

Abstract
Genetic diversity provides a rich repository for understanding the role of proteostasis in the management of the protein fold in human biology. Failure in proteostasis can trigger multiple disease states, affecting both human health and lifespan. Niemann-Pick C1 (NPC1) disease is a rare genetic disorder triggered by mutations in NPC1, a multi-spanning transmembrane protein that is trafficked through the exocytic pathway to late endosomes (LE) and lysosomes (Ly) (LE/Ly) to globally manage cholesterol homeostasis. Defects triggered by >300 NPC1 variants found in the human population inhibit export of NPC1 protein from the endoplasmic reticulum (ER) and/or function in downstream LE/Ly, leading to cholesterol accumulation and onset of neurodegeneration in childhood. We now show that the allosteric inhibitor JG98, that targets the cytosolic Hsp70 chaperone/co-chaperone complex, can significantly improve the trafficking and post-ER protein level of diverse NPC1 variants. Using a new approach to model genetic diversity in human disease, referred to as variation spatial profiling, we show quantitatively how JG98 alters the Hsp70 chaperone/co-chaperone system to adjust the spatial covariance (SCV) tolerance and set-points on an amino acid residue-by-residue basis in NPC1 to differentially regulate variant trafficking, stability, and cholesterol homeostasis, results consistent with the role of BCL2-associated athanogene family co-chaperones in managing the folding status of NPC1 variants. We propose that targeting the cytosolic Hsp70 system by allosteric regulation of its chaperone/co-chaperone based client relationships can be used to adjust the SCV tolerance of proteostasis buffering capacity to provide an approach to mitigate systemic and neurological disease in the NPC1 population.
Introduction
Understanding how genetic diversity in the human population contributes to health and disease in the individual (1) is a problem that is on the leading edge of medical practice (2–4), but remains to be effectively implemented from experimental (drug development), computational (an understanding diversity in the population) and clinical perspectives (5–7). This challenge has led to the need for a high definition (3) or individualized medicine (6, 8) approaches (https://allofus. nih.gov/) that address the diversity in population that make each one of us unique, a diversity that contributes to more than 10 000 familial and somatic rare diseases (9), including cancer (10, 11) and neurodegenerative disorders (1, 12, 13), that have deep genetic and epigenetic origins (14).
Niemann-Pick type C (NPC) is an inherited rare autosomal recessive Mendelian disorder that is characterized by the accumulation of cholesterol and other lipids in the late endosome (LE) and lysosome (Ly) compartments (LE/Ly) (15–17). It is caused by genetic variants in Niemann-Pick C1 (NPC1) or Niemann-Pick C2 (NPC2), with former variants found in 95% of recorded cases. These two proteins are essential for managing the distribution of cholesterol at the plasma membrane through the LE/Ly cholesterol recycling center. There are >300 NPC1 variants recorded in the clinic (https://medgen.medizin.uni-tuebingen.de/NPC-db2/) that result in the accumulation of cholesterol in LE/Ly. Because of the importance of cholesterol homeostasis in the brain (18), cholesterol accumulation can lead to rapid neurological decline with the median age of death as 13 years (19). Currently, there is no effective treatment or cure for NPC1 disease.
NPC1 is a multi-spanning transmembrane (TM) protein responsible for cholesterol efflux from LE/Ly. NPC1 contains three luminal domains (sterol-binding N-terminal luminal domain 1 [SNLD1], middle luminal domain 3 [MLD3], C-terminal luminal domain 5 [CLD5]) and three TM domains (N-terminal TM domain 2 [NTMD2], sterol-binding TM domain 4 [STMD4], C-terminal TM domain 6 [CTMD6]) with 13 helices (Fig. 1A) (20–22). NPC1 utilizes the MLD3 to mediate the transfer of NPC2-bound cholesterol to the SNLD1 of NPC1 (21–23). The most common disease-causing variant, I1061T (10–20% of the NPC1 population), is located in the CLD5. I1061T exhibits similar mRNA levels to that seen with wild-type (WT) NPC1, yet produces a misfolded protein that is accumulated in and targeted for degradation in the endoplasmic reticulum (ER), resulting in a >85% deficiency of LE/Ly-localized NPC1 in all tissues, and thereby creating a loss-of-function condition (24, 25). Overexpression of NPC1-I1061T in NPC1-deficient cells was able to provide a partial correction of the disease phenotype (24). In agreement with these data, it has also been shown that pharmacological treatments that lead to upregulation of NPC1 variant expression, including ryanodine receptor antagonists (26), treatment with oxysterols that bind to and stabilize NPC1 (27), reduced expression of TMEM97 (28), and treatment with histone deacetylase (HDAC) inhibitors (29, 30), partially abrogate the NPC1-I1061T phenotype in heterologous expression systems, in patient fibroblasts and in some cases, mouse models. These findings highlight the general concept that modulation of the folding environment can facilitate disease correction (31–34).
Figure 1.

The impact of Hsp70 allosteric inhibitors on the trafficking and stability of NPC1-I1061T. (A) Domains in the NPC1 protein. (B–D) A series of Hsp70 allosteric inhibitors were tested in U2OS-SRA-shNPC1 cells transfected with NPC1-I1061T. All sample lysates were treated with Endo H and analyzed by SDS-PAGE. The slower migrating band is the Endo HR species representative of the post-ER glycoform of NPC1 protein. The faster migrating band is Endo HS species representative of the immature glycoform of NPC1 protein located in the ER. (E) Quantification of Western-blot results in B–D. TrIdx (left panel) is calculated as Endo HR/(Endo HR + Endo HS) defining the trafficking efficiency of NPC1-I1061T. Fold-change of Endo HR relative to DMSO after JG98 treatment (right panel) represents the change of protein level of the post-ER glycoforms of NPC1 after compound treatment reported as a specific activity of the total μg protein loaded on the gel.
Proteostasis (35) is responsible for differentially maintaining a healthy proteome environment (36–39) under normal homeostatic conditions and in response to folding stress in all cells (40). Protein folding is actively managed, in part, by the heat shock cognate (Hsc) and inducible heat shock protein (Hsp) 70 chaperone/co-cochaperone system (referred to collectively as the ‘Hsp70 system’) that comprises nearly 5% of cytoplasmic protein pool (24, 41, 42). The Hsp70 system consists of a network of proteins mediating nascent protein synthesis, folding, stability, degradation, and manages aggregation states in the cell through autophagic pathways (11, 37, 41–44). Hsp70 is assisted by Hsp40 co-chaperones that stimulate the ATPase activity of Hsp70 (45–49) and nuclear exchange factors (NEFs), such as the BCL2-associated athanogene (BAG) family members, which coordinate adenosine diphosphate (ADP) release from Hsp70 to drive the sequential steps mediating client protein folding (37, 50–52). The Hsp70 system has been recently suggested to impact the stability of NPC1-I1061T (53–56), but it is not clear how this link might be directly targeted with small molecules to rescue NPC1 processing. One possibility is a class of allosteric Hsp70 inhibitors which disrupt the protein-protein interactions between Hsp70 and BAG family co-chaperones that might facilitate the synthesis and folding of NPC1 (50, 57–61). These allosteric inhibitors (41, 42, 60) have been shown to restore normal proteostasis in multiple disease models, for example, Dengue and Zika viruses (62, 63), hepatitis C virus (64), myopathy (65), tauopathy (66) and cancer (58–60).
Herein, we screened a panel of Hsp70 system allosteric inhibitors (11, 41, 50, 51, 58, 60, 62–64) to assess their impact on NPC1-I1061T trafficking. We identified the compound JG98 (11, 60), as a potent trafficking corrector of NPC1-I1061T. JG98 differentially impacts the trafficking defect seen with 58 additional NPC1 variants, suggesting a more universal but variable role of this small molecule on NPC1 maturation imposed by variant diversity in the NPC1 population. To understand how the Hsp70 system manages the properties of diverse NPC1 variants distributed across the entire polypeptide chain, we applied variation spatial profiling (VSP) (1), a Gaussian process regression (GPR)-based machine learning (ML) (GPR-ML) algorithm we have recently developed that utilizes a sparse collection of variants recorded in the world-wide patient population to predict sequence-to-function-to-structure relationships across the entire gene at atomic resolution (1). VSP establishes the phenotypic role of each amino acid residue in polypeptide function as a matrix of weighted proximal spatial interactions dictating function, a new biological principle referred to as spatial covariance (SCV) (1). VSP uses a set of known variant residues and their related functions to predict a quantitative value (referred to as a SCV ‘set-point’) for all residues in the polypeptide chain. These set-point values are associated with an uncertainty that assigns the SCV ‘tolerance’ for all known and unknown variants in globally predicting function across the fold (1). Using VSP of 58 variants, we assigned the differential SCV response of trafficking and post-ER protein levels for each residue in response JG98 (1). SCV relationships reveal that JG98 impacts both the modularity and plasticity of the NPC1 fold to facilitate trafficking. These results are supported by observations showing that correction of the trafficking defect of the I1061T variant occurs in response to silencing of BAG family co-chaperones. Furthermore, we demonstrate that JG98 restores the pathways critical for cholesterol homeostasis that are disrupted in response to I1061T. We propose that SCV tolerance characterized through VSP (1) can be used to define the response of NPC1 residues to the dynamic composition of the Hsp70-NEF (BAG)-client complexes, providing for the first time an individualized approach to leverage the proteostasis program to restore function for the >300 variants contributing to NPC1 disease. In general, SCV principles captured by VSP (1) provide a universal platform to understand the impact of genomic (1) and epigenomic diversity (30) in health and disease of the population to implement application of individualized medicine (3).
Results
Hsp70 system allosteric inhibitors improve trafficking and post-ER level of NPC1-I1016T
NPC1 is a multi-spanning TM protein (Fig. 1A) with a prominent luminal orientation dedicated to the management of cholesterol in the LE/Ly compartment (24, 67, 68). The trafficking of NPC1 can be monitored by assessing the sensitivity or resistance of its 14-N-linked glycans to endoglycosidase H (Endo H) digestion. ER-localized NPC1 glycans are sensitive to Endo H digestion (Endo HS) whereas the glycans on NPC1 molecules exported to the Golgi are modified by Golgi-resident enzymes and become resistant to Endo H digestion (Endo HR), thereby allowing for distinct migration of the retained and exported fractions on SDS-PAGE. ~70% of WT-NPC1 migrates as Endo HR species (Fig. 1B), reflecting its steady-state distribution in post-ER compartments. In contrast, more than 80% of NPC1-I1061T exhibits Endo H sensitivity (Endo HS), demonstrating that this misfolded variant is retained in the ER. Here, we quantify trafficking in two ways. First, while some band laddering is observed, reflecting the variable processing of the 14-N-linked glycans, we use the Endo HR band seen in WT NPC1 (Fig. 1B) as a quantitative measure of ER export for calculating a trafficking index (TrIdx) (Endo HR/(Endo HR + Endo HS)), a metric which reflects the efficiency of ER export. Second, we quantify the fold change in Endo HR band intensity as the amount of Endo HR per mg protein compared with a solvent or mock control, referred to as the ‘absolute Endo HR glycoforms’. Absolute Endo HR glycoforms report directly on the total NPC1 that is transported to the post-ER compartments.
Using this approach, we tested a panel of Hsp70 system allosteric inhibitors (60) for effects on the trafficking and stability of NPC1-I1061T. We tested JG-13, JG-40, JG-98, JG-194, JG-231, JG-234, JG-312 and JG-345, as well as a negative control compound JG258, for effects on TrIdx and levels of absolute Endo HR glycoforms (Fig. 1B–D). Quantitation of the TrIdx (Fig. 1E, left panel) revealed that 5 μm of JG98 and JG194 lead to a striking increase for NPC1-I1061T, resulting in values that are equivalent to or greater than that observed for WT NPC1. We also observed that JG13 and JG98 at 5 μm markedly improved the fold-change in absolute Endo HR glycoforms relative to dimethyl sulfoxide (DMSO) control (Fig. 1E, right panel). Considering both the improvement of TrIdx and absolute Endo HR glycoforms, we use JG98 for further experiments.
Impact of JG98 on NPC1 variants in patient fibroblasts
To validate the corrective properties of JG98 in a physiologically relevant cellular environment, we monitored the impact of JG98 (5 μm) on patient-derived fibroblasts harboring different NPC1 genotypes (Fig. 2A). Consistent with the result in U2OS cells, JG98 improves both the TrIdx and absolute Endo HR glycoforms for I1061T/I1061T fibroblast (Fig. 2B and C, black arrows), though the increase of absolute Endo HR (Fig. 2C, black arrow) is much more prominent than the increase of TrIdx (Fig. 2B, black arrow). Of the 19 genotypes tested, 12 exhibited an increased TrIdx in response to JG98 (Fig. 2B), with 7 of these also exhibiting an increase in absolute Endo HR glycoforms of at least 2-fold (Fig. 2C), including I1061T/I1061T, G673V/I1061T and I1061T/S954L that have significant increase in replicated experiments. This result indicates that JG98 impacts both the trafficking and post-ER protein level for NPC1 in those fibroblasts. In contrast, there were 2 genotypes that exhibited a >2-fold increase in absolute Endo HR in response to JG98 without a significant increase in their respective TrIdx, namely WT/WT and P401T/I1061T (Fig. 2A–C), suggesting that for these genotypes, the primary impact of JG98 is to increase either the expression and/or stability of NPC1 protein. Overall, both TrIdx values (Fig. 2B, right panel) and the absolute Endo HR glycoforms (Fig. 2C, right panel) of most of the variant population are improved by addition of JG98 with more pronounced increase in absolute Endo HR, indicating JG98 is able to increase the NPC1 protein level even if the effects on the improvement of trafficking efficiency assessed by the TrIdx may be limiting. Consistent with this interpretation, an examination of the response of the I1061T homozygous fibroblast to JG98 (Fig. 2D), revealed a dose-dependent increase in the absolute Endo HR glycoforms (Fig. 2E, right panel) which was not observed for the TrIdx (Fig. 2E, left panel). Considering that all genotypes tested, except two, were heterozygous and that many exhibited significant responses to JG98, these data suggest that modulation of the Hsp70 system has a substantial impact on the biogenesis, trafficking and/or potential stability of NPC1.
Figure 2.

Impact of JG98 on NPC1 patient fibroblasts. (A) JG98 was tested on a collection of fibroblasts from patients with different NPC1 genotypes (see Methods). NPC1 was immunoprecipitated from 75 μg of total cell lysate protein, treated with Endo H, eluted from anti-NPC1 beads and separated on a 4–12% Bis-Tris SDS-PAGE. (B) Quantification of TrIdx (left panel) for each fibroblast. Four fibroblasts were tested in triplicates and 1 fibroblast was tested in duplicate. The associated error bar (mean ± SD) and P-value (P < 0.05,*; P > 0.05, ns; Student’s t-test) are indicated. The remaining 14 fibroblasts are reported as singlets. Fibroblasts with different NPC1 genotypes are ordered according to their TrIdx value in DMSO condition. Right panel shows a box and whisker plot for the TrIdx of all the fibroblasts in the DMSO and JG98 condition. P-value (Student’s t-test) is indicated. (C) Quantification of the fold-change of Endo HR species relative to DMSO (left panel). Fibroblasts with different genotypes are arranged according to the fold-change in Endo HR species relative to DMSO. For the fibroblasts that have experimental replicates, error bar (mean ± SD) and P-value (P < 0.05,*; P > 0.05, ns; Student’s t-test) are indicated. Right panel shows a box and whisker plot for the normalized Endo HR species relative to the WT standard run in each gel. P-value (Student’s t-test) is indicated. (D) Analysis of impact of 0.125, 0.25, 0.5, and 1.0 μm JG98 for 24 h fibroblast I1061T/I1061T genotype in duplicate. (E) Quantification of TrIdx (left panel) and the fold change of Endo HR relative to DMSO with JG98 treatment (right panel) for the dose experiment shown in (D) (mean ± SD).
Impact of JG98 on homozygous NPC1 variants in U2OS NPC1 null cell line
Patient fibroblasts are largely heterozygous, have diverse genetic backgrounds and linked to variable natural history of onset and progression of disease. This complex genetic background of each fibroblast makes it difficult to dissect the impact of JG98 on the trafficking of unique NPC1 variant alleles. Therefore, we constructed 58 different patient-derived NPC1 variants spanning the entire polypeptide chain using the NPC1 WT-V sequence (see methods) (29, 55, 69–72) (Fig. 3A). These variants were transiently transfected into NPC1-null U2OS cells we routinely use to assess cholesterol responses (29), for characterization of their baseline trafficking activity, their JG98-responsive TrIdx (Fig. 3B) and the absolute Endo HR response (Fig. 3C) in the homozygous state. This approach provides a direct measure of the contribution of an individual allele on egress from the ER for downstream delivery to the LE/Ly.
Figure 3.
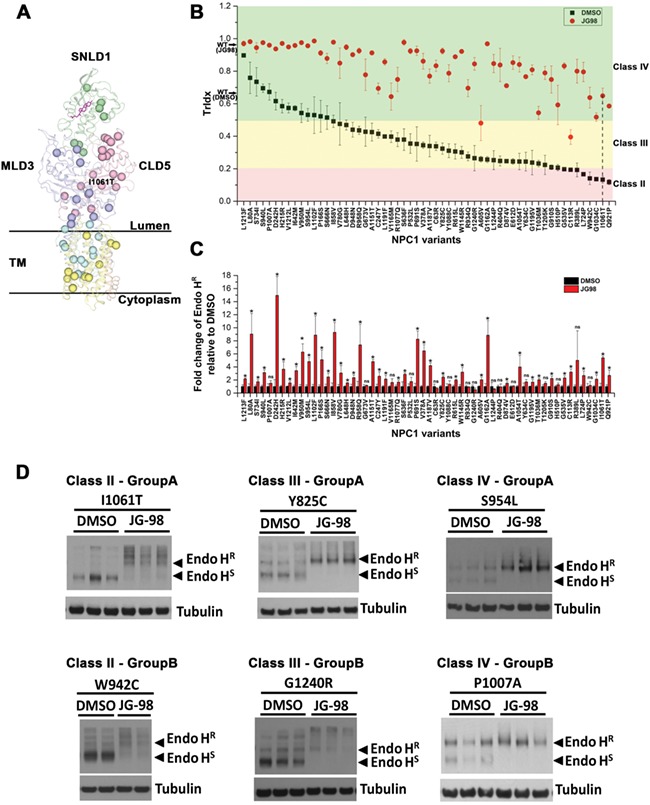
Impact of JG98 on the trafficking and stability of 58 NPC1 variants expressed in U2OS cells. (A) 58 NPC1 variants used in this study are showed as balls in NPC1 structure (20–22). Each domain is labeled. Cholesterol is shown as a stick figure in magenta. (B) Quantification of the TrIdx for all the 58 variants in the absence (black squares) or presence (red circles) of JG98. Error bar is indicated as mean ± SD. Plasmid harboring each of the 58 variants was transfected in U2OS-SRA-shNPC1 cells. After treatment of 5 μm JG98 for 24 h, all sample lysates were treated with Endo H followed by Western blotting. Variants are ordered according to their TrIdx in the DMSO condition. Variants are grouped into different classes based on their TrIdx as indicated by background color (pink—Class II, yellow—Class III, green—Class IV). The P-value associated with each variant when compared with the JG98 treated or untreated condition was <0.05 (Student’s t-test). (C) Quantification of the fold-change of Endo HR relative to DMSO after treatment of JG98 (mean ± SD). Variants are in the same order as (B). P-value for each variant compared with JG98 treated and untreated groups are labeled (P < 0.05,*; P > 0.05, ns; Student’s t-test). (D) Examples of Immunoblots for variants with increased Endo HR (Group A) or no significant change of Endo HR (Group B) in each TrIdx class are shown.
An analysis of the steady-state TrIdx following a standardized protocol involving transient transfection (see Methods) allows us to categorize NPC1 variants into one of four classes (Table 1). Class I includes a single variant that did not produce any NPC1 protein, representative of variants resulting in large protein truncations. Class II represents variants with a severe defect in trafficking efficiency (TrIdx < 0.2), including I1061T variant (Fig. 3B, vertical dash line). Class III represents variants with intermediate trafficking (0.2 < TrIdx < 0.5) (Fig. 3B). Class IV includes variants with >50% of trafficking out of ER, including NPC1-WT that has ~0.65 TrIdx when expressed in U2OS cells (Fig. 3B, black arrow on y-axis). Strikingly, JG98 significantly improves the TrIdx of all the variants (Fig. 3B, red circles) reaching or exceeding the WT-level, indicating that the Hsp70 system can play a prominent role in managing the stability and trafficking of NPC1 variants distributed across the NPC1 polypeptide.
Table 1.
NPC1 variants classification based on TrIdx
| Class II: <20% TrIdx | Class III: 20–50% TrIdx | Class IV: >50% TrIdx |
|---|---|---|
| C113R | C63R | L80 V |
| R389L | C247Y | P166S |
| G535V | V378A | H215R |
| L724P | R404Q | D242H |
| Q921P | H510P | I642M |
| W942C | P532L | S666N |
| G1034C | A605V | S734I |
| I1061T | E612D | S940L |
| R615L | V950M | |
| Y634C | S954L | |
| S636F | P1007A | |
| L648H | L1102F | |
| G673V | V1212L | |
| P691S | L1213F | |
| V780G | ||
| Y825C | ||
| I858V | ||
| D874V | ||
| G910S | ||
| R934Q | ||
| D948N | ||
| R958Q | ||
| T1036M | ||
| A1054T | ||
| R1077Q | ||
| Y1088C | ||
| W1145R | ||
| A1151T | ||
| G1162A | ||
| V1165M | ||
| A1187V | ||
| L1191F | ||
| G1195V | ||
| T1205K | ||
| G1240R | ||
| L1244P |
While the trafficking efficiency (TrIdx) of all the variants expressed in U2OS cells is improved by JG98, there is a more heterogeneous response regarding the level of absolute Endo HR glycoforms recovered in post-ER compartments (Fig. 3C). The absolute level of Endo HR glycoforms are significantly increased for 42 variants while the changes of Endo HR are not significant for 16 of the variants, despite improvement in their TrIdx (Fig. 3C). Therefore, we further classified those variants that were significantly changed for absolute Endo HR as Group A, while those that were not significantly changed for Endo HR were binned as Group B (Table 2). For example, as shown in Figure 3D, I1061T, Y825C and S954L (Group A) show a significant increase of Endo HR band and decrease of Endo HS band. Conversely, W942C, G1240R and P1007A (Group B) only show significant clearance of Endo HS yet without any significant changes on Endo HR band. Taken together, the results suggest that the Hsp70 chaperone system differentially manages the trafficking efficiency (TrIdx) and post-ER (Endo HR) level to impact the diversity in genotype to phenotype transformation for NPC1 variants.
Table 2.
Endo HR response to JG98 in each TrIdx class
| Class II | Class III | Class IV | |||
|---|---|---|---|---|---|
| Group A | Group B | Group A | Group B | Group A | Group B |
| C113R | R389L | C247Y | C63R | L80A | P1007A |
| G535V | W942C | V378A | R404Q | P166S | |
| L724P | A605V | H510P | H215R | ||
| Q921P | R615L | P532L | D242H | ||
| G1034C | S636F | E612D | I642M | ||
| I1061T | L648H | Y634C | S666N | ||
| P691S | G673V | S734I | |||
| V780G | R934Q | S940L | |||
| Y825C | Y1088C | V950M | |||
| I858V | V1165M | S954L | |||
| D874V | T1205K | L1102F | |||
| G910S | G1240R | V1212L | |||
| D948N | L1244P | L1213F | |||
| R958Q | |||||
| T1036M | |||||
| A1054T | |||||
| R1077Q | |||||
| W1145R | |||||
| A1151T | |||||
| G1162A | |||||
| A1187V | |||||
| L1191F | |||||
| G1195V | |||||
Mapping JG98 response across entire NPC1 polypeptide through VSP
To provide comprehensive understanding of how the Hsp70 system manages the trafficking and post-ER stability of the entire NPC1 polypeptide in response to JG98 and how these different functional features correlate with the NPC1 structure reflecting proteostasis sensitive modulation of the biosynthetic protein fold, we applied VSP (1). VSP is based on the input value of a sparse collection of variants, in this case the 58 variants that were experimentally measured, to amplify the insights obtained from these variants to NPC1 function on a residue-by-residue basis across the entire polypeptide fold (1). Briefly, VSP is a GPR-ML approach that builds matrices of relationships utilizing only a sparse collection of variants that can quantitatively define sequence-to-function-to-structure relationships at atomic resolution (1). VSP is based on a new biological principle referred to as SCV (1). VSP uses a set of known residues and their related functions to predict a quantitative value (referred to as a SCV ‘set-point’) for each residue in the polypeptide chain. These set-point values are associated with an uncertainty that assigns the SCV ‘tolerance’ for all known and unknown variants in predicting function (1). GPR-ML analyses allows us to generate ‘phenotype landscapes’ that quantitatively image the known and unknown SCV tolerance set-point of each residue in the NPC1 fold and its cellular physiology (1). These uncharacterized values, predicted from a sparse collection of known, provide a facile interpretation of covarying sequence-to-function-to-structure relationships across the entire NPC1 fold that are not only responsible for NPC1 trafficking but, in this case, its management by the Hsp70 system in response to JG98.
To assign SCV relationships based on VSP (1) defining the tolerance set-point of a variant residue affecting membrane trafficking from the ER in the presence or absence of JG98, we plotted each of the 58 variant residues analyzed according to their assigned sequence position in the NPC1 gene on the x-axis (Fig. 4A, upper panel), its experimentally calculated TrIdx in the DMSO condition on the y-axis (Fig. 4A, upper panel), and its experimentally determined delta (∆) TrIdx response to JG98 on the z-axis (Fig. 4A, upper panel) to computationally build a phenotype landscape predicting the change in SCV tolerance set-point for all NPC1 residues (Fig. 4A, lower panel) (see Materials and Methods) (1). In a parallel approach, we plotted the log2 fold change of the absolute Endo HR glycoform achieved in response to JG98 as the z-axis metric (Fig. 4D, upper panel) to generate a phenotype landscape predicting the change in NPC1 SCV tolerance set-point for total ER export (Fig. 4D, lower panel). Both landscapes achieve significant correlation between measured value and predicted value in the leave-one-out cross validation (Supplementary Material, Fig. S1A and C). The delta (∆) TrIdx prediction accuracy is similar to multivariate linear regression and slightly higher than decision-tree based regression (e.g. Random Forest) (Supplementary Material, Fig. S1B). The absolute Endo HR prediction was insignificant using regression methods other than VSP (Supplementary Material, Fig. S1D). Among these regression methods, VSP approach can explicitly assess prediction uncertainty (or confidence) which allows us to map with confidence the sequence-to-function-to-structure relationships to the entire NPC1 polypeptide (1). The phenotype value predicted for each residue with the highest confidence, which is a quantitative measure of the set-point reflecting the tolerance of the variation for NPC1 fold in response to trafficking from the ER (Fig. 4A and D, lower panels), was mapped onto the NPC1 3D structure (Fig. 4B, C, E and F) (20, 22) to allow more intuitive description of NPC1 trafficking as a ‘functional-structure’ (1).
Figure 4.

Mapping JG98 impact across the entire NPC1 polypeptide using VSP. (A) By analyzing the SCV relationships of 58 NPC1 variants according to their position in primary sequence (x-axis), TrIdx value in DMSO (y-axis) and delta (∆) TrIdx response to JG98 (upper panel), VSP (1) constructs a phenotype landscape (lower panel) that maps delta (∆) TrIdx response for all the residues across the entire NPC1 polypeptide. Color scale shows the predicted delta (∆) TrIdx values with red-orange representing no response, yellow-green representing a low to medium response and blue-cyan representing a high response. Contour lines represent the confidence for the delta (∆) TrIdx response prediction with the top 25% confident region highlighted by bold contour lines (1). (B) The phenotype landscape can be mapped to a structure snapshot of NPC1 (20–22) to show the specific structural features of NPC1 fold that JG98 targets. (C) The top 40% predicted TrIdx responsive regions to JG98 are highlighted by assigning the color for all regions below the 40% threshold TrIdx as gray. (D–F) VSP predicts the phenotype landscape for the absolute Endo HR changes in response to JG98 (D). Log2 transformation of the fold change of Endo HR after JG98 treatment is used as input for VSP training (upper panel) to generate the phenotype landscape (lower panel). The phenotype landscape is mapped onto structure (E). (F) The top 30% predicted Endo HR responsive regions to JG98 are highlighted by assigning the color for all regions below the 30% threshold of Endo HR response as gray.
The phenotype landscape and predicted functional-structure shows that the predicted delta (∆) TrIdx of almost all known and unknown (uncharacterized) variants will be improved by JG98 (Fig. 4A, lower panel, green-cyan-blue; Fig. 4B and C). Particularly, the top 40% high responsive regions (Fig. 4A, lower panel, cyan-blue; Fig. 4C) generally have low TrIdx values in the DMSO condition (Fig. 4A, lower panel, y = ~0.2) and are principally localized to the TM region (Fig. 4C) and the interface between MLD3 and CLD5 domains (Fig. 4C). The SCV relationships of these natural variant residues create SCV ‘intolerance’ and a low set-point for exit from the ER, thus leading to a severe trafficking deficiency and severe disease (1, 73). Intriguingly, JG98 targeting the cytosolic Hsp70 system is able to substantially increase the SCV tolerance set-point for these residues to improve their trafficking efficiency.
In contrast to the phenotype landscape based on TrIdx (Fig. 4A–C), the absolute Endo HR glycoform phenotype landscape was more complex, with both non-responsive regions (Fig. 4D, lower panel, red-orange-yellow) and highly responsive regions (Fig. 4D, lower panel, cyan-blue; Fig. 4F). Most of the highly responsive regions (Fig. 4D, lower panel, cyan-blue, y = ~0.6) have high TrIdx values in DMSO (Fig. 4A, lower panel), reflecting a more tolerated fold for exit from the ER prior to treatment. For example, high tolerance regions responsive to JG98 are found in the SNLD1 domain (Fig. 1A) that forms the cholesterol-binding pocket (Fig. 4F, cholesterol highlighted as magenta). Interestingly, some of the variants at these residues have trafficking levels equivalent or better then WT. The dramatic increase in the level of absolute Endo HR glycoforms in response to JG98 may indicate that the Hsp70 system either increases the expression of these variants or plays a role in regulating their turnover rate in the LE/Ly endomembrane system of fibroblasts. In contrast, most of the residues that have low TrIdx values in the DMSO condition do not show a high response to JG98 for the appearance of absolute Endo HR glycoforms (Fig. 4D, lower panel). One possible explanation is that, although the TrIdx of the variants was improved (Fig. 4A, lower panel), they are unstable and JG98 fail to reverse the pathways leading to degradation. To examine this possibility, the impact of JG98 on stability of select variants was examined by a 4 h cycloheximide (CHX) treatment (Supplementary Material, Figs S2 and S3) for select NPC1 variants representative of different domains of the protein. For 6 of the NPC1 variants tested by transient expression in U2OS cells, P691S (Class III variant) and S954L (Class IV variant) showed a significant increase in the stability of the absolute Endo HR glycoform by JG98 whereas others did not (Supplementary Material, Fig. S2). For the 4 NPC1 variant fibroblasts tested, only I1061T/G673V showed significant increased stability in the absolute Endo HR glycoforms (Supplementary Material, Fig. S3). These data reveal diverse impacts on the trafficking efficiency (TrIdx), post-ER absolute Endo HR glycoform level and the stability of NPC1 variants by Hsp70 system. Indeed, a recent study showed that different NPC1 variants, such as WT/WT, I1061T/I1061T and P1007A/T1036M fibroblasts could be differentially managed by multiple degradation pathways that include the MARCH6 ubiquitin ligase-dependent ER associated degradation (ERAD) and FAM134B ER-phagy pathways (25). I1061T was found predominantly degraded by ER-phagy pathway (25). Combination of JG98 and Bafilomycin A1 (Baf), a compound that inhibits lysosomal acidification and protein degradation, further increases both the TrIdx and absolute Endo HR when compared with JG98 alone (Supplementary Material, Fig. S4). These results highlights the importance of understanding how the balance in different proteostasis pathways manage stability, trafficking and function for diverse variants. Thus, application of a rigorous quantitative formalism such as VSP based on the universal principle of SCV that can link sequence-to-function-to-structure relationships suggests that the Hsp70 system dynamically manages the proteostatic response of different NPC1 domains on a residue-by-residue basis.
Impact of Hsp70 co-chaperone BAG on NPC1-I1061T trafficking
Because JG98 is an allosteric inhibitor that disrupts the protein-protein interactions between Hsp70 and BAG proteins (60), we silenced the major BAG family isoforms (BAG1–3) to test their effects on NPC1-I1061T (Fig. 5A). This experiment is important as a genetic test of the selectivity of JG98 in management of the NPC1 fold. Similar to what we found with the effect of JG98 treatment of I1061T/I1061T fibroblasts, we observed no statistically significant impact of siBAG1 or siBAG2, while siBAG3 had a small impact on TrIdx (Fig. 5B, left panel). In contrast, the value of the absolute Endo HR glycoform of NPC1-I1061T recovered in response to silencing was substantially altered- nearly 3-fold in response to siBAG1, 2, or 3 (Fig. 5B, right panel). These results suggest BAG1–3 could play critical roles in managing either the expression level and/or stabilization of NPC1-I1061T. Consistent with these observations, overexpression of BAG1 and BAG3 WT protein decreased the absolute level of Endo HR NPC1-I1061T (Fig. 5D and E, left panel). The quantitative impact of BAG1 and 3 on NPC1-I1061T is dependent on the BAG domain, as overexpression of the ∆C44 of BAG1 and ∆C450 of BAG3, which are inactive truncated forms (Fig. 5C), stabilized NPC1-I1061T (Fig. 5D and E, left panel). These observations are consistent with the roles of BAG1 and BAG3 in mediating degradation of Hsp70 clients through the ubiquitin proteasome system (UPS) or autophagy-lysosomal pathway, respectively (50–52, 74, 75), possibly linking the Hsp70 function to MARCH6 or FAM134B dependent degradation pathways (25). Interesting, while the overall protein level of NPC1-I1061T is reduced in response to BAG1 and BAG3 overexpression, the trafficking efficiency (TrIdx) is increased (Fig. 5D, long exposure; Fig. 5E, left panel), indicating that JG98, through BAG proteins, may impact both the stability as well as trafficking efficiency relative to the protein load of NPC1 variant in the ER. In contrast to BAG1 and 3, overexpression of BAG2 stabilized NPC1-I1061T, an observation that may relate to its inhibitory role of CHIP to facilitate the chaperone function of Hsp70 for targeting to ERAD (76, 77). Together, these results indicate that the JG98 sensitive BAG co-chaperones (60) can manage the overall SCV tolerance of the fold for biogenesis, stability, trafficking and degradation, all events that may contribute differentially to the impact of a variant in the genotype to phenotype transformation triggering disease.
Figure 5.

Impact of Hsp70 co-chaperone BAG proteins on NPC1-I1061T. (A) Immunoblot analysis Endo H digestion products of NPC1-I1061T after partial silencing of BAGs 1–3 with siRNA. (B) Quantification of (A) for TrIdx (left panel) and fold-change of Endo HR relative to scramble siRNA control (right panel). The error bar (mean ± SD) and P-value (P < 0.05, *; P > 0.05, ns; Student’s t-test) are indicated. (C) Cartoon description of WT and modified BAG1–3 plasmids used for overexpression analysis. BAG1 has a deletion at ΔC44 which creates a truncated form of the protein with the BAG domain disrupted. Similarly, BAG3 has a deletion at ΔC450 which creates a BAG domain truncated form of the protein. (D and E) Immunoblot analysis (D) and quantitation (E) of Endo H digested products of NPC1-I1061T in response to overexpression of BAG proteins described in (C).
JG98 restores cellular pathways for cholesterol uptake and synthesis
It is well-established that deficiency of NPC1 impacts the regulatory gene responses for maintaining intracellular cholesterol homeostasis. For example, the sterol regulatory element-binding protein-2 (SREBP-2) pathway is a master regulator of cholesterol uptake and synthesis (78–80). The decreased transport of cholesterol from LE/Ly to the ER in NPC1 deficient cell increases the activation of SREBP-2 and the expression of target genes such as low density lipoprotein receptor (LDLR) responsible for cholesterol uptake, and 3-hydroxy-3-methyl-glutaryl-CoA synthase (HMGCoA-S) or reductase (HMGCoA-R) critical for cholesterol synthesis (80). Conversely, rebalancing cholesterol homeostasis leading to an increase ER cholesterol reduces SREBP-2 activity (80). Since we are unable to use filipin staining to measure cholesterol homeostasis in the cell due to the fluorescent properties of JG98 (60, 81), the activation of SREBP-2 and the expression of SREBP-2 target genes were used as indirect probes to understand if cholesterol-related pathways were modified in response to JG98.
The precursor form of SREBP-2 (p-SREBP-2) is localized in the ER through its TM domains. Upon activation, p-SREBP-2 (~125 kD) is mobilized from the ER to Golgi where its N-terminal fragment (60–70 kD) is cleaved to yield a soluble cytoplasmic species referred to as m-SREBP-2. Soluble m-SREBP-2 enters the nucleus to activate the transcription of target genes impacting cholesterol homeostasis. As shown in Fig. 6A, both total SREBP-2 (p-SREBP-2 plus m-SREBP-2) and m-SREBP-2 are increased in I1061T/I1061T homozygous human fibroblast compared with WT fibroblasts in the DMSO condition (Fig. 6B), indicating that SREBP-2 is activated by NPC1-I1061T. Treatment of I1061T/I1061T fibroblasts by JG98 decreases both the level of total SREBP-2 and m-SREBP-2 (Fig. 6A and B). Consistent with this result, real-time PCR shows the mRNA level of SREBP-2 is also significantly decreased by JG98 in I1061T/I1061T fibroblast to a level comparable to that of WT/WT fibroblasts (Fig. 6C). Furthermore, the mRNA level of SREBP-2 target genes such as LDLR (Fig. 6D), HMGCoA-S (Fig. 6E) and HMGCoA-R (Fig. 6F) are all increased by NPC1-I1061T and significantly reduced by JG98. These results suggest that JG98 not only improves the TrIdx and the level of Endo HR glycoforms of NPC1-I1061T, but also contributes to restoration of the cellular pathways that control cholesterol uptake and synthesis, possibly in response to restoration of cholesterol homeostasis in the LE/Ly.
Figure 6.

Impact of JG98 on SREBP-2 pathway in homozygous I1061T patient fibroblast. (A) Immunoblot analysis of SREBP-2 in WT/WT and I1061T/I1061T fibroblasts with or without JG98. The p-SREBP-2 and m-SREBP-2 are labeled. (B) Quantification of (A) for total SREBP-2 (p-SREBP-2 plus m-SREBP-2) (left panel) and m-SREBP-2 (right panel). The error bar (mean ± SD) and P-value (P < 0.05, *; P < 0.01, **; Student’s t-test) are indicated. (C–F) mRNA expression measured by real-time PCR for SREBP-2 (C), LDLR (D), HMGCoA S (E) and HMGCoA R (F) in WT/WT and I1061T/I1061T fibroblast with and without JG98. 18 s mRNA was used as an internal housekeeping control. The error bar (mean ± SD) and P-value (P < 0.05, *; P < 0.01, **; P < 0.001,***; Student’s t-test) are indicated.
Discussion
We have provided the first description of the corrective properties of the Hsp70 chaperone/co-chaperone system allosteric inhibitor, JG98 (42, 60), on the trafficking and cholesterol homeostasis of disease-associated variants of NPC1. At steady state, NPC1 variants fall into 1 of 4 categories based on their level of trafficking, including the class II (no trafficking), class III (moderate trafficking) and class IV (WT-like trafficking). Disease-causing class IV mutations likely represent variants with reduced or no activity(s) in the downstream endomembrane trafficking pathways and/or cholesterol management in the LE/Ly. Each of the variant populations were found to significantly, but differentially, respond to JG98. While the observations of improved TrIdx and absolute Endo HR levels following JG98 treatment in U2OS cells are generally consistent with our observations in patient-derived fibroblasts, we did observe that the increase of TrIdx in fibroblasts is smaller than that in U2OS cells for several variants including I1061T. Moreover, we have observed a sustained increase in TrIdx in homozygous I1061T patient-derived fibroblasts upon consecutive low dose treatment with JG98 (unpublished). One possibility could stem from the 2–3-fold lower NPC1 expression seen in primary fibroblasts relative to that seen in transiently transfected U2OS cells. The former required immunoprecipitation of NPC1, thereby possibly imposing a bias for the detection of Endo HS versus Endo HR species. A second possibility is that the primary fibroblast models are more restricted in their JG98-mediated proteostasis reprogramming. A third possibility is that each patient fibroblast carries the burden of its unique genomic diversity that could differentially impact how JG98 impacts NPC1 function; for example, by expressing different drug transporters and/or degradative pathways (25, 33, 82). There could also be different off-target effects in each fibroblast line. Together, these results suggest that the Hsp70 chaperone/co-chaperone system may manage proteome balance impacting multiple pathways influencing trafficking and that the impact of JG98 for NPC1 variants in different patients will not only be dependent on the patient specific variant, but also could be affected by cell and tissue specific modifiers that will need to be addressed in the future.
Due to the fluorescence properties of JG98 (60, 81), we were are unable to monitor potential correlative changes in cholesterol homeostasis in the cell by fluorescent probes such as filipin staining. However, we found that the increase of SREBP-2 activation and target gene expression due to the high cholesterol accumulation in LE/Ly in I1061T patient fibroblast was eliminated by JG98, indirectly indicating from a cholesterol regulation perspective a restoration of cellular cholesterol homeostasis in the cell by JG98. Further studies with non-fluorescence JG98 analogs, additional indirect probes of cholesterol homoeostasis (83), or analyses using Npc1I1061T or Npc1D1005G mouse (68, 84) or cat models (85) that follow clinically relevant metrics of correction of disease phenotype will be required to assess directly the potential functional impact of JG98 in vivo.
A challenge raised by our studies is to understand how a cytosolic chaperone system impacts trafficking of a largely luminal protein such as NPC1. One possibility is that JG98 shifts the functional equilibrium between Hsp70 and the cytosolic ERAD or autophagy-lysosomal degradation pathways favoring productive chaperoning to produce a more facile environment for stability and/or export from the ER based on SCV tolerance driving these relationships. We found that a combination of JG98 and MG132, a proteasome inhibitor that blocks ERAD, is more toxic (lethal) to I1061T transfected cells when compared with either JG98 or MG132 alone (data not shown) indicating that their primary impacts are on different pathways (86). On the other hand, a combination of JG98 and Baf, an autophagy-lysosomal degradation inhibitor, increased both the TrIdx and the absolute Endo HR yield of NPC1-I1061T when compared with JG98 alone. These data suggest that JG98 functions, at least in part, through mechanisms that are distinct from ERAD or autophagy-lysosomal degradation (25) to facilitate NPC1 folding, stability and trafficking. A second possibility is that the JG98-mediated alterations in cytosolic Hsp70 chaperone activity triggers stress response pathways, including the unfolded protein response (UPR), which will alter the level of ER luminal chaperone components that impact NPC1 variant folding. A third possibility is that JG98 is affecting the activity of a yet-to-be identified NPC1 ‘receptor’ for delivery to the COPII ER export machinery for improved delivery to downstream Golgi compartments (87). A fourth possibility is that JG98 is altering the steady state plasticity and selectively of endomembrane trafficking compartments through the activity of coat complexes, Rab GTPases, tether assemblies and SNARE complexes that manage compartmental design and function in a proteostasis sensitive fashion (87–90). These interpretations illustrate the potential complexity of the proteostasis buffering capacity in a given cell type.
Currently, there are two clinical trials for NPC1 disease. One trial involves arimoclomol, a small-molecule Hsp70 co-inducer (53) and a second is VTS-270 (2-hydroxypropyl-beta-cyclodextrin), a bulk phase cholesterol scavenger (94, 95). Arimoclomol has been shown to slow disease progression after 12 months when compared with the placebo control group in subgroup analysis (https://www.orphazyme.com/news-feed/2019/1/30/orphazyme-reports-positive-results-from-full-data-set-of-phase-iiiii-arimoclomol-trial-in-niemann-pick-disease-type-c-npc). VTS-270 has been shown to slow down neurological features of disease progression up to 36 months (94, 95), although a recent clinical trial failed to reveal significant separation between the drug treated group and the placebo group at 52 weeks (https://www.npuk.org/vts-270-phase-2b-3-data-announcement/). Both arimoclomol and VTS-270 were identified by their impact in Npc1−/− mice (53, 96) indicating that at least one of their mechanisms is to clear cholesterol accumulation in a manner independent of NPC1 and therefore not managing the genetic root cause of disease- folding defects in NPC1. Since JG98 has a striking but variable impact on NPC1 trafficking for most variants, it may be possible that a combination approach using either arimoclomol or VTS-270 with JG98 could have either an additive or synergistic impact on cholesterol homeostasis by improving trafficking of NPC1 to LE/ly. A combination approach would be not unlike current efforts to mitigate the disease etiology of cystic fibrosis (CF) where the trafficking and function of cystic fibrosis transmembrane conductance regulator (CFTR) variants can be augmented by a combination of correctors and potentiators to improve disease outcome (97–100).
To address complexity in JG98 responses, we applied the computational formalism afforded by our VSP approach that embraces genetic diversity to understand and explain biology (1). VSP allows us to define the sequence-to-function-to-structure relationships through the new biological principle of SCV (1). SCV is a universal metric that allows us to use a sparse collection of known variants and their functional attributes to predict the tolerance set-point for each amino acid residue in the NPC1 polypeptide through the GPR-ML matrix-based generation of phenotype landscapes. VSP is a powerful tool to deal with the upcoming challenge of understanding the role of genetic diversity in the population (1, 2, 101). Differences reflected in the dynamics of JG98-mediated fold management likely encompass the chemical, biochemical and biophysical features driving assembly and differential stability of the NPC1 polypeptide fold in the different compartments of the endomembrane system, and in different cellular and tissue environments (14, 35). For example, VSP clearly demonstrates that the management of JG98 on TrIdx and absolute Endo HR impacts different features of the NPC1 fold. Moreover, each variant expressing cell may harbor distinct activation states of their heat shock response (HSR) (38), UPR (102, 103) and/or autophagic (25) pathways given that each variant likely presents a distinct folding problem leading to unique tuning of the maladaptive stress response (34). The dynamic temporal and spatial relationships that dictate the SCV tolerance of the fold for a given environment can be used to generate a functional-structure (1) where each residue in the polypeptide can be assigned a quantitative SCV tolerance set-point value based on its uncertainty score in the phenotype landscape. The differences in residue responses suggest that the Hsp70 chaperone system and proteostasis in general is a very dynamic process that tracks the assembly and maintenance of the polypeptide chain on a residue-by-residue basis based on spatial-temporal SCV relationships for each residue in the context of its local kinetic and/or thermodynamic features, not function, a conclusion supported by the known dynamic structural features of the Hsp70 cycle (1, 41, 47). We do not consider this a negative given that proteostasis is the universal machine based on SCV principles that buffers variant evolution in the population (14, 91) and that responds to regulatory pathways that are globally altered by folding stress including HSR (38) and UPR (92). These regulatory pathways balance and rebalance biology in response to folding stress (35, 39, 93) and therefore are logical targets for therapeutic intervention. Our VSP approach suggests that targeting specific protein-protein interactions between Hsp70 and its co-chaperones to adjust the SCV tolerance threshold for a variant residue of interest will reveal how to bin NPC1 variation from a more global perspective using the patient population as the model (1).
In summary, VSP (1), by linking sequence-to-function-to-structure relationships using a sparse collection of variants contributing to NPC1 disease, helps us to understand the genotype to phenotype transformation occurring in response to JG98. Our results indicate that targeting specific protein-protein interactions managed by Hsp70 and its co-chaperones may have a broad therapeutic potential for NPC1 disease, albeit one that will need to be handicapped by knowledge in advance of the sensitivity of each residue in the polypeptide chain to a therapeutic or a therapeutic combination, insights which can be revealed prior to clinical trial by VSP-SCV based analyses (1). We have posited that this new way of thinking alters our perspective of central dogma from a restrictive linear (DNA → RNA → Protein) way of thinking to that of a dynamic VSP-based matrix (SCV [DNA|RNA|Protein]) based on GRP-ML (1), an approach that captures the goal of individualized medicine for implementation in the clinic (1). It is becoming increasingly apparent that both proteostasis, as shown herein, and HDAC sensitive acetylation pathways (29, 30) may provide unanticipated approaches to globally readjust universal SCV relationships found in the population (the many) to provide benefit to genetic diversity in human health and disease in the individual (the one) (1).
Materials and Methods
Reagents
Small molecule Hsp70 modulators (JG compounds) were a generous gift from Jason Gestwicki (San Francisco, CA). Endoglycosidase H (Endo H) was purchased from New England Biolabs (Ipswich, MA). The primary antibodies used were rat monoclonal hNPC-1 created in-house and rat monoclonal anti-tubulin antibody from Abcam (Cambridge, MA). SREPB-2 antibody (No.557037) was from BD Pharmingen™ (San Diego, CA). Benzonase Nuclease and Complete-Mini Protease Inhibitor Cocktail was used for the cell lysis and purchased from Sigma-Aldrich (St. Louis, MO). Cycloheximide was also purchased from Sigma-Aldrich. Bafilomycin A1 (No.ab120497) was purchased from Abcam (Cambridge, MA).
Cell lines and cell culture
Human Wild-Type (GM05659), homozygous NPC1I1061T (GM18453), heterozygous fibroblasts NPC1T1036M/P1007A (GM17912), NPC1G673V/I1061T (GM18398), NPC1V1165M/I1061T (GM18398), NPC1P401T/I1061T (GM17920), NPC1N1165S/R1186H (GM18397) were purchased from Coriell Cell Repositories (Coriell Institute of Medical Research). Additionally, the following heterozygous fibroblasts NPC1I1061T/R1186G (TQNPC5), NPC1I1061T/M1142T (ARNPC9), NPC11036M/S954L (CRNPC67), NPC1R404Q/P1007A (TWRNPC60), NPC1I1061T/D948N (MONPC63), NPC1I1061T/P1007A (MBNPC44), NPC1I1061T/R404Q (TLNPC36), NPC1I1061T/I1094T (TBNPC40), NPC1I1061T/S954L (KWNPC53), NPC1C177Y/V950M (JSNPC49) were courtesy of Denny Porter Lab (NIH, Maryland). The cells were grown in DMEM medium with 2 mm L-glutamine, 10% fetal bovine serum (FBS), 50 units/ml penicillin, and 50 μg/ml streptomycin antibiotics. To examine a large number of NPC1 variants, the cell line U2OS-SRA-shNPC1 was used, in which NPC1 expression is stably silenced (29) The antibiotic puromycin was used for selection. Stable lines were grown in McCoy’s 5A medium (1×: modified) with 5% FBS, 3 μg puromycin and 1 mg/ml G418.
NPC1 expression vector
The cDNA encoding human ΔU3hNPC1-WT construct was kindly provided by Dan Ory (Washington University, St. Louis, MO). The NPC1-WT gene was cloned into the pMIEG3 vector, a murine stem cell virus (MSCV) retrovirus construct that allows the co-expression of GFP and a second gene, in this case NPC1. pMIEG3 plasmid was initially generated from pMSCVneo vector (Clontech) where the neomycin resistance site (Neor) and the murine phosphoglycerate kinase promoter (PPKG), which controls expression of the neomycin resistance for antibiotic selection in eukaryotic cells, was removed. In its place the internal ribosome entry site with enhanced green fluorescent protein gene was generated. The WT construct used in this study had four additional substitution when compared with the current reference NPC1 sequence. The mutations include 387 T > C (Y129Y), 1415 T > C (L472P), 1925 T > C (M642T), and 258 T > C (S863P). These variants were present in the original ΔU3hNPC1-WT construct, which has been previously used in the past as the WT protein control (55, 69–72, 104). This pMIEG3-hNPC1 plasmid was used as a template to generate all other NPC1 variants using the Quick-Change XL Site-directed Mutagenesis Kit (Stratagene, La Jolla, CA).
NPC1 transfection in U2OS-SRA-shNPC1
Cells were seeded at 2.0 × 105 cells/ml in 12-well plates and incubated overnight. In order to express NPC1, transfections was performed using the reagent FuGENE 6 from PROMEGA (Madison, WI) with a 1:4 ratio, 2 μg DNA: 8 μl FuGENE6 in Opti-MEM (1×) + Hepes + sodium bicarbonate + L-glutamine + 5% FBS. Samples were then incubated for 5 h at 37°C and the medium was replaced with normal growth medium. Cells were then incubated for another 48 h and visualized under the fluorescent microscope for GFP-positive transfected cells. These variants were all transiently expressed in the U2OS-SRA-shNPC1 cells.
Hsp70 modulators treatment
After 72 h from initial plating with NPC1 transfection in U2OS-SRA-shNPC1 cells, cells were dosed with either DMSO as small vehicle control, or small molecule JG series Hsp70 allosteric modulators in normal growth media at 5 μm and incubated for 24 h at 37°C. After a total of 96 h, cells washed twice with 1× phosphate buffered saline (PBS) and stored at −80°C until lysis. Patient fibroblasts were treated similarly, except seeded in 6-well plates and grown to confluency before Hsp70 modulator treatment. In order to collect statistically significant data results, each variant and drug treatment condition were performed in triplicate.
Endo H digestion and immunoblotting
Cells were lysed with 50 μl/well of 1× RIPA (150 mm NaCl, 1.0% IGEPAL Ca-360, 0.5% Na-deoxychlorate, 0.1% SDS, 50 mm Tris pH 8.0, 1× protease inhibitors, 1× benzonase) on ice for 30 min. Samples were collected and incubated at 37°C for 20 min, centrifuged at 14 K RPM at 4°C for 20 min. Supernatant was collected and the BCA assay performed. Endo H digestion was performed by addition of 1× glycoprotein denaturing buffer (New England Biolabs) to 20 μg of lysate and incubated at 50°C for 30 min. For each sample 10 000 U/ml Endo H and 1× Glycobuffer (New England Biolabs) was added and incubated overnight at 37°C. Protein samples were separated on 4–12% Bis-Tris BOLT protein gel (invitrogen) and transferred to nitrocellulose membrane. Membranes were probed with NPC1 1:1000 and tubulin 1:25000 diluted in 5% milk solution.
For patient fibroblast samples, fibroblasts were seeded in a 6-well plate and grown until confluent. Once confluent, cells were treated with DMSO or JG-98 at 5 μm for 24 h at 37°C in normal growth media. Cells were then stopped and washed twice with 1× PBS and stored at −80°C until lysis. Following treatment with JG98 or vehicle cells were lysed with 125 μl/well of 1× RIPA (150 mm NaCl, 1.0% IGEPAL Ca-360, 0.5% Na-deoxychlorate, 0.1% SDS, 50 mm Tris pH 8.0, 1× protease inhibitors, 1× benzonase) on ice for 30 min. Samples were collected and then incubated at 37°C for 20 min. Samples were then centrifuged at 14 K RPM at 4°C for 20 min. Supernatant was collected and BCA assay was performed. Based on the BCA Assay, 75 μg of each lysate sample was used for immunoprecipitation with a NPC1 specific antibody. Samples were incubated overnight with 15 μl of NPC1 Ab beads at 4°C with rotation. Lysate samples with beads were centrifuged at 500 RPM at 4°C for 3 min. Supernatant was removed and washed at 10–20× bead volume 2× (50 mm Tris-HCl, pH 7.4; 150 mm NaCl; 1% Triton 100-X). After each wash samples were centrifuged at 500 RPM at 4°C for 3 min and the supernatant discarded. A final wash was performed at 10–20× bead volume 2× (50 mm Tris-HCl, pH 7.4; 150 mm NaCl). Lysate with beads was centrifuged at 500 RPM at 4°C for 3 min and the supernatant discarded. Samples were denatured with 35 μl of 1× Glycoprotein Denaturing buffer (New England Biolabs) at 95°C for 10 min. Denatured samples were centrifuged at 500 RPM at rt for 5 min. Supernatant was collected and 12 μl of sample was prepared for Endo H treatment (New England Biolabs). Samples were incubated with 10 000 U/ml Endo H and 1× Glycobuffer (New England Biolabs) overnight at 37°C. Protein samples were separated on 4–12% Bis-Tris BOLT protein gel (invitrogen) and transferred to nitrocellulose membrane. Membranes were probed with anti-NPC1 antibody at 1:1000 and anti-tubulin antibody at 1:25 000 diluted in 5% milk solution.
Overexpression of BAGS
Additionally, to examine overexpression of BAG 1 and 2 and their effect on NPC1 protein trafficking, a co-transfection was done following the same protocol as described above, but with 1 μg of NPC1 plasmid + 1 μg of BAG1–3 plasmids. Sample preparation was done as described above.
siRNA-mediated silencing
Cells were seeded at 2.0 × 105 cells/ml in 12-well plates and incubated overnight. In order to silence the BAG1–3, Lipofectamine RNAi MAX (Thermofisher) was used to deliver the siRNA. The RNAi MAX and siRNA were diluted in Opti-MEM (1×) + Hepes + sodium bicarbonate + L-glutamine + 5% FBS at 1:100 and 1:50 respectively. Samples were incubated at rt for 20 min and then added to cells and incubated at 37°C for 5 h. Medium was then changed to normal growth medium and incubated for 24 h. Transfection of NPC1 variants was then conducted (a total of 48 h after being seeded) using FuGENE 6 from PROMEGA (Madison, WI). Ratio was 1:4, 2 μg DNA: 8 ul FuGENE6 in Opti-MEM (1×) + Hepes + sodium bicarbonate + L-glutamine + 5% FBS. The 12-well plate was incubated for 5 h and then changed to normal growth medium. Cells were then incubated for another 48 h and visualized under the fluorescent microscope for transfected cells (GFP) every 24 h. Cells were washed twice with 1× PBS and stored at −80°C until lysis. Samples were prepared for immunoblotting as described above.
Cycloheximide chase
U2OS cells were prepared as described above for transfection. After the 48 h, cells were then dosed with either control 5 μm DMSO or compound 5 μm JG98 at final concentration and incubated for 24 h at 37°C. Cells were then treated with CHX (Sigma-Aldrich) time course at 50 μm at time 0 h and 4 h. After a total of 100 h, the plates were stopped and washed with 1× PBS and stored at −80°C until lysis. Samples were then later prepared for western blot analysis as described above. For patient fibroblast samples, fibroblasts were seeded in a 6-well plate and grown until confluent. Once confluent, cells were treated in same manner as described previously.
RNA extraction and real-time PCR
Total RNA was extracted from cells with RNeasy Plus Mini Kit (QIAGEN). The RT-qPCR was performed using the PerfeCTa SYBR Green FastMix (Quantabio) with the CFX96 Real-Time System and C1000 Touch Thermal Cycler (Bio-Rad). Relative expression of each SREBP-2 target gene was estimated by normalization with the expression of 18S mRNA. The SREBP-2 target genes were amplified with specific primers (80). 18S: 5′-AACCCGTTGAACCCCATT-3′, 5′-CCATCCAATCGGTAGTAGCG-3′; HMGCoA S: 5′-AGCAAGTTTCTTTTCATTTCGAGTATC-3′, 5′-GATGTGCTGGACACCAACTTGT-3′; HMGCoA R: 5′-GGGAACCTCGGCCTAATGAA-3′, 5′-CACCACGCTCATGAGTTTCCA-3′; LDLR: 5′-GTCTTGGCACTGGAACTCGT-3′, 5′-CTGGAAATTGCGCTGGAC-3′; SREBP-2: 5′-AACCCGTTGAACCCCATT-3′, 5′-CCATCCAATCGGTAGTAGCG-3′.
VSP analysis
Detailed methods as described in (1). Briefly, to build phenotype landscapes reporting the function for each residue of NPC1, the positions of variants along the primary sequence were normalized to the full length polypeptide and used as x-axis coordinate. The TrIdx of variants in DMSO condition was used as y-axis coordinate. The delta TrIdx or the log2 fold change of Endo HR was used as z-axis. The phenotype landscapes were built based on the Ordinary Kriging (1) module in the software of GS+ (Version 10; Gammadesign software). Leave-one-out cross-validation was performed in GS+ software to evaluate the computational model. The multivariate linear regression and random forest regression were performed in R by using basic lm function or randomForest package respectively. To map the landscape onto NPC1 structure (PDB: 5U74; 3JD8), the prediction value with the highest confidence (i.e. lowest standard deviation) is assigned to each residue as previously described (1). PyMOL was used to generate the structural presentations.
Supplementary Material
Acknowledgements
Support provided by the National Institutes of Health Grants HL095524 and DK051870 to W.E.B. and R01NS059690 to J.E.G. and the Tau Consortium to J.E.G. We thank the continuing support of the Ara Parseghian Medical Research Foundation (APMRF) and Support for Accelerated Research (SOARs) for Samantha Scott and Dr Pei Zhang.
Conflict of Interest statement. The authors declare no competing interests.
Funding
National Institutes of Health (HL095524, DK051870 to W.E.B. R01NS059690 to J.E.G.).
References
- 1. Wang C. and Balch W.E. (2018) Bridging genomics to phenomics at atomic resolution through variation spatial profiling. Cell Rep., 24(2013–2028), e2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hindorff L.A., Bonham V.L., Brody L.C., Ginoza M.E.C., Hutter C.M., Manolio T.A. and Green E.D. (2018) Prioritizing diversity in human genomics research. Nat. Rev. Genet., 19, 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Torkamani A., Andersen K.G., Steinhubl S.R. and Topol E.J. (2017) High-definition medicine. Cell, 170, 828–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Torkamani A., Wineinger N.E. and Topol E.J. (2018) The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet., 19, 581–590. [DOI] [PubMed] [Google Scholar]
- 5. Shendure J., Findlay G.M. and Snyder M.W. (2019) Genomic medicine-progress, pitfalls, and promise. Cell, 177, 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Topol E.J. (2019) High-performance medicine: the convergence of human and artificial intelligence. Nat. Med., 25, 44–56. [DOI] [PubMed] [Google Scholar]
- 7. Vamathevan J., Clark D., Czodrowski P., Dunham I., Ferran E., Lee G., Li B., Madabhushi A., Shah P., Spitzer M. et al. (2019) Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov., 18, 463–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Topol E.J. (2014) Individualized medicine from prewomb to tomb. Cell, 157, 241–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W. et al. (2018) ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res., 46,D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang K.L., Mashl R.J., Wu Y., Ritter D.I., Wang J., Oh C., Paczkowska M., Reynolds S., Wyczalkowski M.A., Oak N. et al. (2018) Pathogenic germline variants in 10,389 adult cancers. Cell, 173, 355–370.e314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moses M.A., Kim Y.S., Rivera-Marquez G.M., Oshima N., Watson M.J., Beebe K.E., Wells C., Lee S., Zuehlke A.D., Shao H. et al. (2018) Targeting the Hsp40/Hsp70 chaperone axis as a novel strategy to treat castration-resistant prostate cancer. Cancer Res., 78, 4022–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jansen I.E., Savage J.E., Watanabe K., Bryois J., Williams D.M., Steinberg S., Sealock J., Karlsson I.K., Hagg S., Athanasiu L. et al. (2019) Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat. Genet., 51, 404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hipp M.S., Kasturi P. and Hartl F.U. (2019) The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol., 20, 421–435. [DOI] [PubMed] [Google Scholar]
- 14. Powers E.T. and Balch W.E. (2013) Diversity in the origins of proteostasis networks--a driver for protein function in evolution. Nat. Rev. Mol. Cell Biol., 14, 237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vance J.E. and Karten B. (2014) Niemann-Pick C disease and mobilization of lysosomal cholesterol by cyclodextrin. J. Lipid Res., 55, 1609–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vanier M.T. (2013) Niemann-Pick diseases. Handb. Clin. Neurol., 113, 1717–1721. [DOI] [PubMed] [Google Scholar]
- 17. Vanier M.T. (2015) Complex lipid trafficking in Niemann-Pick disease type C. J. Inherit. Metab. Dis., 38, 187–199. [DOI] [PubMed] [Google Scholar]
- 18. Vanier M.T. (2010) Niemann-Pick disease type C. Orphanet J. Rare Dis., 5, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bianconi S.E., Hammond D.I., Farhat N.Y., Dang Do A., Jenkins K., Cougnoux A., Martin K. and Porter F.D. (2019) Evaluation of age of death in Niemann-Pick disease, type C: utility of disease support group websites to understand natural history. Mol. Genet. Metab., 126, 466–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li X., Lu F., Trinh M.N., Schmiege P., Seemann J., Wang J. and Blobel G. (2017) 3.3 A structure of Niemann-Pick C1 protein reveals insights into the function of the C-terminal luminal domain in cholesterol transport. Proc. Natl. Acad. Sci. U. S. A., 114, 9116–9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gong X., Qian H., Zhou X., Wu J., Wan T., Cao P., Huang W., Zhao X., Wang X., Wang P. et al. (2016) Structural insights into the Niemann-Pick C1 (NPC1)-mediated cholesterol transfer and Ebola infection. Cell, 165, 1467–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li X., Wang J., Coutavas E., Shi H., Hao Q. and Blobel G. (2016) Structure of human Niemann-Pick C1 protein. Proc. Natl. Acad. Sci. U. S. A., 113, 8212–8217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li X., Saha P., Li J., Blobel G. and Pfeffer S.R. (2016) Clues to the mechanism of cholesterol transfer from the structure of NPC1 middle lumenal domain bound to NPC2. Proc. Natl. Acad. Sci. U. S. A., 113, 10079–10084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gelsthorpe M.E., Baumann N., Millard E., Gale S.E., Langmade S.J., Schaffer J.E. and Ory D.S. (2008) Niemann-Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding. J. Biol. Chem., 283, 8229–8236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schultz M.L., Krus K.L., Kaushik S., Dang D., Chopra R., Qi L., Shakkottai V.G., Cuervo A.M. and Lieberman A.P. (2018) Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6-dependent ERAD. Nat. Commun., 9, 3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yu T., Chung C., Shen D., Xu H. and Lieberman A.P. (2012) Ryanodine receptor antagonists adapt NPC1 proteostasis to ameliorate lipid storage in Niemann-Pick type C disease fibroblasts. Hum. Mol. Genet., 21, 3205–3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ohgane K., Karaki F., Dodo K. and Hashimoto Y. (2013) Discovery of oxysterol-derived pharmacological chaperones for NPC1: implication for the existence of second sterol-binding site. Chem. Biol., 20, 391–402. [DOI] [PubMed] [Google Scholar]
- 28. Ebrahimi-Fakhari D., Wahlster L., Bartz F., Werenbeck-Ueding J., Praggastis M., Zhang J., Joggerst-Thomalla B., Theiss S., Grimm D., Ory D.S. et al. (2016) Reduction of TMEM97 increases NPC1 protein levels and restores cholesterol trafficking in Niemann-Pick type C1 disease cells. Hum. Mol. Genet., 25, 3588–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pipalia N.H., Subramanian K., Mao S., Ralph H., Hutt D.M., Scott S.M., Balch W.E. and Maxfield F.R. (2017) Histone deacetylase inhibitors correct the cholesterol storage defect in most Niemann-Pick C1 mutant cells. J. Lipid Res., 58, 695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Subramanian K., Hutt D.M., Gupta V., Mao S. and Balch W.E. (2019) Correction of Niemann-Pick type C1 disease with the histone deacetylase inhibitor valproic acid. bioRxiv, 724187, doi: https://doi.org/10.1101/724187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Calamini B., Silva M.C., Madoux F., Hutt D.M., Khanna S., Chalfant M.A., Saldanha S.A., Hodder P., Tait B.D., Garza D. et al. (2011) Small-molecule proteostasis regulators for protein conformational diseases. Nat. Chem. Biol., 8, 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hutt D.M., Powers E.T. and Balch W.E. (2009) The proteostasis boundary in misfolding diseases of membrane traffic. FEBS Lett., 583, 2639–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rauniyar N., Subramanian K., Lavallee-Adam M., Martinez-Bartolome S., Balch W.E. and Yates J.R. 3rd. (2015) Quantitative proteomics of human fibroblasts with I1061T mutation in Niemann-Pick C1 (NPC1) protein provides insights into the disease pathogenesis. Mol. Cell. Proteomics, 14, 1734–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roth D.M., Hutt D.M., Tong J., Bouchecareilh M., Wang N., Seeley T., Dekkers J.F., Beekman J.M., Garza D., Drew L. et al. (2014) Modulation of the maladaptive stress response to manage diseases of protein folding. PLoS Biol., 12, e1001998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Balch W.E., Morimoto R.I., Dillin A. and Kelly J.W. (2008) Adapting proteostasis for disease intervention. Science, 319, 916–919. [DOI] [PubMed] [Google Scholar]
- 36. Balch W.E., Roth D.M. and Hutt D.M. (2011) Emergent properties of proteostasis in managing cystic fibrosis. Cold Spring Harb. Perspect. Biol., 3,a004499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Balchin D., Hayer-Hartl M. and Hartl F.U. (2016) In vivo aspects of protein folding and quality control. Science, 353, aac4354. [DOI] [PubMed] [Google Scholar]
- 38. Li J., Labbadia J. and Morimoto R.I. (2017) Rethinking HSF1 in stress, development, and organismal health. Trends Cell Biol., 27, 895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sala A.J., Bott L.C. and Morimoto R.I. (2017) Shaping proteostasis at the cellular, tissue, and organismal level. J. Cell Biol., 216, 1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Labbadia J. and Morimoto R.I. (2015) The biology of proteostasis in aging and disease. Annu. Rev. Biochem., 84, 435–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zuiderweg E.R., Hightower L.E. and Gestwicki J.E. (2017) The remarkable multivalency of the Hsp70 chaperones. Cell Stress Chaperones, 22, 173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gestwicki J.E. and Shao H. (2019) Inhibitors and chemical probes for molecular chaperone networks. J. Biol. Chem., 294, 2151–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Morozova K., Clement C.C., Kaushik S., Stiller B., Arias E., Ahmad A., Rauch J.N., Chatterjee V., Melis C., Scharf B. et al. (2016) Structural and biological interaction of hsc-70 protein with phosphatidylserine in endosomal microautophagy. J. Biol. Chem., 291,18096–18106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pratt W.B., Gestwicki J.E., Osawa Y. and Lieberman A.P. (2015) Targeting Hsp90/Hsp70-based protein quality control for treatment of adult onset neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol., 55, 353–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zarouchlioti C., Parfitt D.A., Li W., Gittings L.M. and Cheetham M.E. (2018) DNAJ proteins in neurodegeneration: essential and protective factors. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci., 373, 20160534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gorenberg E.L. and Chandra S.S. (2017) The role of co-chaperones in synaptic proteostasis and neurodegenerative disease. Front. Neurosci., 11, 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Craig E.A. and Marszalek J. (2017) How Do J-proteins get Hsp70 to Do so many different things? Trends Biochem. Sci., 42, 355–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brehme M. and Voisine C. (2016) Model systems of protein-misfolding diseases reveal chaperone modifiers of proteotoxicity. Dis. Model. Mech., 9, 823–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Alderson T.R., Kim J.H. and Markley J.L. (2016) Dynamical structures of Hsp70 and Hsp70-Hsp40 complexes. Structure, 24, 1014–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rauch J.N. and Gestwicki J.E. (2014) Binding of human nucleotide exchange factors to heat shock protein 70 (Hsp70) generates functionally distinct complexes in vitro. J. Biol. Chem., 289, 1402–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rauch J.N., Zuiderweg E.R. and Gestwicki J.E. (2016) Non-canonical interactions between heat shock cognate protein 70 (Hsc70) and Bcl2-associated anthanogene (BAG) co-chaperones are important for client release. J. Biol. Chem., 291, 19848–19857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hutt D.M., Mishra S.K., Roth D.M., Larsen M.B., Angles F., Frizzell R.A. and Balch W.E. (2018) Silencing of the Hsp70-specific nucleotide-exchange factor BAG3 corrects the F508del-CFTR variant by restoring autophagy. J. Biol. Chem., 293, 13682–13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kirkegaard T., Gray J., Priestman D.A., Wallom K.L., Atkins J., Olsen O.D., Klein A., Drndarski S., Petersen N.H., Ingemann L. et al. (2016) Heat shock protein-based therapy as a potential candidate for treating the sphingolipidoses. Sci. Transl. Med., 8, 355ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schultz M.L., Krus K.L. and Lieberman A.P. (2016) Lysosome and endoplasmic reticulum quality control pathways in Niemann-Pick type C disease. Brain Res., 1649, 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nakasone N., Nakamura Y.S., Higaki K., Oumi N., Ohno K. and Ninomiya H. (2014) Endoplasmic reticulum-associated degradation of Niemann-Pick C1: evidence for the role of heat shock proteins and identification of lysine residues that accept ubiquitin. J. Biol. Chem., 289, 19714–19725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Petersen N.H., Kirkegaard T., Olsen O.D. and Jaattela M. (2010) Connecting Hsp70, sphingolipid metabolism and lysosomal stability. Cell Cycle, 9, 2305–2309. [DOI] [PubMed] [Google Scholar]
- 57. Assimon V.A., Gillies A.T., Rauch J.N. and Gestwicki J.E. (2013) Hsp70 protein complexes as drug targets. Curr. Pharm. Des., 19, 404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li X., Colvin T., Rauch J.N., Acosta-Alvear D., Kampmann M., Dunyak B., Hann B., Aftab B.T., Murnane M., Cho M. et al. (2015) Validation of the Hsp70-Bag3 protein-protein interaction as a potential therapeutic target in cancer. Mol. Cancer Ther., 14, 642–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li X., Srinivasan S.R., Connarn J., Ahmad A., Young Z.T., Kabza A.M., Zuiderweg E.R., Sun D. and Gestwicki J.E. (2013) Analogs of the allosteric heat shock protein 70 (Hsp70) inhibitor, MKT-077, as anti-cancer agents. ACS Med. Chem. Lett., 4, 1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shao H., Li X., Moses M.A., Gilbert L.A., Kalyanaraman C., Young Z.T., Chernova M., Journey S.N., Weissman J.S., Hann B. et al. (2018) Exploration of benzothiazole rhodacyanines as allosteric inhibitors of protein-protein interactions with heat shock protein 70 (Hsp70). J. Med. Chem., 61, 6163–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang A.M., Miyata Y., Klinedinst S., Peng H.M., Chua J.P., Komiyama T., Li X., Morishima Y., Merry D.E., Pratt W.B. et al. (2013) Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat. Chem. Biol., 9, 112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Taguwa S., Maringer K., Li X., Bernal-Rubio D., Rauch J.N., Gestwicki J.E., Andino R., Fernandez-Sesma A. and Frydman J. (2015) Defining Hsp70 subnetworks in dengue virus replication reveals key vulnerability in Flavivirus infection. Cell, 163, 1108–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Taguwa S., Yeh M.T., Rainbolt T.K., Nayak A., Shao H., Gestwicki J.E., Andino R. and Frydman J. (2019) Zika virus dependence on host Hsp70 provides a protective strategy against infection and disease. Cell Rep., 26(906–920), e903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Khachatoorian R., Riahi R., Ganapathy E., Shao H., Wheatley N.M., Sundberg C., Jung C.L., Ruchala P., Dasgupta A., Arumugaswami V. et al. (2016) Allosteric heat shock protein 70 inhibitors block hepatitis C virus assembly. Int. J. Antimicrob. Agents, 47, 289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Meister-Broekema M., Freilich R., Jagadeesan C., Rauch J.N., Bengoechea R., Motley W.W., Kuiper E.F.E., Minoia M., Furtado G.V., van Waarde M. et al. (2018) Myopathy associated BAG3 mutations lead to protein aggregation by stalling Hsp70 networks. Nat. Commun., 9, 5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Young Z.T., Rauch J.N., Assimon V.A., Jinwal U.K., Ahn M., Li X., Dunyak B.M., Ahmad A., Carlson G.A., Srinivasan S.R. et al. (2016) Stabilizing the Hsp70-tau complex promotes turnover in models of tauopathy. Cell Chem. Biol., 23, 992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chung C., Puthanveetil P., Ory D.S. and Lieberman A.P. (2016) Genetic and pharmacological evidence implicates cathepsins in Niemann-Pick C cerebellar degeneration. Hum. Mol. Genet., 25, 1434–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Praggastis M., Tortelli B., Zhang J., Fujiwara H., Sidhu R., Chacko A., Chen Z., Chung C., Lieberman A.P., Sikora J. et al. (2015) A murine Niemann-Pick C1 I1061T knock-in model recapitulates the pathological features of the most prevalent human disease allele. J. Neurosci., 35, 8091–8106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Millard E.E., Srivastava K., Traub L.M., Schaffer J.E. and Ory D.S. (2000) Niemann-Pick type C1 (NPC1) overexpression alters cellular cholesterol homeostasis. J. Biol. Chem., 275, 38445–38451. [DOI] [PubMed] [Google Scholar]
- 70. Pipalia N.H., Cosner C.C., Huang A., Chatterjee A., Bourbon P., Farley N., Helquist P., Wiest O. and Maxfield F.R. (2011) Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc. Natl. Acad. Sci. U. S. A., 108, 5620–5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Watari H., Blanchette-Mackie E.J., Dwyer N.K., Glick J.M., Patel S., Neufeld E.B., Brady R.O., Pentchev P.G. and Strauss J.F. 3rd. (1999) Niemann-Pick C1 protein: obligatory roles for N-terminal domains and lysosomal targeting in cholesterol mobilization. Proc. Natl. Acad. Sci. U. S. A., 96, 805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhang M., Dwyer N.K., Love D.C., Cooney A., Comly M., Neufeld E., Pentchev P.G., Blanchette-Mackie E.J. and Hanover J.A. (2001) Cessation of rapid late endosomal tubulovesicular trafficking in Niemann-Pick type C1 disease. Proc. Natl. Acad. Sci. U. S. A., 98, 4466–4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shammas H., Kuech E.M., Rizk S., Das A.M. and Naim H.Y. (2019) Different Niemann-Pick C1 genotypes generate protein phenotypes that vary in their intracellular processing, trafficking and localization. Sci. Rep., 9, 5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Behl C. (2016) Breaking BAG: the co-chaperone BAG3 in health and disease. Trends Pharmacol. Sci., 37, 672–688. [DOI] [PubMed] [Google Scholar]
- 75. Liang J., Sagum C.A., Bedford M.T., Sidhu S.S., Sudol M., Han Z. and Harty R.N. (2017) Chaperone-mediated autophagy protein BAG3 negatively regulates Ebola and Marburg VP40-mediated egress. PLoS Pathog., 13, e1006132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Arndt V., Daniel C., Nastainczyk W., Alberti S. and Hohfeld J. (2005) BAG-2 acts as an inhibitor of the chaperone-associated ubiquitin ligase CHIP. Mol. Biol. Cell, 16, 5891–5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Schonbuhler B., Schmitt V., Huesmann H., Kern A., Gamerdinger M. and Behl C. (2016) BAG2 interferes with CHIP-mediated ubiquitination of HSP72. Int. J. Mol. Sci., 18, 69, doi:10.3390/ijms18010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Garver W.S., Jelinek D., Oyarzo J.N., Flynn J., Zuckerman M., Krishnan K., Chung B.H. and Heidenreich R.A. (2007) Characterization of liver disease and lipid metabolism in the Niemann-Pick C1 mouse. J. Cell. Biochem., 101,498–516. [DOI] [PubMed] [Google Scholar]
- 79. Garver W.S., Jelinek D., Francis G.A. and Murphy B.D. (2008) The Niemann-Pick C1 gene is downregulated by feedback inhibition of the SREBP pathway in human fibroblasts. J. Lipid Res., 49, 1090–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Eid W., Dauner K., Courtney K.C., Gagnon A., Parks R.J., Sorisky A. and Zha X. (2017) mTORC1 activates SREBP-2 by suppressing cholesterol trafficking to lysosomes in mammalian cells. Proc. Natl. Acad. Sci. U. S. A., 114, 7999–8004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gabai V.L., Yaglom J.A., Wang Y., Meng L., Shao H., Kim G., Colvin T., Gestwicki J. and Sherman M.Y. (2016) Anticancer effects of targeting Hsp70 in tumor stromal cells. Cancer Res., 76, 5926–5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Subramanian K., Rauniyar N., Lavallee-Adam M., Yates J.R. 3rd and Balch W.E. (2017) Quantitative analysis of the proteome response to the histone deacetylase inhibitor (HDACi) Vorinostat in Niemann-Pick type C1 disease. Mol. Cell. Proteomics, 16, 1938–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Li J., Lee P.L. and Pfeffer S.R. (2017) Quantitative measurement of cholesterol in cell populations using flow cytometry and fluorescent perfringolysin O. Methods Mol. Biol., 1583, 85–95. [DOI] [PubMed] [Google Scholar]
- 84. Maue R.A., Burgess R.W., Wang B., Wooley C.M., Seburn K.L., Vanier M.T., Rogers M.A., Chang C.C., Chang T.Y., Harris B.T. et al. (2012) A novel mouse model of Niemann-Pick type C disease carrying a D1005G-Npc1 mutation comparable to commonly observed human mutations. Hum. Mol. Genet., 21, 730–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Somers K.L., Royals M.A., Carstea E.D., Rafi M.A., Wenger D.A. and Thrall M.A. (2003) Mutation analysis of feline Niemann-Pick C1 disease. Mol. Genet. Metab., 79, 99–103. [DOI] [PubMed] [Google Scholar]
- 86. Yaglom J.A., Wang Y., Li A., Li Z., Monti S., Alexandrov I., Lu X. and Sherman M.Y. (2018) Cancer cell responses to Hsp70 inhibitor JG-98: comparison with Hsp90 inhibitors and finding synergistic drug combinations. Sci. Rep., 8, 3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Aridor M. (2018) COPII gets in shape: lessons derived from morphological aspects of early secretion. Traffic, 19, 823–839. [DOI] [PubMed] [Google Scholar]
- 88. Gurkan C., Koulov A.V. and Balch W.E. (2007) An evolutionary perspective on eukaryotic membrane trafficking. Adv. Exp. Med. Biol., 607, 73–83. [DOI] [PubMed] [Google Scholar]
- 89. Gurkan C., Lapp H., Alory C., Su A.I., Hogenesch J.B. and Balch W.E. (2005) Large-scale profiling of Rab GTPase trafficking networks: the membrome. Mol. Biol. Cell, 16, 3847–3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ding J., Segarra V.A., Chen S., Cai H., Lemmon S.K. and Ferro-Novick S. (2016) Auxilin facilitates membrane traffic in the early secretory pathway. Mol. Biol. Cell, 27, 127–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Jarosz D.F., Taipale M. and Lindquist S. (2010) Protein homeostasis and the phenotypic manifestation of genetic diversity: principles and mechanisms. Annu. Rev. Genet., 44, 189–216. [DOI] [PubMed] [Google Scholar]
- 92. Hetz C., Axten J.M. and Patterson J.B. (2019) Pharmacological targeting of the unfolded protein response for disease intervention. Nat. Chem. Biol., 15, 764–775. [DOI] [PubMed] [Google Scholar]
- 93. Klaips C.L., Jayaraj G.G. and Hartl F.U. (2018) Pathways of cellular proteostasis in aging and disease. J. Cell Biol., 217, 51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Farmer C.A., Thurm A., Farhat N., Bianconi S., Keener L.A. and Porter F.D. (2019) Long-term neuropsychological outcomes from an open-label phase I/IIa trial of 2-hydroxypropyl-beta-cyclodextrins (VTS-270) in Niemann-Pick disease, type C1. CNS Drugs, 33, 677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ory D.S., Ottinger E.A., Farhat N.Y., King K.A., Jiang X., Weissfeld L., Berry-Kravis E., Davidson C.D., Bianconi S., Keener L.A. et al. (2017) Intrathecal 2-hydroxypropyl-beta-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: a non-randomised, open-label, phase 1-2 trial. Lancet, 390, 1758–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Davidson J., Molitor E., Moores S., Gale S.E., Subramanian K., Jiang X., Sidhu R., Kell P., Zhang J., Fujiwara H. et al. (2019) 2-Hydroxypropyl-beta-cyclodextrin is the active component in a triple combination formulation for treatment of Niemann-Pick C1 disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids, 1864, 1545–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Oliver K.E., Han S.T., Sorscher E.J. and Cutting G.R. (2017) Transformative therapies for rare CFTR missense alleles. Curr. Opin. Pharmacol., 34, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Taylor-Cousar J.L., Mall M.A., Ramsey B.W., McKone E.F., Tullis E., Marigowda G., McKee C.M., Waltz D., Moskowitz S.M., Savage J. et al. (2019) Clinical development of triple-combination CFTR modulators for cystic fibrosis patients with one or two F508del alleles. ERJ Open Res., 5,00082–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Davies J.C., Moskowitz S.M., Brown C., Horsley A., Mall M.A., McKone E.F., Plant B.J., Prais D., Ramsey B.W., Taylor-Cousar J.L. et al. (2018) VX-659-Tezacaftor-Ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N. Engl. J. Med., 379, 1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Keating D., Marigowda G., Burr L., Daines C., Mall M.A., McKone E.F., Ramsey B.W., Rowe S.M., Sass L.A., Tullis E. et al. (2018) VX-445-Tezacaftor-Ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N. Engl. J. Med., 379, 1612–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Torkamani A. (2018) Drilling for insight: forecasting phenotype from genotype. Trends Genet., 34, 821–822. [DOI] [PubMed] [Google Scholar]
- 102. Gardner B.M., Pincus D., Gotthardt K., Gallagher C.M. and Walter P. (2013) Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol., 5, a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Plate L. and Wiseman R.L. (2017) Regulating secretory proteostasis through the unfolded protein response: from function to therapy. Trends Cell Biol., 27, 722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Watari H., Blanchette-Mackie E.J., Dwyer N.K., Watari M., Neufeld E.B., Patel S., Pentchev P.G. and Strauss J.F. 3rd. (1999) Mutations in the leucine zipper motif and sterol-sensing domain inactivate the Niemann-Pick C1 glycoprotein. J. Biol. Chem., 274, 21861–21866. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.