

Summary:
High-throughput DNA sequencing techniques have enabled diverse approaches for linking DNA sequence to biochemical function. In contrast, assays of protein function have substantial limitations in terms of throughput, automation, and widespread availability. We have adapted an Illumina high-throughput sequencing chip to display an immense diversity of ribosomally-translated proteins and peptides, and then carried out fluorescence-based functional assays directly on this flow cell, demonstrating that a single, widely-available high-throughput platform can perform both sequencing-by-synthesis and protein assays. We quantified the binding of the M2 anti-FLAG antibody to a library of 1.3×104 variant FLAG peptides, exploring non-additive effects of combinations of mutations and discovering a “superFLAG” epitope variant. We also measured the enzymatic activity of 1.56×105 molecular variants of full-length human O6-alkylguanine-DNA alkyltransferase (SNAP-tag). This comprehensive corpus of catalytic rates revealed amino acid interaction networks and cooperativity, linked positive cooperativity to structural proximity, and revealed ubiquitous positively-cooperative interactions with histidine residues.
Keywords: high-throughput, protein array, protein engineering, antibody characterization, protein evolution, in vitro translation, ribosome display, sequencer hacking, superFLAG, SNAP tag
Graphical Abstract
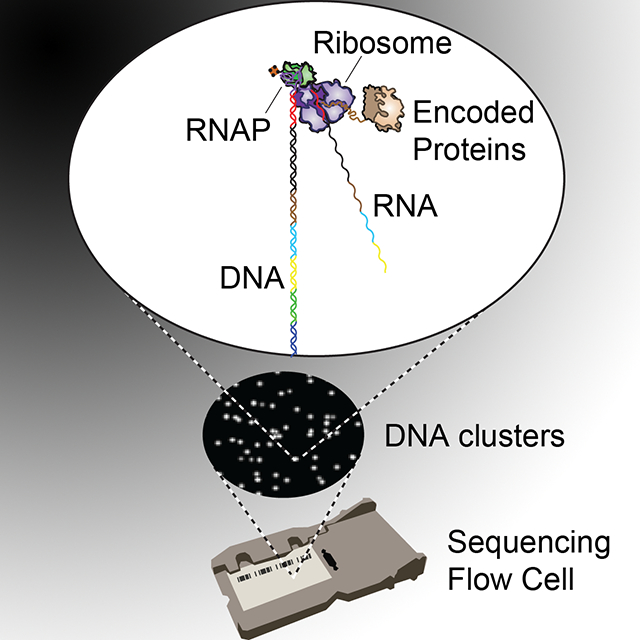
eToc Blurb
By generating a massive peptide/protein array in situ on an Illumina sequencing flow cell with in vitro translation, Layton et al. (2019) demonstrate direct protein assays for enzyme function and antibody-peptide interactions across large libraries (~105) of mutational variants on an automated platform.
Introduction:
High-throughput DNA sequencing technologies have enabled the investigation of diverse biological processes wherein the functional consequences of nucleic acid variation can be linked directly to the abundance and sequence of DNA fragments that are quantified at scale. These applications (e.g. ChIP-seq, ATAC-seq, bisulfite sequencing, Hi-C, bind-n-seq, etc. (Park, 2009, Buenrostro et al., 2013, Krueger et al., 2012, Lieberman-Aiden et al., 2009, Zykovich et al., 2009)) largely define the contemporary methodological foundations of modern functional genomics. In contrast, methods for directly assaying the influence of protein sequence variation on function has remained challenging to similarly scale and disseminate (Kingsmore, 2006). In vitro approaches for high-throughput protein functional measurements have included the quantification of selective enrichment (Larman et al., 2011, Fowler et al., 2010, Gu et al., 2014) of protein particles linked to their encoding nucleic-acids (Levin and Weiss, 2006, Odegrip et al., 2004) and parallelized binding assays on protein and peptide microarrays (Chandra et al., 2011), including arrays generated with in vitro protein translation (He et al., 2008, Ramachandran et al., 2004, Tao and Zhu, 2006) and in situ peptide synthesis (Hilpert et al., 2007, Legutki et al., 2014, Forsström et al., 2014). However, existing implementations have not yet provided the scalability, simplicity, automation, or accessibility necessary for widespread application. The implementation of direct and quantitative assays of protein function with the automation and throughput of a modern high-throughput sequencing platform would greatly expand our ability to develop and test a predictive understanding of the functional impact of coding mutations, to identify and characterize amino acid interaction networks and dependencies, and to learn useful principles and paradigms for rational design of protein function.
Design:
To enable facile, widely deployable, high-throughput, and quantitative protein characterization, we sought to leverage the capabilities and widespread adoption of the now-ubiquitous DNA sequencing flow cells to directly assay protein function at scale. High-throughput flow cell DNA arrays, the core of Illumina sequencing (Bentley et al., 2008), have recently been repurposed for quantitative high-throughput investigation of DNA-protein, RNA-protein, and RNA-RNA interactions across nucleic-acid sequence space. Building on this and other work (Nutiu et al., 2011, Tome et al., 2014, Buenrostro et al., 2014, She et al., 2017, Svensen et al., 2016, Gu et al., 2014) we have reengineered high-throughput DNA sequencing methods to assay protein function across a vast polypeptide sequence space. This approach aims to bring quantitative protein functional investigation to DNA sequencing-scale throughput using a hardware platform and microfluidic chip compatible with fluorescence-based sequencing by synthesis (SBS) methods, demonstrating that a single, widely-available high-throughput platform can, in principle, perform both sequencing-by-synthesis and protein function assays.
To develop methods capable of generating and quantifying our protein array on an SBS-compatible platform, we constructed a flexible, programmable workstation capable of microfluidically interfacing with and imaging sequencing flow cells (Jung et al., 2017, She et al., 2017). The resulting TIRF microscopy platform, based on the automated fluidics and fluorescence microscopy components of an Illumina GAIIx sequencer, interfaces with previously-sequenced (and therefore sequence- and position-indexed) Illumina MiSeq flow cell arrays (She et al., 2017). To generate a protein array, we create a library of engineered DNA constructs that encode for polypeptides of interest (Fig. S1), then sequence this library on an Illumina MiSeq. After moving the sequenced chip to our assay platform, we re-register the cluster positions to their sequences (see methods). We next perform in vitro transcription/translation on chip such that both the transcript and nascent peptide remain associated with their DNA template, producing a tethered protein array. To enable this tethered display, each member of the sequencing library contains DNA sequence elements that allow for 1) prokaryotic in vitro transcription, 2) immobilization of the resulting RNA transcript, 3) translation, and 4) ribosome stall, similar to ribosome display (Lipovsek and Plückthun, 2004) (Fig 1). Fluorescence-based functional assays (e.g. quantifying binding of fluorescently-labeled binding partners or incorporation of fluorescent substrates) may then be conducted directly on this array. Fluorescence images, paired to cluster DNA sequences generated from the sequencing run, are then quantified (see methods) to assay polypeptide binding or other protein function. We call this method Protein display on a Massively Parallel Array, or Prot-MaP.
Fig 1. A high-throughput protein array on a DNA sequencing flow cell.

1.) A polypeptide-encoding DNA library is clustered and sequenced on a MiSeq, then the chip is transferred to an imaging station where 2.) roadblocked transcription and stalled translation takes place in vitro, producing bound polypeptides. Protein functional assays can then be performed on all displayed variants, such as interrogation with a primary antibody and fluorescently-labeled secondary antibody (shown) followed by fluorescence imaging. 3.) Fluorescence images are registered to the sequence information, then quantified and fit.
Results:
As a testbed for this methodology, we characterized the widely-used FLAG peptide/M2 antibody interaction. FLAG peptide (canonically DYKDDDDK) is commonly used for protein labeling, purification, and as a general affinity reagent (Einhauer and Jungbauer, 2001). The consensus sequence profile of the linear peptide epitope of M2 has previously been characterized as DYKxxDxx based on 5 (Srila and Yamabhai, 2013) or 18 (Osada et al., 2009) clones selected from random peptide libraries. We aimed to generate a library of target peptides that would comprehensively probe the contribution of residues in the FLAG peptide to the M2 antibody interaction with a much larger library of variants. We engineered a variant library of 13,154 sequences that encoded all single, double, and triple-combinations of mutant positions, with each position substituted to each of 6 different amino acids – A,K,D,S,F, and L – that represent small, positive, negative, polar, aromatic, and aliphatic substitutions, respectively. The peptide-coding mutant library was produced with microarray synthesis and assembled into a sequenceable library construct with elements enabling transcription, translation, and stable peptide display (Fig. 1, fig. S1, see methods).
After DNA sequencing, 623,075 total clusters encoding FLAG library members were produced with 12,739 of 13,154 (96.8%) programmed variants represented by 8 or more peptide clusters (i.e. nearly complete coverage of this synthetic FLAG library required only ~2.5% of capacity of the flow cell). After peptide generation, M2 antibody was introduced at increasing concentrations and allowed to bind to the peptide, with each concentration followed by detection with a fluorescently-labeled secondary antibody, then imaging (Fig 2A). After image registration and fluorescence quantification, the limit of detection (LoD) for antibody binding was determined for each variant (Fig. 2B, see methods), representing the lowest concentration of antibody that produces detectable binding. This Prot-MaP LoD assay is analogous to a massively multiplexed ELISA whereby primary antibody interactions are probed by secondary detection steps that produce measurable signal.
Fig 2. Binding landscape of FLAG peptide variants to M2 monoclonal antibody.

(A) A representative flow cell image shows fiducial marks and cluster-of-interest positions before the binding assay (top). Experimental images show fluorescent secondary antibody detection of binding across increasing concentrations of M2 anti-FLAG primary antibody (bottom). (B) Quantified fluorescence medians (error bars are SEM) that rise above a background threshold (grey dashed line) are extrapolated (solid) or interpolated (dashed) to estimate the limit of detection (LoD, open squares) for each FLAG variant, including WT FLAG (DYKDDDDK) and negative control (AAADDDDK) as well as superFLAG variant (DYKDEDLL), which, like 2xFLAG (DYKDDDDKGDYKDHD), gives much higher signal than WT. Two different scales are shown (top vs bottom) to accommodate the wide dynamic range of observed signals. Open symbols indicate points below background, closed symbols are above. (C) 6 amino acid mutations, each representing a physicochemical category, are represented by color (if the WT identity is the same, the alternate mutation in parenthesis is made). LoD for M2 anti-FLAG antibody binding to single mutants and (D) double mutants of the canonical (WT) peptide, DYKDDDDK, across the six amino acids at each position. (E) LoD (bottom left) and cooperativity (top right) of double mutants of the D4L base mutant (triple mutants from the WT). Cooperativity = −log(LoDDM/ LoDWT) – (−log(LoDSM1/ LoDWT) + −log(LoDSM2/ LoDWT)), where DM refers to the double mutant and SM1 and SM2 to the two corresponding single mutants. (F) Double mutant cycles. Deviations from the y=x line (grey) indicate non-additivity (cooperativity) (G) Orthogonal investigation of individual peptide variants with a fluorescence-based plate assay. Antibodies bound to variant peptides (see legend) were challenged for 2 hours with varying concentrations of 3xFLAG competitor peptide. Fluorescence values report residual bound M2 antibody.
The single and double mutant affinity landscapes (Fig. 2C–D) of the canonical sequence (DYKDDDDK, “WT”) largely recapitulate the previously reported motif pattern, DYKxxDxx (Srila and Yamabhai, 2013, Osada et al., 2009). However, we observe substantial additional constraint at position D4, with 5 of 6 mutations exhibiting no detectable binding. To investigate mutational constraint at this position further, we asked if detrimental mutations at position 4 could be rescued by mutations at other positions in our higher order mutants. Looking at all double mutants of the only measurable D4 mutant (D4L; triple mutants from WT; Fig. 2E), we observe that several combinations of mutations that include D5E and D7K partially rescue D4L, lowering LoD to near-WT levels (Fig. 2E). Many of these compensating combinations exhibit significant positive cooperativity (e.g. D4L,D5E,D7K), demonstrating the ability of Prot-MaP not only to identify critical residues and motifs, but also to establish cooperative interaction landscapes that can substantially deviate from simple additive models (Fig. 2F).
Interestingly, we also found several FLAG variant sequences that exhibited significantly lower LoD than WT, including a variant we term “superFLAG” (DYKDEDLL) with an extrapolated LoD of 0.14 nM, 7.9-fold lower than WT (Fig. 2B). To confirm these observations, we performed a fluorescence-based plate assay to measure M2 binding competition between immobilized variant peptides and solutions of 3xFLAG high-affinity peptide (Fig 2G). SuperFLAG displays higher avidity to the M2 antibody then all other assayed peptides, including the original DYKDDDDK immunogen to which M2 was raised (Brizzard et al., 1994).
Unlike chemically synthesized peptide arrays, our in vitro transcribed and translated array enables the possibility of generating full-length protein features (He et al., 2008, Ramachandran et al., 2004, Tao and Zhu, 2006). To demonstrate this capability, we investigated the mutational landscape of SNAP-tag, an engineered version of O6-alkylguanine-DNA alkyltransferase, a 181 AA, 20kD human DNA repair enzyme (Sun et al., 2011). The SNAP-tag protein transfers a benzyl group from a derivatized benzylguanine substrate (often substituted with a fluorophore) to its own Cys 145, thereby covalently labeling itself. The simplicity and specificity of this fluorogenic self-labeling reaction has led SNAP-tag to become widely used as a protein labeling tag, and here it provides an elegant model system for investigation of the sequence-function relationship of an enzyme with Prot-MaP (Fig 3A).
Fig 3. High-throughput, on-chip characterization of activity across the mutational landscape of a full-length enzyme.

(A) On-chip SNAP-tag-catalyzed fluorescent self-labeling (B) 7 target amino acid positions (red) and benzylguanine substrate (green) on the structure of SNAP-tag (PDB ID: 3KZZ) (C) Library coverage of all 19 possible amino acid substitutions in single, double, and triple-combinations of the target positions. (D) Quantified fluorescence medians for selected single mutants (error bars are SEM). Variants that rose above the background threshold (grey dashed) were fit with a single exponential. (E) Fit kobs values for single mutants and (F) double mutants across the 20 amino acids. The WT (unmutated) variant is boxed in black. (G) Position 153 is in a loop connecting two domains. (H) Correlation of activity with backbone flexibility (Fuchs et al., 2015) for all 19 amino acid substitutions at position 153. R2=0.77 (p-value = 3×10−6 by permutations). Proline is the least flexible and glycine the most.
To explore the relationship between protein sequence variation and catalysis in high throughput, we examined the functional interrelationship of seven residues previously identified to modulate function without entirely abolishing catalytic activity (Y114, A121, K131, S135, L153, G157, and E159) (Gautier et al., 2008) (Fig. 3B). The DNA sequence encoding this mutated region (residues 114–159, the “mutant region”), was combinatorially assembled from oligos with the aim of generating single-, double-, and triple-mutant combinations of the 7 target positions across all possible 20 amino acid substitutions (fig. S1). Once assembled, the DNA fragment population was bottlenecked (by diluting to a target number of molecules) and reamplified with PCR to obtain multiple identical copies of molecular variants on the array.
After sequencing this library, RNA synthesis and in situ protein generation was performed on the MiSeq flow cell. We then introduced a solution of 200 nM substrate (SNAP-Surface 549) continuously onto the resulting protein array, then measured substrate incorporation over 160 minutes (see methods). A total of 6,751,654 clusters were successfully quantified across all timepoints, and signals were averaged across clusters displaying identical protein variants (> 8 clusters per variant). A total of 156,140 unique variants were assayed, including all possible single mutants across all 20 amino acids (133), 7570 of 7581 possible double mutants, 125,076 of 240,065 possible triple mutants (Fig. 3C), as well as 23,360 other variants. The majority of mutants (118,025/156,140; 75.59%) exhibited no detectable catalytic activity above background levels, while 32,072 variants exhibited detectable activity that could be well-fit by a single exponential to obtain kobs (Fig. 3D, see methods).
We observed large variation in the mutational constraint for each of the 7 targeted amino acids. At one extreme, Y114 is fully constrained to tyrosine across all single and double mutants (Fig. 3E, F). Even Y114F, a conservative substitution that deletes only a hydroxyl moiety, is inactive (Mollwitz et al., 2012) (Fig. 3D), suggesting that the hydrogen bonds that Y114 forms with the benzylguanine substrate are absolutely required for function. While E159 also forms a hydrogen bond with the substrate, a number of mutations to E159 retain measurable activity, suggesting less stringent physicochemical requirements on substrate interactions at this position.
In contrast to these constrained residues, A121 is tolerant to all single mutations. L153 is tolerant to nearly all substitutions, except for proline, which is rotationally constrained in psi and phi Ramachandran angles. By exploring our catalogue of double mutants, we observed that proline is consistently the most detrimental of all the 20 amino acids at position 153 across 119/120 other single mutant backgrounds. These observations led to the hypothesis that backbone flexibility at position 153 (which is in the “hinge” region of the loop connecting helix-loop domain 153–176, including the critical residue E159, with the rest of the protein) may regulate activity via inter-domain geometry and/or dynamics (Fig. 3G). We investigated this possibility by examining the relationship between the observed enzymatic activity and amino acid backbone flexibility (Fuchs et al., 2015) of amino acid substitutions at position 153. We observed a strong relationship (R2=0.77; p-value=3×10−6; Fig. 3H), supporting this hypothesis and highlighting the utility of comprehensive mutational substitutions for testing mechanistic hypotheses.
We next aimed to characterize cooperativity in the interactions between the AAs we perturbed by analyzing double mutant cycles found within our mutational dataset (Fig. 4A). We observe a strong enrichment for physical proximity between highly positively cooperative amino acid pairs, with virtually all strong positively-cooperative interactions occurring at Cα-Cα distances less than 13 Å (Fig. 4B). For example, many of the most positively-cooperative double mutants are between positions 135 and 159 – two AAs that directly physically interact in the crystal structure (PDB ID: 3KZZ).
Fig 4. SNAP-tag mutational analysis.

(A) Comparison of double mutant effects on activity (y-axis) with the sum of single mutant effects (x-axis). Only a few residue position pairs (orange) demonstrate high positive cooperativity (“highly cooperative” refers hereafter to cooperativity <−1.69 (see 4C) where cooperativity = −log(kobs,DM/ kobs,WT) − (−log(kobs,SM1/ kobs,WT) + −log(kobs,SM2/ kobs,WT))). (B) Cumulative number of pairs with Cα-Cα distance within a given distance for highly cooperative (orange), and all target residue pairs (blue). Most highly cooperative interactions occur within 13Å (grey dashed) (C) cooperativity values for all target residue pairs with Cα-Cα distance < 13Å (orange) and > 13Å (blue) (top) and by amino acid (counted for either or both partners) (bottom). Sum of highly cooperative pairs (<−1.69, grey dashed) per amino acid (left). (D) Number of highly cooperative interactions across all amino acid pairs.
Interestingly, histidine appears particularly amenable to cooperative interactions. Across all positional combinations, histidine was far more likely to form a highly-cooperative interaction than any other amino acid (Fig. 4C), and did so fairly uniformly with all possible amino acid partners (Fig. 4D). Histidine can be either positively charged or neutral in different contexts, can have aromatic character, and can function as both a hydrogen bonding donor and acceptor. We hypothesize that histidine may thus function as a “Swiss army knife,” pairing in a multi-functional way with diverse partners to produce positive cooperativity. We anticipate that further investigation with high-throughput methods will provide the opportunity to explore this hypothesis in a variety of protein backgrounds.
Discussion:
Prot-MaP enables the generation of immense mutational datasets for both peptides and full-length functional proteins directly on a sequencing flow cell, allowing high-throughput analysis of mutational effects based on direct biophysical observations of protein function. Large-scale quantitative measurements of peptide-protein interaction can demonstrate and quantify nonadditivity in affinity landscapes and allow the identification of enhanced-affinity interactions (such as superFLAG). Large-scale functional analysis of full-length proteins allows data-driven characterization of functional networks and cooperative interactions of individual amino acids, geometric constraints that enforce amino acid preferences, as well as global observations and hypothesis testing regarding the functionality of specific amino acid species (e.g. histidine’s observed capacity for widespread cooperative interactions).
Compared to selection methods (e.g. phage display) where molecular populations can be measured with sequencing over rounds to infer enrichment factors, Prot-MaP has several advantages. First, Prot-MaP allows for direct fluorometric measurements of protein function (e.g. binding), enabling biophysical characterization and eliminating complexities such as amplification bias and stochastic dropout. Second, whereas selections are sophisticated experiments that often take weeks per round to carry out, a Prot-MaP experiment can be performed in a few hours and is highly amenable to automation. Furthermore, while selection-based methods often start from larger libraries of variants, each variant must be sequenced to relatively high depth to generate quantitative information, whereas every individual cluster (i.e. sequencing read) provides independent information regarding protein function in Prot-Map experiments. Compared to protein and peptide microarrays, Prot-MaP throughput is substantially higher. Prot-MaP can display proteins up to ~200 amino acids in length, while in situ synthesized peptide array platforms are typically limited to ~16aa or less. Also, protein-coding Prot-MaP DNA libraries can be generated with relatively inexpensive and straightforward synthesis and enzymatic manipulation (e.g. microarray synthesis, doped-base DNA synthesis, and PCR assembly), methods that are substantially much more straightforward and less costly than producing individual peptides and proteins for printing on microarrays. Finally, Prot-MaP is in principle extensible to higher throughput sequencing platforms, which would enable billions of parallel measurements. As sequencing throughput grows, we anticipate that Prot-MaP has the potential to keep pace by extension.
Limitations:
ProtMaP does have limitations, including a practical limit of about 1–2 kb on the length of protein-encoding DNA constructs that can be efficiently clustered and sequenced. These limits are inherited from the underlying sequencing platform and are not intrinsic to the display method. Additionally, many proteins will require different in vitro expression conditions and components for efficient production of functional protein (including post-translational modifications). Finally, while binding studies are broadly compatible with fluorescence-based assays, other assays (e.g. for arbitrary enzymatic activities, protein folding, or conformational changes) may necessitate the development of fluorescence-compatible measurement strategies. However, analogous to the many methods built on the foundations of high-throughput sequencing (Morozova and Marra, 2008), we anticipate that the compatibility of Prot-MaP with widely-available high-throughput DNA sequencing platforms may serve as a catalyst for further development of specialized applications. For example, comprehensive mutational mapping of the function of disease-associated proteins could enable a deeper understanding of the linkage between mutations and observed clinical pathogenicity. Prot-MaP may also allow characterization and quantification of proteins in complex biological samples such as blood serum using highly multiplexed affinity assays.
Conclusion:
The implementation of facile high-throughput protein functional analysis on a broadly available sequencing flow cell demonstrates a conceptually straightforward path toward disseminated high-throughput protein functional assays using automated fluorescence imaging hardware currently used in sequencing by synthesis methods. Given the demonstrated power of high-throughput DNA sequencing methods (as applied through functional genomic methods such as ChIP-seq, Hi-C etc.) to provide insights into the relationship between DNA sequence and regulatory function, we anticipate that a similarly ubiquitous platform for high-throughput protein assays could have analogous impact on our ability to dissect biologically relevant relationships between protein sequence, structure, and function.
STAR★Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, William Greenleaf (wjg@stanford.edu)
Method Details
Custom Fluorescence Microscopy Instrument to Interface with MiSeq flow cells (“Imaging Station”)
We developed a custom instrument with all the core functionality of a sequencer that interfaces with a MiSeq flow cell, as well as software methods to align sequence reads to image features (see supplement). This platform has been described previously (She et al., 2017) but briefly, the camera, lasers, z-stage, x-y stage, syringe pump, and objective lens salvaged from Illumina GAIIx instruments were combined with custom laser control electronics, retrofitted temperature control, and a fluidic interface designed to be compatible with the MiSeq flow cell.
Protein Array constructs
To generate a protein array, engineered DNA constructs (Fig. 1, Fig. S1) were constructed to have the elements necessary for sequencing on an unmodified Illumina MiSeq, principally the sequencing adapters P5 and P7 and sequencing primer hybridization sites. After clustering and sequencing on the MiSeq, the sequenced flow cell was moved to our custom platform, where transcription/translation was performed on chip such that both the transcript and nascent peptide remain associated with their DNA template, producing a tethered protein array. Library constructs therefore contain the elements requisite for prokaryotic in vitro transcription and translation, in addition to those that enable tethered display. These include an RNAP promoter and stall sequence that allow generation of a tethered RNA substrate with one-RNAP-per-template as previously described (Buenrostro et al., 2014). To allow generation of polypeptide, constructs contain a Shine-Dalgarno ribosomal binding site to initiate translation followed by a start codon. The polypeptide library of interest is encoded at the N-terminal end of the translated sequence. After this coding region, the open reading frame (ORF) continues with no stop codon via a short linker to a spacer polypeptide (2x ybbR tag (Yin et al., 2005)). A spacer is necessary to display the protein/peptide of interest (POI) away from the ribosome where it can be accessible to interaction and, in the case of longer proteins, fold correctly (Schaffitzel et al., 1999). Sufficient linker sequence is present to ensure that the nascent POI and spacer become accessible outside the ribosome exit tunnel (Hardesty and Kramer, 2000). Next, a ribosome stall sequence that we engineered for maximum stalling efficiency (see Fig. S2) stalls ribosomal progress, allowing robust display of the nascent peptide (Fig. 1). Fluorescence-based functional assays (here, measuring binding of a fluorescently-labeled partner or the enzymatic incorporation of fluorescent substrates) may then be carried out directly on this generated array.
PCR assembly of constructs
All PCR assembly was performed with NEBNext High-Fidelity 2X PCR Master Mix (NEB) using the recommended reaction setup and thermocycling conditions.
Ribosomal Stall Sequence
Highly-stable, efficient ribosomal stalling is critical for generating high-quality peptide arrays. We achieve stalling with a ribosome stall sequence (CJL-PPP) that we rationally designed based on polyproline translational pausing in a maximally efficient context (Starosta et al., 2014). This sequence produces a strong pausing signal in our in vitro translation mix that does not contain Elongation Factor P (EF-P) (which alleviates stalling at the PPP motif) (Ude et al., 2013) nor Release Factor 1 (RF1), which would allow release at the Amber stop codon that immediately follows the polyproline stretch. To further stabilize this display, critical residues from SecM and TnaC that are presumed to promote stalling via direct interaction with the exit tunnel (F150, W155, I156 from SecM (Nakatogawa and Ito, 2002), and W12, D16 from TnaC (Seidelt et al., 2009)) were also transplanted into the CJL-PPP sequence (Fig. S2A). In our (RF-1-free) translation mix, The CJL-PPP stall sequence produces much more efficient display than a simple Amber stop codon with no stall sequence (Fig. S2B).
FLAG Library
Each residue in the 8-amino acid WT FLAG peptide sequence (DYKDDDDK) is substituted with 6 different amino acids (A,K,D,S,F, and L, representing small, positive, negative, polar, aromatic, and aliphatic substitutions, respectively). If the wild-type identity of the residue at that position is already one of the mutation set, a contingency substitution (V,R,E,T,Y, or I) was made in lieu of a redundant mutation. The library consists of all combinations of single, double, and triple substitutions of these 6 mutations at each position using optimal codons for E. coli. 2xFLAG (DYKDDDDKGDYKDHD, the first two FLAG motif repeats of the commonly-used 3xFLAG sequence) was also included in the library. Designs were synthesized on microarray (CustomArray) with flanking primer regions, then assembled by PCR into the full-length protein array construct (Fig. S1)
SNAP-tag Library
Seven amino acid positions in the SNAP-tag sequence chosen for their involvement in substrate specificity (Y114, A121, K131, S135, L153, G157, and E159) (Gautier et al., 2008) were targeted for mutation. Most of these residues, except A121 and L153 are peristeric to the active site. The 138-base mutant region from 114–159, as well as the sequencing primer regions 31 bases upstream and 31 bases downstream, were covered with three overlapping oligos that could be combinatorially assembled. Oligos were obtained (IDT) with all combinations of wild-type and NNK codons at each mutant position. The first primer covered positions 114 and 121, the second 131 and 135, and the third 153, 157, and 159. Assembly reactions for each single (7 reactions), double (21 reactions), and triple (35 reactions) combination of positions were individually assembled (Fig. S1), quantified, and pooled so that the final library representation was 1% single mutants, 5% double mutants, and 94% triple mutants.
Additional constructs spanning the mutant region were also included in the final sequencing library. One encodes the WT region with no mutations, and another NNK codons at all 7 targeted positions. These were spiked in downstream at low percentage, along with the fiducial marks.
Bottlenecking
In order to have each molecular variant represented multiple times on chip, protein array libraries were bottlenecked down to 33fM (FLAG library) and 300fM (SNAP library) in 0.1% tween-20, then reamplified. The ~1kb SNAP library constructs were gel extracted from a 1.5% agarose gel to ensure length homogeneity.
Sequencing Diversity
As with all Illumina sequencing, it is necessary to ensure sufficient sequence diversity in the sequenced region. FLAG peptide constructs were sequenced together with other, unrelated libraries that added sequencing diversity (e.g. ATAC-seq samples). However, because the SNAP-tag constructs are ~1kb in size, a diversity element of matched length (and therefore clustering efficiency) was specifically constructed. This diversity element has randomized N78mer sequence across the entire mutant region, but is otherwise identical to the protein array constructs. Because the SNAP-tag library contains stretches that are essentially homogenous, this diversity element was spiked in at up to 40%.
On-flow cell Polypeptide Array Generation
Libraries were prepared for sequencing on a MiSeq by first precisely quantifying all concentrations with qPCR and/or TapeStation (Agilent), then spiking in fiducial mark constructs into protein array libraries at 0.5–1%. Fiducial marks are sparse library members that serve as hybridization targets for a fluorescent probe; the resulting pattern is used to orient the chip relative to the sequencing data on the imaging station platform. Other library constructs, including the specifically-designed SNAP-tag diversity elements, were also present in the pooled sequencing sample to add sufficient sequence diversity for high-quality sequencing.
Standard amplicon sequencing was performed for FLAG tag peptide variants. For the ~1kb SNAP-tag variant library, a modified “Long Amplicon” sequencing recipe with more clustering cycles was used. In order to make these modifications, the “Amplicon” XML recipe in the MiSeq control software was modified to increase the bridge PCR “Repeat” cycles in “Amplification 1” during initial “OnBoardClusterGeneration” from 24 to 32, as well as the “Repeat” cycles in “Resynthesis” during “PairedEndTurnaround” from 12 to 16.) Sequencing was then performed with custom construct-specific sequencing primers (the read1 primer is the 31-base sequence immediately preceding the mutant region, and the read2 primer is the reverse complement of the 31-base sequence immediately following the mutant region). After sequencing, the sequenced MiSeq flow cell is moved to our custom imaging station.
On the Imaging Station platform, residual primers or read fragments from sequencing are stripped off with 100% formamide at 55°C. Cy3-labeled fiducial mark probes in hybridization buffer (5x SSC, 5mM EDTA, 0.01% Tween-20) are then hybridized to the fiducial marks (60°C for 15 min, 40°C for 12 min). Fiducial mark probes remain hybridized as reference points throughout the experiment.
Though the MiSeq flow cell has clusters along the length of its continuous channel, during the course of sequencing the MiSeq only collects data at a series of tiled locations. Experimental images must be taken at the same tile positions in order to match the imaged clusters to the sequence data. In order to discover these tile locations and program their relative positions on a new instrument, images from the imaging station are registered to the fiducial mark construct locations identified from the sequencing data with cross-correlation. MiSeq flow cells have two imaging surfaces (top and bottom) with a typical throughput of 20–25 million reads total. Because the current implementation of our imaging station only images the bottom of the flow cell and obstructs the objective lens from imaging the last (19th) tile, we typically make 10–12 million individual measurements across bottom tiles 1–18. Therefore, given our requirement for 8 replicates per variant, and that our experimental setup images only 47.3% of the flowcell, the theoretical maximum diversity for the existing setup is approximately (25×106 / 8)*(0.473) = ~1.5M variants – assuming a library of perfectly uniform abundance of each molecular variant). Using a slightly modified imaging infrastructure capable of imaging the whole flowcell, we estimate our throughput would be ~3.1M
Once registered to the sequencing data, tethered RNA transcript is generated on chip as described previously (Buenrostro et al., 2014) by stalling E. coli RNA polymerase on a DNA templates (clusters) with a terminal streptavidin roadblock. Constructs are washed with PBST +MgCl2 (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 2 mM KH2PO4, 0.05% Tween-20, 7mM MgCl2), then prokaryotic ribosomes are initiated on the transcript, which contains a Shine-Dalgarno sequence, with the PURExpress (Shimizu et al., 2014) in vitro translation mix. The PURExpress system is reconstituted from purified components and lacks EF-P (Ude et al., 2013) allowing ribosomes to stall at a polyproline-containing ribomsome stall sequence that we engineered into the library, stably displaying the nascent peptide. We used a custom PURExpress formulation that omits Release Factors 1, 2, and 3 as well as T7 RNA polymerase, CTP, and UTP. Not only are these transcriptional components unnecessary (as the RNA template is already generated and displayed before adding the translation reagents) they interfered with stable nascent peptide display in this system despite the lack of a T7 promoter on our constructs.
Translation was performed for 1 hour at 37°C. After peptide display, the flow cell was washed with PBST +MgCl2. All buffers post-RNA generation, including the translation mix, contain 0.8 U/μL Superase-In RNase inhibitor (Thermo AM2696).
M2 Anti-FLAG Antibody Binding Study
After generation of the peptide array, nonspecific binding sites were blocked with bind buffer (0.8% BSA and 500nM goat IgG in PBST +MgCl2) for 30min at 25°C. All subsequent binding steps were performed at 25°C. 35nM Alexa555-goat anti-mouse secondary antibody (Abcam ab150118) in bind buffer was then incubated for 37 min so that the initial, zero-primary-antibody-concentration point would show any background binding of secondary antibody as baseline. Each experimental concentration of primary antibody (1nM, 3.2nM, 10nM, 32nM, 100nM) was incubated for 45 mins, followed by a 25 min wash with bind buffer, then detected with a 37 min incubation with 35nM Alexa555-goat anti-mouse secondary antibody in bind buffer, and a 25 min wash with bind buffer. After each experimental concentration, tiles on the bottom side of the flow cell were TIRF imaged at 532nM excitation with a 590/104 nm bandpass (Semrock FF01–590/104–25) emission filter, using the same laser power and camera exposure settings across all concentrations.
Continuous flow at 12.5 μL/min was used in lieu of static incubation for low-nM primary Ab concentrations to mitigate depletion.
SNAP-tag Enzyme Activity Assay
SNAP-tag substrate SNAP-surface 549 (NEB #S9112) was dissolved in DMSO to a stock concentration of 1μM, then was diluted immediately before use to a working concentration of 200nM in PBST +MgCl2. After generation of the protein array, the working solution of SNAP substrate was flowed continuously into the flow cell at 12.5μL/min for the experimental incubation times to minimize substrate depletion or product inhibition. At each imaging timepoint, fresh PBST +MgCl2 was flowed in as an imaging buffer, and tiles on the bottom side of the flow cell were TIRF imaged at 532nM excitation and a 590/104 nm bandpass (Semrock FF01–590/104–25) emission filter. (Cumulative reactions times include only incubation time in the presence of substrate, not imaging buffer).
Orthogonal investigation of M2/FLAG peptide variant interactions with a fluorescent plate assay
To test M2 antibody binding to these variant peptides orthogonally from Prot-MaP, an in vitro plate assay was performed. First, several 8-amino acid variants of the FLAG peptide were individually synthesized followed by the context amino acids DHDGS (identical to the 5 downstream amino acids of the on-chip constructs) and a C-terminal PEG-biotin. A neutravidin-coated plate (Pierce 15117) was washed, then blocked with StartingBlock PBS (Thermo 37538). Biotinylated peptides were then bound to the plate by incubating for 30 min at a concentration of 5nM. After washing, primary antibody (M2 anti-FLAG, Sigma F1804) was incubated in the plate for 30 minutes, then washed, then fluorescently-labeled (DyLight 550) goat Anti-Mouse secondary antibody (Abcam ab98713) was incubated for 1 hour, then washed again. In order to observe differential binding, a concentration gradient of 3xFLAG peptide (Sigma F4799) competitor in PBST +MgCl2 was added and allowed to equilibrate for 2 hours, after which the plate was washed and read at 535(25) nm excitation and 580(30) nm emission. (Antibodies and peptides were diluted in StartingBlock PBS + 0.05% Tween-20, all washes and fluorescence measurements were performed with PBST +MgCl2, and all incubations were performed at 4°C with shaking.)
Note in Fig. 2G that, though approximate rank order is maintained between the plate assay and Prot-MaP results, while the D4L rescue mutant (D4L, D5E, D7K) is observed to exhibit nearly WT LoD by Prot-MaP (1.09nM vs 1.05nM, respectively), it shows significantly lower binding than WT in the presence of 3xFLAG competitor in the plate assay. The observed differences may reflect different underlying biophysical interactions between competition in the plate and on-chip. For example, re-binding mechanics of bivalent antibody avidity are likely different between the two assays, given the presence of competitor and differences in peptide surface density between assays.
Quantification and Statistical Analysis
Fluorescence images, paired with cluster sequence identities and positions from the sequencing run, are quantified to measure fluorescent signal, and therefore assay the fluorescence-reported polypeptide or protein function at each cluster.
After imaging, fine registration of the images to the sequencing data is necessary before image quantification due to microscope stage position inaccuracy and, more onerously, potentially non-affine optical aberrations between the sequencing platform and the imaging station. To address this, each image is divided into a grid of sub-images that are individually registered to the data, followed by a fit of the offset positions to a 2D quadratic surface. The resulting continuous offset map is then applied to the data to achieve fine registration of cluster positions to the images (She et al., 2017).
Images are then quantified by fitting small image subtiles to a summation of 2D Gaussians centered at the sub-pixel registered positions of each cluster. Edge effects in quantification are mitigated by discarding fit values close enough to an edge to be affected by out-of-bounds clusters, and using overlapping subtiles to completely cover the image (Buenrostro et al., 2014). The integrated intensity of each Gaussian is reported as the fluorescence value for each cluster.
To correct for small variances in quantified intensity caused by inconsistent focus and/or illumination power fluctuation, median fluorescence values were normalized to the median intensity of the fiducial marks. Protein expression/display efficiency is not directly normalized. Variation in our overall signal from stochastic cluster-to-cluster protein display was minimized by using the median signal from 8 or more clusters encoding identical variants. However, we note that sequence-specific differences in protein expression could be conflated with protein function in interpreting observed signals. We plan to implement translation-level normalization in the future.
Data from quantified images are consolidated across concentrations on a per-cluster basis. The resulting experimental series are then grouped into molecular variants with identical amino acid sequences. Grouping into variants was handled slightly differently for the FLAG and SNAP experiments. For FLAG variants, a 16-base random unique molecular identifier (UMI) was read (as index read 2) and the consensus sequence of the amino acid-coding region determined for each UMI was used. SNAP library constructs did not have a UMI sequence and the raw coding sequence was used. For all variants represented by some minimum number of clusters (here, 8), median fluorescence values were calculated at each experimental timepoint, which were then used for downstream analysis (Fig. S3). Variant medians in this study are aggregated across a single-flow-cell experiment.
M2 Anti-FLAG Analysis
A threshold of detection was defined by a set of negative control peptide sequences. The negative set consists of all variants represented by 8-or-more clusters where residues D1, Y2, and K3 (each individually critical for binding) were all mutated in combination. The threshold was then defined as 3σ above the mean of the fluorescence medians of this negative set. Encouragingly, fluorescent signal from the negative set did not appreciably increase with increasing concentration of primary antibody, so the median value of the negative set across all concentration points was used as a concentration-independent background threshold.
This threshold of detection served as the basis for quantification of variant LoD. Any variant that did not cross the threshold was classified as “negative” (LoD greater than the highest the assay can measure).
For variants with 4-or-more points above threshold, the points above threshold (only) were fit to a line and the intersection of the line with the threshold value was reported as the LoD. For variants with fewer than 4 points above threshold, a linear interpolation was done between the point before and the point after crossing the threshold (Fig. 2B).
SNAP-tag Analysis
A threshold of detection was defined by a set of negative control enzyme variants. The negative set consists of all well-represented variants where all seven targeted residues (Y114, A121, K131, S135, L153, G157, E159) were mutated in combination. The threshold was then defined as 3.3σ above the mean of the negative set medians across concentration points.
All variant means that rose above threshold by the second-to-last timepoint were fit by unconstrained nonlinear least squares to a single exponential:
Quality control (QC) of fits was evaluated by three criteria: (1) R2 > 0.70, (2) Fmax above background threshold (3) Fmin < Fmax < thresholdFmax (see Fig. S4). Variants that did not meet these criteria were excluded from downstream analysis.
Supplementary Material
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal ANTI-FLAG M12 antibody produced in mouse | Sigma | F1804 |
| Goat Anti-Mouse IgG H&L (Alexa Fluor 555) preadsorbed | Abcam | ab150118 |
| Goat Anti-Mouse IgG Fc (DyLight 550) preadsorbed | Abcam | ab98713 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 3X FLAG Peptide | Sigma | F4799 |
| Deposited Data | ||
| FLAG-tag and SNAP-tag high-throughput Prot-MaP mutational datasets | Mendeley Data | doi:10.17632/49rf4xfk8k.1 |
| Software and Algorithms | ||
| Image analysis software | (Buenrostro et al., 2014) | https://doi.org/10.1038/nbt.2880 |
| Hardware control software | GreenleafLab GitHub | https://github.com/GreenleafLab/ImagingStationController |
Highlights.
Generation of a massive peptide/protein array on a sequencing flow cell
Direct measurement of binding/catalysis for ~105 protein mutational variants
Patterns of amino acid mutation and cooperativity were analyzed in high-throughput
Discovery of high-avidity “superFLAG” epitope for the M2 anti-FLAG antibody
Acknowledgements
We thank Gavin Sherlock, Sarah Denny, Winston Becker, and Sandy Klemm for critical reading of the manuscript.
Funding: This work was supported by National Institutes of Health (NIH) Grant R01-GM111990 and a Technology Development Grant by the Beckman Foundation. W.J.G is a Chan Zuckerberg Biohub investigator and acknowledges grants 2017-174468 and 2018-182817 from the Chan Zuckerberg Initiative.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: Stanford University has filed a patent on this work and C.J.L. and W.J.G. are named as co-inventors.
Supplementary References: (Starosta et al., 2014, Nakatogawa and Ito, 2002, Seidelt et al., 2009)
References:
- BENTLEY DR, BALASUBRAMANIAN S, SWERDLOW HP, SMITH GP, MILTON J, BROWN CG, HALL KP, EVERS DJ, BARNES CL & BIGNELL HR 2008. Accurate whole human genome sequencing using reversible terminator chemistry. nature, 456, 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRIZZARD BL, CHUBET RG & VIZARD D 1994. Immunoaffinity purification of FLAG epitope-tagged bacterial alkaline phosphatase using a novel monoclonal antibody and peptide elution. Biotechniques, 16, 730–735. [PubMed] [Google Scholar]
- BUENROSTRO JD, ARAYA CL, CHIRCUS LM, LAYTON CJ, CHANG HY, SNYDER MP & GREENLEAF WJ 2014. Quantitative analysis of RNA-protein interactions on a massively parallel array reveals biophysical and evolutionary landscapes. Nat Biotech, 32, 562–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUENROSTRO JD, GIRESI PG, ZABA LC, CHANG HY & GREENLEAF WJ 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature methods, 10, 1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHANDRA H, REDDY PJ & SRIVASTAVA S 2011. Protein microarrays and novel detection platforms. Expert review of proteomics, 8, 61–79. [DOI] [PubMed] [Google Scholar]
- EINHAUER A & JUNGBAUER A 2001. The FLAG™ peptide, a versatile fusion tag for the purification of recombinant proteins. Journal of biochemical and biophysical methods, 49, 455–465. [DOI] [PubMed] [Google Scholar]
- FORSSTRÖM B, AXNÄS BB, STENGELE K-P, BÜHLER J, ALBERT TJ, RICHMOND TA, HU FJ, NILSSON P, HUDSON EP & ROCKBERG J 2014. Proteome-wide epitope mapping of antibodies using ultra-dense peptide arrays. Molecular & Cellular Proteomics, 13, 1585–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOWLER DM, ARAYA CL, FLEISHMAN SJ, KELLOGG EH, STEPHANY JJ, BAKER D & FIELDS S 2010. High-resolution mapping of protein sequence-function relationships. Nature methods, 7, 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FUCHS JE, WALDNER BJ, HUBER RG, VON GRAFENSTEIN S, KRAMER C & LIEDL KR 2015. Independent Metrics for Protein Backbone and Side-Chain Flexibility: Time Scales and Effects of Ligand Binding. Journal of chemical theory and computation, 11, 851–860. [DOI] [PubMed] [Google Scholar]
- GAUTIER A, JUILLERAT A, HEINIS C, CORRÊA IR, KINDERMANN M, BEAUFILS F & JOHNSSON K 2008. An engineered protein tag for multiprotein labeling in living cells. Chemistry & biology, 15, 128–136. [DOI] [PubMed] [Google Scholar]
- GU L, LI C, AACH J, HILL DE, VIDAL M & CHURCH GM 2014. Multiplex single-molecule interaction profiling of DNA-barcoded proteins. Nature, 515, 554–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARDESTY B & KRAMER G 2000. Folding of a nascent peptide on the ribosome. Progress in nucleic acid research and molecular biology, 66, 41–66. [DOI] [PubMed] [Google Scholar]
- HE M, STOEVESANDT O, PALMER EA, KHAN F, ERICSSON O & TAUSSIG MJ 2008. Printing protein arrays from DNA arrays. Nat Meth, 5, 175–177. [DOI] [PubMed] [Google Scholar]
- HILPERT K, WINKLER DF & HANCOCK RE 2007. Peptide arrays on cellulose support: SPOT synthesis, a time and cost efficient method for synthesis of large numbers of peptides in a parallel and addressable fashion. Nature protocols, 2, 1333–1349. [DOI] [PubMed] [Google Scholar]
- JUNG C, HAWKINS JA, JONES SK, XIAO Y, RYBARSKI JR, DILLARD KE, HUSSMANN J, SAIFUDDIN FA, SAVRAN CA & ELLINGTON AD 2017. Massively Parallel Biophysical Analysis of CRISPR-Cas Complexes on Next Generation Sequencing Chips. Cell, 170, 35–47. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KINGSMORE SF 2006. Multiplexed protein measurement: technologies and applications of protein and antibody arrays. Nature reviews Drug discovery, 5, 310–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRUEGER F, KRECK B, FRANKE A & ANDREWS SR 2012. DNA methylome analysis using short bisulfite sequencing data. Nature methods, 9, 145. [DOI] [PubMed] [Google Scholar]
- LARMAN HB, ZHAO Z, LASERSON U, LI MZ, CICCIA A, GAKIDIS MAM, CHURCH GM, KESARI S, LEPROUST EM, SOLIMINI NL & ELLEDGE SJ 2011. Autoantigen discovery with a synthetic human peptidome. Nat Biotech, 29, 535–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEGUTKI JB, ZHAO Z-G, GREVING M, WOODBURY N, JOHNSTON SA & STAFFORD P 2014. Scalable high-density peptide arrays for comprehensive health monitoring. 5, 4785. [DOI] [PubMed] [Google Scholar]
- LEVIN A & WEISS G 2006. Optimizing the affinity and specificity of proteins with molecular display. Molecular Biosystems, 2, 49–57. [DOI] [PubMed] [Google Scholar]
- LIEBERMAN-AIDEN E, VAN BERKUM NL, WILLIAMS L, IMAKAEV M, RAGOCZY T, TELLING A, AMIT I, LAJOIE BR, SABO PJ, DORSCHNER MO, SANDSTROM R, BERNSTEIN B, BENDER MA, GROUDINE M, GNIRKE A, STAMATOYANNOPOULOS J, MIRNY LA, LANDER ES & DEKKER J 2009. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science, 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIPOVSEK D & PLÜCKTHUN A 2004. In-vitro protein evolution by ribosome display and mRNA display. Journal of Immunological Methods, 290, 51–67. [DOI] [PubMed] [Google Scholar]
- MOLLWITZ B, BRUNK E, SCHMITT S, POJER F, BANNWARTH M, SCHILTZ M, ROTHLISBERGER U & JOHNSSON K 2012. Directed evolution of the suicide protein O 6-alkylguanine-DNA alkyltransferase for increased reactivity results in an alkylated protein with exceptional stability. Biochemistry, 51, 986–994. [DOI] [PubMed] [Google Scholar]
- MOROZOVA O & MARRA MA 2008. Applications of next-generation sequencing technologies in functional genomics. Genomics, 92, 255–264. [DOI] [PubMed] [Google Scholar]
- NAKATOGAWA H & ITO K 2002. The ribosomal exit tunnel functions as a discriminating gate. Cell, 108, 629–636. [DOI] [PubMed] [Google Scholar]
- NUTIU R, FRIEDMAN RC, LUO S, KHREBTUKOVA I, SILVA D, LI R, ZHANG L, SCHROTH GP & BURGE CB 2011. Direct measurement of DNA affinity landscapes on a high-throughput sequencing instrument. Nat Biotech, 29, 659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ODEGRIP R, COOMBER D, ELDRIDGE B, HEDERER R, KUHLMAN PA, ULLMAN C, FITZGERALD K & MCGREGOR D 2004. CIS display: in vitro selection of peptides from libraries of protein–DNA complexes. Proceedings of the National Academy of Sciences of the United States of America, 101, 2806–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OSADA E, SHIMIZU Y, AKBAR BK, KANAMORI T & UEDA T 2009. Epitope mapping using ribosome display in a reconstituted cell-free protein synthesis system. Journal of biochemistry, 145, 693–700. [DOI] [PubMed] [Google Scholar]
- PARK PJ 2009. ChIP–seq: advantages and challenges of a maturing technology. Nature Reviews Genetics, 10, 669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAMACHANDRAN N, HAINSWORTH E, BHULLAR B, EISENSTEIN S, ROSEN B, LAU AY, WALTER JC & LABAER J 2004. Self-Assembling Protein Microarrays. Science, 305, 86–90. [DOI] [PubMed] [Google Scholar]
- SCHAFFITZEL C, HANES J, JERMUTUS L & PLÜCKTHUN A 1999. Ribosome display: an in vitro method for selection and evolution of antibodies from libraries. Journal of immunological methods, 231, 119–135. [DOI] [PubMed] [Google Scholar]
- SEIDELT B, INNIS CA, WILSON DN, GARTMANN M, ARMACHE J-P, VILLA E, TRABUCO LG, BECKER T, MIELKE T & SCHULTEN K 2009. Structural insight into nascent polypeptide chain–mediated translational stalling. Science, 326, 1412–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHE R, CHAKRAVARTY AK, LAYTON CJ, CHIRCUS LM, ANDREASSON JOL, DAMARAJU N, MCMAHON PL, BUENROSTRO JD, JAROSZ DF & GREENLEAF WJ 2017. Comprehensive and quantitative mapping of RNA–protein interactions across a transcribed eukaryotic genome. Proceedings of the National Academy of Sciences, 114, 3619–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIMIZU Y, KURUMA Y, KANAMORI T & UEDA T 2014. The PURE System for Protein Production In: ALEXANDROV K & JOHNSTON WA (eds.) Cell-Free Protein Synthesis: Methods and Protocols. Totowa, NJ: Humana Press. [Google Scholar]
- SRILA W & YAMABHAI M 2013. Identification of Amino Acid Residues Responsible for the Binding to Anti-FLAG™ M2 Antibody Using a Phage Display Combinatorial Peptide Library. Applied Biochemistry and Biotechnology, 171, 583–589. [DOI] [PubMed] [Google Scholar]
- STAROSTA AL, LASSAK J, PEIL L, ATKINSON GC, VIRUMÄE K, TENSON T, REMME J, JUNG K & WILSON DN 2014. Translational stalling at polyproline stretches is modulated by the sequence context upstream of the stall site. Nucleic acids research, 42, 10711–10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUN X, ZHANG A, BAKER B, SUN L, HOWARD A, BUSWELL J, MAUREL D, MASHARINA A, JOHNSSON K & NOREN CJ 2011. Development of SNAP‐ Tag Fluorogenic Probes for Wash‐Free Fluorescence Imaging. ChemBioChem, 12, 2217–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SVENSEN N, PEERSEN OB & JAFFREY SR 2016. Peptide Synthesis on a Next-Generation DNA Sequencing Platform. ChemBioChem, 17, 1628–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAO S-C & ZHU H 2006. Protein chip fabrication by capture of nascent polypeptides. Nat Biotech, 24, 1253–1254. [DOI] [PubMed] [Google Scholar]
- TOME JM, OZER A, PAGANO JM, GHEBA D, SCHROTH GP & LIS JT 2014. Comprehensive analysis of RNA-protein interactions by high-throughput sequencing-RNA affinity profiling. Nature methods, 11, 683–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UDE S, LASSAK J, STAROSTA AL, KRAXENBERGER T, WILSON DN & JUNG K 2013. Translation Elongation Factor EF-P Alleviates Ribosome Stalling at Polyproline Stretches. Science, 339, 82–85. [DOI] [PubMed] [Google Scholar]
- YIN J, STRAIGHT PD, MCLOUGHLIN SM, ZHOU Z, LIN AJ, GOLAN DE, KELLEHER NL, KOLTER R & WALSH CT 2005. Genetically encoded short peptide tag for versatile protein labeling by Sfp phosphopantetheinyl transferase. Proceedings of the National Academy of Sciences of the United States of America, 102, 15815–15820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZYKOVICH A, KORF I & SEGAL DJ 2009. Bind-n-Seq: high-throughput analysis of in vitro protein–DNA interactions using massively parallel sequencing. Nucleic acids research, 37, e151–e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.