D–F), suggests that USP7 inhibitor weakened LSD1‐induced cell migration and invasion depending on LSD1 R838 methylation.
Conclusively, data arising from this study depict a series of molecular events in which CARM1 methylates and stabilizes LSD1 by promoting the recruitment of USP7, providing a direct link between arginine methylation and deubiquitination, leading to the repression of E‐cadherin and activation of vimentin genes by attenuating H3K4me2 and H3K9me2, respectively. Consequently, these reactions result in the promotion of migration and invasion of breast cancer cells, and facilitate cancer metastasis in vivo (Fig 7). Significantly, manifestation of the involvement of the CARM1‐USP7‐LSD1 axis in breast carcinogenesis, and disruption of this axis by inhibitors weakens migration and invasion abilities of MDA‐MB‐231 cells, may provide a basis for the development of a potential therapeutic strategy for breast cancer intervention, through targeting the members of this axis.
Figure 7. A working model.
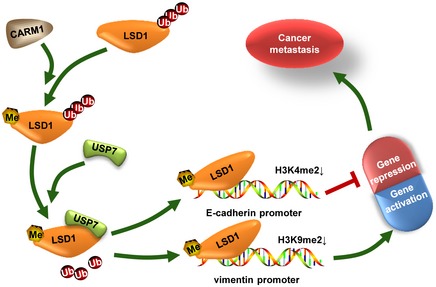
Proposed working model of CARM1‐USP7‐LSD1 axis involved in breast cancer metastasis. CARM1 methylates LSD1, which promotes its binding with and deubiquitylation by USP7, leading to LSD1 stabilization. The stabilization of LSD1 enhances the demethylation of H3K4me2 at E‐cadherin promoter and H3K9me2 at vimentin promoter, resulting in repression of E‐cadherin and activation of vimentin gene expression, respectively, which consequently promotes breast cancer metastasis.
Materials and Methods
Cell culture
All the cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA), where these cell lines were characterized by DNA fingerprinting and isozyme detection. Cells were immediately expanded and frozen such that they could be revived every 3–4 months. Cells were tested every 2 months to ensure negative for mycoplasma contamination with Lonza MycoAlert Mycoplasma Detection Kit (LT07‐218, Lonza, Basel, Switzerland). MCF10A cells were cultured in DMEM/F12 supplemented with 5% horse serum, 20 ng/ml EGF, 0.5 mg/ml hydro cortisone, 100 ng/ml cholera toxin, 10 mg/ml insulin, and 10 U/ml penicillin–streptomycin. MDA‐MB‐231 cells were cultured in L‐15 medium (Sigma‐Aldrich) with 10% FBS. HEK‐293T cells are cultured in DMEM medium (Sigma‐Aldrich) containing 10% FBS. All the cell lines were grown at 37°C with 5% CO2 except MDA‐MB‐231, which was cultured at 37°C without CO2.
Reagents and antibodies
Reagents All the reagents used were purchased from Sangon Biotech (Shanghai) except that listed below (Reagent, Manufacturer, Catalogue Number).
MS049, Selleck, S8147; P5091, Selleck, S7132; MG132, Sigma‐Aldrich, M8699; MG132, Beyotime, Shanghai, China, S1748; cycloheximide (CHX), Sigma‐Aldrich, C104450; EGF, Sigma‐Aldrich, E9644; hydrocortisone, Sangon, A610506; cholera toxin, Sigma‐Aldrich, C8180; insulin, Gibco, Grand Island, NY, USA, 12585‐014; Trypsin (1:250) Powder, Gibco, 27250‐018; hFGF basic, R&D Systems, Minneapolis, MN, USA, P09038; ITS, Sigma‐Aldrich, I3146; vaseline, Sigma‐Aldrich, 16415; and Pentobarbital sodium, Sigma‐Aldrich, P3761. Antibodies (Antibody, Manufacturer, Catalogue Number).
LSD1, Millipore, Billerica, MA, USA, 05‐939; LSD1, Sigma‐Aldrich, L4418; CARM1, Millipore, 09‐818; E‐cadherin (CDH1), BD Biosciences, Lexington, KY, USA, 610181; N‐cadherin, BD Biosciences, 610920; vimentin (VIM), BD Biosciences, 550513; fibronectin (FN), BD Biosciences, 6100770; GAPDH, SunGene, Tianjin, China, KM9002; β‐actin, Sigma‐Aldrich, A5228, A1978; H3K4Me2, Abcam, Cambridge, UK, ab32356; H3K9Me2, Abcam, ab32521; HAUSP/USP7, Abcam, ab108931; FLAG, Abmart, Shanghai, China, M20008; USP9X, Proteintech, 55054‐1‐AP; USP22, Abcam, ab195289; USP28, Abcam, ab126604; Ubiquitin, CST, Boston, MA, USA, #3933; HA, Sigma‐Aldrich, H9658; Anti‐dimethylarginine, asymmetric (ASYM24), Millipore, 07‐414; normal mouse IgG, Santa Cruz Biotechnology, SC‐2025; and normal rabbit IgG, CST, #2729; the secondary antibodies used were goat anti‐mouse and goat anti‐rabbit (ZSGB‐BIO, Beijing, China).
Plasmid construction
All the restriction enzymes used were product of New England BioLabs (NEB. Inc.). The restriction enzymes used included. BamH1‐HF (#R3136), Xho1 (#R0146), Not I‐HF (R3189), Nhe I‐HF (R3131), EcoRI‐HF (R3101), Pme1 (#R0560), Nde1 (#R0111), and T4 DNA Ligase were also an NEB product (#M0202). All these plasmids were confirmed by sequencing in Comate Bioscience (Changchun, China).
Flag‐tagged LSD1 plasmid (constructed by Dr. Jingxin Feng) was used to generate LSD1R108A, LSD1R608A, LSD1R726A, LSD1R838A, LSD1R838K, LSD1R838F, and LSD14 RA; these target DNA fragments were then constructed into the lentiviral expression vector pWPXLd (pWPXLd was a gift from Didier Trono, Addgene plasmid #12258). The bacterial expression plasmids that express a glutathione S‐transferase (GST)‐tagged LSD1 (pGEX‐6P‐1‐LSD1) were made by inserting those target DNA fragments above into the pGEX‐6P‐1 vector in frame with the GST coding sequence.
3× Flag‐CARM1 plasmid was used to generate GST‐tagged CARM1 (pGEX‐6P‐1‐ CARM1).
3× Flag‐tagged USP7 was the gifts from Dr. Pengcheng Ma (Kunming Institute of Zoology, Chinese Academy of Sciences, China). 3× Flag‐tagged USP7 C223S plasmid was kindly provided by Dr. Yongsheng Zhou (Peking University School and Hospital of Stomatology, China).
For the shRNAs, sequences listed below were designed on the BLOCK‐iT RNA Designer online tool (Thermo Fisher) or find on Sigma‐Aldrich official website (https://www.sigmaaldrich.com/life-science/functional-genomics-and-rnai/shrna/individual-genes.html) and then tested in NCBI BLAST system to ensure specificity. The oligonucleotides were synthesized and cloned into the vector pEN‐hH1 (ATCC, 10326368), and the H1 promoter‐shRNA cassette of pEN‐hH1c was subcloned into the destination vector pDSL‐hpUGIP (ATCC, 10326373) through an LR recombination reaction (Gateway® LR Clonase® II Enzyme Mix, Thermo Fisher, 11791‐020). Sequences used are listed below (5′–3′, sense, antisense).
shCtrl, GATCCCCAATTGCCACAACAGGGTCGTGTTCAAGAGACACGACCCTGCCGTGGCAATTTTTTTC, TCGAGAAAAAAATTGCCACAACAGGGTCGTGTCTCTTGAACACGACCCTGCCGTGGCAATTGGG; shLSD1#1, GATCCCCAGGAAGGCTCTTCTAGCAATATTCAAGAGATATTGCTAGAAGAGCCTTCCTTTTTTC, TCGAGAAAAAAGGAAGGCTCTTCTAGCAATATCTCTTGAATATTGCTAGAAGAGCCTTCCTGGG; shLSD1#2, GATCCCCGGAGCTCCTGATTTGACAAAGTTCAAGAGACTTTGTCAAATCAGGAGCTCCTTTTTC, TCGAGAAAAAGGAGCTCCTGATTTGACAAAGTCTCTTGAACTTTGTCAAATCAGGAGCTCCGGG; shCARM1#1, GATCCCCCATGATGCAGGACTACGTGTTCAAGAGACACGTAGTCCTGCATCATGTTTTTC, TCGAGAAAAACATGATGCAGGACTACGTGTCTCTTGAACACGTAGTCCTGCATCATGGGG; shCARM1#2, GATCCCCGGACATGTCTGCTTATTGCTTCAAGAGAGCAATAAGCAGACATGTCCTTTTTC, TCGAGAAAAAGGACATGTCTGCTTATTGCTCTCTTGAAGCAATAAGCAGACATGTCCGGG; shUSP7#1, GATCCCCCCTGGATTTGTGGTTACGTTATTCAAGAGATAACGTAACCACAAATCCAGGTTTTTC, TCGAGAAAAACCTGGATTTGTGGTTACGTTATCTCTTGAATAACGTAACCACAAATCCAGGGGG; shUSP7#2, GATCCCCCGTGGTGTCAAGGTGTACTAATTCAAGAGATTAGTACACCTTGACACCACGTTTTTC, TCGAGAAAAACGTGGTGTCAAGGTGTACTAATCTCTTGAATTAGTACACCTTGACACCACGGGG.
Reverse transcription, PCR, and real‐time PCR analysis
Total RNA was extracted using RNAiso Plus kit (TaKaRa, Code No. 9108) following the manufacturer's instructions. The cDNA was generated with M‐MLV Reverse Transcriptase (Promega, Part No. M170B) as manufacturer's procedure (Part# 9PIM170, Revised 6/05) instructed. RT‐PCR was performed with 2× Taq MasterMix (CWBIOtech, Beijing, Cat.No.CW0682). Real‐time PCR was carried out on Roche LightCycler 480 Instrument I System using LightCycler 480 SYBR Green I Master (Roche, Cat.No.04707516001). GAPDH was used as control in all the experiments. PCRs were run in triplicate for at least three independent repeats. Error bars represent the mean with SD of triplicate experiments. The sequences of PCR primers are listed as follows (5′–3′, sense, antisense).
LSD1: AGCGTCATGGTCTTATCAA, GAAATGTGGCAACTCGTC; CARM1: TCGCCACACCCAACGATTT, GTACTGCACGGCAGAAGACT; GAPDH: ATGACCCCTTCATTGACCTCA, GAGATGATGACCCTTTTGGCT; VIM: GACGCCATCAACACCGAGTT, CTTTGTCGTTGGTTAGCTGGT; CDH1: ATTTTTCCCTCGACACCCGAT, TCCCAGGCGTAGACCAAGA.
Western blotting
Cells were lysed in RIPA lysis buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1% sodium deoxycholate, 1% Triton X‐100, and 0.1% SDS) after washed twice in cold PBS. Protease inhibitor used was purchased from Roche Applied Science (Complete, Mini, EDTA‐free; Protease Inhibitor Cocktail Tablets supplied in EASY packs, Cat.No.4693159001). Protein concentrations were determined by using BCA (bicinchoninic acid) Protein Assay. Protein lysates were subjected to SDS–PAGE, transferred to 0.45‐μm pore size Hydrophobic PVDF Transfer Membrane (Merck Millipore, Cat.No.IPVH00010), detected with appropriate primary antibodies coupled with HRP‐conjugated corresponding secondary antibodies, and visualized with ECL reagent (GE Healthcare, Buckinghamshire, UK). CCD system used to capture chemiluminescent signals was Tanon 5500 high definition low illumination CCD system.
Lentiviral production and infection
Detailed descriptions were presented before 64. The lentivirus packaging vectors used were packaging plasmid psPAX2 (Addgene, Cambridge, MA, USA) and envelop plasmid pMD2.G (Addgene). The transfer vector pDSL‐hpUGIP or pWPXLd together with the previous two plasmid transfected into HEK‐293T cells in a suitable ratio mixed with transfection reagent polyethylenimine (PEI, Sigma‐Aldrich). The supernatant was filtered through a 0.45‐μm filter (Millipore, Temecula, CA, USA) after collected in 48 h, and the concentrated virus was then infected recipient cell lines with 5 mg/ml polybrene.
Wound‐healing, transwell migration, and invasion assays
These experiments were performed as described before 65. For wound‐healing assay, cells were plated in 6‐well plates at a density of 1 × 106 cell/well. Cell layers were scratched by a 10‐μl pipette tip, and the progression of migration was detected and photographed in 20 h. In vitro cell migration and invasion assays were performed using Transwell chambers with 8.0‐μm polyethylene terephthalate membrane (24‐well inserts, Corning BioCoat, Cat.No.354166). For the migration assay, 2 × 104 cells were seeded into the top chambers. For the invasion assay, 2 × 105 cells were added to top chambers coated with Matrigel (BD Biosciences, Cat.No.356234). Basic medium was added to the top chambers, while complete medium was added to the bottom wells to stimulate migration or invasion. After incubation for 20 h (migration) and 48 h (invasion), cells adhered to the lower surface of the membrane were stained with 0.1% crystal violet. Five randomly fields per filter were counted using Photoshop‐CS5 software.
Co‐immunoprecipitation
Detailed protocol has been described previously 19. Briefly, cells were harvested and washed twice in cold PBS, and then lysed in buffer A [20 mM Tris–HCl (pH 8.0), 10 mM NaCl, 1 mM EDTA, and 0.5% NP‐40] plus protease inhibitor cocktail tablet (Roche). Incubate on ice and shock lysates on the vortex shaker 10 times for 30 s each. Microcentrifuge in 12,000 g for 10 min at 4°C, collect the supernatant, and incubate with 2‐5 μg corresponding primary antibodies with gentle shacking overnight at 4°C, followed by adding 20‐40 μl of Pure Proteome Protein A/G Mix Magnetic Beads (Millipore, Cat.No.LSKMAGAG02) for 2–4 h. Wash the beads three times with 1 ml buffer A for 5 min each, resuspend in 40‐60 μl 2× loading buffer, and incubate at 100°C for 8–10 min. Throw beads away and analyze by immunoblotting.
Affinity purification of Flag‐tagged protein
Transfection of plasmid that expressing flag fusion proteins into HEK‐293T cells, PEI reagent was used according to the manufacturer's instructions. Cells were lysed in buffer A mixed with protease inhibitor cocktail tablet (Roche) after 48 h. Add 30–40 μl anti‐Flag affinity gel (Biotool, Kirchberg, Switzerland, Cat.No.B23102) and incubate 4 h at 4°C. Wash the beads three times using 1× Tris saline buffer [50 mM Tris–HCl (pH 7.4) and 150 mM NaCl)] for 5 min each. Then, the Flag‐tagged proteins were eluted by 3× flag peptide (ApexBio, Houston, USA, Cat.No.A6001) for 3 h at 4°C.
GST pull‐down assay
The experiments performed basically as described before 66. GST fusion proteins were expressed in bacteria (BL21) and induced with 0.1 mM isopropyl‐b‐D‐thio‐galactoside (IPTG) for 4 h at 30°C and centrifuge for 5 min at 4°C, 8,000 g. Add 1 ml TEN buffer (20 mM Tris pH 7.4, 0.1 mM EDTA, 100 mM NaCl) into bacteria and sonicate on ice. Collect supernatant by centrifugation in 12,000 g for 5 min at 4°C, incubate with glutathione‐Sepharose 4B beads (GE Healthcare), and then wash three times with TEN buffer. Protein‐adsorbed beads were incubated with flag fusion proteins which purified from HEK‐293T cells. Wash the beads three times with 1 ml TENT buffer (0.5% NP‐40, 20 mM Tris pH 7.4, 0.1 mM EDTA, 300 mM NaCl) for 5 min each, and analyze by immunoblotting.
Generation of antibodies for LSD1 R838 asymmetric dimethylation
The LSD1 R838 asymmetric dimethylation (LSD1 R838 me2as) antibody encompassing the peptide P[R‐(Me)2]QATPGVPAQQSPSM, which corresponds to aa 837–852 of human LSD1 protein, was used to immunize rabbits to generate a polyclonal antibody that detected LSD1 R838 asymmetric dimethylation. The generation of polyclonal antibody was performed in GL Biochem Co., Ltd (Shanghai, China).
In vitro methylation assay and mass spectrometry analysis
Purified enzymes (GST/Flag‐PRMTs) and substrate (LSD1) were incubated in the presence of 3H‐S‐adenosyl‐methionine (3H‐SAM; 15 Ci/mmol; Perkin Elmer, Waltham, MA, USA) for 1 h at 30°C and then analyzed by SDS–PAGE. The gel was dried for 1 h at 60°C followed by autoradiography for about a month or so at −80°C. Flag‐LSD1 protein was purified from HEK‐293T cells and followed by SDS–PAGE. The gel was stained by Coomassie Brilliant Blue (CBB). The flag‐LSD1 band was excised and analyzed by liquid chromatography‐tandem mass spectrometry (LC‐MS/MS) performed in APTBIO (Shanghai, China).
Chromatin Immunoprecipitation (ChIP)
The chromatin immunoprecipitation was performed as described in Feng et al 19. The primers used in ChIP are listed below (5′–3′, sense, antisense). CDH1 promoter: AGGGTCACCGCGTCTATG, CTTCCGCAAGCTCACAGG;
VIM promoter: TGACTTCCACCAGGGT, CGTCCGTAGTTTCAAAAT.
Human breast cancer specimens and immunohistochemistry
Human breast cancer tissue specimen microarrays were purchased from Outdo Biotech (Shanghai, China). The microarrays we used were HBre‐Duc070CS‐01, HBreD030PG02, and HBreD030CS02. Briefly, tissue specimen microarrays were deparaffinized and rehydrated. Antigen retrieval was then done by heating the tissues in 10 mM citrate buffer (pH 6.0) for 15 min at 95°C. Endogenous peroxidase activity was blocked with 3% H2O2 (DAKO system), and tissues were blocked with 10% goat serum followed by incubation with Corresponding primary antibody in a suitable dilution ratio for 2 h and then incubation with secondary antibodies (DAKO) for 30 min. The immunostaining was developed with 3,3′‐diaminobenzidine. Finally, tissues were counterstained with hematoxylin. Data were analyzed by doctors of the Pathology Department, The First Hospital of Jilin University, China. IHC score was evaluated according to Feng et al 19.
In vivo mouse lung metastasis assay
Detailed descriptions were presented before 67. Briefly, stable cells MM‐231‐shLSD1‐WT, MM‐231‐shLSD1‐R838A, MM‐231‐shLSD1‐R838K, and MM‐231‐shLSD1‐R838F (1 × 106 cells suspended with 150 μl of PBS) were injected into the tail veins of female BALB/c nude mice (HFK Bioscience, Beijing) at 5 weeks old. 7 weeks later, the mice were sacrificed with euthanasia, and lungs were fixed in paraformaldehyde solutions before counting the number of metastatic lesions. Then embedded lungs in paraffin and performed with H&E staining. All animal experiments were approved by the Animal Care Committee of the Northeast Normal University, China.
Statistical analysis
Unless specified, data are presented as mean ± SD. Student's t‐test (two‐tailed) was used to determine the statistical significance of differences between groups. The proteins expression level in human breast cancer samples was analyzed by chi‐square test. P < 0.05 was considered statistically significant. Statistical analysis was carried out using GraphPad Prism 5 Software (GraphPad Software, La Jolla, CA, USA).
Author contributions
Conception and design by JL, JL, and YZ; Development of methodology by JL; Acquisition of data by JL, JF, LL, JJ, CL, LL, NZ, and DD; Computational analysis by JL; Writing by BH, JL, and JL. Administrative, technical or material support by LL and ZL; Study supervision by YZ, JL, and BH.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Appendix and Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank Dr. Pengcheng Ma (Kunming Institute of Zoology, Chinese Academy of Sciences, China) and Dr. Yongsheng Zhou (Peking University School and Hospital of Stomatology, China) for providing the plasmids mentioned in Supplementary Information. This work was supported by the grants from the National Natural Science Foundation of China (Grant numbers: 31571317, 31570718, 31771335, 31770825 and 31870765) and the Science and Technology Development Project of Jilin Province (Grant numbers: 20180101232JC and 20180101234JC).
EMBO Reports (2020) 21: e48597
Contributor Information
Yu Zhang, Email: zhangy288@nenu.edu.cn.
Jun Lu, Email: luj809@nenu.edu.cn.
References
- 1. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119: 941–953 [DOI] [PubMed] [Google Scholar]
- 2. Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, Buettner R, Schule R (2005) LSD1 demethylates repressive histone marks to promote androgen‐receptor‐dependent transcription. Nature 437: 436–439 [DOI] [PubMed] [Google Scholar]
- 3. Nair SS, Nair BC, Cortez V, Chakravarty D, Metzger E, Schule R, Brann DW, Tekmal RR, Vadlamudi RK (2010) PELP1 is a reader of histone H3 methylation that facilitates oestrogen receptor‐alpha target gene activation by regulating lysine demethylase 1 specificity. EMBO Rep 11: 438–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Laurent B, Ruitu L, Murn J, Hempel K, Ferrao R, Xiang Y, Liu S, Garcia BA, Wu H, Wu F et al (2015) A specific LSD1/KDM1A isoform regulates neuronal differentiation through H3K9 demethylation. Mol Cell 57: 957–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, Opravil S, Shiekhattar R, Bedford MT, Jenuwein T et al (2007) p53 is regulated by the lysine demethylase LSD1. Nature 449: 105–108 [DOI] [PubMed] [Google Scholar]
- 6. Zhang J, Yuan B, Zhang F, Xiong L, Wu J, Pradhan S, Wang Y (2011) Cyclophosphamide perturbs cytosine methylation in Jurkat‐T cells through LSD1‐mediated stabilization of DNMT1 protein. Chem Res Toxicol 24: 2040–2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lv T, Yuan D, Miao X, Lv Y, Zhan P, Shen X, Song Y (2012) Over‐expression of LSD1 promotes proliferation, migration and invasion in non‐small cell lung cancer. PLoS ONE 7: e35065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang M, Liu X, Guo J, Weng X, Jiang G, Wang Z, He L (2015) Inhibition of LSD1 by Pargyline inhibited process of EMT and delayed progression of prostate cancer in vivo . Biochem Biophys Res Comm 467: 310–315 [DOI] [PubMed] [Google Scholar]
- 9. Ding J, Zhang ZM, Xia Y, Liao GQ, Pan Y, Liu S, Zhang Y, Yan ZS (2013) LSD1‐mediated epigenetic modification contributes to proliferation and metastasis of colon cancer. Br J Cancer 109: 994–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Serce N, Gnatzy A, Steiner S, Lorenzen H, Kirfel J, Buettner R (2012) Elevated expression of LSD1 (Lysine‐specific demethylase 1) during tumour progression from pre‐invasive to invasive ductal carcinoma of the breast. BMC Clin Pathol 12: 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nagasawa S, Sedukhina AS, Nakagawa Y, Maeda I, Kubota M, Ohnuma S, Tsugawa K, Ohta T, Roche‐Molina M, Bernal JA et al (2015) LSD1 overexpression is associated with poor prognosis in basal‐like breast cancer, and sensitivity to PARP inhibition. PLoS ONE 10: e0118002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lim S, Janzer A, Becker A, Zimmer A, Schule R, Buettner R, Kirfel J (2010) Lysine‐specific demethylase 1 (LSD1) is highly expressed in ER‐negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 31: 512–520 [DOI] [PubMed] [Google Scholar]
- 13. Zhou A, Lin K, Zhang S, Chen Y, Zhang N, Xue J, Wang Z, Aldape KD, Xie K, Woodgett JR et al (2016) Nuclear GSK3beta promotes tumorigenesis by phosphorylating KDM1A and inducing its deubiquitylation by USP22. Nat Cell Biol 18: 954–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yi L, Cui Y, Xu Q, Jiang Y (2016) Stabilization of LSD1 by deubiquitinating enzyme USP7 promotes glioblastoma cell tumorigenesis and metastasis through suppression of the p53 signaling pathway. Oncol Rep 36: 2935–2945 [DOI] [PubMed] [Google Scholar]
- 15. Liu W, Zhang Q, Fang Y, Wang Y (2018) The deubiquitinase USP38 affects cellular functions through interacting with LSD1. Biol Res 51: 53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Han X, Gui B, Xiong C, Zhao L, Liang J, Sun L, Yang X, Yu W, Si W, Yan R et al (2014) Destabilizing LSD1 by Jade‐2 promotes neurogenesis: an antibraking system in neural development. Mol Cell 55: 482–494 [DOI] [PubMed] [Google Scholar]
- 17. Piao L, Suzuki T, Dohmae N, Nakamura Y, Hamamoto R (2015) SUV39H2 methylates and stabilizes LSD1 by inhibiting polyubiquitination in human cancer cells. Oncotarget 6: 16939–16950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu Y, Wang Y, Yang XH, Kang T, Zhao Y, Wang C, Evers BM, Zhou BP (2013) The deubiquitinase USP28 stabilizes LSD1 and confers stem‐cell‐like traits to breast cancer cells. Cell Rep 5: 224–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Feng J, Li L, Zhang N, Liu J, Zhang L, Gao H, Wang G, Li Y, Zhang Y, Li X et al (2017) Androgen and AR contribute to breast cancer development and metastasis: an insight of mechanisms. Oncogene 36: 2775–2790 [DOI] [PubMed] [Google Scholar]
- 20. Feng J, Xu G, Liu J, Zhang N, Li L, Ji J, Zhang J, Zhang L, Wang G, Wang X et al (2016) Phosphorylation of LSD1 at Ser112 is crucial for its function in induction of EMT and metastasis in breast cancer. Breast Cancer Res Treat 159: 443–456 [DOI] [PubMed] [Google Scholar]
- 21. Lin Y, Wu Y, Li J, Dong C, Ye X, Chi YI, Evers BM, Zhou BP (2010) The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine‐specific demethylase 1. EMBO J 29: 1803–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y (2005) Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell 19: 857–864 [DOI] [PubMed] [Google Scholar]
- 23. Cardozo T, Pagano M (2004) The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol 5: 739 [DOI] [PubMed] [Google Scholar]
- 24. Sowa ME, Bennett EJ, Gygi SP, Harper JW (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138: 389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reyes‐Turcu FE, Ventii KH, Wilkinson KD (2009) Regulation and cellular roles of ubiquitin‐specific deubiquitinating enzymes. Annu Rev Biochem 78: 363–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paik WK, Paik DC, Kim S (2007) Historical review: the field of protein methylation. Trends Biochem Sci 32: 146–152 [DOI] [PubMed] [Google Scholar]
- 27. Cheung N, Chan LC, Thompson A, Cleary ML, So CWE (2007) Protein arginine‐methyltransferase‐dependent oncogenesis. Nat Cell Biol 9: 1208 [DOI] [PubMed] [Google Scholar]
- 28. Yang Y, Bedford MT (2013) Protein arginine methyltransferases and cancer. Nat Rev Cancer 13: 37 [DOI] [PubMed] [Google Scholar]
- 29. Yao R, Jiang H, Ma Y, Wang L, Wang L, Du J, Hou P, Gao Y, Zhao L, Wang G et al (2014) PRMT7 induces epithelial‐to‐mesenchymal transition and promotes metastasis in breast cancer. Can Res 74: 5656–5667 [DOI] [PubMed] [Google Scholar]
- 30. Frietze S, Lupien M, Silver PA, Brown M (2008) CARM1 regulates estrogen‐stimulated breast cancer growth through up‐regulation of E2F1. Can Res 68: 301–306 [DOI] [PubMed] [Google Scholar]
- 31. Mann M, Cortez V, Vadlamudi R (2013) PELP1 oncogenic functions involve CARM1 regulation. Carcinogenesis 34: 1468–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cheng H, Qin Y, Fan H, Su P, Zhang X, Zhang H, Zhou G (2013) Overexpression of CARM1 in breast cancer is correlated with poorly characterized clinicopathologic parameters and molecular subtypes. Diagn Pathol 8: 129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Davis MB, Liu X, Wang S, Reeves J, Khramtsov A, Huo D, Olopade OI (2013) Expression and sub‐cellular localization of an epigenetic regulator, co‐activator arginine methyltransferase 1 (CARM1), is associated with specific breast cancer subtypes and ethnicity. Mol Cancer 12: 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang L, Zhao Z, Meyer MB, Saha S, Yu M, Guo A, Wisinski KB, Huang W, Cai W, Pike JW et al (2014) CARM1 methylates chromatin remodeling factor BAF155 to enhance tumor progression and metastasis. Cancer Cell 25: 21–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu F, Ma F, Wang Y, Hao L, Zeng H, Jia C, Wang Y, Liu P, Ong IM, Li B et al (2017) PKM2 methylation by CARM1 activates aerobic glycolysis to promote tumorigenesis. Nat Cell Biol 19: 1358–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luo H, Shenoy AK, Li X, Jin Y, Jin L, Cai Q, Tang M, Liu Y, Chen H, Reisman D et al (2016) MOF acetylates the histone demethylase LSD1 to suppress epithelial‐to‐mesenchymal transition. Cell Rep 15: 2665–2678 [DOI] [PubMed] [Google Scholar]
- 37. Nam HJ, Boo K, Kim D, Han DH, Choe HK, Kim CR, Sun W, Kim H, Kim K, Lee H et al (2014) Phosphorylation of LSD1 by PKCalpha is crucial for circadian rhythmicity and phase resetting. Mol Cell 53: 791–805 [DOI] [PubMed] [Google Scholar]
- 38. Peng B, Wang J, Hu Y, Zhao H, Hou W, Zhao H, Wang H, Liao J, Xu X (2015) Modulation of LSD1 phosphorylation by CK2/WIP1 regulates RNF168‐dependent 53BP1 recruitment in response to DNA damage. Nucleic Acids Res 43: 5936–5947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma L, Ma S, Zhao G, Yang L, Zhang P, Yi Q, Cheng S (2016) miR‐708/LSD1 axis regulates the proliferation and invasion of breast cancer cells. Cancer Med 5: 684–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Derr RS, van Hoesel AQ, Benard A, Goossens‐Beumer IJ, Sajet A, Dekker‐Ensink NG, de Kruijf EM, Bastiaannet E, Smit VT, van de Velde CJ (2014) High nuclear expression levels of histone‐modifying enzymes LSD1, HDAC2 and SIRT1 in tumor cells correlate with decreased survival and increased relapse in breast cancer patients. BMC Cancer 14: 604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W, Liang J, Sun L, Yang X, Shi L (2009) LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 138: 660–672 [DOI] [PubMed] [Google Scholar]
- 42. Bennesch MA, Segala G, Wider D, Picard D (2016) LSD1 engages a corepressor complex for the activation of the estrogen receptor alpha by estrogen and cAMP. Nucleic Acids Res 44: 8655–8670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kuhn P, Chumanov R, Wang Y, Ge Y, Burgess RR, Xu W (2011) Automethylation of CARM1 allows coupling of transcription and mRNA splicing. Nucleic Acids Res 39: 2717–2726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang L, Charoensuksai P, Watson NJ, Wang X, Zhao Z, Coriano CG, Kerr LR, Xu W (2013) CARM1 automethylation is controlled at the level of alternative splicing. Nucleic Acids Res 41: 6870–6880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shirley SH, Rundhaug JE, Tian J, Cullinan‐Ammann N, Lambertz I, Conti CJ, Fuchs‐Young R (2009) Transcriptional regulation of estrogen receptor‐α by p53 in human breast cancer cells. Can Res 69: 3405–3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee Y‐H, Bedford MT, Stallcup MR (2011) Regulated recruitment of tumor suppressor BRCA1 to the p21 gene by coactivator methylation. Genes Dev 25: 176–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. O'Brien KB, Alberich‐Jordà M, Yadav N, Kocher O, DiRuscio A, Ebralidze A, Levantini E, Sng NJ, Bhasin M, Caron T (2010) CARM1 is required for proper control of proliferation and differentiation of pulmonary epithelial cells. Development 137: 2147–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang YP, Zhou W, Wang J, Huang X, Zuo Y, Wang TS, Gao X, Xu YY, Zou SW, Liu YB et al (2016) Arginine methylation of MDH1 by CARM1 inhibits glutamine metabolism and suppresses pancreatic cancer. Mol Cell 64: 673–687 [DOI] [PubMed] [Google Scholar]
- 49. Al‐Dhaheri M, Wu J, Skliris GP, Li J, Higashimato K, Wang Y, White KP, Lambert P, Zhu Y, Murphy L et al (2011) CARM1 is an important determinant of ERalpha‐dependent breast cancer cell differentiation and proliferation in breast cancer cells. Can Res 71: 2118–2128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Carascossa S, Dudek P, Cenni B, Briand PA, Picard D (2010) CARM1 mediates the ligand‐independent and tamoxifen‐resistant activation of the estrogen receptor alpha by cAMP. Genes Dev 24: 708–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bao J, Di Lorenzo A, Lin K, Lu Y, Zhong Y, Sebastian MM, Muller WJ, Yang Y, Bedford MT (2018) Mouse models of overexpression reveal distinct oncogenic roles for different type I protein arginine methyltransferases. Can Res 79: 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Metzger E, Yin N, Wissmann M, Kunowska N, Fischer K, Friedrichs N, Patnaik D, Higgins JM, Potier N, Scheidtmann K‐H (2008) Phosphorylation of histone H3 at threonine 11 establishes a novel chromatin mark for transcriptional regulation. Nat Cell Biol 10: 53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Metzger E, Imhof A, Patel D, Kahl P, Hoffmeyer K, Friedrichs N, Müller JM, Greschik H, Kirfel J, Ji S (2010) Phosphorylation of histone H3T6 by PKCβ I controls demethylation at histone H3K4. Nature 464: 792 [DOI] [PubMed] [Google Scholar]
- 54. Amente S, Lania L, Majello B (2013) The histone LSD1 demethylase in stemness and cancer transcription programs. Biochem Biophys Acta 1829: 981–986 [DOI] [PubMed] [Google Scholar]
- 55. Shi Y, Sawada J‐i, Sui G, Affar EB, Whetstine JR, Lan F, Ogawa H, Luke MP‐S, Nakatani Y, Shi Y (2003) Coordinated histone modifications mediated by a CtBP co‐repressor complex. Nature 422: 735 [DOI] [PubMed] [Google Scholar]
- 56. Garcia‐Bassets I, Kwon Y‐S, Telese F, Prefontaine GG, Hutt KR, Cheng CS, Ju B‐G, Ohgi KA, Wang J, Escoubet‐Lozach L (2007) Histone methylation‐dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell 128: 505–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Perillo B, Ombra MN, Bertoni A, Cuozzo C, Sacchetti S, Sasso A, Chiariotti L, Malorni A, Abbondanza C, Avvedimento EV (2008) DNA oxidation as triggered by H3K9me2 demethylation drives estrogen‐induced gene expression. Science 319: 202–206 [DOI] [PubMed] [Google Scholar]
- 58. Wang J, Telese F, Tan Y, Li W, Jin C, He X, Basnet H, Ma Q, Merkurjev D, Zhu X et al (2015) LSD1n is an H4K20 demethylase regulating memory formation via transcriptional elongation control. Nat Neurosci 18: 1256–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Domínguez JM, Fuertes A, Orozco L, del Monte‐Millán M, Delgado E, Medina M (2012) Evidence for irreversible inhibition of glycogen synthase kinase‐3β by tideglusib. J Biol Chem 287: 893–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang Q, Ma S, Song N, Li X, Liu L, Yang S, Ding X, Shan L, Zhou X, Su D (2016) Stabilization of histone demethylase PHF8 by USP7 promotes breast carcinogenesis. J Clin Invest 126: 2205–2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang C, Lu J, Zhang Q‐W, Zhao W, Guo J‐H, Liu S‐L, Wu Y‐L, Jiang B, Gao F‐H (2016) USP7 promotes cell proliferation through the stabilization of Ki‐67 protein in non‐small cell lung cancer cells. Int J Biochem Cell Biol 79: 209–221 [DOI] [PubMed] [Google Scholar]
- 62. Wang L, Zeng H, Wang Q, Zhao Z, Boyer TG, Bian X, Xu W (2015) MED12 methylation by CARM1 sensitizes human breast cancer cells to chemotherapy drugs. Sci Adv 1: e1500463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shen Y, Szewczyk MM, Eram MS, Smil D, Kaniskan HU, Ferreira de Freitas R, Senisterra G, Li F, Schapira M, Brown PJ (2016) Discovery of a potent, selective, and cell‐active dual inhibitor of protein arginine methyltransferase 4 and protein arginine methyltransferase 6. J Med Chem 59: 9124–9139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liang Q, Li L, Zhang J, Lei Y, Wang L, Liu D‐X, Feng J, Hou P, Yao R, Zhang Y (2013) CDK5 is essential for TGF‐β1‐induced epithelial‐mesenchymal transition and breast cancer progression. Sci Rep 3: 2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li L, Liu DX, Zhang N, Liang Q, Feng J, Yao M, Liu J, Li X, Zhang Y, Lu J (2015) SHON, a novel secreted protein, regulates epithelial–mesenchymal transition through transforming growth factor‐β signaling in human breast cancer cells. Int J Cancer 136: 1285–1295 [DOI] [PubMed] [Google Scholar]
- 66. Zhao L, Zhang Y, Gao Y, Geng P, Lu Y, Liu X, Yao R, Hou P, Liu D, Lu J (2015) JMJD3 promotes SAHF formation in senescent WI38 cells by triggering an interplay between demethylation and phosphorylation of RB protein. Cell Death Differ 22: 1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li Z, Hou P, Fan D, Dong M, Ma M, Li H, Yao R, Li Y, Wang G, Geng P et al (2017) The degradation of EZH2 mediated by lncRNA ANCR attenuated the invasion and metastasis of breast cancer. Cell Death Differ 24: 59–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Appendix and Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6