

Summary
Mutations and inactivation of phosphatase and tensin homolog deleted from chromosome ten (PTEN) are observed in 15-25% cases of human T-cell acute lymphoblastic leukemia (T-ALL). Pten deletion induces myeloproliferative disorders (MPD), acute myeloid leukemia (AML), and/or T-ALL in mice. Previous studies attributed Pten-loss-related hematopoietic defects and leukemogenesis to excessive activation of PI3K/AKT/mTOR signaling. Although inhibition of this signal dramatically suppresses the growth of PTEN-null T-ALL cells in vitro, treatment with inhibitors of this pathway do not cause a complete remission in vivo. Here we report that focal adhesion kinase (Fak), a protein substrate of Pten, also contributes to T-ALL development in Pten-null mice. Inactivation of FAK signaling pathway by either genetic or pharmacologic methods significantly sensitizes both murine and human PTEN-null T-ALL cells to PI3K/AKT/mTOR inhibition when cultured in vitro on feeder layer cells or a matrix and in vivo.
Introduction
PTEN mutations are detected in 15-25% of T-ALL patients (Gutierrez et al., 2009; Jotta et al., 2010; Zuurbier et al., 2012). Inactivation of PTEN and constitutive activation of its downstream signaling pathways are associated with early treatment failure, drug resistance, and poor prognosis (Jotta et al., 2010; Piovan et al., 2013). Thus, it should be useful to completely elucidate the roles of the PTEN downstream signaling pathways in the pathogenesis of T-ALL for development of less toxic targeted therapies for this fatal disease.
PTEN possesses both lipid and protein phosphatase activities (Lee et al., 1999; Tamura et al., 1998). It represses growth factor/cytokine-stimulated cell growth and survival by repressing PI3K/AKT/mTOR signaling through dephosphorylation of phosphatidylinositol (3,4,5)-trisphosphate (PIP3) (Stambolic et al., 1998). It also dephosphorylates FAK, thus inhibiting integrin-induced adhesion and migration by suppressing the turnover of focal adhesions and cytoskeletal reorganization (Gu et al., 1998; Guo and Giancotti, 2004; Tamura et al., 1998). It was reported that T-ALL development in Pten-null mice could be repressed by inactivation of the PI3K/Akt/mTor signaling (Kalaitzidis et al., 2012; Subramaniam et al., 2012). Preclinical studies showed that PI3K/AKT/mTOR inhibitors induce apoptosis and repress the growth of T-ALL cell lines and primary human T-ALL cells, suggesting that this signaling pathway might be a promising target for T-ALL therapy (Evangelisti et al., 2011; Subramaniam et al., 2012). However, PI3K/AKT/mTOR inhibition did not cause complete remission in vivo (Rodon et al., 2013), suggesting that there might be an additional survival signal which compensates for inhibition of PI3K/AKT signaling in PTEN-null T-ALL. As an important target of PTEN, the role of FAK has not been studied in the etiology and progression of T-ALL induced by PTEN mutations.
FAK is a non-receptor tyrosine kinase. In response to integrin and/or chemokine stimulation, activated FAK promotes cell adhesion, spreading, motility, proliferation and anchorage-dependent survival by inducing the activation of its downstream signals, including pl30Cas/Rac/CDC42, Rho/Gap/RhoA, ERK/MAPK, Wnt/β-catenin and NF-κB (Desgrosellier and Cheresh, 2010; Guan, 2010; Mitra and Schlaepfer, 2006; Schaller, 2010). Dysregulation of FAK has been detected in multiple cancers, including breast, brain, prostate and liver cancer (Guan, 2010; Mitra and Schlaepfer, 2006; Pylayeva et al., 2009). In these cancers, the activity of FAK generally correlates with increased tumor malignancy, drug resistance and metastasis (Golubovskaya, 2014; Lechertier and Hodivala-Dilke, 2012). FAK also plays an essential role in regulating the self-renewal of cancer stem cells (Guan, 2010). Thus, FAK has been suggested to be a potentially advantageous target for cancer therapy (Golubovskaya, 2014).
Despite the liquid nature of blood and the peripheral circulation of leukemic cells (LCs), the majority of leukemia-initiating cells (LICs) reside in the bone marrow, spleen, and other organs where they adhere to their niches; this could provide in vivo signals to activate FAK in LCs (Konopleva and Jordan, 2011). Clinically, increased FAK activity is correlated with high blast cell counts and shorter survival times (Despeaux et al., 2011; Recher et al., 2004). We speculated that signals emanating from niches might stimulate FAK activation in LCs, which is normally restricted by PTEN. In PTEN-mutant T-ALL cells, activated FAK may play a critical role in disease initiation, progression and the development of drug resistance by providing a survival signal in LCs parallel to PI3K/AKT/mTOR signaling. To test this hypothesis, we inactivated FAK by genetic and pharmacologic methods to study the functions of FAK in PTEN-null T-ALL cells.
Results
The Activity of Fak is Significantly Increased in Pten−/− Thymocytes
Loss of Pten causes MPD, AML, and/or T-ALL in Mx-1-Cre+Ptenfx/fx mice (Pten−/−), in which Pten is deleted in nearly 100% of adult hematopoietic cells (HCs) after 3-5 polyinosine-polycytidine (pI:C) injections (Yilmaz et al., 2006; Zhang et al., 2006). Repression of PI3K/Akt/mTor signaling by inhibitors or genetic deletion largely suppresses the defects of HSCs and leukemia development, but none of these inhibitors or deletions can completely block leukemia development in Pten−/− mice (Kalaitzidis et al., 2012; Yilmaz et al., 2006). Thus, it is possible that another potential downstream target(s) of Pten which can be activated in vivo compensates for PI3K/Akt/mTor inhibition in the development of leukemia in Pten−/− mice. Pten dephosphorylates both the lipid substrate PIP3, which is activated by PI3K, and protein targets such as Fak (Tamura et al., 1998), which is activated by adhesion signals (Mitra and Schlaepfer, 2006). We predicted that FAK might mediate such compensatory signaling. To test this hypothesis, we determined the expression of Fak in wild-type (WT) hematopoietic stem/progenitor cells from bone marrow (BM) and differentiated HCs from spleen (SP) and thymus (TH) (Fig.1A). Fak was expressed at low levels in Lin−Sca1+c-Kit+ cells (LSK), and very low in progenitor cells (CMP, GMP, MEP, and CLP), myeloid cells (Gr1+Mac1+), macrophages (F4/80+), B-cells (B220+), and erythroid lineage cells (Ter119+). Fak was highly expressed in platelet precursors (CD41+), as well as CD4−CD8−, CD4+CD8+, CD4+CD8−, and CD4−CD8+ T-lymphocytes. Although the expression of Fak was comparable between WT and Pten−/−, Pten deletion enhanced the activation of both Akt and Fak in thymocytes, as shown by the increased levels of pAkt/pS6k, and pFak/pSrc, respectively. Neither Cre expression by itself nor pI:C treatment activated either signal (Fig.1B). In the four subtypes of thymocytes (Fig.1C,D; Fig.S1), the expression of Fak was not changed by Pten deletion. However, Pten deletion enhanced Fak activation in all thymocytes, especially in CD4+CD8− cells, which consist of T-ALL cells in Pten−/− mice (Zhang et al., 2011). These data suggest that hyper-activated Fak in CD4+CD8− cells may contribute to T-ALL development in Pten−/− mice.
Figure 1. Akt and Fak Signals are Highly Activated in Pten−/− Thymocytes.
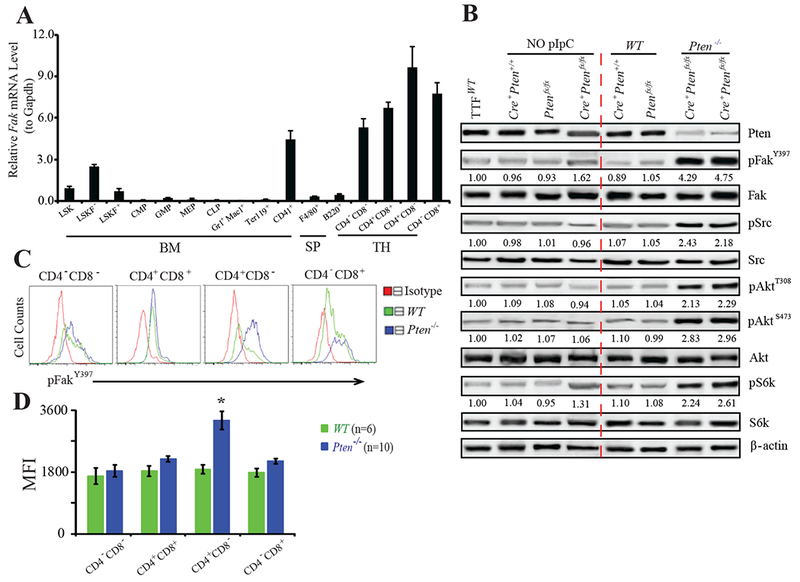
(A) Fak expression in Lin−Sca1+c-Kit+ (LSK), LSKFlk2−, LSKFlk2+, CMP, GMP, MEP, CLP, Gr1+Mac1+, Ter119+, and CD41+ cells isolated from BM; F4/80+, B220+ cells from SP; and CD4−CD8−, CD4+CD8+, CD4+CD8−, CD4−CD8+ cells from TH of WT mice. (B) Fak and Akt activities in WT and Pten−/− thymocytes. Controls: thymocytes from mice without pI:C injection; WT tail tip fibroblasts (TTF). Images representative of three independent experiments. Quantification (Multi Gauge 3.0) relative to WT control after normalized to loading controls. (C&D) pFakY397 in T-lymphocytes measured by FACS (C), and quantified by mean fluorescence intensity (MFI) (D). Data shown are Mean ± S.E.M. * compared to WT, P< 0.001, by Student’s t-test.
Fak Deletion Significantly Delays T-ALL Development but does not Affect MPD in Pten−/− Mice
Mx-1-Cre+Ptenfx/fx mice were crossed with Fakfx/fx mice to generate Mx-1-Cre+Ptenfx/fxFakfx/fx mice to study the role of Fak in hematopoietic defects in Pten−/− mice. Initially, the mice received five pI:C injections as reported (Yilmaz et al., 2006; Zhang et al., 2006). The hematopoietic alterations among Mx-1-Cre+Ptenfx/fx, Mx-1-Cre+Fakfx/fx, Mx-1-Cre+Ptenfx/fxFakfx/+, and Mx-1-Cre+Ptenfx/fxFakfx/fx mice were analyzed 20-25 days after pI:C treatments. Mx-1-Cre+, Mx-1-Cre+Ptenfx/+Fakfx/+, Mx-1-Cre+Ptenfx/+, Mx-1-Cre+Fakfx/+ or Ptenfx/fxFakfx/fx mice from the same or a comparable litter were used as WT controls (no phenotypic differences among these mice, data not shown). Consistent with previous reports, Mx-1-Cre+Fakfx/fx mice (Fak−/− hereafter) had no significant hematopoietic defects (Fig.2 E, F, G, H; Fig.3 A, B, C;Fig.S2 A, B) except for a small but significant increase in platelet numbers (Fig.S2 C) (Vemula et al., 2010). Twenty to 25 days after 5×pI:C injections, both Mx-1-Cre+Ptenfx/fx or Mx-1-Cre+Ptenfx/fxFakfx/+ mice (Pten−/− hereafter, no differences between them) and Mx-1-Cre+Ptenfx/fxFakfx/fx mice (Pten−/−Fak−/− hereafter) developed MPD, as shown by a significant increase in Gr-1+Mac-1+ cells in BM, PB, and SP. The percentage of B220+ and Gr1+Mac1+ cells (Fig.S2 A, B) or nucleated cells in PB (Fig.S2 F, G) were not affected by Fak deletion. Interestingly, compared to Pten−/− mice, there was a mild, but statistically significant impact on white blood cell counts (WBC), platelets and red blood cell counts (RBC) (Fig.S2 C), and a subtle increased survival, though without statistical significance, in Pten−/−Fak−/− mice (Fig.S2 D). These data suggest that Fak is less essential for MPD development in Pten−/− mice.
Figure 2. Fak Deletion Significantly Delays the Development of T-ALL in Pten−/− Mice.
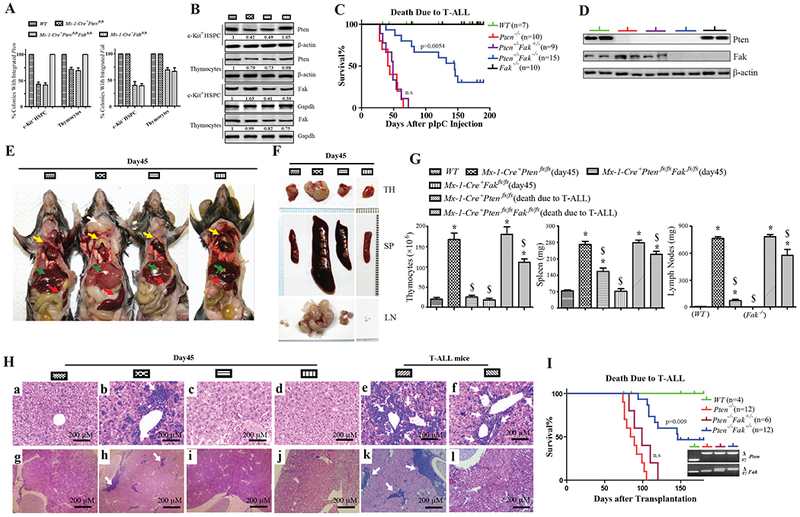
(A&B) Pten and Fak induced deletion efficiency (A), and Pten and Fak protein (B) in c-Kit+ HSPCs and thymocytes on day 10 treated with single pI:C. (C) Kaplan-Meier survival curve of indicated mice with a single pI:C injection; compared to Pten−/−, P=0.0054. (D) Pten and Fak protein in thymocytes recovered from moribund T-ALL mice. (E&F) Phenotypes on day 45 after single pI:C injection. Anatomic mice images: white arrow, lymph nodes; yellow arrows, thymuses; green arrows, livers; and red arrows, spleens (E). TH, SP, and LN (F). Images shown are representative of five independent experiments. (G&H) Phenotypes on day 45 and moribund T-ALL mice after single pI:C injection. Thymocyte number, SP and LN weight: data shown are Mean ± S.D. * compared to WT, $ compared to Pten−/−, P<0.001, by 2-way ANOVA tests (G). Images of H&E staining for lymphocytes infiltrated into livers and kidneys (H). (I) Kaplan-Meier survival curve of recipient mice (CD45.2+) transplanted with CD3+c-Kitlow cells from BM of indicated mice (CD45.1+) on day 10 after 5×pI:C injections. Compared to Pten−/−-recipient mice, P= 0.009. Inset: donor cell genotype immediately prior to transplantation.
Figure 3. Fak Deletion Partially Restores Apoptosis of Pten−/− Thymocytes but does not Affect the Survival or Proliferation of Pten-null T-ALL Cells.
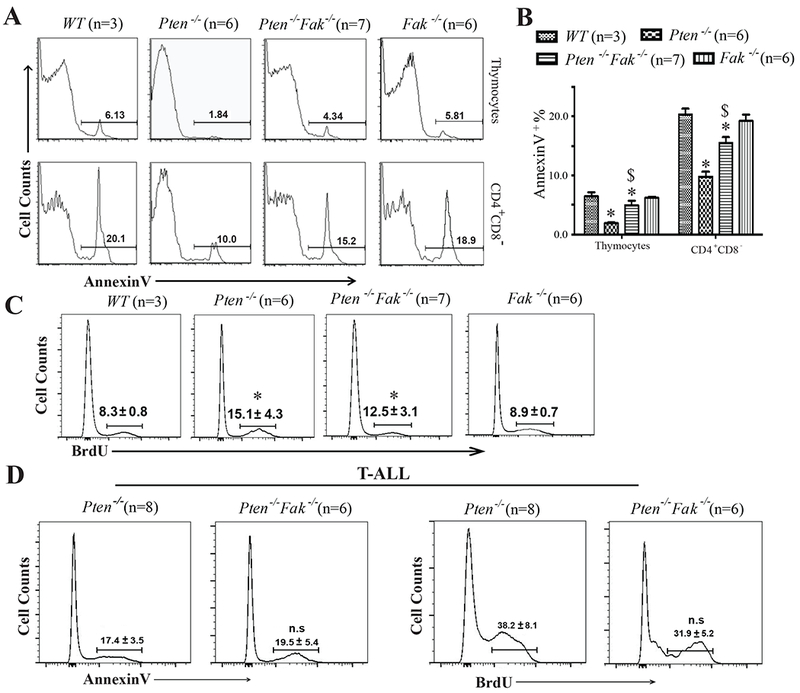
(A&B) Apoptosis by AnnexinV staining: total thymocytes and CD4+CD8− T-lymphocytes before T-ALL development after a single pI:C injection. (C) BrdU pulse-labelled thymocytes prior to T-ALL development. (D) Apoptosis and proliferation analysis of T-ALL cells from Pten−/− or Pten−/− Fak−/− mice. Data shown are Mean ± S.D. * compared to WT, $ compared to Pten−/−, P<0.001; n.s, no significant difference, by 2-way ANOVA tests.
Without pI:C treatment, Mx-1-Cre+Ptenfx/fx mice develop T-ALL 3-4 months after birth, but rarely develop MPD (Zhang et al., 2011). VEC-Cre+Ptenfx/fx mice, in which Pten is deleted in about 40% of HCs, also develop only T-ALL (Guo et al., 2008). Thus, to study the role of Fak in T-ALL induced by Pten deletion we used a reduced regimen of pI:C to induce deletion in a fraction of HCs . We injected WT, Pten−/−, Pten−/−Fak−/−, and Fak−/− mice once with 25 mg/kg pI:C. Ten days after injection, c-Kit+ HSPCs and thymocytes were isolated. We used colony PCR to determine the efficiency of Pten and Fak deletion in these cells. This pI:C regimen induced Pten deletion in 55% of c-Kit+ HSPCs and approximately 25% of thymocytes, both in Pten−/− and Pten−/−Fak−/− mice. Fak was co-deleted with Pten in Pten−/−Fak−/− thymocytes (Fig.2 A). This result was confirmed by measuring protein levels of Pten and Fak (Fig.2 B). Thus, a reduced regimen of pI:C induced both Pten and Fak deletion to a lesser degree than with 5×pI:C treatments. A relatively limited Pten deletion may enable sufficient time for Pten-null T-ALL cells accumulation, in contrast to death from MPD after complete Pten deletion in mice (Zhang et al., 2006).
After a single pI:C injection, all Pten−/− mice died of T-ALL, diagnosed by measuring CD4+CD8mid/−CD45high LCs (Guo et al., 2008) in BM and SP (Fig.S2E), within 70 days; some Pten−/−Fak−/− mice began dying of T-ALL on day 50, but most Pten−/−Fak−/− mice survived significantly longer than Pten−/− mice. About 25% of Pten−/−Fak−/− mice were leukemia-free on day 200 post-pI:C treatment. Fak−/− mice are grossly normal. We confirmed Pten and Fak deletion in T-ALL cells recovered from moribund mice (Fig.2 C, D).
On day 45 after a single pI:C treatment, most Pten−/− had obvious disease phenotypes, including significantly enlarged lymph nodes (LNs), TH and SP compared to Pten−/−Fak−/− mice. Pten−/− mice had pale-colored livers, which are typically observed during leukemia development (Fig.2 E, F, G). Pten−/− mice had significantly greater LC infiltration into both livers and kidneys (Fig.2 H: b, h), and showed considerable destruction of splenic histologic structure compared to WT or Pten−/−Fak−/− mice (Fig.S2 H); At this stage, Pten−/−Fak−/− mice did not show such a high degree of disease phenotypes, having larger SP and LNs than WT or Fak−/− mice (Fig.2 E, F, G), very low liver or kidney infiltration (Fig.2H: c, i), less destruction of splenic histologic structure (Fig.S2 H). After T-ALL developed, though still smaller than Pten−/− mice, Pten−/−Fak−/− mice had significantly enlarged SP, LNs, TH (Fig.2G) and high infiltration of LCs into livers and kidneys (Fig.2H:f, l), as well as significant structural destruction of the SP (Fig.S2 H). To test whether delayed T-ALL development with Fak deletion was cell intrinsic, we transplanted CD3+c-Kitlow cells, which are enriched for T-ALL initiating cells (Guo et al., 2008), isolated from the BM of Pten−/− or Pten−/−Fak−/− mice (CD45.1+) on day 10 after 5×pI:C treatments into lethally-irradiated recipient mice (CD45.2+). After transplantation, all of the Pten−/−-recipient mice died of T-ALL within 125 days; only about 55% of Pten−/−Fak−/−-recipient mice died of T-ALL within 150 days, with the remainder of these mice surviving over 180 days (Fig.2 I). These data suggest that Fak deletion delays T-ALL development in Pten−/− mice.
Fak Deletion Partially Restores Apoptosis in Pten−/− T-lymphocytes, but not in Pten−/− T-ALL Cells.
To investigate the mechanisms by which Fak deletion delays T-ALL development in Pten−/− mice, we analyzed thymocyte apoptosis and proliferation on day 10 after 5×pI:C injection. Pten−/− mice had fewer apoptotic thymocytes than WT. Increased apoptosis in Pten−/−Fak−/− thymocytes, especially CD4+CD8− cells, suggests that Fak contributes to the enhanced survival of Pten−/− thymocytes (Fig.3 A, B). However, combined Pten and Fak deletion had no significant effect on apoptosis in CD4−CD8−, CD4+CD8+ and CD4−CD8+ T lymphocytes (Fig.S3 A, B). Fak deletion did not change the proliferation of Pten-null cells, which was higher than WT and Fak−/− controls (Fig.3 C). Consistent with this, there was no significant cell-cycle difference between Pten−/− and Pten−/−Fak−/− thymocytes, but both had more cycling cells than did controls (Fig.S3 C). After T-ALL development, Pten−/−Fak−/− LCs did not differ from Pten−/− LCs in apoptosis or proliferation (Fig.3 D). These data suggest that Fak deletion delays T-ALL development in Pten−/− mice by partially restoring apoptosis in thymocytes, especially in CD4+CD8− T-lymphocytes, but this effect disappears after T-ALL development.
Fak Promotes a PI3K/Akt/mTOR/Mcl-1-Independent Survival Signal in Pten−/− Thymocytes and T-ALL Cells By Enhancing the NF-κB/Bcl-2/Bcl-xL Pathway
To study the molecular mechanisms whereby Fak mediated thymocyte apoptosis, we tested survival-related signals in Pten−/− and Pten−/−Fak−/− thymocytes freshly recovered from mice on day 10 after 5×pI:C treatment. Activation of Akt and Fak signaling in Pten−/− thymocytes was verified by increased levels of p-Akt/p-Gsk3β and pFAK/p-Src/p-P130Cas/p-Paxillin, respectively. Fak deletion reduced the level of the latter but not the former (Fig.4A). Over-activated PI3K/Akt/mTor signaling provides a sustained survival signal by up-regulating Mcl-1 in AML, T-ALL and other types of cancer cells (Hsieh et al., 2010; Inuzuka et al., 2011; Jebahi et al., 2014). Consistent with these studies, a significant increase of Mcl-1 was detected in Pten−/− thymocytes compared to WT (Fig.4 A). This was not affected by Fak deletion. Comparable Mcl-1 mRNA levels among WT, Pten−/− and Pten−/−Fak−/− thymocytes (Fig.4 D) supported the idea that Mcl-1 is regulated by PI3K/Akt/mTor through a post-transcriptional mechanism (Hsieh et al., 2010; Inuzuka et al., 2011).
Figure 4. Two Parallel Survival Signaling Pathways, PI3K/Akt/mTOR/Mcl-1 and Fak/NF-κB/Bcl-xL/Bcl-2, in Pten−/− Thymocytes and T-ALL Cells.
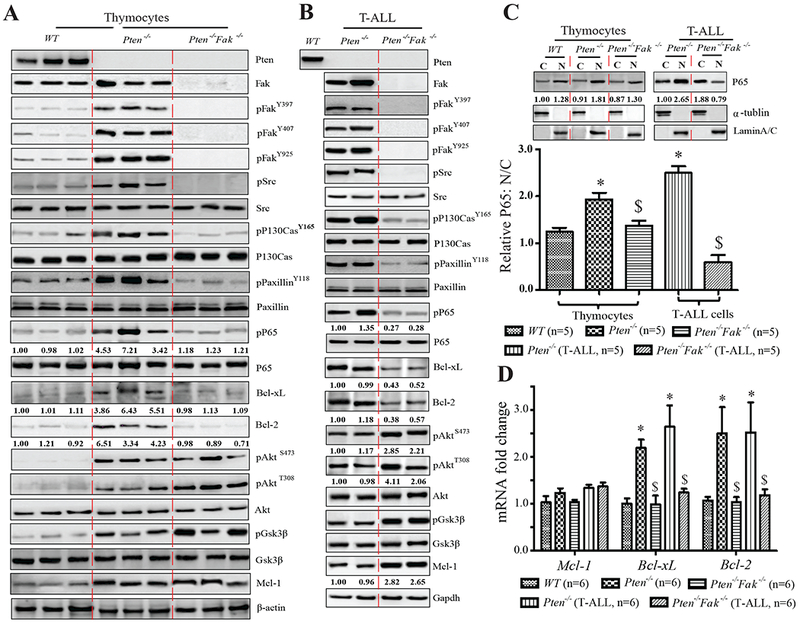
(A&B) pFak, pSrc, pP130Cas, pPaxillin, pP65, pAkt, pGsk3β, Bcl-xL, Bcl-2, and Mcl-1 expression in thymocytes before T-ALL development (A) and T-ALL cells (B). (C) Nuclear (N) and cytoplasmic (C) P65 in thymocytes (left) or T-ALL cells (right). Images shown representative of five independent experiments. (D) mRNA levels of Mcl-1, Bcl-xL, and Bcl-2 in thymocytes before T-ALL development and T-ALL cells, with WT as controls. Data shown are Mean ± S.D. * compared to WT, $ compared to Pten−/−, P<0.001, determined by 2-way ANOVA tests.
NF-κB mediates a critical survival signal in many types of cancer by enhancing the expression of several key survival genes, such as Bcl-2 and Bcl-xL (Karin, 2006). To determine whether NF-κB mediates a Fak-stimulated PI3K/AKT-independent survival signal in Pten−/− thymocytes, we measured NF-κB activity in these cells by assessing whole cell pP65 and nuclear P65. NF-κB activation was greatly enhanced in Pten−/− thymocytes compared to WT. However, in Pten−/−Fak−/− thymocytes, NF-κB activity was reduced compared to Pten−/− (Fig.4 A, C), suggesting Fak-dependent NF-κB activation in Pten−/− cells. As a consequence, mRNA and protein levels of Bcl-xL and Bcl-2, known downstream target genes of NF-κB, were up-regulated in Pten−/− thymocytes, and repressed by Fak deletion (Fig.4 A, D). The comparable expression of other known NF-κB target genes such as cIAP1/2, Cyclin D1/2 and Cyclin A2 in WT and Pten-null thymocytes suggested that not all NF-κB target genes were induced by Pten deletion. The expression of p57, p21, p27 and p-Rb, all negative cell cycle regulators, were not altered in Pten-null cells (Fig.S4 A, B). Enhanced c-Myc and p-H3 (a mitotic marker) in both Pten−/− and Pten−/−Fak−/− thymocytes might explain the increased proliferation of Pten-null cells compared to WT (Fig.3 C; Fig.S4 A, B). Moreover, Pyk2 activity, a homolog of Fak with a potential compensatory effect for Fak deletion, and Jnk1/2, another potential survival pathway to neutralize the inactivation of NF-κB (Volk et al., 2014), were measured. There was a negligible difference between Pten−/− and Pten−/−Fak−/− thymocytes, yet p-Jnk1/2 was at a higher level than WT (Fig.S4 B).
Although Fak deletion significantly enhanced survival, most Pten−/−Fak−/− mice still developed T-ALL after a single pI:C treatment (Fig.2 C). Inactivation of NF-κB/Bcl-xL/Bcl-2 signaling in Pten−/−Fak−/− T-ALL cells might be compensated by Fak-independent signal(s). We recovered T-ALL cells from both Pten−/− and Pten−/−Fak−/− mice to test this possibility. Pten−/−Fak−/− cells had lower NF-κB activity and reduced Bcl-2/Bcl-xL than Pten−/− (Fig.4 B, C). Interestingly, we found significantly enhanced PI3K/Akt/mTOR signaling and further increased Mcl-1 in Pten−/−Fak−/− compared to Pten−/− T-ALL cells (Fig.4 B). This indicated that further enhancement of PI3K/Akt/mTOR/Mcl-1 signaling could compensate for inactivated Fak/NF-κB/Bcl-xL/Bcl-2 signaling in Pten−/−Fak−/− cells to promote T-ALL development and progression. These data suggest that PI3K/Akt/mTOR/Mcl-1 and Fak/NF-κB/Bcl-xL/Bcl-2 mediate two parallel survival signaling pathways, which are normally restricted by Pten, in Pten-null thymocytes and T-ALL cells.
Fak Deletion Restores the Sensitivity of Pten-null Thymocytes and T-ALL Cells to PI3K/mTOR/Mcl-1 Inhibitors in vitro
When cultured in suspension without feeder cells or ECM-coating (hereafter as “suspension”), both Pten−/− and Pten−/−Fak−/− thymocytes and T-ALL cells were sensitive to a pan-PI3K inhibitor, LY294002 (LY), mTOR inhibitors, KU0063794 (KU)/Rapamycin (RA), and Mcl-1 inhibitor (MI); no difference was observed between Pten−/− and Pten−/−Fak−/− cells (Fig.5 A, B: leftmost panels). Because Fak is activated by extracellular matrix (ECM) signals (Mitra and Schlaepfer, 2006), we speculated that Fak might not be activated in suspension culture. We measured Fak activation in freshly-recovered and suspension-cultured Pten−/− thymocytes and T-ALL cells. Compared to freshly-recovered cells, Fak activation was much lower in suspension-cultured cells. Grown on OP9 feeder layer cells (OP9), or on MatriGel (MG)-, collagen (CLG)-, or fibronectin (FN)-coated plates, Fak could be activated to a level comparable to freshly-recovered cells (Fig.S5 A, B). Fak activation by OP9, or MG/CLG/FN-coated plates enabled Pten−/− thymocytes and T-ALL cells to become less sensitive to PI3K/mTOR/Mcl-1 inhibitors, which were titrated to optimize treatment concentrations (Fig.S5 C), although they were still more sensitive to these inhibitors than WT cells. Pten−/−Fak−/− demonstrated increased death compared to Pten−/− cells upon PI3K/Akt signal inhibition by LY, KU, RA, or MI. Consistent with the genetic inactivation of Fak, suppression of both PI3K/Akt and Fak signals (by FAK inhibitor PF573228, PF) with combined pharmacological inhibitors, LY+PF, caused more death in Pten−/− cells than with individual inhibitor LY (Fig.5 A, B;Fig.S5 D, E). To test whether the increased death induced by Fak deletion was due to its kinase activity, we overexpressed FakWT or FakY397F (kinase-null) in Pten−/−Fak−/− thymocytes and T-ALL cells. Expression of FakWT, but not FakY397F, increased survival of Pten−/−Fak−/− cells treated with LY, KU, RA, or MI (Fig.S5 F;Fig.5 C-E). These data suggest that Fak activation by OP9, or ECM-coated plates increases the resistance of Pten−/− thymocytes and T-ALL cells to PI3K/Akt/mTOR/Mcl-1 inhibition.
Figure 5. Inhibition of Fak/NF-κB Signaling Sensitizes Pten−/− Thymocytes and T-ALL Cells to PI3K/mTOR/Mcl-1 Inhibition in vitro and in vivo.
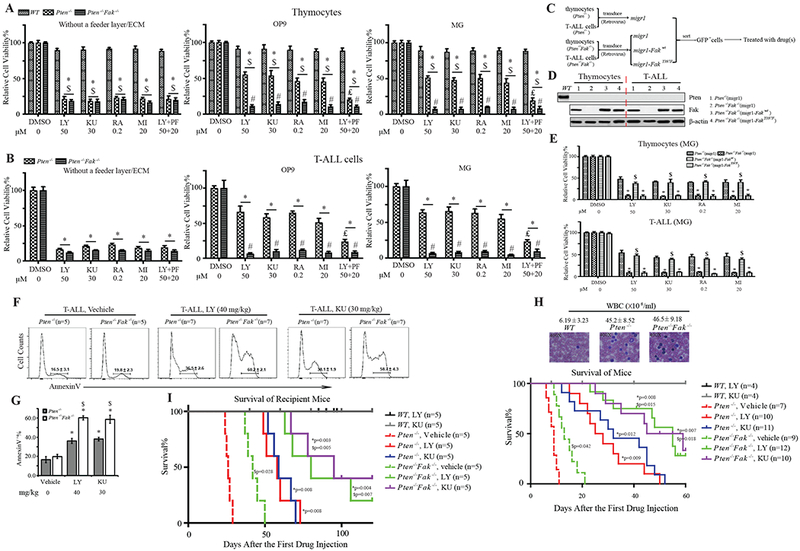
(A&B) Sensitivity of thymocytes (A) and T-ALL cells (B) to DMSO (DO), LY, KU, RA, MI, and LY+PF when cultured in suspension, on OP9 feeder cells, or on MG-coated plates, respectively; WT, control. Data shown are Mean ± S.D. * compared to DMSO, $ compared to WT, # compared to Pten−/−, ₤ compared to LY treatment, P<0.001, determined by 2-way ANOVA. (C-E) Sensitivity of Fak-overexpressing Pten−/−Fak−/− thymocytes and T-ALL cells to DO, LY, KU, RA, or MI when cultured on MG-coated plates. Experimental procedure (C), Pten and Fak protein in transduced cells (D). Sensitivity of transduced cells on MG-coated plates (E), using Migr1 (empty vector)-transduced cells as controls. Data shown are Mean ± S.D. * compared to Migr1-Pten−/−, $ compared to migr1-Pten−/−Fak−/−, P<0.001, determined by 2-way ANOVA. (F&G) Apoptosis of T-ALL cells from leukemic Pten−/− and Pten−/−Fak−/− mice on day 10 with vehicle, LY, or KU treatment (F), statistical quantification of AnnexinV+ cells (G). Data shown are Mean ± S.E.M. * compared to WT, $ compared to Pten−/−, P<0.001, determined by Student’s t-test. (H) Kaplan-Meier survival curve for Pten−/− and Pten−/−Fak−/− T-ALL mice treated with vehicle, LY, or KU; WT mice treated with LY or KU to evaluate treatment toxicity. Upper panel: WBC counts and Giemsa staining of PB of Pten−/− and Pten−/−Fak−/− T-ALL mice, using WT as control. (I) Kaplan-Meier survival curve of recipient mice (CD45.2+) transplanted with CD3+c-Kitlow cells isolated from BM of Pten−/−, or Pten−/−Fak−/− T-ALL, or WT mice (CD45.1+) treated with vehicle, LY, or KU. * compared to corresponding vehicle treatment; $ compared to Pten−/− mice with corresponding vehicle or LY or KU treatment.
Fak Inactivation Increases the Sensitivity of Pten-null T-ALL Cells to Apoptosis Induced by PI3K/mTOR Inhibitors in vivo
We transplanted CD3+c-Kitlow cells from Pten−/− T-ALL mice (CD45.1+) into lethally-irradiated recipient mice (CD45.2+). Beginning on day 14 after transplantation, we treated recipient mice with KU at 30 mg/kg, PF at 50 mg/kg, alone or together, every other day until analysis or death of the animal (maximum: 15 injections). Either KU or PF alone significantly enhanced the survival of recipient mice compared to vehicle treatment. KU+PF treatment further increased the survival compared to either single inhibitor treatment (Fig.S5 G). We next tested whether Fak inhibition could enhance the therapeutic effects of PI3K/Akt/mTOR inhibitors on Pten-null T-ALL mice. Immediately following T-ALL development, diagnosed by >30% blasts (Fig.S2 F, G) and 35,000-60,000 μl−1 WBC (Fig.5 H) in their PB, we treated Pten−/−, Pten−/−Fak−/− mice with LY at 40 mg/kg (Hu et al., 2002; Nagata et al., 2004) or KU at 30 mg/kg every other day until analysis or a maximum 15 injections. After five drug treatments, we analyzed T-ALL cell apoptosis. Pten−/− T-ALL cells underwent apoptosis to a greater extent with LY or KU than with vehicle treatment. Pten−/−Fak−/− T-ALL cells underwent apoptosis to an even greater extent than Pten−/− (Fig.5 F, G). Pten−/− mice survived longer with LY or KU compared to vehicle treatment; Pten−/−Fak−/− mice survived even longer than Pten−/−, and about 25% were still alive more than 60 days after the first drug treatment. To rule out the treatment toxicity , we treated WT mice with LY or KU at the same time; no WT mice died (Fig.5 H). To determine whether the role of Fak in Pten-null T-ALL was cell-intrinsic, we transplanted CD3+c-Kitlow cells from either Pten−/− or Pten−/−Fak−/− T-ALL, or WT mice (CD45.1+) into lethally-irradiated recipient mice (CD45.2+). Recipient mice were treated with vehicle, LY or KU from day 14 post-transplantation every other day until analysis or maximum 15 injections. LY and KU enhanced the survival of Pten−/−-recipient mice compared to vehicle treatment. Pten−/−Fak−/−-recipient mice survived longer than Pten−/−-recipient mice, whether treated with vehicle, LY, or KU. With LY or KU treatment, 25% or 40% of Pten−/−Fak−/−-recipient mice survived more than 120 days after the first drug treatment, respectively (Fig.5 I). To further rule out in vivo toxicity of LY, we treated Pten−/− and Pten−/−Fak−/− transgenic or transplanted T-ALL mice with another pan-PI3K inhibitor, GDC-0941 (GDC) (Folkes et al., 2008). Similar results were observed in treatments with GDC as with LY (Fig.S5 H, I). These data suggest that Fak deletion makes Pten-null T-ALL cells more sensitive to PI3K/AKT/mTOR inhibition in vivo.
FAK/NF-κB/Bc1-xL/Bc1-2 and PI3K/AKT/mTOR/Mcl-l Provide two Independent Survival Signals in PTEN-mutant Human T-ALL Cell Lines
When cultured on MG-coated plates, both FAK and AKT signals were activated in PTEN-null human T-ALL cell lines (JURKAT, CCRF-CEM, MOLT-4, and LOUCY), but not in PTEN-WT cell lines (CUTLL1, HPB-ALL, or KOPT-K1) (Fig.6A). This result was confirmed in JURKAT and HBP-ALL cells, when grown on OP9 cells, or on CLG/FN-coated plates (Fig.S6 A). Pten regulates the activity of Fak in NIH 3T3 cells (Tamura et al., 1998). To test whether FAK activation was also regulated by PTEN in human T-ALL cells, we expressed PTEN-WT, PTENC124S, PTENY138L, and PTENG129E in JURKAT cells cultured on MG-coated plates. PTENWT repressed the activities of both AKT and FAK; PTENC124S, a phosphatase-null mutant, did not inhibit either signal; PTENY138L, a protein phosphatase-inactive mutant, suppressed only AKT activity; the lipid phosphatase-null mutant, PTENG129E, restrained only FAK activity (Fig.6 B). Immunoprecipitation using PTEN or FAK antibodies in these transduced JURKAT cells demonstrated that PTEN could bind to FAK in a phosphatase activity-independent manner (Fig.6 C). The interaction of endogenous PTEN and FAK was verified in HPB-ALL cells (Fig.6 D). Though PTEN can bind FAK in cells, it is possible that PTEN dephosphorylated FAK indirectly. These data suggest that the activities of both AKT and FAK are regulated by PTEN in human T-ALL cells.
Figure 6. PI3K/AKT/mTOR/MCL-1 and FAK/NF-κB/BCL-xL/BCL-2 Represent Two Parallel Survival Signaling Pathways in PTEN-mutant Human T-ALL Cell Lines.
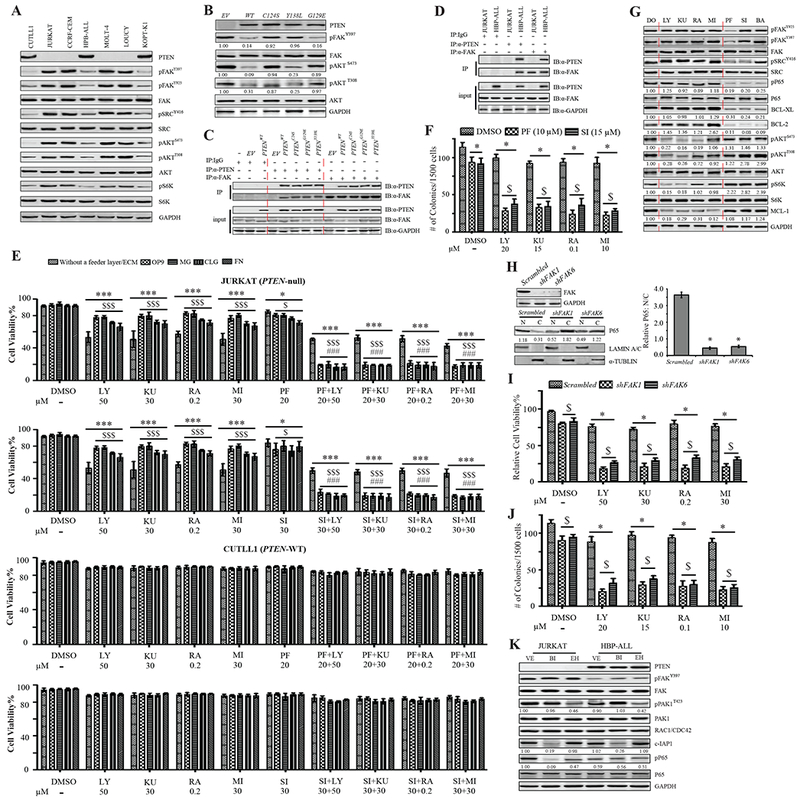
(A) PTEN, pFAK, pSRC, pAKT and pS6K expression in human T-ALL cell lines, CUTLL1, JURKAT, CCRF-CEM, HPB-ALL, MOLT-4, LOUCY, and KOPT-K1, cultured on MG-coated plates. (B) PTEN, pFAK and pAKT in PTENWT, or PTENC124S, PTENG129E, or PTENY138L-transduced JURKAT cells cultured on MG-coated plates; control: empty vector (EV) transduced cells. (C&D) The binding of PTEN and FAK in transduced JURKAT cells (C) and HBP-ALL cells (D). (E) Viability of human T-ALL cell lines in suspension, on OP9, or on MG/CLG/FN-coated plates, treated by DMSO (DO), LY, KU, RA, MI, PF, or LY+PF, KU+PF, RA+PF, MI+PF. Data shown are Mean ± S.D. ***, * compared to DO treatment, and $$$, $ compared to suspension culture, P<0.001, <0.05, respectively; ### compared to single inhibitor treatment, P<0.001. (F) Colony number of JURKAT cells treated with DO, LY, KU, RA, or MI, alone or in combination with PF or SI. Data shown are Mean ± S.D. * compared DO treatment, $ compared to single inhibitor treatment, P<0.001. (G) pFAK, pSRC, pP65, pAKT, pS6K, BCL-xL, BCL-2 and MCL-1 expression in JURKAT cells cultured on MG-coated plates treated by DO, LY, KU, RA, MI, PF, SI, or BA, respectively. (H) Nuclear (N) and cytoplasmic (C) P65 in JURKAT cells with knockdown of FAK by shRNA. Data shown are Mean ± S.D. * compared to scrambled, P<0.001. (I&J) Viability, cultured on MG-coated plates (I) and colony forming ability (J) of JURKAT cells to FAK inhibition by shRNA, treated with DO, LY, KU, RA, or MI. Data shown are Mean ± S.D. * compared to DO treatment, $ compared to scrambled, P<0.001. (K) The levels of pFAK, pPAK1, pP65, and the expression of PTEN, RAC1/CDC42, c-IAP1 in JURKAT and HBP-ALL cells. Images shown in western blot are representative of three to five independent experiments. All statistical values were determined by 2-way ANOVA.
To explore the effect of AKT and/or FAK signal inhibition on human T-ALL cell lines cultured in suspension, or on OP9, or MG/CLG/FN-coated plates, we treated the cells with PI3K/mTOR/MCL-1 and FAK/SRC inhibitors, which were titrated to optimize concentrations (Fig.S6 B, C), alone, or in combination. In suspension culture the survival of JURKAT (PTEN-null) cells was highly restrained by PI3K, mTOR, or MCL-1 inhibitors, but there was no further restrained effect with combined inhibition of both AKT and FAK signaling. When cultured on OP9 or MG/CLG/FN-coated plates, JURKAT cells were less sensitive to AKT signaling inhibition; which was further suppressed with combined AKT and FAK signaling inhibition. However, CUTLL1 (PTEN-WT) cells did not respond to the inhibition of AKT, FAK alone, or both (Fig.6 E). We confirmed this result with other PTEN-null (CCRF-CEM, MOLT-4, LOUCY) and PTEN-WT (HPB-ALL, KOPT-K1) human T-ALL cell lines (Fig.S6 D). The colony-forming ability of JURKAT cells was more dramatically reduced by the combined inhibition of AKT and FAK signaling compared to the inhibition of either alone (Fig.6 F). To study the molecular mechanisms of survival signals in PTEN-null human T-ALL cells, we treated JURKAT cells with PI3K/AKT/mTOR/MCL-1 inhibitors (LY, KU, RA, MI), or FAK signal inhibitors (PF, SRC inhibitor (SI)) cultured on MG-coated plates. Inhibition of PI3K/AKT signal did not change the expression of BCL-xL or BCL-2, but significantly reduced MCL-1. Inhibition of FAK signal reduced the expression of both BCL-xL and BCL-2; so did the restriction of NF-κB by Bay 11-7085 (BA). Similar to what we observed in Pten−/−Fak−/− T-ALL mouse cells, enhanced AKT signaling and further increased MCL-1 were also detected when FAK activity was suppressed in JURKAT cells (Fig.6 G).
As observed with pharmacologic inhibitors treatment, shRNA restriction of FAK, suppressed NF-κB activity, as shown by reduced protein level (Fig.6 H) and P65 nuclear localization (Fig.S6 E). Suppression of FAK by shRNA made JURKAT cells more sensitive to the inhibition of PI3K/AKT signal by LY, KU, RA, or MI treatment, displaying reduced viability on MG-coated plates (Fig.6 I) or OP9 (Fig.S6 F), and colony forming ability (Fig.6 J).
Activated FAK promotes NF-κB activation in a Rho/Rac-GTPase- or RIP1-dependent manner in cancers (Kamarajan et al., 2010; Tong and Tergaonkar, 2014). To determine which pathway mediated the activated Fak promoting NF-κB activity in Pten-null T-ALL cells, we tested the expression and activity of Rac1/Cdc42 (assessed by p-Pak1), which were not affected by Fak deletion (Fig.S4 B). Rac GTPase inhibitor, EHop-016 (EH), failed to repress NF-κB activity in JURKAT (PTEN-null) cells. However, a Smac mimetic (inhibiting NF-κB activity by repressing c-IAP1/2-mediated RIP1 ubiquitination (Wang et al., 2008)), birinapant (BI), restrained NF-κB activity in JURKAT cells. Neither inhibitor changed NF-κB activity in HBP-ALL (PTEN-WT) cells (Fig.6 K). This indicated that FAK activated NF-κB through RIP1, not Rho/Rac in PTEN-null T-ALL cells. To address whether the reduction in BCL-xL and BCL-2 expression was caused by inhibition of NF-κB, we treated JURKAT, KOPT-K1, and CCRF-CEM with BA (inhibiting NF-κB) or ABT-737 (BCL-xL/BCL-2 inhibitor) on MG-coated plates. In PTEN-mutant cell lines, JURKAT and CCRF-CEM, inhibition of NF-κB reduced BCL-xL/BCL-2, but inhibition of BCL-xL/BCL-2 did not affect NF-κB activity, as measured by pP65 and nuclear P65 (Fig.S7 A, B). This indicated that BCL-xL and BCL-2 were regulated by NF-κB in PTEN-null human T-ALL cell lines.
PTEN-mutant Human Primary T-ALL Cells are more Sensitive to Combined Treatment Using PI3K/mTOR and FAK Inhibitors than to Treatment with Either Inhibitor Alone
We collected and cultured primary T-ALL patient samples and CD3+ lymphocytes from healthy subjects on MG-coated plates, and assessed the activation of AKT and FAK signals. In two of three PTEN positive samples, patients 1 and 4, neither signal was activated. All PTEN-negative samples and one PTEN positive (patient 7) showed increased activation of both AKT and FAK signals (Fig.7 A). We considered that patient 7 might contain a loss-of-function PTEN-mutation, which is commonly associated with human T-ALL (Zuurbier et al., 2012), but this could not be determined due to limited cell availability. Cells were treated with PI3K/mTOR, FAK inhibitors alone, or in combination. The specific inhibitory activities of these chemicals on primary T-ALL cells were verified by western blot (Fig.7 B). Unlike healthy CD3+ cells, the survival of all primary T-ALL cells could be restricted to some extent by either PI3K/mTOR or FAK inhibition; however, PTEN-negative and patient 7 cells were significantly more sensitive to combined treatment (LY+PF/KU+PF) than to treatment with either single inhibitor, but PTEN-WT or healthy controls were not (Fig.7 C). These data suggest that simultaneous inhibition of both PI3K/AKT/mTOR and FAK/NF-κB signaling might represent a more effective treatment strategy for PTEN-null T-ALL.
Figure 7. PTEN-mutant Human Primary T-ALL Cells are more Sensitive to Combined Treatment Using PI3K/mTOR and FAK Inhibitors than to Either Inhibitor Alone.
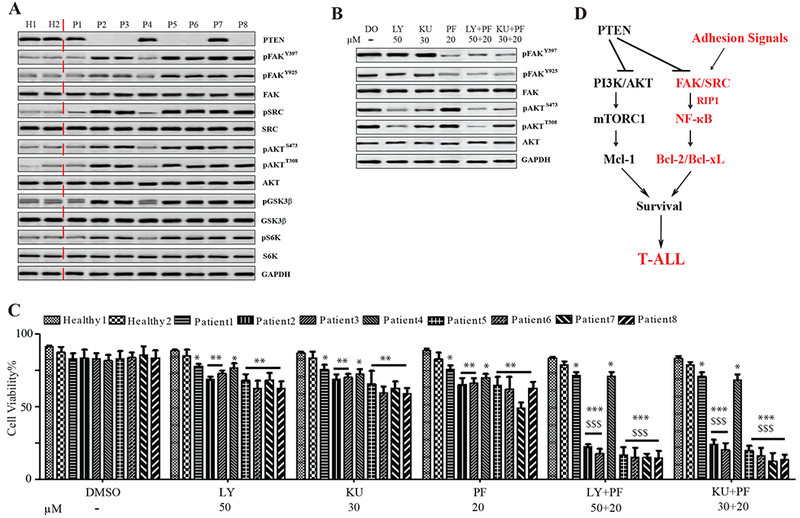
(A) PTEN, pFAK, pSRC, pAKT, pGSK3β and pS6K expression in primary human T-ALL cells cultured on MG-coated plates; controls: healthy CD3+ lymphocytes. (B) pFAK and pAKT in patient 2 samples (PTEN-null) treated with DMSO (DO), LY, KU, PF, or LY+PF, KU+PF. Images representative of three independent experiments. (C) Primary human T-ALL cell viability after treatment with DO, LY, KU, PF, or LY+PF, KU+PF; controls: healthy CD3+ lymphocytes. Data shown are Mean ± S.D. ***, **, * compared to healthy controls, P<0.001, 0.01, 0.05; $$$ compared to single inhibitor treatment, P<0.001, determined by 2-way ANOVA. (D) Schematic model of two parallel survival signals in PTEN-mutant T-ALL cells.
Discussion
In PTEN-null, but not in PTEN-WT murine and human T-ALL cells, both AKT and FAK signals are activated in vivo or when cultured on a feeder layer or ECM-coated plates. We determined that PI3K/AKT acts through mTOR, and FAK works via NF-κB, up-regulating distinct survival genes, MCL-1 and BCL-2/BCL-xL, respectively. Thus, two parallel survival signaling pathways are stimulated in PTEN-mutant T-ALL cells (Fig.7 D). Inhibition of PI3K/AKT/mTOR signaling induces apoptosis and represses the growth of T-ALL cells, which sensitizes drug-resistant T-ALL cells to conventional chemotherapy (Evangelisti et al., 2011; Piovan et al., 2013; Subramaniam et al., 2012). These inhibitors are currently in clinical trials in T-ALL patients, but no complete remission was reported with these inhibitors treatment (Rodon et al., 2013). We found that suppression of FAK further enhanced the inhibitory effects of PI3K/AKT signal restriction on PTEN-null T-ALL cells. FAK inhibitors are also in clinical trials for treatment of solid tumors and have been approved as relatively safe and promising drugs (Golubovskaya, 2014). Our study provides a strong rationale to treat PTEN-mutant T-ALL by co-inhibition of both AKT and FAK-stimulated parallel survival signals.
Both the lipid and protein phosphatase activities of PTEN were well demonstrated in earlier studies (Lee et al., 1999; Tamura et al., 1998). However, the pathophysiologic role downstream of PTEN’s protein phosphatase activity has been underestimated in leukemogenesis because inactivation of its phosphatase or constitutive activation of its lipid substrates is sufficient to induce tumors in mice (Kharas et al., 2010; Myers et al., 1998; Wang et al., 2010). Nevertheless, transgenic over-expression of active PI3K or AKT fails to faithfully replicate the Pten−/− phenotype, suggesting a PI3K/AKT-independent function of PTEN (Blanco-Aparicio et al., 2007). Several direct substrates of PTEN’s protein phosphatase, such as IRS1 and CREB, have been defined (Gu et al., 2011; Shi et al., 2014; Tibarewal et al., 2012). We determined that the ECM activates FAK in PTEN-mutant T-ALL cells, providing an additional layer of survival/proliferation signaling to promote the initiation and progression of leukemia, together with PI3K/AKT signaling. PTEN-mutant T-ALL cells are sensitive to inhibition of non-canonical PI3K signaling, both when cultured on MS5-DL feeder cells and in vivo (Subramaniam et al., 2012). Consistent with this report, we found that PI3K/AKT inhibitors are effective in killing PTEN-null but not PTEN-WT T-ALL cells both in vitro and in vivo. However, PTEN-null T-ALL cells are relatively resistant to these inhibitors when grown on stromal feeder cells or ECM-coated plates compared to suspension culture. Further study suggested that inactivation of both FAK and PI3K/AKT signals might provide a better treatment for PTEN-null T-ALL than inhibition of PI3K/AKT alone. Furthermore, in our study of primary human T-ALL cells, we began with 14 samples, of which 8 were able to grow on MG-coated plates. Of these 8 samples, 6 of them (75%) showed PTEN inactivation. Given that PTEN mutations are generally reported in 15-25% of T-ALL patients (Gutierrez et al., 2009; Jotta et al., 2010; Zuurbier et al., 2012), we believe that MG-coated culture selectively favors the growth of PTEN-null T-ALL cells.
Both MCL-1 and BCL-xL/BCL-2 are highly expressed in T-ALL cells, and they functionally compensate for each other to support LCs survival. Simultaneously inhibiting both MCL-1 and BCL-2/BCL-xL shows an additive effect in killing T-ALL LCs (Inuzuka et al., 2011). The activated AKT signal increases MCL-1 (Hsieh et al., 2010; Inuzuka et al., 2011; Jebahi et al., 2014), and we demonstrated that FAK/NF-κB signal activated by ECM of niches enhances BCL-xL/BCL-2 in PTEN-null T-ALL cells. Further studies of these signaling mediators could produce favorable drug target(s) for T-ALL treatment.
Experimental Procedures
Mice and drug treatments:
Fakfx/fx mice (Stock Number: 009967-UCD) were purchased from Mutant Mice Regional Resource Center. Mx-1-Cre+Ptenfx/+ mice were maintained in our laboratory (Zhang et al., 2011). Drug treatments were conducted as indicated. (See Supplementary Information)
T-ALL transplantation and in vivo treatments:
CD3+c-KitLow cells were isolated from Pten−/− or Pten−/−Fak−/− T-ALL, or WT mice (CD45.1+) and transplanted into lethally-irradiated recipient mice (CD45.2+) together with support BM cells (CD45.2+). Each mouse received 1×104 T-ALL cells and 2×105 support BM cells. Drug treatments were conducted as indicated. (See Supplementary Information).
Flow cytometry and antibodies:
Bone marrow, spleens, thymuses and peripheral blood were collected after the mice had been sacrificed. MNC were isolated from these tissues after red blood cell lysis. Cells were suspended in FACS buffer (1×PBS supplemented with 2% FBS) at a concentration of 1×107 cells/ml and aliquotted into flow cytometry tubes (100 μl per tube) for antibody staining. Antibodies used in this study were purchased from eBioscience Inc. (See Supplementary Information).
Supplementary Material
Significance.
We determined that focal adhesion kinase (FAK), another downstream target of PTEN, promotes a survival signal parallel to PBK/AKT/mTOR in PTEN-null T-ALL cells. Our data suggest that targeting both PI3K/AKT/mTOR and FAK may provide a more effective therapeutic strategy against T-ALL in patients with PTEN mutations.
Acknowledgements
We thank the staff of the Department of Comparative Medicine of Loyola University Chicago for animal care services. We gratefully received T-ALL cell lines from Drs. Adolfo Ferrando (Columbia University) and Yan Liu (Indiana University). Drs. Wei Qiu (Loyola University Chicago), Filippo Giancotti (Memorial Sloan-Kettering Cancer Center), and Nick Leslie (Heriot-Watt University) donated plasmids.
This work was supported by National Institutes of Health grant R01HL095896 to J.Z, R01HL087188 to N.Z-L. and P01CA154778 to M.N.
Footnotes
Disclosure of Potential Conflicts of Interest
The authors declare no conflicting financial interests.
References
- Blanco-Aparicio C, Renner O, Leal JF, and Carnero A (2007). PTEN, more than the AKT pathway. Carcinogenesis 28, 1379–1386. [DOI] [PubMed] [Google Scholar]
- Desgrosellier JS, and Cheresh DA (2010). Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 10, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despeaux M, Labat E, Gadelorge M, Prade N, Bertrand J, Demur C, Recher C, Bonnevialle P, Payrastre B, Bourin P, and Racaud-Sultan C (2011). Critical features of FAK-expressing AML bone marrow microenvironment through leukemia stem cell hijacking of mesenchymal stromal cells. Leukemia 25, 1789–1793. [DOI] [PubMed] [Google Scholar]
- Evangelisti C, Ricci F, Tazzari P, Chiarini F, Battistelli M, Falcieri E, Ognibene A, Pagliaro P, Cocco L, McCubrey JA, and Martelli AM (2011). Preclinical testing of the Akt inhibitor triciribine in T-cell acute lymphoblastic leukemia. J Cell Physiol 226, 822–831. [DOI] [PubMed] [Google Scholar]
- Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, et al. (2008). The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-t hieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem 51, 5522–5532. [DOI] [PubMed] [Google Scholar]
- Golubovskaya VM (2014). Targeting FAK in human cancer: from finding to first clinical trials. Frontiers in bioscience 19, 687–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Tamura M, and Yamada KM (1998). Tumor suppressor PTEN inhibits integrin- and growth factor-mediated mitogen-activated protein (MAP) kinase signaling pathways. J Cell Biol 143, 1375–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu T, Zhang Z, Wang J, Guo J, Shen WH, and Yin Y (2011). CREB is a novel nuclear target of PTEN phosphatase. Cancer Res 71, 2821–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JL (2010). Integrin signaling through FAK in the regulation of mammary stem cells and breast cancer. IUBMB Life 62, 268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, and Giancotti FG (2004). Integrin signalling during tumour progression. Nat Rev Mol Cell Biol 5, 816–826. [DOI] [PubMed] [Google Scholar]
- Guo W, Lasky JL, Chang CJ, Mosessian S, Lewis X, Xiao Y, Yeh JE, Chen JY, Iruela-Arispe ML, Varella-Garcia M, and Wu H (2008). Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature 453, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y, Dahlberg S, Neuberg D, Moreau LA, Winter SS, et al. (2009). High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood 114, 647–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AC, Costa M, Zollo O, Davis C, Feldman ME, Testa JR, Meyuhas O, Shokat KM, and Ruggero D (2010). Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell 17, 249–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Hofmann J, Lu Y, Mills GB, and Jaffe RB (2002). Inhibition of phosphatidylinositol 3’-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res 62, 1087–1092. [PubMed] [Google Scholar]
- Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, et al. (2011). SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 471, 104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jebahi A, Villedieu M, Petigny-Lechartier C, Brotin E, Louis MH, Abeilard E, Giffard F, Guercio M, Briand M, Gauduchon P, et al. (2014). PI3K/mTOR dual inhibitor NVP-BEZ235 decreases Mcl-1 expression and sensitizes ovarian carcinoma cells to Bcl-xL-targeting strategies, provided that Bim expression is induced. Cancer Lett 348, 38–49. [DOI] [PubMed] [Google Scholar]
- Jotta PY, Ganazza MA, Silva A, Viana MB, da Silva MJ, Zambaldi LJ, Barata JT, Brandalise SR, and Yunes JA (2010). Negative prognostic impact of PTEN mutation in pediatric T-cell acute lymphoblastic leukemia. Leukemia 24, 239–242. [DOI] [PubMed] [Google Scholar]
- Kalaitzidis D, Sykes SM, Wang Z, Punt N, Tang Y, Ragu C, Sinha AU, Lane SW, Souza AL, Clish CB, et al. (2012). mTOR complex 1 plays critical roles in hematopoiesis and Pten-loss-evoked leukemogenesis. Cell Stem Cell 11, 429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamarajan P, Bunek J, Lin Y, Nunez G, and Kapila YL (2010). Receptor-interacting protein shuttles between cell death and survival signaling pathways. Mol Biol Cell 21, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M (2006). Nuclear factor-kappaB in cancer development and progression. Nature 441, 431–436. [DOI] [PubMed] [Google Scholar]
- Kharas MG, Okabe R, Ganis JJ, Gozo M, Khandan T, Paktinat M, Gilliland DG, and Gritsman K (2010). Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 115, 1406–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopleva MY, and Jordan CT (2011). Leukemia stem cells and microenvironment: biology and therapeutic targeting. J Clin Oncol 29, 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechertier T, and Hodivala-Dilke K (2012). Focal adhesion kinase and tumour angiogenesis. J Pathol 226, 404–412. [DOI] [PubMed] [Google Scholar]
- Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, and Pavletich NP (1999). Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 99, 323–334. [DOI] [PubMed] [Google Scholar]
- Mitra SK, and Schlaepfer DD (2006). Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol 18, 516–523. [DOI] [PubMed] [Google Scholar]
- Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP, and Tonks NK (1998). The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci U S A 95, 13513–13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, et al. (2004). PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 6, 117–127. [DOI] [PubMed] [Google Scholar]
- Piovan E, Yu J, Tosello V, Herranz D, Ambesi-Impiombato A, Da Silva AC, Sanchez-Martin M, Perez-Garcia A, Rigo I, Castillo M, et al. (2013). Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell 24, 766–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva Y, Gillen KM, Gerald W, Beggs HE, Reichardt LF, and Giancotti FG (2009). Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J Clin Invest 119, 252–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recher C, Ysebaert L, Beyne-Rauzy O, Mansat-De Mas V, Ruidavets JB, Cariven P, Demur C, Payrastre B, Laurent G, and Racaud-Sultan C (2004). Expression of focal adhesion kinase in acute myeloid leukemia is associated with enhanced blast migration, increased cellularity, and poor prognosis. Cancer Res 64, 3191–3197. [DOI] [PubMed] [Google Scholar]
- Rodon J, Dienstmann R, Serra V, and Tabernero J (2013). Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol 10, 143–153. [DOI] [PubMed] [Google Scholar]
- Schaller MD (2010). Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J Cell Sci 123, 1007–1013. [DOI] [PubMed] [Google Scholar]
- Shi Y, Wang J, Chandarlapaty S, Cross J, Thompson C, Rosen N, and Jiang X (2014). PTEN is a protein tyrosine phosphatase for IRS1. Nat Struct Mol Biol 21, 522–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, and Mak TW (1998). Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95, 29–39. [DOI] [PubMed] [Google Scholar]
- Subramaniam PS, Whye DW, Efimenko E, Chen J, Tosello V, De Keersmaecker K, Kashishian A, Thompson MA, Castillo M, Cordon-Cardo C, et al. (2012). Targeting nonclassical oncogenes for therapy in T-ALL. Cancer Cell 21, 459–472. [DOI] [PubMed] [Google Scholar]
- Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, and Yamada KM (1998). Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 280, 1614–1617. [DOI] [PubMed] [Google Scholar]
- Tibarewal P, Zilidis G, Spinelli L, Schurch N, Maccario H, Gray A, Perera NM, Davidson L, Barton GJ, and Leslie NR (2012). PTEN protein phosphatase activity correlates with control of gene expression and invasion, a tumor-suppressing phenotype, but not with AKT activity. Sci Signal 5, ra18. [DOI] [PubMed] [Google Scholar]
- Tong L, and Tergaonkar V (2014). Rho protein GTPases and their interactions with NFkappaB: crossroads of inflammation and matrix biology. Bioscience reports 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemula S, Ramdas B, Hanneman P, Martin J, Beggs HE, and Kapur R (2010). Essential role for focal adhesion kinase in regulating stress hematopoiesis. Blood 116, 4103–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk A, Li J, Xin J, You D, Zhang J, Liu X, Xiao Y, Breslin P, Li Z, Wei W, et al. (2014). Co-inhibition of NF-kappaB and JNK is synergistic in TNF-expressing human AML. J Exp Med 211, 1093–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Karikomi M, Naidu S, Rajmohan R, Caserta E, Chen HZ, Rawahneh M, Moffitt J, Stephens JA, Fernandez SA, et al. (2010). Allele-specific tumor spectrum in pten knockin mice. Proc Natl Acad Sci U S A 107, 5142–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Du F, and Wang X (2008). TNF-alpha induces two distinct caspase-8 activation pathways. Cell 133, 693–703. [DOI] [PubMed] [Google Scholar]
- Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, and Morrison SJ (2006). Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 441, 475–482. [DOI] [PubMed] [Google Scholar]
- Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, Haug JS, Rupp D, Porter-Westpfahl KS, Wiedemann LM, et al. (2006). PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 441, 518–522. [DOI] [PubMed] [Google Scholar]
- Zhang J, Xiao Y, Guo Y, Breslin P, Zhang S, Wei W, Zhang Z, and Zhang J (2011). Differential requirements for c-Myc in chronic hematopoietic hyperplasia and acute hematopoietic malignancies in Pten-null mice. Leukemia 25, 1857–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuurbier L, Petricoin EF 3rd, Vuerhard MJ, Calvert V, Kooi C, Buijs-Gladdines JG, Smits WK, Sonneveld E, Veerman AJ, Kamps WA, et al. (2012). The significance of PTEN and AKT aberrations in pediatric T-cell acute lymphoblastic leukemia. Haematologica 97, 1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.