

Abstract
TMEM16A, a Ca2+‐activated Cl− channel, is known to modulate the excitability of various types of cells; however, its function in central neurons is largely unknown. Here, we show the specific expression of TMEM16A in the medial habenula (mHb) via RNAscope in situ hybridization, immunohistochemistry, and electrophysiology. When TMEM16A is ablated in the mHb cholinergic neurons (TMEM16A cKO mice), the slope of after‐hyperpolarization of spontaneous action potentials decreases and the firing frequency is reduced. Reduced mHb activity also decreases the activity of the interpeduncular nucleus (IPN). Moreover, TMEM16A cKO mice display anxiogenic behaviors and deficits in social interaction without despair‐like phenotypes or cognitive dysfunctions. Finally, chemogenetic inhibition of mHb cholinergic neurons using the DREADD (Designer Receptors Exclusively Activated by Designer Drugs) approach reveals similar behavioral phenotypes to those of TMEM16A cKO mice. We conclude that TMEM16A plays a key role in anxiety‐related behaviors regulated by mHb cholinergic neurons and could be a potential therapeutic target against anxiety‐related disorders.
Keywords: anxiety, cholinergic neurons, medial habenula, social interaction, TMEM16A
Subject Categories: Neuroscience
The TMEM16A chloride channel has an excitatory role in cholinergic neurons of the medial habenula (mHb). Mice deficient for TMEM16A in mHb cholinergic neurons display anxiety‐related behaviors.
Introduction
Anxiety is a physiological state characterized by cognitive, emotional, and behavioral responses to various stressors. It is manifested as various somatic symptoms of tension and nervous behaviors that may arise from the perception of uncontrollable or unavoidable threats 1. Since anxiety disorders are associated with the inability to cope with these potential threats, it is necessary to determine therapeutic targets for effective treatment of anxiety‐related symptoms. Therefore, the neural circuits and molecular mechanisms of anxiety disorders must be ascertained.
The amygdala, innervated by multiple input pathways, has been established as one of the major regions involved in anxiety 2. The serotonin circuits, including the dorsal raphe nucleus (DRN) and bed nucleus of the stria terminalis, have also been implicated in anxiety‐related disorders 3. In addition to these circuits, the medial habenula–interpeduncular nucleus (mHb‐IPN) pathway represents an essential circuit that signals heightened anxiety induced by nicotine withdrawal 4, 5. However, it is yet to be addressed whether the mHb‐IPN circuit is directly involved in anxiety per se or it is only activated under the conditions of drug addiction or in withdrawal states.
The mHb is a subregion of the habenula, a bilateral epithalamic nucleus that contributes to aversive state expression by linking the forebrain areas to the midbrain monoaminergic centers 6, 7. It has been known that the mHb is involved in diverse brain functions, such as nicotine addiction, contextual fear memory, anhedonia, and anxiety 4, 8, 9, 10. Interestingly, the mHb neurons exhibit spontaneous action potential firing, which has been shown to be increased by nicotine 4, 11, 12, 13. However, neither the functional role of key regulators (e.g., ion channels) nor the underlying mechanisms of activity alterations in the mHb neurons have been characterized.
The activity of calcium‐activated chloride channels (CaCCs) has been reported in various types of cells. They exert diverse roles, including epithelial secretion of electrolytes and smooth muscle cell contraction 14. In the peripheral nervous system in particular, CaCCs have been ascribed to setting the membrane potential and membrane depolarization via chloride efflux 15, 16. Since the identification of TMEM16A (also called anoctamin‐1) as a CaCC 17, 18, 19, TMEM16A and TMEM16B are accepted as CaCCs. TMEM16A and TMEM16B have functional similarities and differences. Both share the same anionic selectivity and show a strong outward rectification, and they are activated by both membrane depolarization and cytosolic Ca2+ elevation 20. However, these channels display distinct kinetics for activation and deactivation, and have differential affinity for Ca2+ and contrasting response to phosphatidylinositol 4,5‐bisphosphate 21, 22, 23.
In the central nervous system (CNS), the functional role of TMEM16B has been characterized in various types of neurons in different brain areas—olfactory neurons, photoreceptor cells, hippocampal CA1 pyramidal neurons, thalamocortical neurons, and inferior olive neurons 24, 25, 26, 27, 28. In contrast, the role of TMEM16A in the CNS has not been elucidated, although a few studies have demonstrated its function in sensory neurons 15, 29, 30, 31, 32. The presence of CaCC activity in the mHb neurons was first reported more than two decades ago 11, 33, and the mRNA expression of TMEM16A in mHb was also recently reported 13, 34. However, the functional role of TMEM16A in the mHb is still unknown. In this study, we identified TMEM16A as a CaCC in the mHb cholinergic neurons. Using electrophysiological, biochemical, genetic, and chemogenetic approaches, as well as behavioral studies, we demonstrated that TMEM16A deficiency in the mHb has anxiogenic effects.
Results
Electrophysiological examination of the CaCC activity in mHb neurons
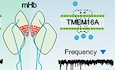
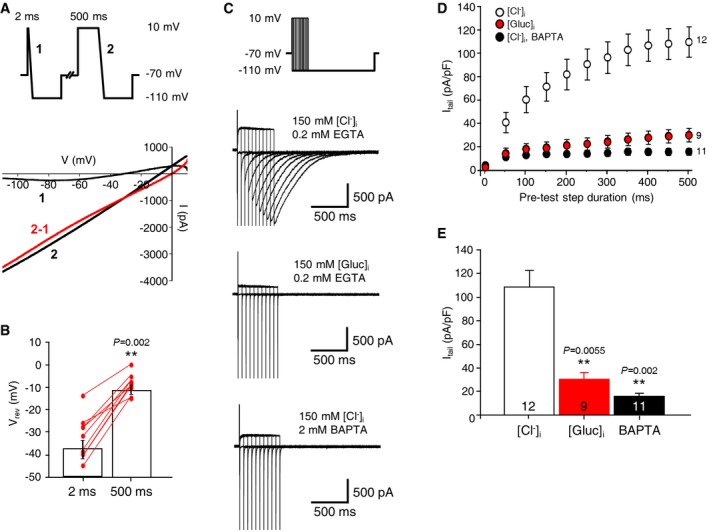
To examine the activity of CaCCs in the mHb in acute brain slices, we measured the currents elicited by a voltage step protocol with a high‐chloride (150 mM) pipette solution 26. The holding potential was stepped from −70 to +10 mV for 2 or 500 ms followed by a 100‐ms ramp to −110 mV (Fig 1A, top panel). A long depolarization step (500 ms) evoked large chloride currents compared to the negligible ones caused by a short step (2 ms) (Fig 1A, bottom panel). Injection of a long depolarization voltage step displayed a linear I–V curve, and it also shifted the reversal potential for chloride to the positive direction (Figs 1B and EV1A). In addition, the voltage pulse protocol of variable durations elicited the inward tail currents, as shown in Fig 1C (top trace). The amplitude and duration of the tail currents were positively correlated with the duration of the injected voltage steps. Chloride replacement with gluconate or the presence of a high‐affinity calcium chelator, BAPTA (2 mM), in the pipette solution largely abolished the tail currents (middle and bottom traces in Fig 1C−E). Furthermore, pre‐incubation (2−5 min) in chloride channel blockers (NPPB, DIDS, or NFA) also greatly reduced the tail currents of the mHb neurons (Fig EV1B) 14. These results strongly suggested the presence of CaCCs in the mHb neurons.
Figure 1. Functional expression of CaCCs in mHb neurons.
- The reversal potential of currents was positively shifted when a long voltage ramp pulse followed the depolarizing steps. (top panel) The voltage protocol used for the experiment. (bottom panel) Representative traces of currents elicited by voltage ramp pulses are shown. (top panel) The red line represents the arithmetical difference between currents elicited by 500‐ms (2) and 2‐ms pulses (1).
- The summary bar graph represents the mean values of the reversal potentials of currents. (nine cells from three mice, paired Student's t‐test; data are presented as mean values ± s.e.m).
- Tail currents elicited by the hyperpolarizing voltage steps following the depolarizing steps of variable durations. Representative traces of currents elicited by the voltage protocol with a pipette solution containing 150 mM Cl− and 0.2 mM EGTA (top panel), 150 mM gluconate (middle panel), or 2 mM BAPTA (bottom panel).
- A summary graph of averaged tail current amplitudes as a function of the depolarizing step duration (the numbers on the right indicate the numbers of recorded cells, data are presented as mean values ± s.e.m.).
- A bar graph of averaged values of the tail current amplitudes summarizing the results of (C) at 500 ms. (The numbers inside each bar indicate the numbers of recorded cells from at least three mice, unpaired Student's t‐test; data are presented as mean values ± s.e.m.).
Figure EV1. CaCC blockers inhibited CaCC‐mediated tail currents of the mHb neurons.
- The averaged I–V curve of currents elicited by the voltage ramp following a 500‐ms depolarizing step after subtracting currents elicited by the voltage ramp following a 2‐ms depolarizing step as shown in Fig 1A (all data are presented as mean values ± s.e.m.; n = 9).
- (top panel) Tail currents elicited by a −100‐mV hyperpolarizing voltage step were blocked by CaCC blockers. Representative traces showed the currents elicited by hyperpolarizing voltage step following the 500‐ms depolarizing step before and during applying DIDS (100 μM), NPPB (100 μM), or NFA (100 μM). (bottom panel) A summary bar graph for the inhibitory effects of CaCC blockers on the tail currents (top panel). Numbers inside bars indicate the number of patched cells. (unpaired Student's t‐test).
TMEM16A is expressed in medial habenula
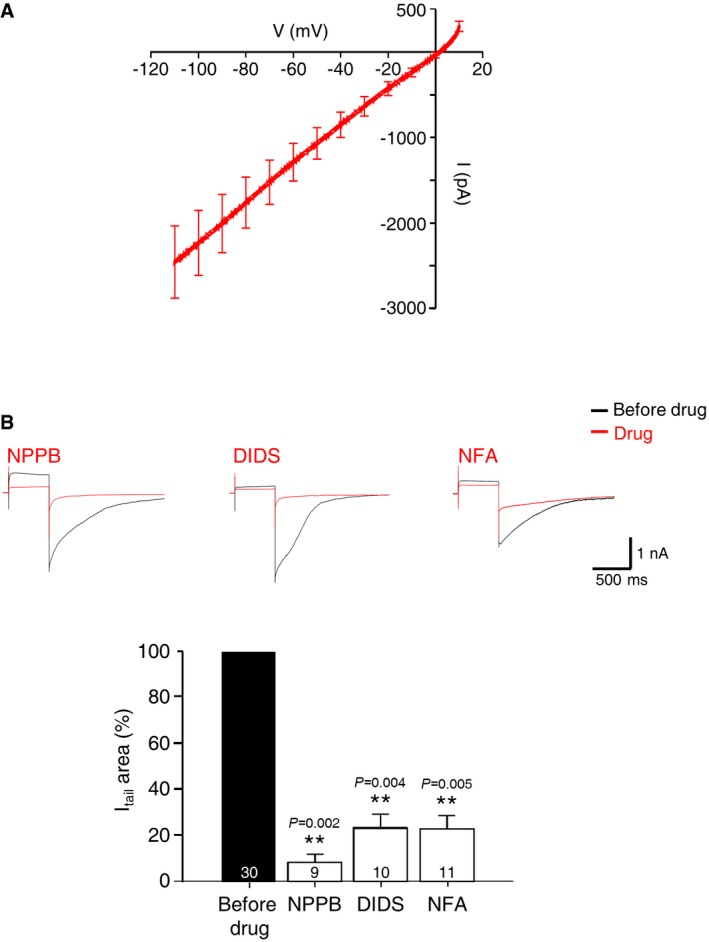
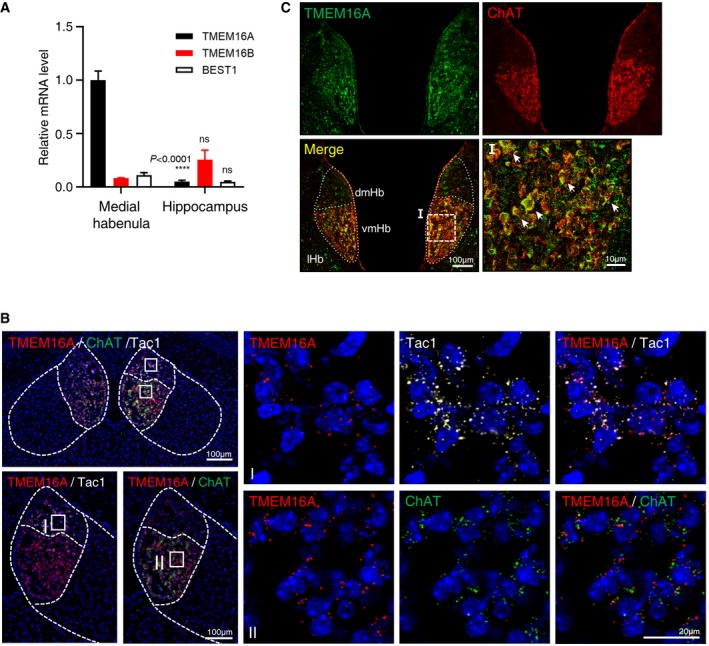
Next, we attempted to identify the CaCCs in mHb neurons using quantitative real‐time PCR with cDNA pools prepared from dissected mHb tissue samples, using specific sets of primers for TMEM16A, TMEM16B, and BEST1, which is a different type of CaCC 26, 35. As shown in Fig 2A, TMEM16A expression was high in the mHb compared to that of TMEM16B and BEST1. This result is consistent with that of previous studies showing TMEM16A mRNA expression in the mHb and poor activity of TMEM16A in the hippocampus 13, 26, 34. To verify the cellular expression of TMEM16A in the mHb, we performed RNAscope in situ hybridization (ISH). As shown in Fig 2B, the TMEM16A signal was restricted to the mHb, with no signal in the lateral habenula (lHb). The TMEM16A signal was broadly present in the mHb, overlapping with the signal of choline acetyltransferase (ChAT), a cholinergic neuronal marker in the ventral part of the mHb, and Tac1 (tachykinin precursor 1), a substance P‐positive neuronal marker in the dorsal part of the mHb 7. This expression pattern is also consistent with the images of the TMEM16A signals observed in the Allen Brain Atlas database (http://mouse.brain-map.org/; Fig EV2A). In addition, immunohistochemistry with anti‐TMEM16A and anti‐ChAT antibodies in coronal slices containing the mHb revealed that TMEM16A colocalized with ChAT in the ventral part of the mHb (Fig EV2B), which is consistent with the RNAscope in situ data (Fig 2C). In contrast, TMEM16A expression was not detected in four other brain regions (cortex, hippocampus, amygdala, and thalamus) (Fig EV2C). Taken together, these data strongly suggested that TMEM16A is specifically expressed in the mHb neurons.
Figure 2. Specific expression of TMEM16A in the mHb neurons.
- A summary bar graph showing the quantitative real‐time PCR data for TMEM16A, TMEM16B, and BEST1 with cDNA samples prepared from the mHb and hippocampus of adult mouse brains (N = 3, n = 3 each, unpaired Student's t‐test, data are presented as mean values ± s.e.m.).
- RNAscope ISH showed an apparent expression of TMEM16A mRNA colocalized with ChAT and Tac1 signals in the mHb compared to the ones in lHb. Enlarged images showed that signals of TMEM16A and Tac1 in the dorsal mHb or ChAT in the ventral mHb reside together at the single‐cell level.
- Immunohistochemical staining of 40‐μm coronal brain sections with anti‐TMEM16A antibody. In the mHb, TMEM16A signals colocalize with the cholinergic marker ChAT (arrows). High magnifications of the indicated areas are shown in the merged panel.
Figure EV2. Specific expression of TMEM16A in the mHb neurons.
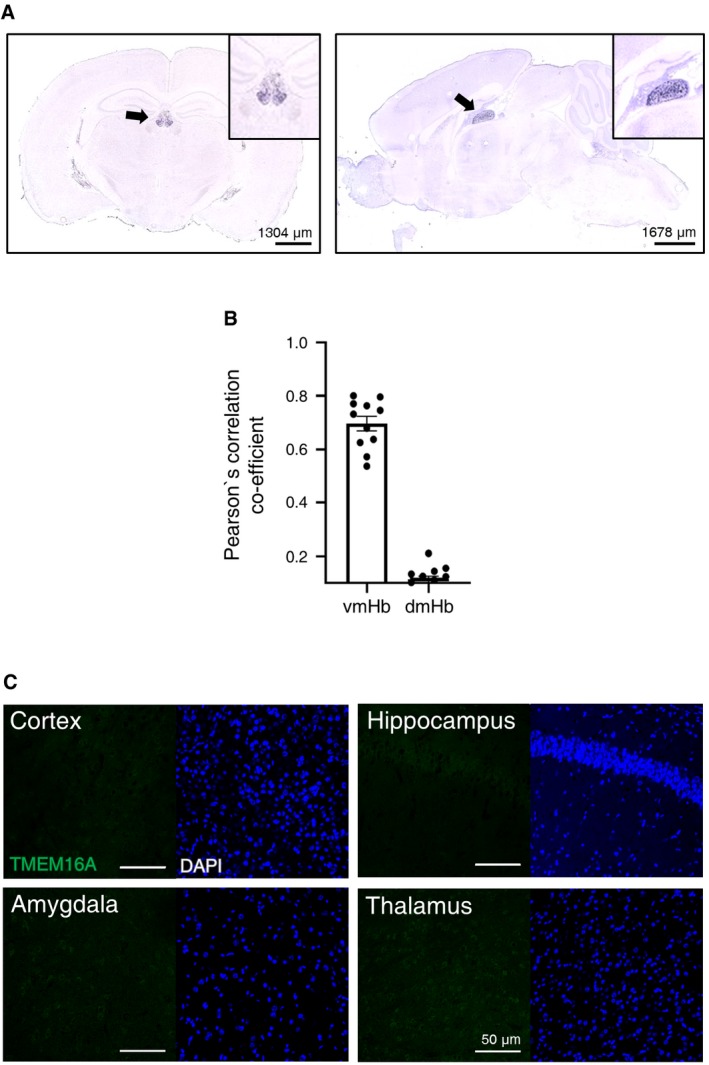
- Images showing expression of TMEM16A by ISH on brain slices (left panel—coronal section and right panel—sagittal section) with specific TMEM16A probes from the Allen Brain Atlas (http://portal.brain-map.org/). Insets are enlarged images of the mHb. Arrows indicate mHb.
- Quantitative analysis of the colocalization between TMEM16A and ChAT signals using Pearson's correlation co‐efficient in 11 images. Data are presented as mean values ± s.e.m.
- Immunohistochemical staining of different brain areas with anti‐TMEM16A antibody showing the absence of TMEM16A signals in the cortex, amygdala, hippocampus, and thalamus. Note that immunohistochemistry experiments were performed under the same conditions shown in Fig 2C. DAPI staining is shown in blue.
TMEM16A deficiency in the cholinergic neurons reduced the spontaneous firing in the mHb
Next, we used the Cre/lox genetic strategy to delete TMEM16A specifically in the cholinergic neurons of the mHb by crossing ChAT‐Cre and TMEM16A floxed mice (Fig EV3A) 15. Immunohistochemistry with an anti‐TMEM16A antibody showed the absence of TMEM16A signals in the ventral part of the mHb in these conditional knockout (cKO) mice compared to the littermate controls (Fig 3A). To rule out the possibility of TMEM16A expression in cholinergic neurons in other brain regions, we also examined the TMEM16A expression in ChAT‐cre(+)/tdTomato mice 36. The immunohistochemical results showed that TMEM16A solely overlapped with ChAT(+) cells in the mHb, indicating that all defects observed in these cKO mice can be attributed to defects in the mHb (Fig EV3B).
Figure EV3. Conditional knockout mice of TMEM16A in the mHb.
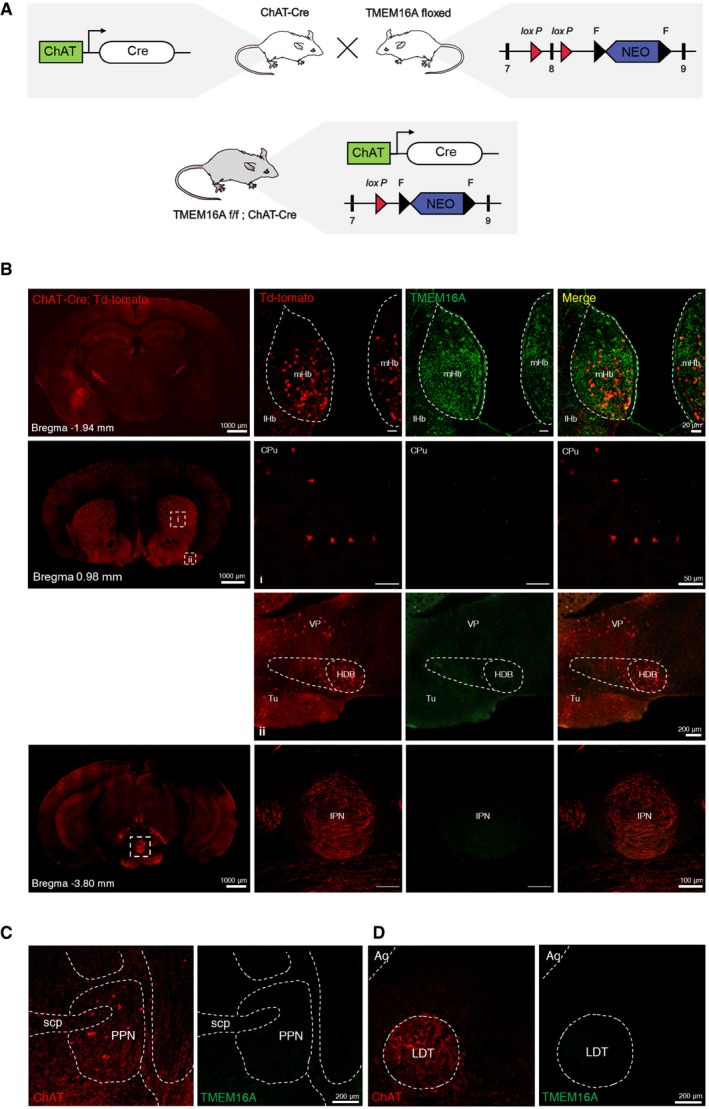
-
AA schematic diagram showing the strategy for generating the TMEM16A cKO mouse.
-
BImmunohistochemical staining on 40‐μm coronal brain sections of the ChAT‐cre(+)::tdTomato mouse using an anti‐TMEM16A antibody. The TMEM16A signals in the mHb of ChAT‐cre(+)::tdTomato mice overlap with tdTomato signals (top panel). Note that other brain regions with intense ChAT(+) signals lack the TMEM16A signal.
-
C, DImmunohistochemical staining on 40‐μm coronal brain sections containing the (C) pedunculopontine nucleus (PPN) and (D) laterodorsal tegmental nucleus (LDT) of the naive mouse using anti‐TMEM16A and anti‐ChAT antibody. Note that there is no TMEM16A signal colocalized with intense ChAT(+) signals.
Figure 3. TMEM16A deficiency reduced the frequency of spontaneous firing of the mHb neurons.
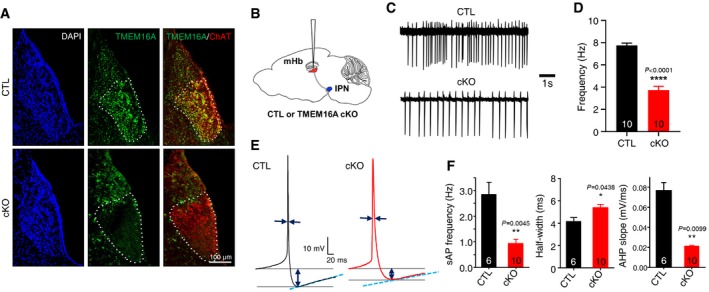
- Immunohistochemical staining of 40‐μm coronal brain sections with an anti‐TMEM16A antibody. The TMEM16A signals in the mHb of TMEM16A cKO mice were compared to the ones of littermate controls. TMEM16A colocalized with the cholinergic marker ChAT in the mHb. The specific deletion of TMEM16A in the mHb is evident.
- An illustration of a brain slice (sagittal view) showing the site of electrophysiological recordings.
- Representative traces demonstrating the spontaneous activity of the mHb neurons in a cell‐attached mode from TMEM16A cKO mice compared to the one from littermate controls.
- A summary bar graph showing the frequencies of spontaneous firing of the mHb neurons in TMEM16A cKO and littermate controls (the numbers inside each bar indicate the numbers of recorded cells from 4 mice each, paired Student's t‐test, data are presented as mean values ± s.e.m.).
- Representative averaged traces of sAPs of the mHb neurons with the pipette solution (40 mM Cl− and 0.5 mM EGTA) from TMEM16A cKO mice and littermate controls.
- Summary bar graphs showing the frequency, half‐width, and AHP slope of the sAPs of mHb neurons from TMEM16A cKO mice and littermate controls (the numbers inside each bar indicate the numbers of recorded cells, unpaired Student's t‐test, data are presented as mean values ± s.e.m.).
Using these TMEM16A cKO mice, we examined the contribution of TMEM16A in the spontaneous firing of the mHb neurons (Fig 3B−D). First, we performed cell‐attached recordings of the mHb neurons to avoid disrupting the intracellular milieu. The neurons in the ventral part of the mHb from control mice displayed spontaneous firings at the range of 5.5−10.4 Hz 11, whereas the mHb neurons from TMEM16A cKO mice showed reduced frequency of spontaneous firing. This result indicated that TMEM16A plays an excitatory role in the mHb neurons. To confirm this finding, we also tested the effect of T16Ainh‐A01, a specific inhibitor of TMEM16A, on the spontaneous firing of mHb neurons (Fig EV4A). T16Ainh‐A01 (50 μM) was bath‐applied to the recording of mHb neurons to study whether TMEM16A was active in the mHb 11, 13, 37. The frequency of spontaneous firings of the mHb neurons was significantly reduced by T16Ainh‐A01 (the averaged frequency was 3.5 ± 0.6 Hz and 1.9 ± 0.6 Hz before and during T16Ainh‐A01 application, respectively; Fig EV4A). In addition, another TMEM16A inhibitor, CaCCinh‐A01 (30 μM), also reduced the frequency of spontaneous firing of the mHb neurons (Fig EV4B). The excitatory role of TMEM16A has been reported in dorsal root ganglia, where [Cl−]in is relatively higher than that in typical neurons due to the lack of KCC2 expression 15, 38. In addition, activation of the GABAA receptors in the mHb increases the firing of action potential 39. Consistent with previous reports 39, 40, when we performed immunohistochemistry with a specific antibody against KCC2, no KCC2 signal was detected in the mHb (Fig EV4C). Therefore, the excitatory role of TMEM16A in the mHb may result from the relatively higher [Cl−]in due to lack of KCC2 expression in this structure.
Figure EV4. TMEM16A functions as an excitatory channel in the mHb cholinergic neurons.
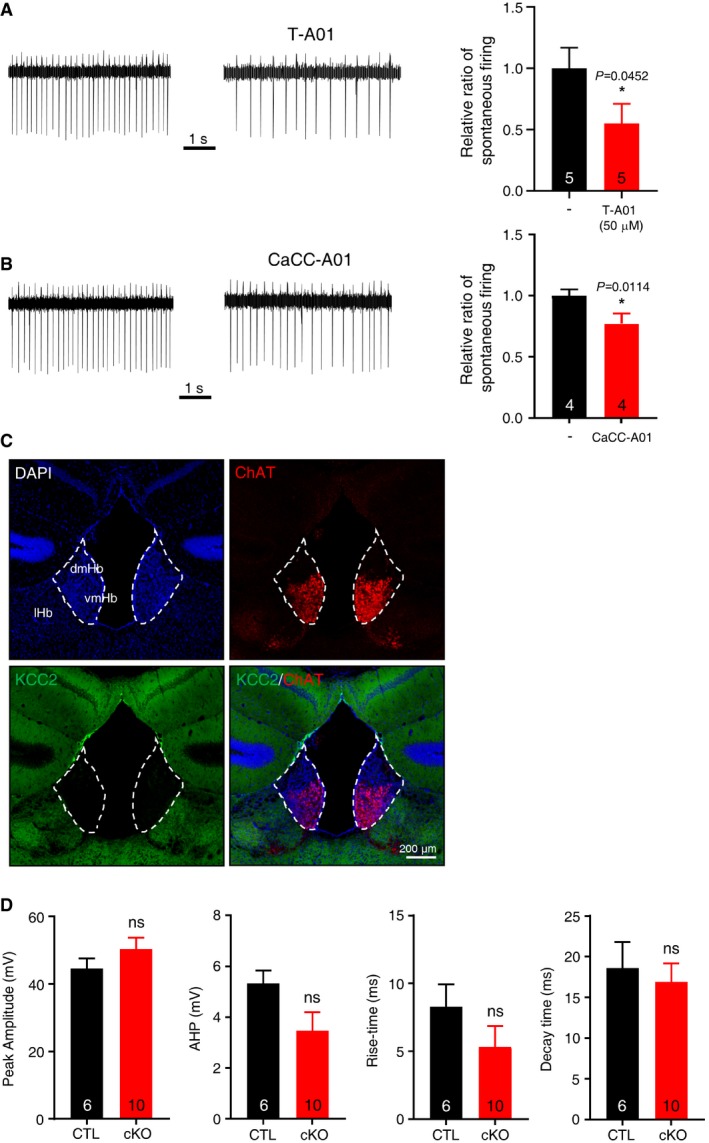
-
A, B(left panel) Representative traces demonstrating that the spontaneous activity of the mHb neurons in cell‐attached recordings was reduced in T‐A01 (A) and CaCC‐A01 (B). (right panel) Summary bar graphs showing the normalized frequencies of spontaneous firing of the mHb neurons before and during application of T‐A01 (A) or CaCC‐A01 (B). Data are presented as mean values ± s.e.m. *P < 0.05.
-
CIHC on mHb with anti‐KCC2 antibody displayed lack of KCC2 signal in mHb.
-
DSummary bar graphs showing the peak amplitude, AHP amplitude, rise time (10–90%), and decay time (10–90%) of the sAPs of mHb neurons from TMEM16A cKO and littermate controls.
Next, we examined how TMEM16A regulates the spontaneous action potentials (sAPs) of mHb neurons in a whole‐cell current‐clamp mode (Figs 3E and F, and EV4D). The mHb neurons from TMEM16A cKO mice showed a reduced firing frequency; TMEM16A ablation decreased the slope of after‐hyperpolarization (AHP) and increased the half‐width duration of the action potential compared to those of the littermate controls (Fig 3E and F). There was no difference in the resting membrane potential and cell capacitance of the mHb neurons between TMEM16A cKO and control mice (Vrmp: cKO—−42.37 ± 1.57 mV and CTL—−42.27 ± 1.00 mV, P = 1; Cm: cKO—11.92 ± 0.68 pF and CTL—11.49 ± 0.90 pF, P > 0.01) 41. Therefore, we concluded that TMEM16A regulates the excitability of the mHb neurons by contributing to the decay slope of AHP.
Reduced activity of IPN neurons in TMEM16A cKO mice
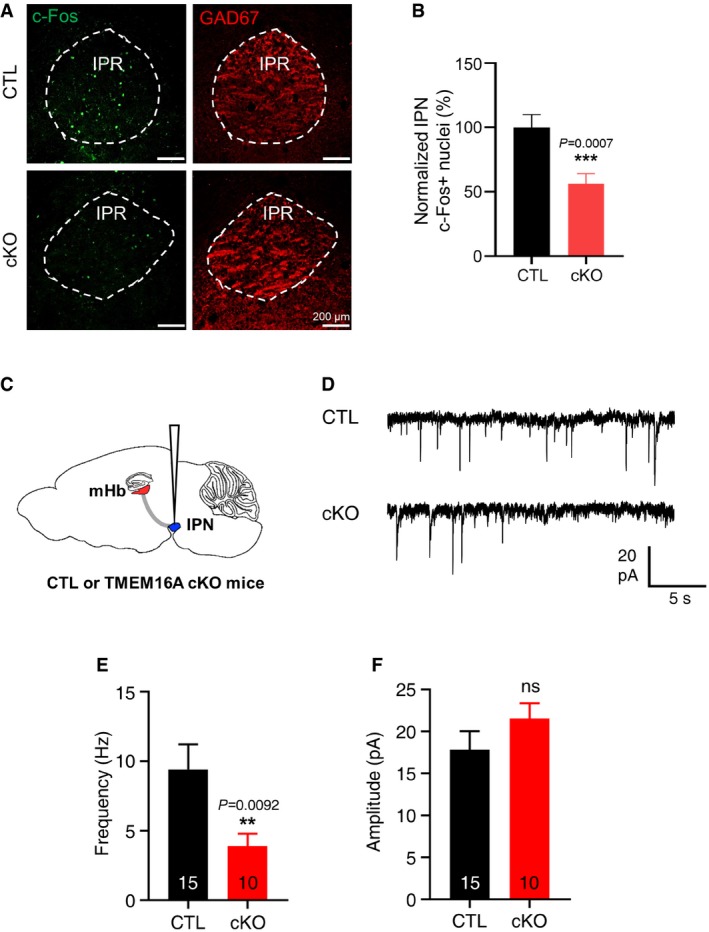
Because TMEM16A regulates the spontaneous firings of mHb neurons as shown above, we hypothesized that modulating the TMEM16A activity in the mHb could affect the neuronal firings of the IPN, a major downstream target of the mHb 42. Therefore, we examined the neuronal activity of the IPN in TMEM16A cKO and control mice by staining for c‐Fos, an immediate early gene product (Fig 4A and B). The results showed that TMEM16A cKO mice had a reduced number of c‐Fos(+) cells compared to that in controls. In addition, we also measured the spontaneous excitatory postsynaptic currents (sEPSCs) of the IPN neurons (Fig 4C and D). The frequency, but not the amplitude, of sEPSCs of neurons in the medial part of the IPN was significantly lower in TMEM16A cKO mice than that in littermate controls (Fig 4E and F). These data indicated that reduced mHb inputs resulted in reduced neuronal activity in the IPN of TMEM16A cKO mice. Therefore, we concluded that TMEM16A cKO mice have a reduced neuronal activity in the mHb‐IPN circuit. This led us to investigate the phenotypic implications of the reduced activity of this circuit in TMEM16A cKO mice.
Figure 4. TMEM16A deficiency reduced the frequency of spontaneous firing of the IPN neurons.
-
AImmunohistochemical images of the IPN region from control and TMEM16A cKO mice. The c‐Fos signal was greatly reduced in TMEM16A cKO mice. Scale bar = 200 μm.
-
BA summary bar graph showing reduced c‐Fos expression in the IPN in TMEM16A cKO mice. (N CTL = 7 (n = 42), N cKO = 6 (n = 36), unpaired Student's t‐test, data are presented as mean values ± s.e.m.).
-
CAn illustration of a brain slice (sagittal view) to show the site of electrophysiological recordings.
-
DRepresentative traces of sEPSCs of neurons in the medial part of the IPN showing a reduced frequency in TMEM16A cKO mice compared to that of littermate controls.
-
E, FSummary bar graphs of the frequency and amplitude of the sEPSCs of IPN neurons in TMEM16A cKO (cKO) mice and littermate controls (CTL). Data are presented as mean values ± s.e.m. (paired Student's t‐test). The numbers inside each bar indicate the numbers of recorded cells.
TMEM16A cKO mice showed anxiogenic behaviors
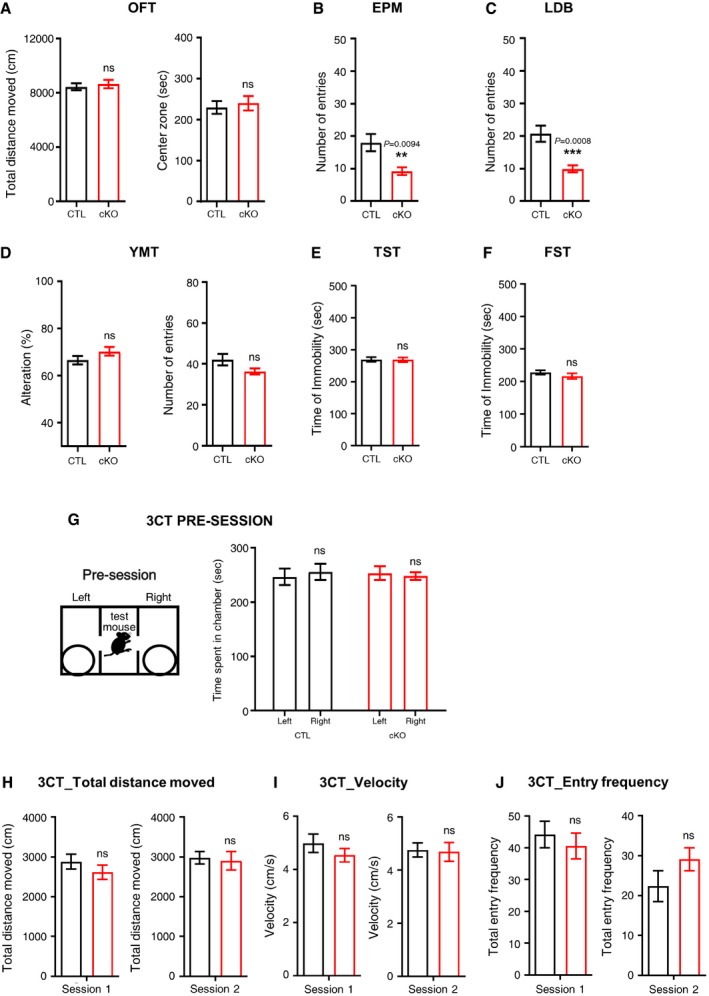
In addition to the functional significance of TMEM16A and its contribution to the neuronal mechanism in the mHb neurons, we studied whether the mHb‐related phenotypes were affected by the cholinergic neuron‐specific depletion of TMEM16A. General locomotion was assessed in an open field, and the total distance moved and time spent in the center of the chamber were similar between TMEM16A cKO mice and littermate controls (Fig EV5A).
Figure EV5. TMEM16A cKO mice displayed no differences in various basal behaviors.
-
AOpen‐field test of the CTL and the cKO group showed similar patterns in locomotor activity.
-
BY‐maze test for both groups revealed normal cognitive functions.
-
CDuring the TST, the duration of immobility was similar between the CTL and cKO groups.
-
DDuring the FST, the duration of immobility was similar between the CTL and cKO groups.
-
EIn the EPM test, the number of entries to the open arms of cKO mice was reduced compared to that of CTL mice.
-
FThe number of entries to the light zone was reduced in cKO in the LDB test.
-
GThe time spent in each chamber was presented in the pre‐session.
-
H–JIn the 3CT task, the total distance (H), velocity (I), and total entry frequency (J) of the CTL and the cKO groups during both sessions were similar. (for all 3CT, n CTL = 12, n cKO = 14) Unpaired Student's t‐test; data are presented as mean values ± s.e.m, **P < 0.01, ***P < 0.001.
To investigate whether anxiety‐related behaviors were altered in TMEM16A cKO mice, we used the elevated plus maze (EPM) and the light–dark box (LDB) tasks 43, 44. We found that TMEM16A cKO mice displayed significantly reduced the reentering time and number in open arms compared to that in littermate control mice in the EPM task (Figs 5A and EV5B). In the LDB task, TMEM16A cKO mice displayed significantly reduced time and number in the light area than the littermate controls (Figs 5B and EV5C). The results of these two tasks indicate that TMEM16A cKO mice display anxiogenic behaviors.
Figure 5. TMEM16A cKO mice displayed anxiety‐like behaviors and impairments in social interaction.
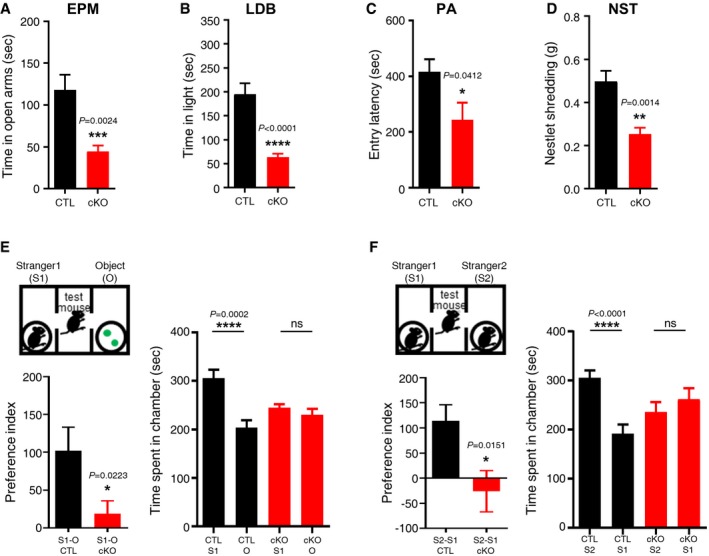
- In the EPM task, the time spent in the open arms was significantly decreased in cKO mice compared to that in the control (CTL) group (n CTL = 20, n cKO = 12, unpaired Student's t‐test).
- In the LDB task, the time spent in the light zone by cKO mice was significantly decreased compared to that spent by the CTL mice (n CTL = 20, n cKO = 12, unpaired Student's t‐test).
- In the PA task, the entry latency of cKO mice was significantly decreased compared to that of the CTL mice (n CTL = 20, n cKO = 12, unpaired Student's t‐test).
- Nestlet shredding behaviors of the cKO group were also decreased compared to those of the CTL group. The weight of the shredded nestlets of the cKO group was significantly lower than that of the CTL group (n CTL = 20, n cKO = 12, unpaired Student's t‐test).
- In the 3‐chamber test, the cKO mice exhibited a decreased interest for the stranger mouse (S1) against objects (O) compared to the CTL group (n CTL = 12, n cKO = 14, unpaired Student's t‐test).
- When cKO mice were exposed to a new stranger mouse (S2) against the familiar S1 mouse, they also showed a decreased interest for the S2 mouse compared to that of the CTL group. The preference index showed the decreased sociability of the cKO group in both sessions (n CTL = 12, n cKO = 14, unpaired Student's t‐test).
In addition, we performed the passive avoidance (PA) test since the behavioral response of PA has been reported to be affected by anxiety 45, 46. As shown in Fig 5C, TMEM16A cKO mice showed a significantly reduced entry latency from the light chamber to the dark chamber compared to that of the littermate controls. Since the PA task is also relevant to the evaluation of cognitive functions 45, we performed the Y‐maze task, a cognitive‐behavioral test, to investigate whether TMEM16A cKO mice displayed signs of cognitive impairment 47. Interestingly, this task revealed similar results between TMEM16A cKO and littermate controls, supporting the fact that cognitive functions are not altered in TMEM16A cKO mice (Fig EV5D). Overall, our results indicated that TMEM16A cKO mice displayed anxiogenic behavior more than littermate controls without cognitive impairments.
Depression is also closely associated with anxiety in human and rodent models 48. The forced swimming task (FST) and tail suspension task (TST) were performed to examine whether TMEM16A cKO mice had despair‐like phenotypes. The results showed that there was no difference in the duration of immobility between the TMEM16A cKO mice and the littermate controls (Fig EV5E and F). These results led us to conclude that TMEM16A cKO mice displayed anxiogenic behaviors without despair‐like symptoms, as found in other studies of mutant mice 49.
A close relationship has been described between levels of anxiety and social behaviors, not only in human but also in rodents 50, 51. Therefore, we also performed the nestlet shredding task (NST) and the 3‐chamber social interaction task (3‐CT) to examine the sociality‐related behaviors in TMEM16A cKO mice. In the NST, TMEM16A cKO mice showed less shredding activity than that of the littermate controls (Fig 5D). In the 3‐CT, both TMEM16A cKO and littermate control mice spent almost equal time either on the left or on the right side of the central chamber before the session started (Fig EV5G–J). In the sociability session, when a stranger mouse (S1) and plastic objects were placed in each chamber, TMEM16A cKO mice showed no preference between the two chambers, while littermate controls displayed a clear preference for the stranger mouse (S1) compared to the plastic objects (session 1, Fig 5E). In the social novelty session, TMEM16A cKO mice showed a decreased interest in the novel stranger mouse (S2) over the familiar one (S1) compared to littermate controls, which displayed more interest in the new partner (session 2, Fig 5F). The preference index—the difference between the two values of the time spent in each chamber—was significantly decreased in the TMEM16A cKO compared to littermate controls (Fig 5E and F). The number of c‐Fos(+) cells in the IPN of TMEM16A cKO mice was still reduced compared to that in littermate controls even after the behavioral experiments (Appendix Fig S1).
To further confirm the specific role of TMEM16A in the mHb neurons, we expressed Cre recombinase under hSyn promoter (neuron‐specific promoter) by stereotaxic injection of adeno‐associated virus (AAV‐hSyn‐cre) into the mHb of homozygotic TMEM16A floxed (f/f) mice (AAV‐hSyn‐cre::TMEM16A mice, Appendix Fig S2A). Three weeks after the AAV injection, proper AAV injections and loss of TMEM16A expression in the mHb were confirmed by fluorescent imaging (Appendix Fig S2B). The reduced c‐fos activity in the IPN of AAV‐hSyn‐cre::TMEM16A mice was comparable with the reduced mHb‐IPN circuit activity in TMEM16A cKO mice (Appendix Fig S2C and D). In the behavioral experiments, compared to mice that received control AAV (AAV‐hSyn‐mCherry), AAV‐hSyn‐cre::TMEM16A mice exhibited the same behavioral alterations as did the TMEM16A cKO mice (Appendix Fig S2E–K). These results indicated that silencing TMEM16A in the mHb not only during development but also at adult stages induced the same behavioral phenotypes. Taken together, the results of our behavioral tests indicated that TMEM16A cKO mice displayed anxiogenic behaviors and impaired social behaviors.
Chemogenetic inhibition of the cholinergic neurons in the mHb induced anxiogenic behaviors
As demonstrated above, the reduced activity of the cholinergic neurons in the mHb via TMEM16A silencing caused anxiogenic behaviors and impaired social interactions. To examine whether the specific inhibition of mHb activity directly mediated anxiogenic phenotypes, we used DREADD (Designer Receptors exclusively activated by Designer Drugs) technology 52. To reduce the activity of the cholinergic neurons in the mHb, we injected AAV‐hSyn‐DIO‐hM4D(Gi)‐mCherry, an inhibitory DREADD, into the mHb of adult ChAT‐cre mice (ChAT‐cre::hM4D(Gi)) and confirmed its expression in the mHb by fluorescent imaging (Fig 6A and Appendix Fig S3A). Two to three weeks after the virus injection, we first examined the effect of DREADD on the mHb activity (Fig 6B and C). The frequency of spontaneous firing was significantly reduced when clozapine‐N‐oxide (CNO, 10 μM), a selective ligand for hM4D(Gi), was bath‐applied. This suggested that DREADD‐mediated inhibition (or relative silencing) of the excitability of cholinergic neurons in the mHb can be achieved with CNO treatment, and this technology can be used in combination with behavioral tests (Fig 6D–I).
Figure 6. DREADD‐mediated inhibition of the mHb neurons in mice revealed anxiety‐like behaviors and impairment in social interactions.
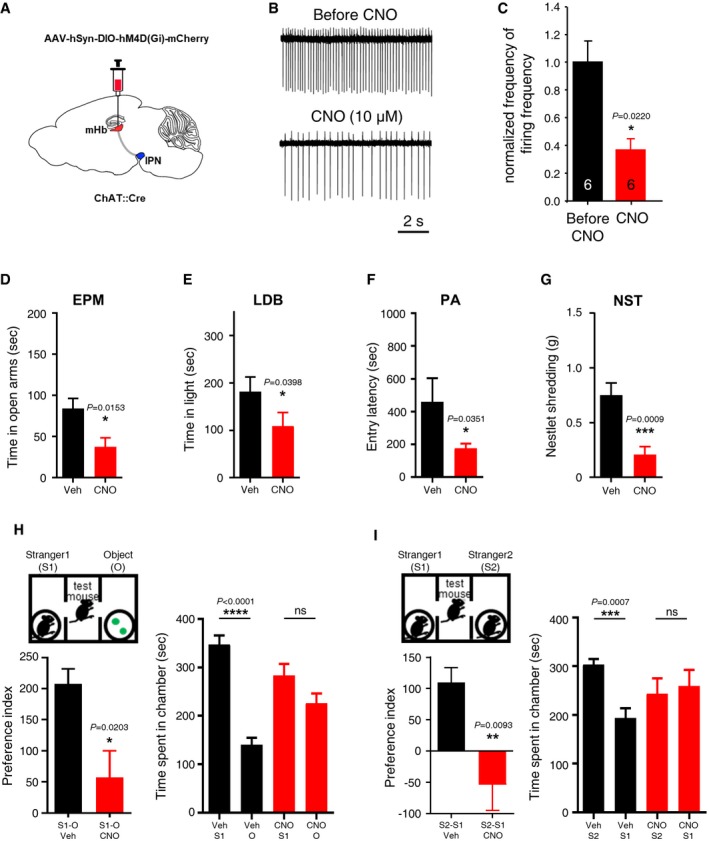
- An illustration of a brain slice (sagittal view) of ChAT(+)‐cre mouse showing the site of AAV‐hM4D(Gi)‐mCherry injection.
- Representative traces showing that the spontaneous firing of mHb neurons from ChAT(+)‐cre::hM4D(Gi) mice were reduced when CNO (10 μM) was applied in the ACSF.
- A summary bar graph showing the reduction of normalized frequencies of spontaneous firing of the mHb neurons transfected with AAV‐hM4Di‐mCherry before and during CNO treatment (the numbers inside each bar indicate the numbers of recorded cells).
- In the EPM task, the time spent in the open arms of CNO‐treated ChAT(+)‐cre::hM4D(Gi) mice was significantly decreased compared to that of the PBS‐treated controls (CTL). (n CTL = 6, n cKO = 9, unpaired Student's t‐test).
- In the LDB task, the time spent in the light zone by CNO‐treated ChAT(+)‐cre::hM4D(Gi) mice was significantly decreased compared to that by the PBS‐treated control mice. (n CTL = 6, n cKO = 9, unpaired Student's t‐test).
- In the PA task, the entry latency of CNO‐treated mice was significantly decreased compared to that of the CTL group. (n CTL = 6, n cKO = 9, unpaired Student's t‐test).
- Nestlet shredding behaviors of the CNO‐treated ChAT(+)‐cre::hM4D(Gi) mice were also decreased. (n CTL = 6, n cKO = 9, unpaired Student's t‐test;).
- In the 3‐chamber test, the subject mouse of the PBS‐treated mice and the CNO‐treated mice explored freely in the test chambers. The time spent in each chamber during two sessions was measured. The CNO‐treated ChAT(+)‐cre::hM4D(Gi) mice exhibited a decreased interest for a stranger mouse (S1) against objects (O) compared to the PBS‐treated group (left two panels) (n CTL = 6, n cKO = 9, unpaired Student's t‐test).
- When CNO‐treated ChAT(+)‐cre::hM4D(Gi) mice were exposed to a new stranger mouse (S2) against the familiar S1 mouse, they also showed a decreased interest for the S2 mouse compared to the PBS‐treated control group (right two panels). The preference index showed decreased sociability of the CNO‐treated group in both sessions (n CTL = 6, n cKO = 9, unpaired Student's t‐test).
In the behavioral experiments, we found that there was no difference in the general locomotion between the CNO‐treated and PBS‐treated groups (Appendix Fig S3B). In the EPM task, the CNO‐treated ChAT‐cre::hM4D(Gi) mice had significantly decreased reentering time and number in the open arms compared to that of the PBS‐treated ChAT‐cre::hM4D(Gi) mice (Fig 6D and Appendix Fig S3C). In the LDB task, the CNO‐treated ChAT‐cre::hM4D(Gi) mice had significantly decreased time and number in the light area than the PBS‐treated ChAT‐cre::hM4D(Gi) mice (Fig 6E and Appendix Fig S3D). These results are consistent with the anxiogenic phenotype of TMEM16A cKO mice described above (Figs 5A and EV5B, and Figs 5B and EV5C).
In the PA task, CNO‐treated ChAT‐cre::hM4D(Gi) mice showed a significantly decreased entry latency compared to that of PBS‐treated ChAT‐cre::hM4D(Gi) mice, despite the condition of shock punishment in the dark zone (Fig 6F). These data are consistent with the results of the featured phenotypes in TMEM16A cKO mice (Fig 5C). In the NST, CNO‐treated ChAT‐cre::hM4D(Gi) mice showed less shredding activity than that of the PBS‐treated controls (Fig 6G). In the 3CT, CNO‐treated ChAT‐cre::hM4D(Gi) mice displayed social behavioral deficits similar to the ones observed in TMEM16A cKO mice (Fig 6H and I). Both PBS‐treated and CNO‐treated ChAT‐cre::hM4D(Gi) mice showed no preference for either side of the central chamber in the 3‐CT (Appendix Fig S3E). These data are consistent with the results of the featured phenotypes in TMEM16A cKO mice (Fig 5E and F). In summary, DREADD‐mediated inhibition of the cholinergic neurons in the mouse mHb resulted in anxiogenic phenotypes, similar to those observed in TMEM16A cKO mice.
Discussion
TMEM16A, a CaCC, has been shown to contribute to setting the membrane potential and controlling excitability of diverse cell types 20. Although TMEM16A has been shown to play important functions in peripheral sensory neurons 15, 16, 31, 32, its role in central neurons had not been previously examined. In this study, we found CaCC electrophysiological activity in the mHb neurons using intracellular Ca2+ chelation, chloride replacement with gluconate, and CaCC inhibitors (Figs 1 and EV1). We also demonstrated a specific expression of mRNA and protein TMEM16A in the mHb with quantitative real‐time PCR, RNAscope ISH, and immunohistochemistry (Figs 2 and EV2).
It was noticeable that mHb neurons in which TMEM16A was genetically deleted (TMEM16A cKO mice) had a reduced firing frequency compared to that in control mHb neurons (Fig 3C and D). In addition, we also showed that TMEM16A inhibitors (Tinh‐A01 and CaCCinh‐A01) reduced the frequency of spontaneous firing of the mHb neurons (Fig EV4A and B). These data strongly suggest that TMEM16A is functionally active and plays an excitatory role in the mHb neurons. This is consistent with our previous finding that TMEM16A contributes to depolarization of the dorsal root ganglia 15, 16, 31. In general, activation of the chloride channels can depolarize or hyperpolarize the membrane potential of the cells depending on the [Cl−]i. Therefore, the depolarizing effect of TMEM16A in the mHb neurons may result from the elevated [Cl−]i, due to lack of KCC2 expression (Fig EV4C).
Based on our findings, it is likely that TMEM16A contributes to the decay slope of AHP and this may be the underlying mechanism of TMEM16A‐regulated mHb activity (Fig 3E and F). Therefore, TMEM16A‐ablated cholinergic neurons in the mHb require more time to reinitiate the next firing of action potentials. These phenomena may account for the reduced cholinergic activity of the mHb‐IPN pathway in TMEM16A cKO mice (Fig 4).
In the behavioral experiments using TMEM16A cKO and ChAT‐cre::hM4D(Gi) mice, the reduced activity of the cholinergic neurons in the mHb mediated anxiogenic behaviors (Figs 5 and 6). Previously, the increased signaling of the nicotinic acetylcholine receptor in the mHb cholinergic neurons was reported to regulate nicotine withdrawal‐induced anxiety behavior 5. In addition, it has been recently shown that mHb activation via the posterior septal input induces anxiolysis 53. In this study, we clearly showed that the reduced activity of the mHb cholinergic neurons directly mediates the anxiogenic behaviors (Figs 5A−C, and 6D−F).
Interestingly, Otsu et al 53 reported that stimulating the posterior septal input to the mHb enhanced locomotor activity. Therefore, one could have expected that reducing the mHb activity would cause a decrease in locomotor activity. However, our results showed that TMEM16A cKO mice or CNO‐treated ChAT‐Cre::hM4D(Gi) mice displayed no change in locomotor activity compared to the control mice (Fig EV5A, and Appendix Figs S2E and S3B). Although previous reports have shown that anxiogenic behaviors could be expressed without changes in the locomotor activity 54, we believe that the relationship between locomotor activity and mHb activity needs to be studied further in detail to explain this discrepancy.
The level of anxiety strongly affects the social behaviors 50, 51. Based on our results, it is noteworthy that the functional reduction of the mHb‐IPN circuit induced impairments in social behaviors (Figs 5D–F, and 6G–I). Although the lHb has been implicated in social behaviors 55, 56, 57, the involvement of the mHb in social behaviors has been largely unknown. A previous report showed that Wnt‐Cre and Wnt‐GAL4 double transgenic mice, which have abnormal mHb projections to the IPN, exhibited decreased social interaction and nesting behaviors 58. These observations along with our results suggest that the mHb‐IPN circuit is an important brain region implicated in social behaviors. The DRN has also been shown to be involved in social behaviors and anxiety 59, 60, 61, 62, and it receives afferent inputs from the IPN. Therefore, we suggest that the mHb‐IPN‐DRN pathway may be directly involved in social behaviors, a notion that should be further investigated. In addition, we showed that TMEM16A is specifically expressed in the mHb. Therefore, we propose that TMEM16A could be a novel target for therapeutics against anxiety‐related disorders. It is well known that TMEM16A is activated by Ca2+ influx through activation of the Ca2+‐permeable channels or increased Ca2+ release elicited from the intracellular stores 17, 18, 19. The mHb expresses various membrane proteins (e.g., nicotinic acetylcholine receptor subunits α3/β4 and somatostatin receptor 2) that can increase the [Ca2+]i 13, 34. Thus, nicotine or somatostatin may activate their specific receptors to affect TMEM16A function and regulate the excitability of the mHb cholinergic neurons, which may result in behavioral modifications. Since the activities of TMEM16A can be regulated via protein–protein interactions 63, 64, it will be worth examining whether and how TMEM16A‐interacting proteins may regulate the neuronal excitability of the mHb.
Materials and Methods
Animals
Male C57BL/6 mice, 6–9 weeks old, were used for all experiments. Animal care and handling were performed according to the guidelines of the Institutional Animal Care and Use Committee (IACUC) at the Korea Institute of Science and Technology (Seoul, Korea) and the Korea University (Seoul, Korea). We did not predetermine the sample sizes using statistical methods, but our sample sizes were similar to those previously reported. Furthermore, we did not perform randomization for blinded experiments in this animal study. The mHb‐specific TMEM16A‐knockout mice were bred by crossing ChAT‐Cre (+) (Jackson Laboratory #6410) with floxed TMEM16A f/f mice 15.
Chemicals
Bicuculline methobromide, CGP55845 hydrochloride, D‐AP5, and CNQX were purchased from Tocris, and niflumic acid (NFA), T16Ainh‐A01, CaCCinh‐A01, 5‐nitro‐2‐(3‐phenylpropylamino)benzoic acid (NPPB), disodium 4,4′‐diisothiocyanatostilbene‐2,2′‐disulfonate (DIDS), and clozapine‐N‐oxide (CNO) were purchased from Sigma‐Aldrich. Other reagents were purchased from Sigma‐Aldrich or Calbiochem.
Genotyping for ChAT‐Cre and TMEM16A f/f mice
ChAT‐Cre(+)/TMEM16A f/f mice were genotyped using PCR, and ChAT‐Cre(‐)/TMEM16A f/f mice were used as controls. The primer sequences were as follows: TMEM16A: forward—5′TACTCAGCTGCATAC‐CCTCT3′; reverse—5′ GGCCATAAACACAGAGAAGA‐3′; for ChAT‐Cre: forward—5′ GGTCTCCTTGTGGA GTGGGAGT‐3′; and reverse—5′CGGCAAACGGACAGAAGCATT‐3′.
Real‐time PCR
Microdissection of the mHb was performed as previously described 65. The mHb regions were microdissected with a surgical blade from the coronal slices (100 μm thickness) of frozen brains on a cryostat (CM3050S, Leica) at −20°C. Total RNA was isolated from the mHb and hippocampus using an RNA Purification Kit (GeneAll), according to the manufacturer's instructions. The cDNAs were synthesized from 500 ng total RNA and reverse transcription was performed using SensiFAST™ cDNA Synthesis Kit (Bioline), according to the manufacturer's instructions. Real‐time PCR was performed using SensiFAST™ Probe Hi‐ROX Kit (BIOLINE, London, UK). Primer sets for TMEM16A, TMEM16B, BEST1, and GAPDH were purchased at IDT (PrimeTime qPCR Assays). GAPDH was used as a reference gene. The 2−ΔΔCt method was used to calculate the fold changes in gene expression. All experiments were repeated at least three times.
Real‐time PCR primers and probes
| Gene | Sequence |
|---|---|
| TMEM16A | |
| Forward primer | 5′‐GTGGAGTCATACTGGAACATGTAG‐3′ |
| Probe | 5′‐/56‐FAM/AGAATCCAG/ZEN/AAACAAAGAGACCGACAAGG/3IABkFQ/‐3′ |
| Reverse primer | 5′‐GCCAGAGTCTTAGAGAAGTCAC‐3′ |
| TMEM16B | |
| Forward primer | 5′‐GTGGAGTCATACTGGAACATGTAG‐3′ |
| Probe | 5′‐/56‐FAM/AGAAACATG/ZEN/GTAGCCCACAGAGCC/3IABkFQ/‐3′ |
| Reverse primer | 5′‐CCACCGTCTTCTTTTCCATCT‐3′ |
| BEST1 | |
| Forward primer | 5′‐GGCATCACTCATAGAGAACAGG‐3′ |
| Probe | 5′‐/56‐FAM/AGCATGCAG/ZEN/AGTCCTATCCCTACAGG/3IABkFQ/‐3′ |
| Reverse primer | 5′‐CCCCACAGAACATCTTCAAC‐3′ |
| GAPDH | |
| Forward primer | 5′‐GTGGAGTCATACTGGAACATGTAG‐3′ |
| Probe | 5′‐/56‐FAM/TGCAAATGG/ZEN/CAGCCCTGGTG/3IABkFQ/‐3′ |
| Reverse primer | 5′‐AATGGTGAAGGTCGGTGTG‐3′ |
DREADD‐mediated mHb inhibition with CNO
AAV‐hSyn‐DIO‐hM4D(Gi)‐mCherry (Addgene #44362, serotype 2) were injected into the mHb area of anesthetized adult male ChAT‐cre mice. For the behavioral experiments, CNO (0.5 mg/kg in PBS) or PBS control solution was injected intraperitoneally 2–3 weeks after the stereotaxic injection. For the electrophysiological study, CNO was dissolved in artificial cerebrospinal fluid (ACSF) solution (final concentration 10 μM).
Stereotaxic virus injections
Virus injections were performed in deeply anesthetized, 6‐ to 9‐week‐old mice, placed in a stereotaxic frame (Kopf Instruments). Anesthesia was induced with tribromoethanol (Avertin®, 2,2,2‐tribromoethanol in 2‐methyl 2‐butanol) via an intraperitoneal injection. Briefly, the scalp was opened and two holes were drilled in the skull (−1.8 mm AP from bregma, ±0.5 mm ML). AAV‐hSyn‐mCherry‐cre (UNC vector core, CV‐17053‐AV2), AAV‐hSyn‐mCherry (UNC vector core, CV‐17105‐AV2), or AAV‐hSyn‐DIO‐hM4D(Gi)‐mCherry was bilaterally injected (1 μl per side) into the mHb area (2.7 mm DV from the dura) through a glass microdispenser (VWR/SP Scientific) with a syringe pump (KD Scientific) that infused the virus at a speed of 0.2 μl/min. The microdispenser was left in place for 2 min before and 2 min after the injection. To examine the function of TMEM16A in mHb and modulate the activity of cholinergic mHb neurons, we used AAV‐hSyn‐Cre with homozygotic TMEM16A floxed (f/f) mice and AAV‐hSyn‐DIO‐hM4D(Gi)‐mCherry with ChAT‐Cre mice, respectively. Verification of proper viral injections was examined by fluorescent signals of mCherry expression from AAVs after behavioral experiments.
RNAscope ISH
The RNAscope ISH was performed as follows. Briefly, 8‐week‐old mice were sacrificed and the isolated brains were frozen immediately. Subsequently, the brains were embedded in the optimum cutting temperature compound and sectioned at 14 μm thickness with the coronal axis through the habenula region. Frozen sections were thaw‐mounted onto Superfrost Plus Microscope Slides (Fisher Scientific #12‐550‐15). The RNAscope kit is commercially available from Advanced Cell Diagnostics (ACD, Hayward, CA). The sequence of the ChAT (Cat No. 408731‐C2) and TMEM16A (Cat No. 417201) mRNA target probe was manufactured by ACD. The sections were fixed in 4% paraformaldehyde (PFA) for 10 min; dehydrated in 50%, 75%, 95%, and 100% EtOH for 5 min; and finally air‐dried. Tissues were then treated with pre‐treatment solution for 10 min at room temperature (RT). For RNA detection, the sections were incubated with TMEM16A and ChAT probes for 2 h at 40°C, and then, the different amplifier solutions were added to the tissue following the method from the RNAscope Fluorescent Multiplex Kit (320293). Each amplifier was removed by buffer washing for 2 min at RT. The signals were developed using Alexa Fluor 488 for TMEM16A and Alexa Fluor 550 for ChAT. Hoechst 33343 was used for the nuclei staining. The tissue was washed, and the slides were mounted and viewed with an LSM 700 microscope (Zeiss).
Fluorescence immunohistochemistry and image collection
Mouse brain tissues were obtained following intracardiac perfusion with 4% PFA in PBS solution, and 40‐μm‐thick frozen sections were prepared using a cryostat. The sections were mounted on the slides and permeabilized with 0.5% Triton X‐100 in PBS for 20 min at RT, followed by blocking with 10% donkey serum and 0.1% Triton X‐100 in PBS for 1 h at RT. Tissues were incubated overnight at 4°C with primary antibodies. The following antibodies were used in this study: rabbit anti‐TMEM16A (Abcam cat# ab64085); goat polyclonal anti‐ChAT (Merck Millipore, cat# AB144); mouse anti‐KCC2 (Thermo fisher, cat# MA5‐27610); rabbit polyclonal anti‐c‐Fos (Merck Millipore, cat# ABE457); and mouse anti‐GAD67 (Merck Millipore, cat# MAB5406). For detection, suitable fluorescence (Alexa Fluor)‐tagged secondary antibodies (Molecular Probes, Eugene, OR) were used and the tissues were counterstained with DAPI.
Brain slice preparation and electrophysiology
Mouse brains were rapidly removed and placed into ice‐cold oxygenated cutting solution containing (in mM) choline chloride, 125; KCl, 2.5; NaH2PO4, 1.25; NaHCO3, 25; CaCl2, 0.5; MgSO4, 2; and D‐glucose, 10 (pH 7.2–7.4 was adjusted with NMDG). Coronal brain slices (250–300 μm thick for mHb or 180 μm thick for IPN) containing the mHb or IPN were obtained using a vibratome. The composition of the ACSF (in mM) was as follows: NaCl, 125; KCl, 2.5; NaH2PO4, 1.25; NaHCO3, 26; CaCl2, 1.5; MgCl2, 1.5; and D‐glucose, 10 (pH 7.3−7.4 was adjusted with 1 M HCl). Slices were visualized using an Olympus BX51WI microscope equipped with epifluorescence. Whole‐cell patch clamping was performed on cells in the ventral part of the mHb. Patch pipettes had a resistance of 6–9 MΩ. The standard pipette solution contained (in mM) potassium gluconate, 110; KCl, 37; MgCl2, 1; EGTA, 0.5; HEPES, 10; MgATP, 3; and NaGTP, 0.3 (pH 7.2 was adjusted with NMDG). All experiments were performed in the presence of antagonists of AMPA‐Rs (50 μM D‐AP5), NMDA‐Rs (10 μM CNQX), GABAA‐Rs (10 μM bicuculline), and GABAB‐Rs (10 μM CGP55845) to block glutamatergic and GABAergic synaptic transmissions. Recordings were filtered at 2 kHz and digitized at 10 kHz. In the current‐clamp experiments, minimal hyperpolarizing currents were injected into the mHb neurons to maintain stable baseline membrane potential after making a whole‐cell configuration and they were kept unaltered during the recording. Recordings in mHb neurons were included in the study only when they had stable sAP firings. Data were collected with a MultiClamp 700B Amplifier (Molecular Devices) using Clampex 10 data acquisition software (Molecular Devices) and were digitized with Digidata 1322A or 1550A (Molecular Devices). All experiments were performed at RT.
Voltage step protocol for tail current electrophysiology
The voltage protocol used in the tail current recordings was as follows: depolarizing voltage steps to 10 mV of variable duration (from 2 to 500 ms, with 50‐ms increments) were applied from the holding potential of −70 mV. These depolarizing voltage steps were followed by a hyperpolarizing step to −110 mV of 2.5 s duration (Fig 1A). For determination of the reversal potential of the tail current, ramp voltage pulses descending from 40 to −150 mV were applied for 1 s after a 2‐ms or 500‐ms depolarizing voltage step to 10 mV from the holding potential of −70 mV.
Spontaneous action potentials
From whole‐cell recordings, the averaged sAPs were calculated from a 30‐s segment in the Clampfit software (Molecular Probe) and several features (e.g., frequency, half‐width, and AHP slope) were measured from the averaged sAPs (Figs 3E and F, and EV4D). Cell‐attached recordings were performed to measure spontaneous firing to see the effects of TMEM16A inhibitors (Fig EV4A and B) and CNO (Fig 6B and C). Patch pipettes were filled with ACSF. The lag time of our perfusion system (bath application) was ~ 15 s, and firing frequency was calculated before and 30–60 s after application of drugs.
IPN sEPSCs recording
Coronal slices (180 μm) containing the IPN were prepared to obtain sEPSC recordings from TMEM16A cKO mice and their littermate controls. The pipette solution contained (in mM) CsCH3SO3, 117; EGTA, 0.4; NaCl, 2.8; TEA‐Cl, 5; HEPES, 20; spermine, 0.1; MgATP, 4; and NaGTP, 0.2 (pH 7.2−7.4 was adjusted with 1 M NaOH) 4. Data were analyzed using the template search function in Clampfit (Molecular Devices).
Behavioral tests
Open‐field test
For the analysis of the locomotor activity of mice, an open‐field test (OFT) was performed using white acrylic behavioral boxes with a square arena (40 × 40 cm) and 40 cm of white wall. The center zone was determined as a 20 × 20 cm square in the center of the box. Mice were placed in the box facing the wall. Tests were conducted in a soundproofing room with dim light of 10 lux. Mice were allowed to freely explore the open field for 30 min, and their movements were recorded. Locomotor activities, such as total distance moved and duration in the center zone, were measured using the EthoVision 3.0 software (Noldus). After every session, the behavioral box was cleaned with 70% ethanol, washed with distilled water, and wiped with paper towels.
Elevated plus maze test
To check the anxiety level of mice, an EPM test was conducted with the plus‐maze apparatus composed of two white open arms and two black closed arms (45 × 10 cm each). Tests were conducted in a soundproofing room with dim light of 10 lux. The closed arms had walls, while the open arms had no walls and the height was about 45 cm. Mice were placed in the center zone of the EPM box facing the open arms. During the 10‐min test trial, the time spent in both arms and the entries to each arm were video recorded and analyzed manually. The time spent in each arm was recorded only when all four paws were inside the arm.
Light and dark box test
The apparatus consisted of two acrylic chambers (27 × 27 × 27 cm white room and 18 × 27 × 27 cm black room) connected by a small opening. The black chamber had black walls and ground and was covered with a lid, while the white chamber had transparent walls with a white ground. Tests were conducted in a soundproofing room with dim light of 10 lux. Mice were placed in the dark chamber and allowed to freely explore the two chambers for 10 min. During a 10‐min video recording, the time spent in both rooms and the entries to each room were recorded and analyzed. Entries were recorded only when all four paws were inside a room. The two chambers were cleaned with 70% ethyl alcohol and dried between trials.
Passive avoidance test
Each animal was gently placed in the brightly lit compartment of the PA test apparatus; after 1 min, the guillotine door was opened and the animal was allowed to enter the dark compartment. The latency with which the animal entered the dark chamber was recorded. Once the animal entered with all four paws to the next chamber, the guillotine door was closed and a foot shock (0.45 mV, 2 s) was immediately delivered to the grid floor of the darkroom. After 1 day, each animal was gently placed in the light compartment, and after 1 min, the door was opened. In the absence of electric foot shocks, the step‐through latency (s) was recorded as an indicator of inhibitory avoidance behavior.
Tail suspension test
Mice were suspended by the end of their tail with adhesive tape about 50 cm above the floor of a white acrylic box (35 × 35 × 50 cm) immediately before the experiments (the adhesive tapes were hooked on a rod attached on the ceiling of the TST box). Tests were conducted in a soundproofing room with dim light of 10 lux. The movement of mice was recorded for 10 min. The duration of immobility was defined as the time with the absence of any movement, and it was scored over a 10‐min test session manually by an observer.
Forced swimming test
FST for mice consists of a single 10‐min exposure. Each mouse was individually forced to swim in a 2‐l acrylic beaker (height: 250 mm; width: 150 mm) containing water at a temperature of approximately 25°C for 10 min. The mice were unable to escape or rest by touching the bottom of the beaker. The time taken by the animal to show signs of struggling, the swimming time, and the duration of immobility were measured using the EthoVision 3.0 software (Noldus). After every session, the behavioral box was cleaned with 70% ethanol and the water was changed.
Nestlet shredding test
Pre‐weighed nestlets (5 × 5 cm) were placed into a transparent polycarbonate cage (19 × 29 × 13 cm) with a single mouse for 60 min. The unshredded remainder of the nestlet was weighed to calculate the amount of the shredded nestlet.
Three‐chamber test
The test mice were acclimated in the 3‐chamber test (3CT) chamber for 10 min. After the acclimation, test mice were placed in the middle chamber and two plastic caps and a stranger (S1) mouse were placed into the metal grid enclosure on each side. In the first session, the doors were opened simultaneously for 10 min allowing the test mouse to freely explore the three chambers. In the second session, S1 was moved to the other side and a new stranger mouse (S2) was placed in the chamber previously occupied by S1. Ten more minutes was given to the test mouse. The time spent in each chamber, the number of entries into each chamber, and the total distance traveled were analyzed using EthoVision XT 11.5.
Y‐maze test
During the Y‐maze test (YMT), the mice were placed in the center of the Y‐maze and were allowed to freely explore the Y‐maze for 10 min. The time spent in each arm and the number of arm entries were recorded and analyzed in order to calculate the percentage of alternation. An entry was counted only when all four limbs were within an arm.
Statistical analysis
Statistical analysis was performed using Origin software, version 7.0 (Origin Lab cooperation). Sample size choice was based on previous studies and was not predetermined by a statistical method. No randomization method was used. Data distribution was assumed to be normal, but this was not formally tested. Two‐way ANOVA was used in the morphological and electrophysiological studies to analyze more than two parameters. Numerical data are presented as means ± standard error of the mean (s.e.m). The variances were similar between the groups that were being compared. The statistical significance of the data was assessed by unpaired or paired Student's t‐test. The significance level was set at P < 0.05, P < 0.01, P < 0.001, or P < 0.0001.
Author contributions
C‐HC, SL, AK, KR, OY, Y‐SL, H‐GJ, DYL, EY, and EMH performed the experiments. C‐HC, SL, AK, BL, HK, UO, H‐II, EMH, and J‐YP interpreted the data. C‐HC, SL, AK, H‐II, EMH, and J‐YP designed the study, analyzed the data, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
This work was supported by the Bio‐Synergy Research Project (NRF‐2017M3A9C4092979 to J‐YP) and the Basic Science Research Program (NRF‐2016M3C7A1904149 and NRF‐2017M3C7A1079694 to J‐YP) from the National Research Foundation in Korea. This work was also supported by the National Research Council of Science & Technology of the Korean government (No.CRC‐15‐04‐KIST to H‐II) and KBRI basic research program through Korea Brain Research Institute funded by Ministry of Science and ICT(19‐BR‐03‐02 to EMH).
EMBO Reports (2020) 21: e48097
Contributor Information
Heh‐In Im, Email: him@kist.re.kr.
Eun Mi Hwang, Email: emhwang@kist.re.kr.
Jae‐Yong Park, Email: jaeyong68@korea.ac.kr.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Gellman MD, Turner JR (2013) Encyclopedia of behavioral medicine. New York, NY: Springer; [Google Scholar]
- 2. Janak PH, Tye KM (2015) From circuits to behaviour in the amygdala. Nature 517: 284–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marcinkiewcz CA, Mazzone CM, D'Agostino G, Halladay LR, Hardaway JA, DiBerto JF, Navarro M, Burnham N, Cristiano C, Dorrier CE et al (2016) Serotonin engages an anxiety and fear‐promoting circuit in the extended amygdala. Nature 537: 97–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhao‐Shea R, DeGroot SR, Liu L, Vallaster M, Pang X, Su Q, Gao G, Rando OJ, Martin GE, George O et al (2015) Increased CRF signalling in a ventral tegmental area‐interpeduncular nucleus‐medial habenula circuit induces anxiety during nicotine withdrawal. Nat Commun 6: 6770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pang X, Liu L, Ngolab J, Zhao‐Shea R, McIntosh JM, Gardner PD, Tapper AR (2016) Habenula cholinergic neurons regulate anxiety during nicotine withdrawal via nicotinic acetylcholine receptors. Neuropharmacology 107: 294–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hikosaka O (2010) The habenula: from stress evasion to value‐based decision‐making. Nat Rev Neurosci 11: 503–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boulos LJ, Darcq E, Kieffer BL (2017) Translating the habenula‐from rodents to humans. Biol Psychiatry 81: 296–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yamaguchi T, Danjo T, Pastan I, Hikida T, Nakanishi S (2013) Distinct roles of segregated transmission of the septo‐habenular pathway in anxiety and fear. Neuron 78: 537–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Soria‐Gomez E, Busquets‐Garcia A, Hu F, Mehidi A, Cannich A, Roux L, Louit I, Alonso L, Wiesner T, Georges F et al (2015) Habenular CB1 receptors control the expression of aversive memories. Neuron 88: 306–313 [DOI] [PubMed] [Google Scholar]
- 10. Han S, Yang SH, Kim JY, Mo S, Yang E, Song KM, Ham BJ, Mechawar N, Turecki G, Lee HW et al (2017) Down‐regulation of cholinergic signaling in the habenula induces anhedonia‐like behavior. Sci Rep 7: 900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McCormick DA, Prince DA (1987) Acetylcholine causes rapid nicotinic excitation in the medial habenular nucleus of guinea pig, in vitro . J Neurosci 7: 742–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Salas R, Sturm R, Boulter J, De Biasi M (2009) Nicotinic receptors in the habenulo‐interpeduncular system are necessary for nicotine withdrawal in mice. J Neurosci 29: 3014–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gorlich A, Antolin‐Fontes B, Ables JL, Frahm S, Slimak MA, Dougherty JD, Ibanez‐Tallon I (2013) Reexposure to nicotine during withdrawal increases the pacemaking activity of cholinergic habenular neurons. Proc Natl Acad Sci USA 110: 17077–17082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hartzell C, Putzier I, Arreola J (2005) Calcium‐activated chloride channels. Annu Rev Physiol 67: 719–758 [DOI] [PubMed] [Google Scholar]
- 15. Cho H, Yang YD, Lee J, Lee B, Kim T, Jang Y, Back SK, Na HS, Harfe BD, Wang F et al (2012) The calcium‐activated chloride channel anoctamin 1 acts as a heat sensor in nociceptive neurons. Nat Neurosci 15: 1015–1021 [DOI] [PubMed] [Google Scholar]
- 16. Salzer I, Gantumur E, Yousuf A, Boehm S (2016) Control of sensory neuron excitability by serotonin involves 5HT2C receptors and Ca (2 + )‐activated chloride channels. Neuropharmacology 110: 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra‐Moran O, Galietta LJ (2008) TMEM16A, a membrane protein associated with calcium‐dependent chloride channel activity. Science 322: 590–594 [DOI] [PubMed] [Google Scholar]
- 18. Schroeder BC, Cheng T, Jan YN, Jan LY (2008) Expression cloning of TMEM16A as a calcium‐activated chloride channel subunit. Cell 134: 1019–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM et al (2008) TMEM16A confers receptor‐activated calcium‐dependent chloride conductance. Nature 455: 1210–1215 [DOI] [PubMed] [Google Scholar]
- 20. Pedemonte N, Galietta LJ (2014) Structure and function of TMEM16 proteins (anoctamins). Physiol Rev 94: 419–459 [DOI] [PubMed] [Google Scholar]
- 21. Pifferi S, Dibattista M, Menini A (2009) TMEM16B induces chloride currents activated by calcium in mammalian cells. Pflugers Arch 458: 1023–1038 [DOI] [PubMed] [Google Scholar]
- 22. Scudieri P, Sondo E, Caci E, Ravazzolo R, Galietta LJ (2013) TMEM16A‐TMEM16B chimaeras to investigate the structure‐function relationship of calcium‐activated chloride channels. Biochem J 452: 443–455 [DOI] [PubMed] [Google Scholar]
- 23. Ta CM, Acheson KE, Rorsman NJG, Jongkind RC, Tammaro P (2017) Contrasting effects of phosphatidylinositol 4,5‐bisphosphate on cloned TMEM16A and TMEM16B channels. Br J Pharmacol 174: 2984–2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stohr H, Heisig JB, Benz PM, Schoberl S, Milenkovic VM, Strauss O, Aartsen WM, Wijnholds J, Weber BH, Schulz HL (2009) TMEM16B, a novel protein with calcium‐dependent chloride channel activity, associates with a presynaptic protein complex in photoreceptor terminals. J Neurosci 29: 6809–6818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Billig GM, Pal B, Fidzinski P, Jentsch TJ (2011) Ca2+‐activated Cl− currents are dispensable for olfaction. Nat Neurosci 14: 763–769 [DOI] [PubMed] [Google Scholar]
- 26. Huang WC, Xiao S, Huang F, Harfe BD, Jan YN, Jan LY (2012) Calcium‐activated chloride channels (CaCCs) regulate action potential and synaptic response in hippocampal neurons. Neuron 74: 179–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ha GE, Lee J, Kwak H, Song K, Kwon J, Jung SY, Hong J, Chang GE, Hwang EM, Shin HS et al (2016) The Ca(2 + )‐activated chloride channel anoctamin‐2 mediates spike‐frequency adaptation and regulates sensory transmission in thalamocortical neurons. Nat Commun 7: 13791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang Y, Zhang Z, Xiao S, Tien J, Le S, Le T, Jan LY, Yang H (2017) Inferior olivary TMEM16B mediates cerebellar motor learning. Neuron 95: 1103–1111 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dauner K, Mobus C, Frings S, Mohrlen F (2013) Targeted expression of anoctamin calcium‐activated chloride channels in rod photoreceptor terminals of the rodent retina. Invest Ophthalmol Vis Sci 54: 3126–3136 [DOI] [PubMed] [Google Scholar]
- 30. Amjad A, Hernandez‐Clavijo A, Pifferi S, Maurya DK, Boccaccio A, Franzot J, Rock J, Menini A (2015) Conditional knockout of TMEM16A/anoctamin1 abolishes the calcium‐activated chloride current in mouse vomeronasal sensory neurons. J Gen Physiol 145: 285–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takayama Y, Uta D, Furue H, Tominaga M (2015) Pain‐enhancing mechanism through interaction between TRPV1 and anoctamin 1 in sensory neurons. Proc Natl Acad Sci USA 112: 5213–5218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang XD, Lee JH, Lv P, Chen WC, Kim HJ, Wei D, Wang W, Sihn CR, Doyle KJ, Rock JR et al (2015) Etiology of distinct membrane excitability in pre‐ and posthearing auditory neurons relies on activity of Cl− channel TMEM16A. Proc Natl Acad Sci USA 112: 2575–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mulle C, Choquet D, Korn H, Changeux JP (1992) Calcium influx through nicotinic receptor in rat central neurons: its relevance to cellular regulation. Neuron 8: 135–143 [DOI] [PubMed] [Google Scholar]
- 34. Quina LA, Wang S, Ng L, Turner EE (2009) Brn3a and Nurr1 mediate a gene regulatory pathway for habenula development. J Neurosci 29: 14309–14322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boudes M, Sar C, Menigoz A, Hilaire C, Pequignot MO, Kozlenkov A, Marmorstein A, Carroll P, Valmier J, Scamps F (2009) Best1 is a gene regulated by nerve injury and required for Ca2+‐activated Cl− current expression in axotomized sensory neurons. J Neurosci 29: 10063–10071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR et al (2010) A robust and high‐throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13: 133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Namkung W, Phuan PW, Verkman AS (2011) TMEM16A inhibitors reveal TMEM16A as a minor component of calcium‐activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem 286: 2365–2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kanaka C, Ohno K, Okabe A, Kuriyama K, Itoh T, Fukuda A, Sato K (2001) The differential expression patterns of messenger RNAs encoding K‐Cl cotransporters (KCC1,2) and Na‐K‐2Cl cotransporter (NKCC1) in the rat nervous system. Neuroscience 104: 933–946 [DOI] [PubMed] [Google Scholar]
- 39. Kim U, Chung LY (2007) Dual GABAergic synaptic response of fast excitation and slow inhibition in the medial habenula of rat epithalamus. J Neurophysiol 98: 1323–1332 [DOI] [PubMed] [Google Scholar]
- 40. Wang DG, Gong N, Luo B, Xu TL (2006) Absence of GABA type A signaling in adult medial habenular neurons. Neuroscience 141: 133–141 [DOI] [PubMed] [Google Scholar]
- 41. Goldberg JA, Wilson CJ (2005) Control of spontaneous firing patterns by the selective coupling of calcium currents to calcium‐activated potassium currents in striatal cholinergic interneurons. J Neurosci 25: 10230–10238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McLaughlin I, Dani JA, De Biasi M (2017) The medial habenula and interpeduncular nucleus circuitry is critical in addiction, anxiety, and mood regulation. J Neurochem 142(Suppl 2): 130–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kumar V, Bhat ZA, Kumar D (2013) Animal models of anxiety: a comprehensive review. J Pharmacol Toxicol Methods 68: 175–183 [DOI] [PubMed] [Google Scholar]
- 44. Calhoon GG, Tye KM (2015) Resolving the neural circuits of anxiety. Nat Neurosci 18: 1394–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tzavara ET, Bymaster FP, Felder CC, Wade M, Gomeza J, Wess J, McKinzie DL, Nomikos GG (2003) Dysregulated hippocampal acetylcholine neurotransmission and impaired cognition in M2, M4 and M2/M4 muscarinic receptor knockout mice. Mol Psychiatry 8: 673–679 [DOI] [PubMed] [Google Scholar]
- 46. Dubrovina NI, Tomilenko RA (2007) Characteristics of extinction of a conditioned passive avoidance reflex in mice with different levels of anxiety. Neurosci Behav Physiol 37: 27–32 [DOI] [PubMed] [Google Scholar]
- 47. Kraeuter AK, Guest PC, Sarnyai Z (2019) The Y‐maze for assessment of spatial working and reference memory in mice. Methods Mol Biol 1916: 105–111 [DOI] [PubMed] [Google Scholar]
- 48. Cryan JF, Holmes A (2005) The ascent of mouse: advances in modelling human depression and anxiety. Nat Rev Drug Discov 4: 775–790 [DOI] [PubMed] [Google Scholar]
- 49. Tsujimura A, Matsuki M, Takao K, Yamanishi K, Miyakawa T, Hashimoto‐Gotoh T (2008) Mice lacking the kf‐1 gene exhibit increased anxiety‐ but not despair‐like behavior. Front Behav Neurosci 2: 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li X, Morrow D, Witkin JM (2006) Decreases in nestlet shredding of mice by serotonin uptake inhibitors: comparison with marble burying. Life Sci 78: 1933–1939 [DOI] [PubMed] [Google Scholar]
- 51. Wu T, Luo Y, Broster LS, Gu R, Luo YJ (2013) The impact of anxiety on social decision‐making: behavioral and electrodermal findings. Soc Neurosci 8: 11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Urban DJ, Roth BL (2015) DREADDs (designer receptors exclusively activated by designer drugs): chemogenetic tools with therapeutic utility. Annu Rev Pharmacol Toxicol 55: 399–417 [DOI] [PubMed] [Google Scholar]
- 53. Otsu Y, Lecca S, Pietrajtis K, Rousseau CV, Marcaggi P, Dugue GP, Mailhes‐Hamon C, Mameli M, Diana MA (2018) Functional principles of posterior septal inputs to the medial habenula. Cell Rep 22: 693–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang HT, Huang Y, Masood A, Stolinski LR, Li Y, Zhang L, Dlaboga D, Jin SL, Conti M, O'Donnell JM (2008) Anxiogenic‐like behavioral phenotype of mice deficient in phosphodiesterase 4B (PDE4B). Neuropsychopharmacology 33: 1611–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ahern M, Goodell DJ, Adams J, Bland ST (2016) Brain regional differences in social encounter‐induced Fos expression in male and female rats after post‐weaning social isolation. Brain Res 1630: 120–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Flanigan M, Aleyasin H, Takahashi A, Golden SA, Russo SJ (2017) An emerging role for the lateral habenula in aggressive behavior. Pharmacol Biochem Behav 162: 79–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Benekareddy M, Stachniak TJ, Bruns A, Knoflach F, von Kienlin M, Kunnecke B, Ghosh A (2018) Identification of a corticohabenular circuit regulating socially directed behavior. Biol Psychiatry 83: 607–617 [DOI] [PubMed] [Google Scholar]
- 58. Nakajima M, Mori H, Nishikawa C, Tsuruta M, Okuyama S, Furukawa Y (2013) Psychiatric disorder‐related abnormal behavior and habenulointerpeduncular pathway defects in Wnt1‐cre and Wnt1‐GAL4 double transgenic mice. J Neurochem 124: 241–249 [DOI] [PubMed] [Google Scholar]
- 59. Marchand ER, Riley JN, Moore RY (1980) Interpeduncular nucleus afferents in the rat. Brain Res 193: 339–352 [DOI] [PubMed] [Google Scholar]
- 60. Cooper MA, Huhman KL (2007) Corticotropin‐releasing factor receptors in the dorsal raphe nucleus modulate social behavior in Syrian hamsters. Psychopharmacology 194: 297–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lowry CA, Hale MW, Evans AK, Heerkens J, Staub DR, Gasser PJ, Shekhar A (2008) Serotonergic systems, anxiety, and affective disorder: focus on the dorsomedial part of the dorsal raphe nucleus. Ann N Y Acad Sci 1148: 86–94 [DOI] [PubMed] [Google Scholar]
- 62. Luo J, Feng Q, Wei L, Luo M (2017) Optogenetic activation of dorsal raphe neurons rescues the autistic‐like social deficits in Shank3 knockout mice. Cell Res 27: 950–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Perez‐Cornejo P, Gokhale A, Duran C, Cui Y, Xiao Q, Hartzell HC, Faundez V (2012) Anoctamin 1 (Tmem16A) Ca2+‐activated chloride channel stoichiometrically interacts with an ezrin‐radixin‐moesin network. Proc Natl Acad Sci USA 109: 10376–10381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lee YS, Lee JK, Bae Y, Lee BS, Kim E, Cho CH, Ryoo K, Yoo J, Kim CH, Yi GS et al (2016) Suppression of 14‐3‐3gamma‐mediated surface expression of ANO1 inhibits cancer progression of glioblastoma cells. Sci Rep 6: 26413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lee S, Woo J, Kim YS, Im HI (2015) Integrated miRNA‐mRNA analysis in the habenula nuclei of mice intravenously self‐administering nicotine. Sci Rep 5: 12909 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.