

Abstract
An improved understanding of the origin of the electrocatalytic activity is of importance to the rational design of highly efficient electrocatalysts for the hydrogen evolution reaction. Here, an ambipolar single‐crystal tungsten diselenide (WSe2) semiconductor is employed as a model system where the conductance and carrier of WSe2 can be individually tuned by external electric fields. The field‐tuned electrochemical microcell is fabricated based on the single‐crystal WSe2 and the catalytic activity of the WSe2 microcell is measured versus the external electric field. Results show that WSe2 with electrons serving as the dominant carrier yields much higher activity than WSe2 with holes serving as the dominant carrier even both systems exhibit similar conductance. The catalytic activity enhancement can be characterized by the Tafel slope decrease from 138 to 104 mV per decade, while the electron area concentration increases from 0.64 × 1012 to 1.72 × 1012 cm−2. To further understand the underlying mechanism, the Gibbs free energy and charge distribution for adsorbed hydrogen on WSe2 versus the area charge concentration is systematically computed, which is in line with experiments. This comprehensive study not only sheds light on the mechanism underlying the electrocatalysis processes, but also offers a strategy to achieve higher electrocatalytic activity.
Keywords: ambipolar carrier, density function theory, electrochemical microcells, hydrogen evolution, model catalysis
An ambipolar single‐crystal tungsten diselenide (WSe2) semiconductor electrocatalyst is employed as a benchmark system for interfacial model catalysis. By tuning the external electric field, the conductance and carrier of ambipolar WSe2 can be individually modulated. Combining the experimental and theoretical study, it is demonstrated that the electron carrier plays a key role in the hydrogen evolution reaction.
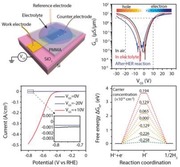
1. Introduction
Fossil fuel, as a dominant energy supply, gives rise to environmental pollution and leads to climate change. Therefore, the development of clean and renewable energy is the key way to meet the increasing global energy requirement and to resolve the environmental pollution caused by the overuse of fossil fuels.1, 2, 3 Hydrogen has been considered as a promising green energy carrier due to its highest energy density and pollution‐free and carbon‐free production.4, 5, 6 Generally, the splitting of water by electrocatalysts is regarded as a promising way for hydrogen generation.7 The key to realizing this reaction is to find an efficient and robust electrocatalyst which can effectively lower the reaction barrier and result in highly efficient utilization of electric.8
One key reaction in water splitting is the hydrogen evolution reaction (HER),5, 9, 10 which includes three reactions:
| (1) |
| (2) |
| (3) |
There are two pathways for generating molecular hydrogen, following either Volmer–Heyrovsky reaction or Volmer–Tafel reaction.11 Therefore, the Volmer reaction is the key step in HER and it is strongly dependent on the electron transfer, the density of active sites, and the Gibbs free energy of adsorbed atomic hydrogen.12, 13, 14, 15 To improve these factors, many strategies have been developed, such as improving the catalyst's conductance by introducing a conductive network, increasing the number of active sites by making nanostructure, and modifying materials through element doping or compositing.3, 4, 7, 16, 17, 18 Among them, the enhancement mechanism through improving conductivity is still unclear, possibly due to the complicated model system with varying factors like active site or nanostructures. Recently, it has been reported that reducing the resistance of catalyst by phase changing or introduction of external field can enhance the catalytic activity as both strategies can facilitate electron injection onto the active sites.19, 20, 21 Because the external electric field can modify the resistance of semiconductor and the carrier type and concentration, it is hard to distinguish whether the enhanced HER performance is due to enhanced conductance or due to increased electron concentration. Revealing this mechanism not only can further help to understand the enhancement mechanism, but also can help to rationally design of new electrocatalyst with high activity.
However, distinguishing the role of conductance and carrier on the powder‐like catalyst is very complicated due to factors such as the uncontrolled defects, nanostructures, and conductance. As an alternative, the high‐quality single crystal flake is an ideal model electrocatalyst for fundamental study.22 Although the conductance of unipolar semiconductor (n‐type or p‐type semiconductor) can be tuned by external electric field,23 the dominant carrier type could hardly switch between electron and hole. In this study, the ambipolar single‐crystal WSe2 semiconductor is employed as a model electrocatalyst because the conductance of WSe2 could be effectively tuned, and more importantly, its dominant carrier can be easily switched between hole and electron by external electric field.24, 25 Therefore, the influence on catalytic activity by conductance or by carrier can be effectively isolated. The ambipolar WSe2‐based electrochemical microcell is constructed. By tuning the external electric field, the catalytic activity can be in situ estimated under the same conductance with the opposite carrier type. This is a good platform to investigate the role of conductance and carrier in the catalytic reaction. The results show that with the similar conductance, WSe2 with electrons as the dominant carrier exhibits much higher catalytic activity than that of WSe2 with holes as the dominant carrier. With increasing electron concentration, the catalytic activity is enhanced, as demonstrated by the decrease of Tafel slope from 138 to 104 mV per decade and the decrease of overpotential at 10 mA cm−2 from 0.37 to 0.28 V. These results directly demonstrates that the electron carrier concentration plays a more important role in HER. Further, the WSe2 with different area charge concentration has been studied by using density‐functional‐theory (DFT) computation. The Gibbs free energy and the charge distribution have been computed, both being in line with our experiment.
2. Results and Discussion
The WSe2 flake was transferred on SiO2/Si substrate by mechanical exfoliation. The thickness of WSe2 was identified according to the optical contrast and AFM morphology measurement. Our previous study demonstrates that the electrical property of WSe2 is strongly dependent on its thickness, and the WSe2 flake with 12 layers has the best ambipolar electrical property with the highest carrier mobility.24 Therefore, in this study, the WSe2 flakes with a thickness of 12 layers were selected. The standard electron beam lithography process and followed electron beam evaporation were carried to form the electrical connections. To archive enough space to form an electrolyte droplet, the electrodes are extended to more than 1 cm, locating near the edge of the substrate. Then, PMMA (A11 concentration) was coated and expose the reaction window on the WSe2 flake by electron beam lithography. It should be noted that the surface of the sample has been fully covered by PMMA except the exposed window. The fabrication schema and corresponding optical images are shown in Figure 1 a,b. Figure 1c displays the AFM morphology of the exposed window, where the thickness of the covered PMMA is about 2.3 µm. The high quality of the WSe2 can be demonstrated by the atomic pattern inset in Figure 1c which is required by lateral force microscopy. The micro‐size topographic AFM image is shown in Figure S1a (Supporting Information). There are no PMMA residues and no discontinuities (edges or steps) on the WSe2 surface, indicating that the catalytic activity should stem from WSe2 basal plane, rather than from the discontinuities (edges or steps) or the defects. Figure 1d shows the transfer characterization of the WSe2 transistor where the gate voltage is giving through the back conductive silicon. It is clearly seen that the WSe2 transistor exhibits the ambipolar transport behavior, where the hole is the dominant carrier for V GS < 0 V and the electron is the dominant carrier for V GS > 0 V.24 Further, the transport curve shows a symmetric characterization in both hole and electron branches, indicating the balanced hole and electron conductance. After introducing the 0.5 m H2SO4 electrolyte, the source‐drain conductance and the gate leakage as a function of gate voltage have no obvious change, suggesting WSe2 is stable in the electrolyte showing symmetric conductance. The ambipolar transport behavior of WSe2 is a platform to study the role of the carrier in surface catalysis since WSe2 could have a similar conductance but with opposite carrier type.
Figure 1.
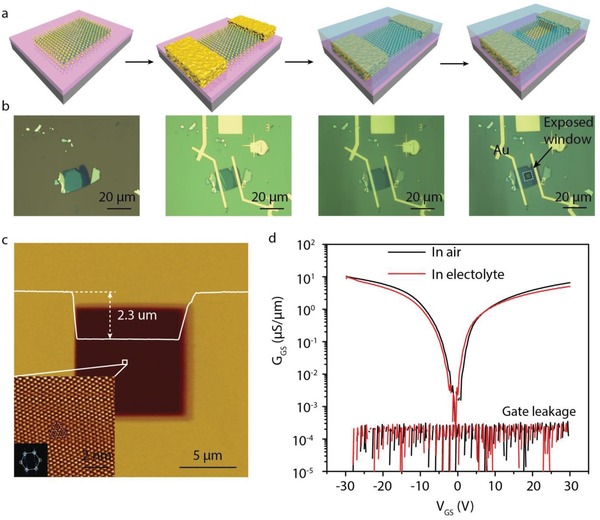
The device fabrication on WSe2 basal plane. a) A schematic diagram of the fabrication process and b) a realistic optical image of WSe2 device. c) The AFM height image of the protected PMMA layer, where the window is exposed by EBL only on the WSe2 basal plane. The inset shows the low pass filtered atomic pattern acquired from the center region by lateral force microscopy. d) The electrical performance of WSe2 device in the air and an electrolyte. The electrolyte is 0.5 mol L−1 H2SO4 solution.
To explore the role of the carrier in surface catalysis, the ambipolar WSe2 transistor with symmetric conductance was employed to construct an electric field tuned electrochemical microcell, as shown in Figure 2 a. In this setup, the surface catalysis was assessed by measuring the water splitting property through HER with employing the WSe2 as the electrocatalyst. HER is carried out using a three‐electrode configuration with Pt wire as the counterelectrode, micro‐Ag/AgCl electrode as the reference electrode, and the electrode connected with the WSe2 flake as the working electrode.26 Figure 2b shows the real image of the setup. The transfer curves of the WSe2 transistor at different conditions (in the air, in the electrolyte and after HER) are shown in Figure 2c. The HER polarization curves (Figure 2d) were measured at different gate voltage corresponding to different carrier envelope. As seen, the WSe2 flake exhibits a similar conductance (≈3 µS µm−1) at V GS = +10 and −20 V but with electron and hole as a dominant carrier, respectively. It is surprising that the polarization current at V GS = +10 V can reach about 0.5 A cm−2 (when the potential is −0.8 V vs RHE), which is three orders higher than the polarization current at V GS = −20 V. This implies that the conductance of catalysts may not be the key factor in catalytic reaction. The lower polarization current at V GS = 0 V is much lower since WSe2 is an insulator. The blue line in Figure 2c shows the transport curve after these HER measurements which directly confirm the stability of WSe2, further confirming that the WSe2 flake at V GS = +10 and −20 V exhibits the similar conductance but different carrier type. Considering that the solution gate from the counterelectrode has little influence on the electrochemical catalytic reaction,21 the enhanced catalytic activity should be originated from the material's property. The topographic AFM image and Raman spectra of WSe2 prior to and after the reaction is shown in Figure S1 (Supporting Information). As seen, there are no obvious changes in morphology and Raman spectra, indicating that the WSe2 basal plane is stable.
Figure 2.
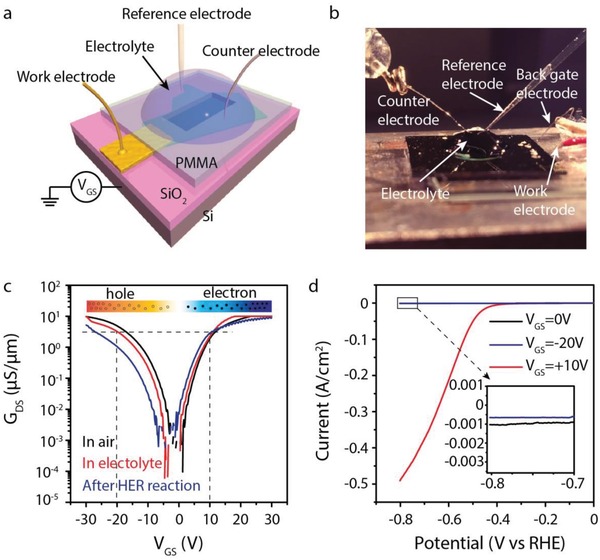
The electrocatalytic performance in electron and hole branches on the ambipolar WSe2 device. a) A schematic of the setup showing a single gold pad connected with a WSe2 flake used as the working electrode. b) Photography of the electrochemical microcell. c) The electrical transfer properties of the WSe2 device in the air, in the electrolyte and after HER reaction, respectively. d) Typical polarization curves measured for the WSe2 device when the gate voltage is 0, −20, and +10 V, respectively. The electrocatalytic measurement is in 0.5 m H2SO4 with a scan ratio of 5 mV s−1. The inset is the corresponding Tafel plot.
The polarization curves of the WSe2 flake at different gate voltages are shown in Figure 3 . The gate voltages are tuned as the sequence of 0, +5, +10, +15, +20, 0, and −20 V. The polarization curves of V GS = 0 V are overlapped excluding the variation of WSe2 material during the measurement. After all HER measurements, the transfer curve of the WSe2 transistor in electrolyte still exhibits the symmetric conductance character, meaning the above HER measurements are performed under symmetric transport condition. The carrier concentration could be calculated by n = C 0(V GS − V 0)/e, where n is the carrier concentration, C 0 is the capacity, V 0 is the neutral point, and e is the electron charge.27 In this device, the V 0 is about −4 V, possible due to the intrinsic dopant in the WSe2 crystal. Figure 3b shows the Tafel curves which are fitted according to Tafel equation: η = b log(j) + a, where η is the potential, b is the Tafel slope, and j is current density, respectively. The carrier area concentration, conductance, Tafel slope, and overpotential are summarized in Table 1 . As seen, the polarization current is largely boosted with the increasing of gate voltage from 0 to +20 V. The overpotentials calculated at the polarization current of 10 mA cm−2 are 0.37, 0.32, 0.30, and 0.28 V when gate voltages are +5, +10, +15, and +20 V, respectively. Even WSe2 at V GS = −20 V exhibits a higher conductance (0.932 µS µm−1) compared with WSe2 at V GS = +5 V (conductance is 0.174 µS µm−1), the polarization current at V GS = −20 V is still much lower than that at V GS = +5 V, which shows a similar phenomenon with the previous device in Figure 2. During tuning the gate voltage, the carrier concentration and also the conductance are both tuned. However, the conductance of WSe2 increases from 0.174 to 4.695 µS µm−1 when the gate voltage increases from +5 to +20 V. Considering that when the resistance is smaller than 10–100 kΩ mm (corresponding to that conductance is higher than 0.01–0.1 µS µm−1), the variation of resistance or conductance gives no obvious influence on catalytic activity.20, 26 Thus, we can propose that the varied catalytic activity (Tafel slope or overpotential) is due to the increased area electron concentration rather than the improved conductance. The results also suggest that the electron carrier is the key factor during electrocatalytic reaction rather than the conductivity, and that synthesis of electron‐rich electrocatalyst is preferred. Although the external field modulation may not lead to the highest catalytic performance, combination with other strategies, e.g., introducing point defects via doping heteroatoms28, 29 may further enhance the catalytic performance.
Figure 3.

Gate voltage‐dependent electrocatalytic activity on the ambipolar WSe2 device. a) The polarization curves measured under different gate voltages, and the inset shows the electrical transport property of WSe2 device after all measurements. b) The Tafel plots from (a).
Table 1.
The carrier concentration (per unit area), conductance, Tafel slope, and overpotential of WSe2 electrocatalysts under different gate voltage
| V GS [V] | N [×1012 cm−2] | G [µS µm−1] | Tafel slope [mV per decade] | Overpotential (at 10 mA cm−2, V vs RHE) |
|---|---|---|---|---|
| 0 | −0.29 | 2.393e−3 | – | – |
| 5 | −0.64 | 0.174 | 138 | 0.37 |
| 10 | −1.00 | 1.595 | 119 | 0.32 |
| 15 | −1.36 | 3.271 | 114 | 0.30 |
| 20 | −1.72 | 4.695 | 104 | 0.28 |
| −20 | +1.15 | 0.932 | – | – |
“−”: electron carrier; “+”: hole carrier.
The in situ HER measurements are all made under the same condition, for example, the same contact property and the same surface structure. The experimental results indicate that WSe2 with electrons as the dominant carrier exhibits much higher catalytic activity than that of WSe2 with holes as the dominant carrier when they have similar conductance. To further understand the underlying mechanism of enhanced HER activity by extra electron or hole, DFT calculation is carried out to evaluate the Gibbs free energy of a hydrogen atom on a charged WSe2 sheet, a known factor for determining the HER activity. For seeking a starting configuration of the most stable adsorption site, we examine all possible initial positions for H adsorption with symmetry considerations. Figure S2 (Supporting Information) depicts the top and side views of three optimized configurations. The corresponding formation energy is calculated by , where is the total energy of the H atom adsorbed on the WSe2 sheet, E H is the energy of the H atom, and is the energy of the WSe2 sheet. As seen, the hydrogen atom adsorbed on the top of six‐ring and near the Se atom (Figure S2c, Supporting Information) is the favorable energy state with the lowest formation energy. Based on the most stable adsorption structure, Figure 4 a–c shows the optimized WSe2 sheet with an adsorbed H at different carrier area concentration (n) of 6.45 × 1013, 0.00, and −2.58 × 1013 cm−2, respectively. The negative carrier means the carrier is an electron, while the positive carrier means the carrier is a hole. The corresponding carrier concentration varies from 1.94 × 1013 to −2.58 × 1013 cm−2, calculated based on the area of the model system (1.55 × 10−14 cm2). When the extra carrier of the system is zero, the preferential H position is near a Se atom, where a Se—H bond is formed. Interestingly, the position of the adsorbed H with respect to the Se atom depends on its charge state. In the neutral and negative charge states, the H is located at the antibonding site, as shown in Figure 4b,c. The Se—H bond is aligned with a W—Se bond, pointing toward the surface spacing.
Figure 4.

DFT calculation results of hydrogen atom adsorbed on charged WSe2 sheet with respect to the different carrier concentration (per unit area n). The associated adsorption sites at n = a) 0.65 × 1013 cm−2, b) neutral, and c) −2.58 × 1013 cm−2. d) The Gibbs free‐energy diagram of hydrogen evolution reaction. The blue, purple and green balls represent Se, W, and H atoms, respectively. The n > 0 corresponds to the added hole in the system, while n < 0 corresponds to the added electron in the system.
The preferential position of the adsorbed H in the positive charge state is different, where the adsorbed H moves toward the top of a Se atom (Figure 4a). A similar adsorption site of H was reported in MoS2.28, 29 Generally, a good HER catalyst should feature value near zero, which allows optimal adsorption/desorption kinetics. Further atomic insight into the carrier‐induced HER activity differences is obtained from the computed adsorption Gibbs free energy of the bonded hydrogen atom. The calculated free energy is illustrated in Figure 4d. In the cases of the extra holes, the values are too positive, and the H* species cannot be adsorbed onto the catalyst surface, and thus they are also unsuitable for HER. Compared with the pristine WSe2 structure without an extra carrier, the extra electrons lead to more negative values, indicating a more favorable adsorption process for the H* species. When the extra electron area concentration is −2.58 × 1013 cm−2, the catalyst exhibits a desired value of 0.033 eV, which is closer to zero than that of the well‐known Pt‐based catalyst ( = 0.09 eV).30, 31, 32 The basic trend of our theoretical results is qualitatively consistent with that of experimental results, even though the specific values are not exactly the same. We examine the effects of spin polarization and the use of different pseudopotential in our DFT calculations. As shown in Figures S4 and S5 (Supporting Information), neither the spin nor the GBRV pseudopotential can have much influence on the variation trends of the free energy, indicating the reliability of the current DFT calculation results. To gain a deeper understanding of the experimental results, we also examine the role of defect sites in the catalytic reaction (see Figures S6 and S7 in the Supporting Information). Both W point defects and edge defects can decrease the Gibbs free energy to a certain level, depending on the position relative to the defect site, whereas the Se vacancy can increase the Gibbs free energy. The opposing effects of the defects on the Gibbs free energy suggest that the defects alone cannot explain the experimental results of increased HER activity. Alternatively, we find that the HER activity is very localized to the adsorption site, suggesting that the system area in the DFT calculation plays a decisive role in the electron area concentration. Besides the system size of 4 × 4 × 1 supercell, we also consider a larger supercell size with 8 × 8 × 1 unit cells (193 atoms) to illustrate this point. As shown in Figure S8 (Supporting Information), the larger supercell results in reduced electron area concentration to nearly one eighth. It is known that Gibbs free energy ΔG H* is a commonly used descriptor for characterizing the catalytic activity of H atom adsorption. Some other important descriptors for understanding the structure‐activity‐selectivity relationships include the electronic level, i.e., electron charge, valance band position, etc.28
The difference in the H atomic position and the interaction between H atom and the WSe2 sheet cause distinct charge transfer behaviors. To correlate this charge transfer behavior with the HER activity, we construct the charge‐difference plot, where the charge difference is calculated by . Here, is the total charge density of the H atom adsorbed on the WSe2 sheet, ρH is the charge density of the H atom, and is the charge density of the WSe2 sheet.33 Figure 5 a–d present the WSe2 sheet adsorbed with one H atom with respect to different carrier area concentration 6.45 × 1013, 3.22 × 1013, 0.00, and −2.58 × 1013 cm−2, respectively. Similar to the reported MoS2 for HER process,16, 26, 34 because of the very high level of 3p orbital with respect to the H 1s orbital, the H adsorption on the basal plane of pristine WSe2 is too weak ( = 1.68 eV), leading to a poor HER performance. As shown in Figure 5a–d, when the extra carrier is a hole, the hydrogen atom is surrounded by the electron depletion region, as denoted by Figure 4e, which is unfavorable for HER activity. With the extra electron increasing from 0.00 to −2.58 × 1013 cm−2, it will strengthen the adsorption and change the charge state of H atom from electron depletion to electron accumulation, which offsets the energy level for enhancing the H adsorption and HER activity. However, with the area concentration of extra electron further increasing, the further enhanced adsorption energy would render the desorption process difficult, and thus deteriorate the HER performance. In other words, the extra electron carrier would contribute to the charge redistribution with the decrease of adsorption energy. A suitable amount of electrons would lead to favorable adsorption/desorption of H atoms for high‐performance HER. To illustrate in detail the nature of charge transfer behavior, the plane averaged carrier concentration along the out‐of‐plane direction is shown in Figure 5e. The plane averaged carrier concentration is calculated by , where is plane averaged charge density of the combined H–WSe2 system, σH(z) and are, respectively, the plane averaged charge density of the isolated H atom and WSe2 layer, which are calculated by fixing the atomic positions of the corresponding components in the combined system. The positive and negative values indicate charge loss and gain, respectively. It is clear that the extra electron contributes to a smoother distribution of charge, which attributes to the higher mobility of extra electron. However, the hole carrier causes a more localized charge transfer. Both the charge depletion and charge accumulation constitute the charge redistribution behavior, and ultimately, affect the hydrogen adsorption. The DFT calculation demonstrates that the hole carrier or overly introduced electron carrier can induce too positive or too negative Gibbs free energy, neither favorable to the adsorption and desorption of hydrogen atom. An appropriate area concentration of electron carrier would give rise to optimal Gibbs free energy, closer to the value of Pt.
Figure 5.

The charge difference plot of the hydrogen atom adsorbed on the charged WSe2 sheet with different carrier concentration (per unit area n): a) n = 6.45 × 1013 cm−2; b) n = 3.22 × 1013 cm−2; c) n = 0.00 cm−2; and d) n = −2.58 × 1013 cm−2. The isovalue is 0.0025 e Bohr−3. The yellow region indicates electron accumulation, and the cyan region indicates electron depletion, respectively. The H atom is highlighted by a black circle. e) The plane averaged carrier concentration along the out‐of‐plane direction when the WSe2 has a different charge density.
3. Conclusion
In summary, the role of the carrier on catalytic activity has been systemically studied from experiment and DFT calculation. The ambipolar WSe2‐based electrochemical microcell provides the platform to estimate the in situ catalytic activity under the same condition (defect, active site, nanostructure, etc.). With varying the external field, the dominant carrier in WSe2 can switch between electron and hole, while the conductance of WSe2 can be tuned as well. The effect of the electron carrier, hole carrier, and conductance in catalysis reaction has been carefully discussed. The results show that with the similar conductance, WSe2 with the electrons as the dominant carrier shows much higher catalytic activity than that with the holes as the dominant carrier. With increasing the gate voltage in electron dominated WSe2, the catalytic activity can be further enhanced with the Tafel slope decreasing from 138 to 104 mV per decade, attributing to the increased electron concentration rather than the conductance. Furthermore, the Gibbs free energies of adsorbed atomic hydrogen on WSe2 with different electron and hole area concentrations are calculated. The results show that of electron dominated WSe2 is much lower than that of hole dominated WSe2. Moreover, the can be lowered to 0.033 from 1.68 eV by injecting electrons into pristine WSe2 with an electron area concentration of 2.58 × 1013 cm−2. The results show that the electron carrier plays a more important role in HER, rather than the conductance, which provides a new strategy to rationally design highly efficient electrocatalysts.
4. Experimental Section
Device Fabrication and Measurement: The WSe2 flakes were mechanically exfoliated from a bulk crystal onto a SiO2/Si substrate with a size of 2 cm × 2 cm. The WSe2 flake with a thickness of 12 layers, which exhibits the highest carrier mobility and ambipolar behavior,24 is selected. The source and drain electrodes were patterned with electron beam lithography. Then 10 nm Ti/40 nm Au layers were evaporated by electron beam deposition with a depositing speed of 0.2 Å s−1. Before evaporation, the sample was kept overnight under the high vacuum in the electron beam deposition system. To have enough space to form an electrolyte droplet, the source and drain electrodes were extended to near the edge of the substrate with a length of about 1 cm. A second electron beam lithography process was employed to expose the basal plane of WSe2. To fully cover the WSe2 edges and the electrodes, PMMA with a concentration of 11% in anisole was spin‐coated on the sample and then a conductive polymer was coated on its surface to eliminate the charge effect during electron beam lithography. More attention was devoted to ensuring that no gold electrode and WSe2 edge were exposed to the electrolyte. After opening the window on the WSe2 flake, the sample was baked at 180 °C for 10 min. The electrical transport property of WSe2 transistor was carried by Keithley 4200 SCS, and the field tuned electrochemical reaction was performed on electrochemical workstation combined with Keithley 4200 SCS. Linear sweep voltammetry with a scan rate of 5 mV s−1 was conducted in 0.5 m H2SO4 solution, using a Pt wire as the counterelectrode. The micro‐reference electrode was calibrated for the reversible hydrogen potential, where E(vs RHE) = E(vs Ag/AgCl) + 0.197 V. Before measurement, the H2SO4 solution was degassed using pure Ar gas.
Computation: DFT calculations were carried out using the Hartwigsen–Goedeker–Hutter norm‐conserving pseudopotentials35 and the Perdew–Burke–Ernzerhof (PBE) exchange‐correlation functional36 as implemented in the Quantum ESPRESSO package.37, 38 The spin‐polarized calculation was employed,39 and the vdW + DF2 functional was selected to account for van der Waals (vdW) interactions.40 The model system is a 4 × 4 supercell of WSe2 (with 48 atoms) and a single hydrogen atom adsorbed on the surface. In the direction vertical to the surface, the supercell extends with a vacuum space for 30 Å. The Brillouin zone is sampled using a 3 × 3 × 1 Monkhorst–Pack k‐point grid. Atomic positions are optimized until the maximum force on all atoms is less than 0.001 a.u. Cutoffs of 40 and 160 Ry were chosen for the wave function and the electronic density, respectively. For the computational scheme based on pseudopotential and periodic slab geometry, it is challenging to deal with the charged system when imposing the periodic boundary condition in the surface normal direction.41, 42 In the case of the system with the extra carrier, an effective screening medium method (ESM)42, 43 was utilized to treat effectively charged slabs. ESM screens the electronic charge of a polarized/charged medium along one perpendicular direction by introducing a classical charge model and a local relative permittivity into the DFT calculation framework. In this condition, excess or deficit charge is accommodated on one side of a slab, and the image charge is automatically induced in the medium. Thus, this permits calculation by using open boundary conditions. Lastly, for DFT calculation with a larger supercell with 8 × 8 × 1 unit cells (193 atoms), the Gamma point was adopted.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
Z.W. and H.H.W. contributed equally to this work. This research was supported by grants from the Danish National Research Foundation, AUFF‐NOVA project from Aarhus Universitets Forskningsfond, and EU H2020RISE 2016‐MNR4S Cell project. H. H. Wu acknowledges the financial support from the Natural Science Foundations of China (51901013). X. C. Zeng was supported by UNL Holland Computing Center. Z. Wang was supported by Fundamental Research Funds for the Central Universities, China (YJ201893), and State Key Lab of Advanced Metals and Materials, China (Grant No. 2019‐Z03).
Wang Z., Wu H.‐H., Li Q., Besenbacher F., Li Y., Zeng X. C., Dong M., Reversing Interfacial Catalysis of Ambipolar WSe2 Single Crystal. Adv. Sci. 2020, 7, 1901382 10.1002/advs.201901382
Contributor Information
Xiao Cheng Zeng, Email: xzeng1@unl.edu.
Mingdong Dong, Email: dong@inano.au.dk.
References
- 1. Nellist M. R., Laskowski F. A. L., Qiu J., Hajibabaei H., Sivula K., Hamann T. W., Boettcher S. W., Nat. Energy 2018, 3, 46. [Google Scholar]
- 2. Song B., Jin S., Joule 2017, 1, 220. [Google Scholar]
- 3. Li H., Tsai C., Koh A. L., Cai L., Contryman A. W., Fragapane A. H., Zhao J., Han H. S., Manoharan H. C., Abild‐Pedersen F., Nørskov J. K., Zheng X., Nat. Mater. 2016, 15, 48. [DOI] [PubMed] [Google Scholar]
- 4. Hu J., Huang B., Zhang C., Wang Z., An Y., Zhou D., Lin H., Leung M. K. H., Yang S., Energy Environ. Sci. 2017, 10, 593. [Google Scholar]
- 5. Merki D., Hu X., Energy Environ. Sci. 2011, 4, 3878. [Google Scholar]
- 6. Xu H., Cheng D., Cao D., Zeng X. C., Nat. Catal. 2018, 1, 339. [Google Scholar]
- 7. Miao J., Xiao F.‐X., Yang H. B., Khoo S. Y., Chen J., Fan Z., Hsu Y.‐Y., Chen H. M., Zhang H., Liu B., Sci. Adv. 2015, 1, e1500259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Voiry D., Shin H. S., Loh K. P., Chhowalla M., Nat. Rev. Chem. 2018, 2, 0105. [Google Scholar]
- 9. Xie J., Zhang H., Li S., Wang R., Sun X., Zhou M., Zhou J., Lou X. W., Xie Y., Adv. Mater. 2013, 25, 5807. [DOI] [PubMed] [Google Scholar]
- 10. Voiry D., Yang J., Chhowalla M., Adv. Mater. 2016, 28, 6197. [DOI] [PubMed] [Google Scholar]
- 11. Zhao G., Rui K., Dou S. X., Sun W., Adv. Funct. Mater. 2018, 28, 1803291. [Google Scholar]
- 12. Su T., Shao Q., Qin Z., Guo Z., Wu Z., ACS Catal. 2018, 8, 2253. [Google Scholar]
- 13. Ying Y., Yumin Z., Tangling G., Tai Y., Xinghong Z., Jiecai H., Xianjie W., Zhihua Z., Ping X., Peng Z., Xingzhong C., Bo S., Song J., Adv. Mater. 2017, 29, 1700311. [Google Scholar]
- 14. Wang Z., Li Q., Besenbacher F., Dong M., Adv. Mater. 2016, 28, 10224. [DOI] [PubMed] [Google Scholar]
- 15. Liu Q., Fang Q., Chu W., Wan Y., Li X., Xu W., Habib M., Tao S., Zhou Y., Liu D., Xiang T., Khalil A., Wu X., Chhowalla M., Ajayan P. M., Song L., Chem. Mater. 2017, 29, 4738. [Google Scholar]
- 16. Deng J., Li H., Wang S., Ding D., Chen M., Liu C., Tian Z., Novoselov K. S., Ma C., Deng D., Bao X., Nat. Commun. 2017, 8, 14430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pető J., Ollár T., Vancsó P., Popov Z. I., Magda G. Z., Dobrik G., Hwang C., Sorokin P. B., Tapasztó L., Nat. Chem. 2018, 10, 1246. [DOI] [PubMed] [Google Scholar]
- 18. Luo Z., Ouyang Y., Zhang H., Xiao M., Ge J., Jiang Z., Wang J., Tang D., Cao X., Liu C., Xing W., Nat. Commun. 2018, 9, 2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Junhui W., Mengyu Y., Kangning Z., Xiaobin L., Peiyao W., Xuelei P., Wei Y., Liqiang M., Adv. Mater. 2017, 29, 1604464. [Google Scholar]
- 20. Voiry D., Fullon R., Yang J., de Carvalho Castro e Silva C., Kappera R., Bozkurt I., Kaplan D., Lagos M. J., Batson P. E., Gupta G., Aditya Mohite D., Dong L., Er D., Shenoy V. B., Asefa T., Chhowalla M., Nat. Mater. 2016, 15, 1003. [DOI] [PubMed] [Google Scholar]
- 21. Yan M., Pan X., Wang P., Chen F., He L., Jiang G., Wang J., Liu J. Z., Xu X., Liao X., Yang J., Mai L., Nano Lett. 2017, 17, 4109. [DOI] [PubMed] [Google Scholar]
- 22. Guo D., Shibuya R., Akiba C., Saji S., Kondo T., Nakamura J., Science 2016, 351, 361. [DOI] [PubMed] [Google Scholar]
- 23. Kim C., Moon I., Lee D., Choi M. S., Ahmed F., Nam S., Cho Y., Shin H.‐J., Park S., Yoo W. J., ACS Nano 2017, 11, 1588. [DOI] [PubMed] [Google Scholar]
- 24. Wang Z., Li Q., Chen Y., Cui B., Li Y., Besenbacher F., Dong M., NPG Asia Mater. 2018, 10, 703. [Google Scholar]
- 25. Zhou C., Zhao Y., Raju S., Wang Y., Lin Z., Chan M., Chai Y., Adv. Funct. Mater. 2016, 26, 4223. [Google Scholar]
- 26. Wang Z., Li Q., Xu H., Dahl‐Petersen C., Yang Q., Cheng D., Cao D., Besenbacher F., Lauritsen J. V., Helveg S., Dong M., Nano Energy 2018, 49, 634. [Google Scholar]
- 27. Wang Z., Wu H.‐H., Li Q., Besenbacher F., Zeng X. C., Dong M., Nanoscale 2018, 10, 18178. [DOI] [PubMed] [Google Scholar]
- 28. Vancsó P., Popov Z. I., Pető J., Ollár T., Dobrik G., Pap J. S., Hwang C., Sorokin P. B., Tapasztó L., ACS Energy Lett. 2019, 4, 1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shi Y., Zhou Y., Yang D. R., Xu W. X., Wang C., Wang F. B., Xu J. J., Xia X. H., Chen H. Y., J. Am. Chem. Soc. 2017, 139, 15479. [DOI] [PubMed] [Google Scholar]
- 30. Nørskov J. K., Bligaard T., Logadottir A., Kitchin J. R., Chen J. G., Pandelov S., Stimming U., J. Electrochem. Soc. 2005, 152, J23. [Google Scholar]
- 31. Pei Z., Gu J., Wang Y., Tang Z., Liu Z., Huang Y., Huang Y., Zhao J., Chen Z., Zhi C., ACS Nano 2017, 11, 6004. [DOI] [PubMed] [Google Scholar]
- 32. Yu S., Rao Y.‐C., Wu H.‐H., Duan X.‐M., Phys. Chem. Chem. Phys. 2018, 20, 27970. [DOI] [PubMed] [Google Scholar]
- 33. Tang W., Sanville E., Henkelman G., J. Phys. Condens. Matter 2009, 21, 084204. [DOI] [PubMed] [Google Scholar]
- 34. Tsai C., Abild‐Pedersen F., Nørskov J. K., Nano Lett. 2014, 14, 1381. [DOI] [PubMed] [Google Scholar]
- 35. Hartwigsen C., Goedecker S., Hutter J., Phys. Rev. B 1998, 58, 3641. [Google Scholar]
- 36. Perdew J. P., Burke K., Ernzerhof M., Phys. Rev. Lett. 1996, 77, 3865. [DOI] [PubMed] [Google Scholar]
- 37. Giannozzi P., Andreussi O., Brumme T., Bunau O., Nardelli M. B., Calandra M., Car R., Cavazzoni C., Ceresoli D., Cococcioni M., Colonna N., Carnimeo I., Corso A. D., Gironcoli S. d., Delugas P., DiStasio J. R. A., Ferretti A., Floris A., Fratesi G., Fugallo G., Gebauer R., Gerstmann U., Giustino F., Gorni T., Jia J., Kawamura M., Ko H. Y., Kokalj A., Küçükbenli E., Lazzeri M., Marsili M., Marzari N., Mauri F., Nguyen N. L., Nguyen H. V., Otero‐de‐la‐Roza A., Paulatto L., Poncé S., Rocca D., Sabatini R., Santra B., Schlipf M., Seitsonen A. P., Smogunov A., Timrov I., Thonhauser T., Umari P., Vast N., Wu X., Baroni S., J. Phys. Condens. Matter 2017, 29, 465901. [DOI] [PubMed] [Google Scholar]
- 38. Paolo G., Stefano B., Nicola B., Matteo C., Roberto C., Carlo C., Davide C., Guido L. C., Matteo C., Ismaila D., Andrea Dal C., de Stefano G., Stefano F., Guido F., Ralph G., Uwe G., Christos G., Anton K., Michele L., Layla M.‐S., Nicola M., Francesco M., Riccardo M., Stefano P., Alfredo P., Lorenzo P., Carlo S., Sandro S., Gabriele S., Ari P. S., Alexander S., Paolo U., Renata M. W., J. Phys. Condens. Matter 2009, 21, 395502.21832390 [Google Scholar]
- 39. Wu H.‐H., Huang H., Zhong J., Yu S., Zhang Q., Zeng X. C.. Nanoscale 2019, 11, 12210. [DOI] [PubMed] [Google Scholar]
- 40. Grimme S., J. Comput. Chem. 2006, 27, 1787. [DOI] [PubMed] [Google Scholar]
- 41. Makov G., Payne M. C., Phys. Rev. B 1995, 51, 4014. [DOI] [PubMed] [Google Scholar]
- 42. Otani M., Sugino O., Phys. Rev. B 2006, 73, 115407. [Google Scholar]
- 43. Hamada I., Morikawa Y., J. Phys. Chem. C 2008, 112, 10889. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information