

Abstract
We previously identified five single nucleotide polymorphisms (SNPs) at four susceptibility loci for diffuse large B-cell lymphoma (DLBCL) in individuals of European ancestry through a large genome-wide association study (GWAS). To further elucidate genetic susceptibility to DLBCL, we sought to validate two loci at 3q13.33 and 3p24.1 that were suggestive in the original GWAS with additional genotyping. In the meta-analysis (5662 cases and 9237 controls) of the four original GWAS discovery scans and three replication studies, the 3q13.33 locus (rs9831894; minor allele frequency [MAF] = 0.40) was associated with DLBCL risk [odds ratio (OR) = 0.83, P = 3.62 × 10−13]. rs9831894 is in linkage disequilibrium (LD) with additional variants that are part of a super-enhancer that physically interacts with promoters of CD86 and ILDR1. In the meta-analysis (5510 cases and 12 817 controls) of the four GWAS discovery scans and four replication studies, the 3p24.1 locus (rs6773363; MAF = 0.45) was also associated with DLBCL risk (OR = 1.20, P = 2.31 × 10−12). This SNP is 29 426-bp upstream of the nearest gene EOMES and in LD with additional SNPs that are part of a highly lineage-specific and tumor-acquired super-enhancer that shows long-range interaction with AZI2 promoter. These loci provide additional evidence for the role of immune function in the etiology of DLBCL, the most common lymphoma subtype.
Introduction
With an aggressive clinical course, diffuse large B-cell lymphoma (DLBCL) is the most common non-Hodgkin lymphoma (NHL) subtype, accounting for ~ 30% of adult NHL (1, 2). Family history of lymphoma has been consistently associated with risk of developing lymphoma (3), and in a large pooled study from the International Lymphoma Epidemiology Consortium (InterLymph), risk of DLBCL was associated with a family history of both NHL [odds ratio (OR) = 1.8, 95% confidence intervals (CI) 1.5–2.3] and Hodgkin lymphoma (OR = 2.1, 95%CI 1.4–3.2) (4). In a population-based registry study, first-degree relatives of DLBCL cases had a 9.8-fold (95%CI 3.1–31) increased risk of DLBCL (5).
In the first genome-wide association study (GWAS) of DLBCL of European ancestry, we identified (6) five independent single nucleotide polymorphisms (SNPs) in four loci at 6p25.3 (EXOC2), 6p21.33 (HLA-B), 2p23.3 (NCOA1) and 8q24.21 (near PVT1 and MYC). Three of these SNPs (6p25.3, 6p21.33 and 8q24.21) were also significantly associated with DLBCL in an East Asian population (7). We estimate that common SNPs, both established and unknown, are likely to explain ~ 16% of the variance in DLBCL risk (6). To further elucidate genetic susceptibility to DLBCL, we sought to validate two loci at 3q13.33 and 3p24.1 that were suggestive in our original GWAS (6). Based on in silico bioinformatics analysis, both were confirmed as DLBCL risk loci tagged by rs9831894 and rs6773363, respectively. These two risk loci were located at super-enhancers that physically interacted with genes involved in immune response.
Results
In a meta-analysis of the four original GWAS scans, rs9831894 (minor allele frequency [MAF] = 0.40) at 3q13.33 was associated with DLBCL risk (OR = 0.84, P = 4.52 × 10−9). The SNP replicated in an independent set of cases and controls (OR = 0.80, P = 4.17 × 10−5) with the combined discovery and replication showing an inverse association with DLBCL risk (OR = 0.83, P = 3.62 × 10−13) (Table 1).
Table 1.
Association of new loci with risk of DLBCL
| SNP | Nearest gene | Position | Stage | Study | i/g | Information content | Controls (N) | Cases (N) | Effect allele | ref allele | Effect allele MAF, controls | Effect allele MAF, cases | OR | P | Phet | I2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs9831894 | CD86 | 121 800 487 | Discovery | NCI | i | 0.99 | 6221 | 2660 | C | A | 0.40 | 0.35 | 0.81 | 3.08 × 10−9 | ||
| GELA | g | 1 | 525 | 549 | C | A | 0.39 | 0.37 | 0.94 | 0.473 | ||||||
| MAYO | g | 1 | 172 | 393 | C | A | 0.39 | 0.35 | 0.82 | 0.151 | ||||||
| UCSF2 | g | 1 | 748 | 254 | C | A | 0.38 | 0.37 | 0.94 | 0.593 | ||||||
| META (6) | 7666 | 3856 | 0.84 | 4.52 × 10 –9 | 0.30 | 18.3 | ||||||||||
| Replication | MAYO | g | 1 | 1053 | 782 | C | A | 0.40 | 0.34 | 0.79 | 4.57 × 10−4 | |||||
| MDA | g | 1 | 370 | 371 | C | A | 0.39 | 0.33 | 0.78 | 0.022 | ||||||
| MSKCC | g | 1 | 148 | 653 | C | A | 0.33 | 0.32 | 0.93 | 0.617 | ||||||
| META | 1571 | 1806 | 0.80 | 4.17 × 10 –5 | 0.54 | 0 | ||||||||||
| Combined discovery and replication | 9237 | 5662 | 0.83 | 3.62 × 10 –13 | 0.51 | 0 | ||||||||||
| rs6773363 | EOMES | 27 793 632 | Discovery | NCI | i | 0.98 | 6220 | 2660 | C | T | 0.45 | 0.49 | 1.16 | 2.34 × 10−5 | ||
| GELA | i | 0.98 | 525 | 548 | C | T | 0.45 | 0.45 | 0.99 | 0.917 | ||||||
| MAYO | i | 0.98 | 171 | 393 | C | T | 0.47 | 0.51 | 1.22 | 0.129 | ||||||
| UCSF2 | i | 0.94 | 747 | 253 | C | T | 0.42 | 0.52 | 1.48 | 1.57 × 10−4 | ||||||
| META (6) | 7663 | 3854 | 1.17 | 3.68 × 10 –7 | 0.03 | 65.24 | ||||||||||
| Replication | NCI | g | 1 | 3349 | 196 | C | T | 0.47 | 0.48 | 1.05 | 0.67 | |||||
| MSKCC | g | 1 | 372 | 428 | C | T | 0.45 | 0.54 | 1.41 | 2.01 × 10−3 | ||||||
| MDA | g | 1 | 374 | 373 | C | T | 0.45 | 0.51 | 1.30 | 0.01 | ||||||
| MAYO | g | 1 | 1059 | 659 | C | T | 0.47 | 0.53 | 1.31 | 1.59 × 10–4 | ||||||
| META | 5154 | 1656 | 1.27 | 3.78 × 10 −7 | 0.22 | 32.05 | ||||||||||
| Combined discovery and replication | 12 817 | 5510 | 1.20 | 2.31 × 10 −12 | 0.03 | 53.76 |
MAF, minor allele frequency; i/g, imputed/genotyped; and Phet, P-value for heterogeneity
In eQTL analyses with GTEx data, compared to the reference alleles, alternative alleles at sentinel SNP rs9831894 and three linked SNPs were associated with increased CD86 expression in the testis, but no association was detected in the HapMap lymphoblastoid cell lines (Supplementary Table S1). We used the R package coloc to perform the colocalization analysis of GWAS signals and GTEx eQTL signals in testis, which provided strong evidence for a shared causal variant (posterior probability PP.H4 = 0.996) underlying the association with both DLBCL risk and CD86 expression variation at this locus.
To identify the potential causal variants at the 3q13.33 locus, we intersected rs9831894 and the variant rs28876421 in linkage disequilibrium (LD, r2 ≥ 0.5) with publicly available epigenetic data generated by chromatin immunoprecipitation sequencing (ChIP-seq), DNase I digestion and sequencing (DNase-seq) and transposase-accessible chromatin using sequencing (ATAC-seq). While rs28876421 showed no overlap with the epigenetic marks, rs9831894 overlapped epigenetic marks in multiple samples, in a super-enhancer identified in lymphoblastoid cell lines, DLBCL and high-grade B-cell lymphoma (Fig. 1,Supplementary Fig. S1). The enhancer spanning GWAS SNP rs9831894 resided in an open chromatin region in GM12878 with DNase-seq/ATAC-seq signal and showed binding of 24 TFs in GM12878 (Supplementary Fig. S1) and two TFs [IRF4 and its cofactor basic leucine zipper ATF-like transcription factor (BATF)] in DLBCL cell lines (Supplementary Fig. S1). IRF4 and BATF are known to play a role in transcriptional regulation of activated B-cell-like DLBCL (8, 9). The alternative C allele at rs9831894 was predicted to disrupt CBX5 and MYBL2 binding motif (Supplementary Fig. S2).
Figure 1.
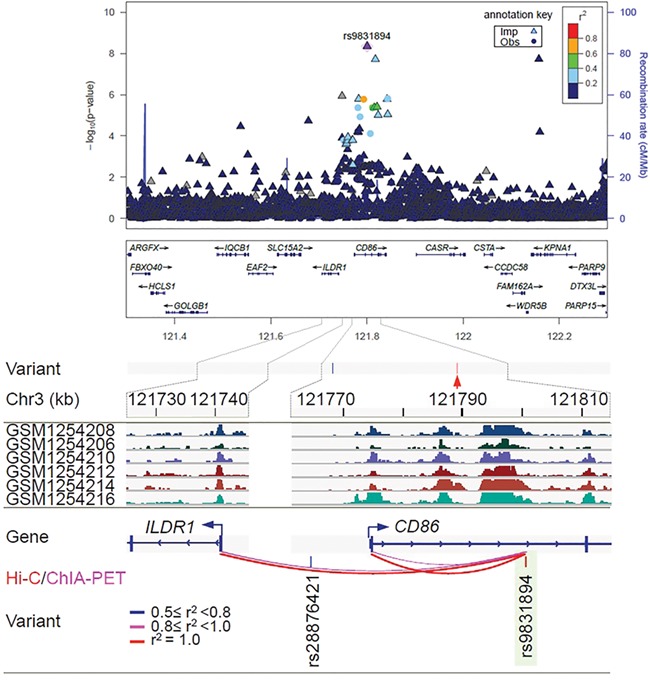
Regional association plot and epigenetic features of rs9831894 at independent loci associated with the risk of diffuse large B-cell lymphoma (DLBCL). At the top of the figure is the regional association plot of rs9831894; −log10 association P-values from the discovery log-additive genetic model for all SNPs in the region (dots and triangles). The lead SNPs are shown in purple. Recombination rates estimated from 1000 Genomes are plotted in blue. The SNPs surrounding the most significant SNP are color coded to reflect their correlation with that SNP. Pairwise r2 values are from 1000 Genomes European data. Locations of recombination hotspots are depicted by peaks corresponding to the rate of recombination (blue vertical lines). The rest of figure shows the epigenetic features: Epigenetic features at the 3q13.33 risk locus. Tag SNP rs9831894 (red arrow) has only one linked variant (r2 ≥ 0.5). The GWAS SNP rs9831894 is in a super-enhancer in DLBCL patient biopsies. The region spanning rs9831894 (highlighted in green) showed looping interactions with the promoters of CD86 and ILDR1.
Active enhancers can transcribe unstable nontranscripts, namely enhancer RNAs (eRNAs) that are believed to play a role in mediating enhancer-promoter interaction. To find evidence for the activity of this super-enhancer, we used a catalogue of 43 011 putative transcriptional enhancers supported by the FANTOM cap analysis of gene expression data (10). The analysis identified eRNAs transcribed from a 212-bp region (121800469–121 800 680 bp) within the super-enhancer that covers the GWAS sentinel SNP rs9831894 (at 121800487 bp) in both B and T cells. This enhancer was also found to transcribe in GM12878 (11). In GM12878, this enhancer, like the ILDR1 and CD86 promoter it interacts with (see below), showed the binding of RNA polymerase II (RNAPII), P300, RAD21, MED1 and multiple TFs, as typically seen for a transcribed enhancer. Collectively, our analysis revealed the possible mechanistic roles of rs9831894 and in the etiology of DLBCL. Finally, we used three chromatin interaction datasets to infer the target gene(s) for the super-enhancer, including promoter capture Hi-C (CHi-C) data in 17 primary blood cell types, Hi-C and chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) with RNAPII data in GM12878. Within the super-enhancer, the rs9831894-carrying-enhancer region interacted with the promoters of CD86 and immunoglobulin-like domain containing receptor 1 (ILDR1), which was supported by ChIA-PET (six supporting read pairs) and Hi-C data (false discovery rate [FDR]= 7.51 × 10−4) (Fig. 1).
In the meta-analysis of the original discovery GWAS scans, rs6773363 (MAF = 0.45) at 3p24.1 showed a suggestive (but not genome-wide significant) association with DLBCL risk (OR = 1.17, P = 3.68 × 10−7). The SNP replicated in an independent set of cases and controls (OR = 1.27, P = 3.78 × 10−7) with the combined discovery and replication showing a genome-wide significant positive association with DLBCL risk (OR = 1.20, P = 2.31 × 10−12) (Table 1).
In eQTL analyses, rs6773363 alleles were not associated with gene expression in the HapMap lymphoblastoid cell lines or other tissues (Supplementary Table S1). Using two additional lists of eQTLs identified in whole blood (12, 13), we identified the linked SNP rs12497690 (the last SNP shown in Fig. 2) as an eQTL association with EOMES expression (P = 1.16 × 10−9). Located within an intergenic region, rs12497690 overlapped none of the epigenetic marks. At 3p24.1, 43 of the 49 correlated variants (r2 ≥ 0.5) starting from rs34269949, including tag SNP rs6773363, did not overlap any of the active marks in B-cells and DLBCL (Supplementary Fig. S3). Four (rs2371108, rs2371109, rs2887944 and rs9866625) with r2 > 0.7 with rs6773363 lie in an 11.6-kb super-enhancer with extensive H3K27ac occupancy (Fig. 2). This super-enhancer was identified in biopsies from DLBCL (14), high-grade B-cell lymphoma (15), mantle cell lymphoma (15, 16) and small lymphocytic lymphoma patients (15), although generally less enriched with H3K27ac in the latter two (Fig. 2,Supplementary Fig. S3), but depleted in the sorted malignant B cells (CD19+) from nine follicular lymphoma patient lymph node biopsies (17) (data not shown). In addition, none of the 11 DLBCL cell lines (14 datasets) and only one of the nine mantle cell lymphoma cell lines (JVM2) had the super-enhancer. The presence of this super-enhancer in the patient biopsies but not in cell lines is not unexpected, as long-term cell culturing is known to alter the epigenome (18, 19).
Figure 2.
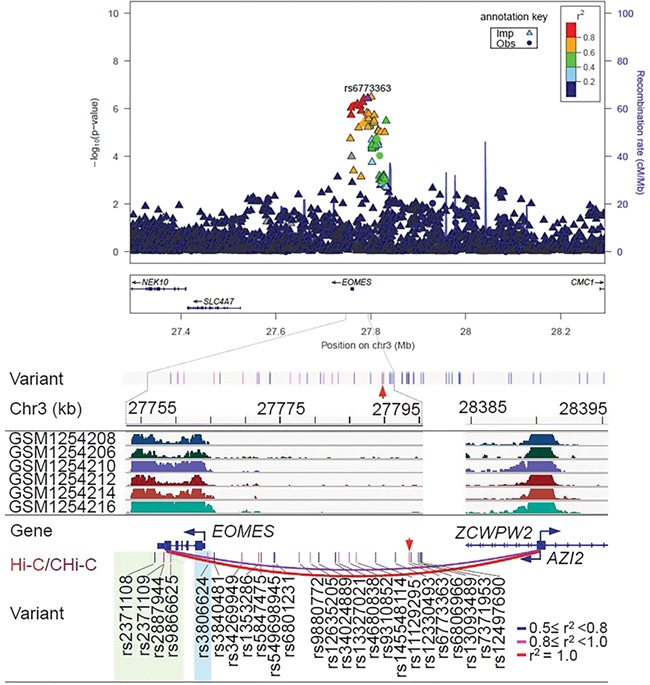
Regional association plot and epigenetic features of rs6773363 at independent loci associated with the risk of diffuse large B cell lymphoma (DLBCL). At the top of the figure is the regional association plot of rs6773363; −log10 association P-values from the discovery log-additive genetic model for all SNPs in the region (dots and triangles). The lead SNPs are shown in purple. Recombination rates estimated from 1000 genomes are plotted in blue. The SNPs surrounding the most significant SNP are color-coded to reflect their correlation with that SNP. Pairwise r2 values are from 1000 Genomes European data. Locations of recombination hotspots are depicted by peaks corresponding to the rate of recombination (blue vertical lines). The rest of figure shows the epigenetic features: Epigenetic features at the 3p24.1 risk locus. This 74 961-bp risk locus contains 51 variants (r2 ≥ 0.5), with red arrow indicating the tag SNP rs6773363. For simplicity, only 25 of the variants were displayed (bottom panel). Four variants (highlighted in green, r2 > 0.7) overlapped an 11.6-kb super-enhancer in DLBCL. The 2.3-kb region spanning the four SNPs physically interacted with AZI2. Promoter, which was identified by Hi-C in GM12878 and by CHi-C in 15 blood cell types, including activated total CD4+, erythroblasts, fetal thymus, macrophages M0, macrophages M1, macrophages M2, megakaryocytes, monocytes, naïve CD4+, naïve CD8+, naïve B, nonactivated total CD4+, total CD4+, total CD8+ and total B cells. In addition, rs3806624 (highlighted in light blue), located 417 bp upstream of the transcription start site of EOMES, is in the edge of the super-enhancer with lower levels of H3K27ac.
To better understand the chromatin dynamics in this region, we examined the 127 reference epigenomes, of which 98 had H3K27ac data (20), a catalog of super-enhancers identified in 86 tissue and cell types (21), and H3K27ac profiles from 19 lymphoblastoid cell lines and seven primary B-cell samples. Only H1 BMP4-derived mesendoderm cultured cells (EID: E004) and primary natural killer cells from peripheral blood (E046) showed this super-enhancer. Rather, in the 127 epigenomes, 99 had a broad H3K27me3 signal extending over 10–26.6 kb (e.g. CD14+ monocyte reference E124), often forming two distinct subdomains (up to 3.6 and 4.1 kb in size) together with H3K4me1, H3K4me3 or both. These analyses suggested the presence of an activated, poised or repressed chromatin state in this broad region depending on cell type. For example, the primary B-cells are in a poised chromatin state within the two subdomains, showing DNase-seq signal, H3K4me1/3 and H3K27me3, and in a repressed state elsewhere with H3K27me3 alone. Therefore, this super-enhancer is highly lineage-specific and tumor-acquired, reflecting a phenomenon observed in many other cancer types.
Using ATAC-seq data from 14 blood cell types (22), we found that SNP rs2371108 is located within an open chromatin region in nine blood cell types (Supplementary Fig. S4). The alternative T allele at this SNP was predicted to disrupt the binding motifs for six TFs, including FOX3, GLI1, KLF3, PKNX2, ZIC1 and ZIC3 (Supplementary Fig. S4).
Lastly, we used Hi-C data in GM12878 and CHi-C data from 17 blood cell types to identify the potential target gene(s) for the 2.3-kb region that represents part of the super-enhancer in DLBCL and harbors the four linked SNPs with r2 > 0.7 (highlighted in green, Fig. 2). The strongest long-range interactions were revealed with the region covering the promoters of 5-azacytidine induced 2 (AZI2) and ZCWPW2 located ~ 630 kb away in GM12878 (supported by 40 pairs of reads, FDR = 0) and in 15 of the 17 blood cell types (CHiCAGO scores = 5.3–22.9, except neutrophils and endothelial precursors), most notably in total B, naive B and all six types of T cells. To support the inferred physical interaction, CHi-C data from both GM12878 and Jurkat cell lines suggested the interaction between the same enhancer region harboring rheumatoid arthritis associated SNPs with the promoter of AZI2 (23). AZI2 is involved in the activation of the NF-κB signaling pathway (24). GWAS SNP rs6773363 is also in LD with rs3806624 (r2 = 0.94), which is located −417 bp upstream of the transcription start site of eomesodermin (EOMES), an autoimmune disease-associated transcription factor. SNP rs3806624 is located in an open chromatin region in hematopoietic stem cell (Supplementary Fig. S4). As indicated by the ChromHMM profile, rs3806624 overlaps a bivalent enhancer in B-cells enriched with H3K4me1/3 and H3K27me3; it also overlaps the super-enhancer in some of the DLBCL and high-grade B-cell lymphoma patients that shows interaction with the promoter of AZI2. Finally, we calculated the enrichment of DLBCL SNPs in H3K27ac peaks across different cell types, which indicates that DLBCL risk variants are enriched in enhancers from DLBCL patients, H1 BMP4-derived mesendoderm and three types of blood cells (CD3+, CD4+ and CD56+) (P < 0.01, Supplementary Fig. S5).
Discussion
GWAS-identified risk variants are highly enriched in gene regulatory regions, particularly within enhancers, of disease-relevant cell types (21, 25). The molecular mechanisms by which they perturb target gene expression often involve alteration of TF recruitment, enhancer-promoter looping interaction and higher order chromatin structure (26, 27). In this large meta-analysis, we were able to identify two novel SNPs that are associated with DLBCL risk. At both risk loci, we mapped the candidate casual variants to lineage-restricted super-enhancers that showed long-range interactions with genes involved in immune response. The rs9831894-defined risk locus at 3q13.33 interacted with CD86 and ILDR1. CD86 is a member of the immunoglobulin superfamily that encodes a type I membrane protein, is expressed by antigen-presenting cells and is the ligand for cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and CD28 antigen, two proteins at the cell surface of T-cells. Binding of CD86 with CTLA-4 negatively regulates T-cell activation and diminishes the immune response, while binding of CD86 with CD28 antigen is a costimulatory signal for activation of T-cells (28, 29). Costimulation through CD86 can lead to proliferation and secretion of antibodies that may help lymphomas to evade immune surveillance (30). Moreover, stimulation through CD86 can modulate the humoral response by transducing positive and negative signals in B-cells, which may control the progression of B-cell lymphomas (30). Loss of CD86 expression in DLBCL samples has been associated with decreased tumor-infiltrating lymphocytes and subsequently shorter relapse-free survival (31).
The rs67733633-tagged risk locus 3p24.1 resides near EOMES and interacts with AZI2. EOMES is a member of the T-box gene family (32), and is a key regulator in cell-mediated immunity and CD8+ T-cell differentiation, which is involved in defense against viral infections (33). Inherited lymphoproliferative disorders associated with autoimmunity have demonstrated that EOMES is crucial for lymphoproliferation due to Fas-deficiency (34–36). Extranodal natural killer/T (NK/T)-cell and peripheral T-cell lymphomas have shown overexpression of EOMES (37). EOMES was also found to be differentially methylated in specific cell lineages and stages of hematopoietic cascade, whose expression was inversely correlated with methylation of 5′ untranslated region, suggesting that this gene is also regulated by DNA methylation (38). Aberrant methylation of transcription factor genes, such as EOMES, is frequently observed in DLBCL and might have a functional role during tumorigenesis (38). AZI2 contributes to the activation of NF-κB signaling pathway and antiviral innate immunity. Interestingly, this DLBCL risk locus also harbors SNPs associated with another two B-cell malignancies, chronic lymphocytic leukemia (rs9880772, r2 = 0.90) and Hodgkin lymphoma (rs3806624, r2 = 0.91) (39), and with rheumatoid arthritis (rs3806624, r2 = 0.91) (23). The regulatory SNPs are predicted to target EOMES and AZI2 promoter in these diseases. The identification of pleiotropic risk locus suggests a shared genetic susceptibility at 3p24.1 and common gene targets involved in immune response and risk of several B-cell malignancies and rheumatoid arthritis.
We identified two or more candidate regulatory variants at each of the two risk loci. The establishment of disease causality at single variant level has been particularly challenging (40). Further work is needed to verify which of the linked regulatory variants are causal in DLBCL, for example, by examining allele-specific protein binding/histone modifications or by sequentially deleting the enhancer region via CRISPR/Cas9 systems (26, 41). Future studies in DLBCL also need to consider molecular subtypes, such as cell of origin. In summary, in this follow-up analysis of our initial GWAS, we have identified two additional loci associated with risk of DLBCL. In particular, we identified key immune-related genes targeted by the two risk loci and the binding of several immune-related TFs to the super-enhancer at 3q13.33 locus. These loci provide additional evidence for the role of dysregulation of immune function in susceptibility to DLBCL.
Materials and Methods
This analysis builds off a previously published GWAS (6). Briefly, the prior GWAS study consisted of four scans (3857 cases and 7666 controls) of European ancestry conducted on different genotyping platforms (Supplementary Table S2). We imputed common SNPs for each study on the basis of 1000 Genomes Project release version 3 using IMPUTE2, conducted a meta-analysis, followed by additional genotyping of nine promising SNPs in 1359 cases and 4557 controls. Each genotyping array underwent rigorous quality-control metrics as previously detailed (6).
rs9831894 was the most significant SNP at the 3q13.33 locus (P = 4.52 × 10−9); however, the SNP failed design with Taqman and was unable to be analyzed in the replication stage. Here, we were able to genotype this SNP using the Sequenom platform on an independent set of 1806 DLBCL cases and 1571 controls from studies at Mayo Clinic, MD Anderson, and Memorial Sloan Kettering (Supplementary Fig. S6). For the 3p24.1 locus, rs6773363 failed to be replicated in the original study; however, we later found that this was due to an analytic coding error in one of the four replication studies, where the reference allele and risk allele were switched. Here, we corrected the error and conducted genotyping on an additional 659 DLBCL cases and 1059 controls from the Mayo Clinic, enlarging the sample size of the independent replication to 1656 cases and 5154 controls (Supplementary Table S3, Supplementary Fig. S6).
Logistic regression was used to estimate ORs, using the additive model and adjusting for age and gender (and significant eigenvectors in the discovery set). Meta-analysis of the discovery and replication was conducted using the fixed-effects inverse variance method based on the β estimates and standard errors from each study.
To understand the possible functional roles of the two risk loci, we performed in silico analyses of lists of eQTL data from GTEx release V7 (42), and two additional lists of eQTLs identified by RNA-seq of whole blood. The first list is from 463 cases of major depressive disorder and 459 healthy individuals of European ancestry (12). The case/control structure introduces no noticeable bias in eQTL detection. The second list is from 2116 healthy adults in four Dutch cohorts (13). To understand whether a single casual variant exists at the 3q13.33 risk locus, we performed colocalization analysis of GWAS summary statistics and GTEx eQTL signals using the coloc.abf function in the R package coloc (43). This function uses Bayes test to estimate the posterior probability (PP.H4) of a shared casual variant associated with the two traits. We included the common variants between GWAS and CD86 eQTLs within the 400-kb region centered on the tag SNP rs9831894 in the test. PP.H4 ≥ 0.8 was considered significant (43). We also analyzed chromatin accessibility, chromatin interaction and ChIP-seq data (Supplemental Table S4, more details below).
We extracted 48 and 1 variants from the 1000 Genomes Project phase 3 release (r2 ≥ 0.5, EUR ethnic group) that were in LD with rs6773363 and rs9831894, respectively. A majority (92.2%) of the variants were in intergenic regions, with the remainder in introns or three prime untranslated regions, suggesting that their roles in the etiology of DLBCL are likely regulatory. To test this hypothesis, we examined the overlap between the 51 variants and a collection of publicly available DNase-seq, ATAC-seq and ChIP-seq data and further identified the potential target genes using chromatin interaction data from Hi-C, CHi-C and ChIA-PET with RNAPII.
Chromatin accessibility and ChIP-seq data in lymphoblastoid cell lines, primary B-cells, and five types of B-cell lymphomas (Supplemental Table S4), ATAC-seq data for 14 blood cell types from healthy donors, as well as the 127 reference epigenomes were obtained from publicly available data. For histone modifications, we focused on those that preferentially occur in promoters (H3 lysine 4 trimethylation, H3K4me3), enhancers (H3 lysine 4 monomethylation and lysine 27 acetylation, H3K4me1 and H3K27ac) or repressed regions (H3 lysine 27 trimethylation, H3K27me3). The 127 reference epigenome data were generated by the Roadmap and ENCODE epigenomics projects (20, 44) and downloaded from http://egg2.wustl.edu/roadmap/data/byFileType. GM12878 DNase-seq data and ChIP-seq data for the four histone modifications, chromatin regulators (CHD1, CHD2 and EP300), as well as for 74 TFs and DNA-binding proteins, were from the ENCODE project (44), which are available at http://hgdownload.cse.ucsc.edu/goldenpath/hg19/encodeDCC/. GM12878 ATAC-seq data (GSM1155959) was from Buenrostro et al. (45). In addition, H3K4me1, H3K4me3 and H3K27ac ChIP-seq data from 19 lymphoblastoid cell lines (GSE50893) (46) were used. B-cell epigenetic data (Supplemental Table S4) included DNase-seq data (1 dataset) and ChIP-seq data for CREBBP (1 dataset), H3K4me1 (2 datasets), H3K4me3 (2 datasets), H3K27ac (7 datasets) and H3K27me3 (3 datasets). The paired-end ATAC-seq data (sra files) for 14 blood cell types from healthy donors were downloaded from NCBI Gene Expression Omnibus under the accession GSE74912 (22). For the same donor and cell type, data from replicates were combined. Finally, we used two DNase-seq datasets and 38 ChIP-seq datasets for BRD4, H3K4me3, H3K27me3, and TFs and DNA-binding proteins in DLBCL cell lines, as well as 55 H3K27ac datasets from lymphoma cell lines and patient samples (Supplemental Table S4). ATAC-seq, DNase-seq and ChIP-seq data were processed with the Hi-ChIP pipeline (47). In brief, reads were mapped to the hg19 reference genome using Burrows–Wheeler Alignment tool (BWA, v0.7.10) (48). For paired-end reads, only properly mapped pairs with one or both ends uniquely mapped (mapping quality score ≥20) were retained; for single-end reads, only uniquely mapped reads with a minimum mapping quality score of 20 were kept. Duplicates were removed using Picard MarkDuplicates command (http://broadinstitute.github.io/picard/). Peaks were called using model-based analysis of ChIP-Seq (MACS, v2.0.10) (49), with the parameter settings ‘-f BAM -g hs --keep-dup all -q 0.01 --nomodel’. To identify whether variants were present in peak regions, the two tag SNPs and their linked SNPs/INDELs from the 1000 Genomes Project (r2 ≥ 0.5, EUR ethnic group) were intersected with the above epigenetic marks using BEDTools (50).
To identify whether the alterative allele alters a TF binding motif, we used the Find Individual Motif Occurrences (FIMO) program (P = 1 × 10−4) to scan the 100-bp sequence spanning each SNP (51). The position weight matrix was compiled from four TF motif databases that include JASPAR (http://jaspar.binf.ku.dk/html/DOWNLOAD/JASPAR_CORE/pfm/nonredundant/), ENCODE motifs (http://compbio.mit.edu/encode-motifs/) (52), UniPROBE (http://thebrain.bwh.harvard.edu/uniprobe/downloads.php/) (53) and HOCOMOCO (http://hocomoco.autosome.ru/downloads) (54).
To calculate the enrichment of SNPs in H3K27ac peaks across different cell types, we performed a permutation test with 100 000 iterations, as described by (55) with minor modifications. We used the four SNPs on the DLBCL H3K27ac peak at the 3p24.1 risk locus and the two SNPs at the 3q13.33 risk locus. The same number of EUR MAF-matched SNPs was randomly sampled from the 1000 Genomes Project phase 3 release, excluding those in the coding exons and in the TSS +/−2 kb regions. The sampled SNPs were intersected with H3K27ac peaks from DLBCL patient biopsy and 98 of the 127 reference epigenomes with H3K27ac data. The P-value was estimated as the number of permutations with overlap greater than or equal to the observed value plus one divided by 100 001.
To understand the dynamics of super-enhancers identified in both risk loci, we compared the H3K27ac peaks in lymphoma with those from the 127 reference epigenomes (20) and with a catalog of super-enhancers from 86 cell and tissue types (21). As eRNAs are proposed to reflect enhancer activity, we further compared the enhancers to a list of 43 011 putative transcriptional enhancers derived from the FANTOM cap analysis of gene expression data across 808 human samples (10). Three chromatin interaction datasets were used to infer the potential target genes for the risk variants. Of those, GM12878 Hi-C reads (MboI, GSE63525) (56) were mapped to the hg19 reference genome using Bowtie 2 (57) and significant interactions were identified with HOMER (http://homer.ucsd.edu/homer/interactions/) using a 5-kb bin size and FDR ≤ 0.01. For promoter CHi-C data in 17 primary blood cell types (58), regions of significant interactions with CHiCAGO scores ≥5 were downloaded from https://osf.io/u8tzp/. Finally, a list of RNAPII-mediated chromatin interaction regions identified by ChIA-PET in GM12878 were downloaded from Gene Expression Omnibus (GSM1872887) (59).
Supplementary Material
Acknowledgements
Dr G.K. was supported by the National Institutes of Health grant, R25 CA92049 (Mayo Cancer Genetic Epidemiology Training Program). Replication genotyping was supported by R01 CA200703 (to JRC), the Center for Translational and Public Health Genomics (to X.W.), MD Anderson’s Cancer Center Support Grant P30CA016672, MSKCC Core grant NIH P30CA008748, the Lymphoma Genetics Research Fund (KO), and the Intramural Research Program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH. The original GWAS was supported by the Intramural Research Program, Division of Cancer Epidemiology and Genetics, NCI, NHI. Individual study support is included in the Supplementary Materials.
Conflicts of Interest statement
There are no conflicts of interest.
Authorship Contributions
Organized and designed the study: G.K., M.A.T.H., J.V., S.I.B., H.G., J.M., S.S.W., A.N., S.L.S., X.W., K.E.S., C.F.S., N.R., S.J.C. and J.R.C.
Conducted and supervised de novo genotyping of samples: G.K., M.A.T.H., J.V., Y.Y., S.L.S., X.W. and J.R.C.
Contributed to the design and execution of statistical analysis: G.K., M.A.T.H., J.V., S.I.B., S.L.S., X.W. and J.R.C.
Contributed to the bioinformatics analysis: H.Y., V.S. and Y.A.
Wrote the first draft of the manuscript: G.K., M.A.T.H., J.V., S.S.W., A.N., S.L.S., K.E.S., C.F.S. and J.R.C.
Conducted the epidemiological studies or contributed samples or data: M.A.T.H., J.V., S.I.B., H.G., J.M., S.S.W., A.M., A.R.B.W., Q.L., M.M., R.D.J., L.R.T., M.P.P., C.M.V., D.A., A.Z.J., S.C., Y.Z., K.O., G.S., X.W., K.E.S., G.S., C.F.S., S.L.S., N.R., S.J.C. and J.R.C.
All authors contributed to the writing of the manuscript.
REFERENCES
- 1. Beham-Schmid C. (2017) Aggressive lymphoma 2016: revision of the WHO classification. Memo, 10, 248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Swerdlow S.H., Campo E., Pileri S.A., Harris N.L., Stein H., Siebert R., Advani R., Ghielmini M., Salles G.A., Zelenetz A.D. et al. (2016) The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood, 127, 2375–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cerhan J.R. and Slager S.L. (2015) Familial predisposition and genetic risk factors for lymphoma. Blood, 126, 2265–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morton L.M., Slager S.L., Cerhan J.R., Wang S.S., Vajdic C.M., Skibola C.F., Bracci P.M., de Sanjose S., Smedby K.E., Chiu B.C. et al. (2014) Etiologic heterogeneity among non-Hodgkin lymphoma subtypes: the InterLymph non-Hodgkin lymphoma subtypes project. J. Natl. Cancer Inst. Monogr., 2014, 130–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goldin L.R., Bjorkholm M., Kristinsson S.Y., Turesson I. and Landgren O. (2009) Highly increased familial risks for specific lymphoma subtypes. Br. J. Haematol., 146, 91–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cerhan J.R., Berndt S.I., Vijai J., Ghesquieres H., McKay J., Wang S.S., Wang Z., Yeager M., Conde L., de Bakker P.I. et al. (2014) Genome-wide association study identifies multiple susceptibility loci for diffuse large B cell lymphoma. Nat. Genet., 46, 1233–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bassig B.A., Cerhan J.R., Au W.-Y., Kim H.N., Sangrajrang S., Hu W., Tse J., Berndt S.I., Zheng T., Zhang H. et al. (2015) Genetic susceptibility to diffuse large B-cell lymphoma in a pooled study of three eastern Asian populations. Eur. J. Haematol., 95, 442–448. [DOI] [PubMed] [Google Scholar]
- 8. Hallek M., Cheson B.D., Catovsky D., Caligaris-Cappio F., Dighiero G., Dohner H., Hillmen P., Keating M., Montserrat E., Chiorazzi N. et al. (2018) iwCLL guidelines for diagnosis, indications for treatment, response assessment and supportive management of CLL. Blood, 131, 2745–2760. [DOI] [PubMed] [Google Scholar]
- 9. Care M.A., Cocco M., Laye J.P., Barnes N., Huang Y., Wang M., Barrans S., Du M., Jack A., Westhead D.R. et al. (2014) SPIB and BATF provide alternate determinants of IRF4 occupancy in diffuse large B-cell lymphoma linked to disease heterogeneity. Nucleic Acids Res., 42, 7591–7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Andersson R., Gebhard C., Miguel-Escalada I., Hoof I., Bornholdt J., Boyd M., Chen Y., Zhao X., Schmidl C., Suzuki T. et al. (2014) An atlas of active enhancers across human cell types and tissues. Nature, 507, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F. et al. (2012) Landscape of transcription in human cells. Nature, 489, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Battle A., Mostafavi S., Zhu X., Potash J.B., Weissman M.M., McCormick C., Haudenschild C.D., Beckman K.B., Shi J., Mei R. et al. (2014) Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res., 24, 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhernakova D.V., Deelen P., Vermaat M., van Iterson M., van Galen M., Arindrarto W., van 't Hof P., Mei H., van Dijk F., Westra H.J. et al. (2017) Identification of context-dependent expression quantitative trait loci in whole blood. Nat. Genet., 49, 139–145. [DOI] [PubMed] [Google Scholar]
- 14. Chapuy B., McKeown M.R., Lin C.Y., Monti S., Roemer M.G., Qi J., Rahl P.B., Sun H.H., Yeda K.T., Doench J.G. et al. (2013) Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell, 24, 777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ryan R.J.H., Drier Y., Whitton H., Cotton M.J., Kaur J., Issner R., Gillespie S., Epstein C.B., Nardi V., Sohani A.R. et al. (2015) Detection of enhancer-associated rearrangements reveals mechanisms of oncogene dysregulation in B-cell lymphoma. Cancer Discov., 5, 1058–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ryan R.J.H., Petrovic J., Rausch D.M., Zhou Y., Lareau C.A., Kluk M.J., Christie A.L., Lee W.Y., Tarjan D.R., Guo B. et al. (2017) A B cell regulome links notch to downstream oncogenic pathways in small B cell lymphomas. Cell Rep., 21, 784–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koues O.I., Kowalewski R.A., Chang L.W., Pyfrom S.C., Schmidt J.A., Luo H., Sandoval L.E., Hughes T.B., Bednarski J.J., Cashen A.F. et al. (2015) Enhancer sequence variants and transcription-factor deregulation synergize to construct pathogenic regulatory circuits in B-cell lymphoma. Immunity, 42, 186–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sharma N.L., Massie C.E., Ramos-Montoya A., Zecchini V., Scott H.E., Lamb A.D., MacArthur S., Stark R., Warren A.Y., Mills I.G. et al. (2013) The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell, 23, 35–47. [DOI] [PubMed] [Google Scholar]
- 19. Zhu J., Adli M., Zou J.Y., Verstappen G., Coyne M., Zhang X., Durham T., Miri M., Deshpande V., De Jager P.L. et al. (2013) Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell, 152, 642–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roadmap Epigenomics Consortium, Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J. et al. (2015) Integrative analysis of 111 reference human epigenomes. Nature, 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-Andre V., Sigova A.A., Hoke H.A. and Young R.A. (2013) Super-enhancers in the control of cell identity and disease. Cell, 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Corces M.R., Buenrostro J.D., Wu B., Greenside P.G., Chan S.M., Koenig J.L., Snyder M.P., Pritchard J.K., Kundaje A., Greenleaf W.J. et al. (2016) Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet., 48, 1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martin P., McGovern A., Orozco G., Duffus K., Yarwood A., Schoenfelder S., Cooper N.J., Barton A., Wallace C., Fraser P. et al. (2015) Capture Hi-C reveals novel candidate genes and complex long-range interactions with related autoimmune risk loci. Nat. Commun., 6, 10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fujita F., Taniguchi Y., Kato T., Narita Y., Furuya A., Ogawa T., Sakurai H., Joh T., Itoh M., Delhase M. et al. (2003) Identification of NAP1, a regulatory subunit of IkappaB kinase-related kinases that potentiates NF-kappaB signaling. Mol. Cell. Biol., 23, 7780–7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Farh K.K., Marson A., Zhu J., Kleinewietfeld M., Housley W.J., Beik S., Shoresh N., Whitton H., Ryan R.J., Shishkin A.A. et al. (2015) Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature, 518, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kandaswamy R., Sava G.P., Speedy H.E., Bea S., Martin-Subero J.I., Studd J.B., Migliorini G., Law P.J., Puente X.S., Martin-Garcia D. et al. (2016) Genetic predisposition to chronic lymphocytic leukemia is mediated by a BMF super-enhancer polymorphism. Cell Rep., 16, 2061–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schmiedel B.J., Seumois G., Samaniego-Castruita D., Cayford J., Schulten V., Chavez L., Ay F., Sette A., Peters B. and Vijayanand P. (2016) 17q21 asthma-risk variants switch CTCF binding and regulate IL-2 production by T cells. Nat. Commun., 7, 13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Girard T., Gaucher D., El-Far M., Breton G. and Sekaly R.P. (2014) CD80 and CD86 IgC domains are important for quaternary structure, receptor binding and co-signaling function. Immunol. Lett., 161, 65–75. [DOI] [PubMed] [Google Scholar]
- 29. Van Gool S.W., Vandenberghe P., de Boer M. and Ceuppens J.L. (1996) CD80, CD86 and CD40 provide accessory signals in a multiple-step T-cell activation model. Immunol. Rev., 153, 47–83. [DOI] [PubMed] [Google Scholar]
- 30. Suvas S., Singh V., Sahdev S., Vohra H. and Agrewala J.N. (2002) Distinct role of CD80 and CD86 in the regulation of the activation of B cell and B cell lymphoma. J. Biol. Chem., 277, 7766–7775. [DOI] [PubMed] [Google Scholar]
- 31. Stopeck A.T., Gessner A., Miller T.P., Hersh E.M., Johnson C.S., Cui H., Frutiger Y. and Grogan T.M. (2000) Loss of B7.2 (CD86) and intracellular adhesion molecule 1 (CD54) expression is associated with decreased tumor-infiltrating T lymphocytes in diffuse B-cell large-cell lymphoma. Clin. Cancer Res., 6, 3904–3909. [PubMed] [Google Scholar]
- 32. Yi C.H., Terrett J.A., Li Q.Y., Ellington K., Packham E.A., Armstrong-Buisseret L., McClure P., Slingsby T. and Brook J.D. (1999) Identification, mapping, and phylogenomic analysis of four new human members of the T-box gene family: EOMES, TBX6, TBX18, and TBX19. Genomics, 55, 10–20. [DOI] [PubMed] [Google Scholar]
- 33. Pearce E.L., Mullen A.C., Martins G.A., Krawczyk C.M., Hutchins A.S., Zediak V.P., Banica M., DiCioccio C.B., Gross D.A., Mao C.A. et al. (2003) Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science, 302, 1041–1043. [DOI] [PubMed] [Google Scholar]
- 34. Drappa J., Vaishnaw A.K., Sullivan K.E., Chu J.L. and Elkon K.B. (1996) Fas gene mutations in the Canale-smith syndrome, an inherited lymphoproliferative disorder associated with autoimmunity. N. Engl. J. Med., 335, 1643–1649. [DOI] [PubMed] [Google Scholar]
- 35. Fisher G.H., Rosenberg F.J., Straus S.E., Dale J.K., Middleton L.A., Lin A.Y., Strober W., Lenardo M.J. and Puck J.M. (1995) Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell, 81, 935–946. [DOI] [PubMed] [Google Scholar]
- 36. Kinjyo I., Gordon S.M., Intlekofer A.M., Dowdell K., Mooney E.C., Caricchio R., Grupp S.A., Teachey D.T., Rao V.K., Lindsten T. et al. (2010) Cutting edge: Lymphoproliferation caused by Fas deficiency is dependent on the transcription factor eomesodermin. J. Immunol., 185, 7151–7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang S., Li T., Zhang B., Nong L. and Aozasa K. (2011) Transcription factors engaged in development of NK cells are commonly expressed in nasal NK/T-cell lymphomas. Hum. Pathol., 42, 1319–1328. [DOI] [PubMed] [Google Scholar]
- 38. Ivascu C., Wasserkort R., Lesche R., Dong J., Stein H., Thiel A. and Eckhardt F. (2007) DNA methylation profiling of transcription factor genes in normal lymphocyte development and lymphomas. Int. J. Biochem. Cell Biol., 39, 1523–1538. [DOI] [PubMed] [Google Scholar]
- 39. Law P.J., Sud A., Mitchell J.S., Henrion M., Orlando G., Lenive O., Broderick P., Speedy H.E., Johnson D.C., Kaiser M. et al. (2017) Genome-wide association analysis of chronic lymphocytic leukaemia, Hodgkin lymphoma and multiple myeloma identifies pleiotropic risk loci. Sci. Rep., 7, 41071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Flister M.J., Tsaih S.W., O'Meara C.C., Endres B., Hoffman M.J., Geurts A.M., Dwinell M.R., Lazar J., Jacob H.J. and Moreno C. (2013) Identifying multiple causative genes at a single GWAS locus. Genome Res., 23, 1996–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sanjana N.E., Wright J., Zheng K., Shalem O., Fontanillas P., Joung J., Cheng C., Regev A. and Zhang F. (2016) High-resolution interrogation of functional elements in the noncoding genome. Science, 353, 1545–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.GTEx Consortium (2017) Genetic effects on gene expression across human tissues. Nature, 550, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Giambartolomei C., Vukcevic D., Schadt E.E., Franke L., Hingorani A.D., Wallace C. and Plagnol V. (2014) Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet., 10, e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y. and Greenleaf W.J. (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods, 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kasowski M., Kyriazopoulou-Panagiotopoulou S., Grubert F., Zaugg J.B., Kundaje A., Liu Y., Boyle A.P., Zhang Q.C., Zakharia F., Spacek D.V. et al. (2013) Extensive variation in chromatin states across humans. Science, 342, 750–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yan H., Evans J., Kalmbach M., Moore R., Middha S., Luban S., Wang L., Bhagwate A., Li Y., Sun Z. et al. (2014) HiChIP: a high-throughput pipeline for integrative analysis of ChIP-Seq data. BMC Bioinformatics, 15, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li H. and Durbin R. (2009) Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol., 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Quinlan A.R. and Hall I.M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Grant C.E., Bailey T.L. and Noble W.S. (2011) FIMO: scanning for occurrences of a given motif. Bioinformatics, 27, 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kheradpour P. and Kellis M. (2014) Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res., 42, 2976–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hume M.A., Barrera L.A., Gisselbrecht S.S. and Bulyk M.L. (2015) UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res., 43, D117–D122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kulakovskiy I.V., Vorontsov I.E., Yevshin I.S., Soboleva A.V., Kasianov A.S., Ashoor H., Ba-Alawi W., Bajic V.B., Medvedeva Y.A., Kolpakov F.A. et al. (2016) HOCOMOCO: expansion and enhancement of the collection of transcription factor binding sites models. Nucleic Acids Res., 44, D116–D125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Parker S.C., Stitzel M.L., Taylor D.L., Orozco J.M., Erdos M.R., Akiyama J.A., van Bueren K.L., Chines P.S., Narisu N., Program N.C.S. et al. (2013) Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc. Natl. Acad. Sci. U. S. A., 110, 17921–17926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S. et al. (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Langmead B. and Salzberg S.L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Javierre B.M., Burren O.S., Wilder S.P., Kreuzhuber R., Hill S.M., Sewitz S., Cairns J., Wingett S.W., Varnai C., Thiecke M.J. et al. (2016) Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell, 167, 1369–1384.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tang Z., Luo O.J., Li X., Zheng M., Zhu J.J., Szalaj P., Trzaskoma P., Magalska A., Wlodarczyk J., Ruszczycki B. et al. (2015) CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell, 163, 1611–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.