

Abstract
Glycogen storage disease type 1a (GSD Ia) is an inborn error of metabolism caused by mutations in the G6PC gene, encoding the catalytic subunit of glucose-6-phosphatase. Early symptoms include severe fasting intolerance, failure to thrive and hepatomegaly, biochemically associated with nonketotic hypoglycemia, fasting hyperlactidemia, hyperuricemia and hyperlipidemia. Dietary management is the cornerstone of treatment aiming at maintaining euglycemia, prevention of secondary metabolic perturbations and long-term complications, including liver (hepatocellular adenomas and carcinomas), kidney and bone disease (hypovitaminosis D and osteoporosis). As impaired vitamin A homeostasis also associates with similar symptoms and is coordinated by the liver, we here analysed whether vitamin A metabolism is affected in GSD Ia patients and liver-specific G6pc−/− knock-out mice. Serum levels of retinol and retinol binding protein 4 (RBP4) were significantly increased in both GSD Ia patients and L-G6pc−/− mice. In contrast, hepatic retinol levels were significantly reduced in L-G6pc−/− mice, while hepatic retinyl palmitate (vitamin A storage form) and RBP4 levels were not altered. Transcript and protein analyses indicate an enhanced production of retinol and reduced conversion the retinoic acids (unchanged LRAT, Pnpla2/ATGL and Pnpla3 up, Cyp26a1 down) in L-G6pc−/− mice. Aberrant expression of genes involved in vitamin A metabolism was associated with reduced basal messenger RNA levels of markers of inflammation (Cd68, Tnfα, Nos2, Il-6) and fibrosis (Col1a1, Acta2, Tgfβ, Timp1) in livers of L-G6pc−/− mice. In conclusion, GSD Ia is associated with elevated serum retinol and RBP4 levels, which may contribute to disease symptoms, including osteoporosis and hepatic steatosis.
Introduction
Glycogen storage disease type 1a (GSD Ia or von Gierke disease) is an autosomal recessive inherited disorder of carbohydrate metabolism. Mutations in G6PC, encoding the catalytic subunit of glucose-6-phosphatase (G6PC), limit the production of glucose from glucose-6-phosphate (G6P) leading to hepatic glycogen accumulation and life-threatening hypoglycemia in times of inadequate dietary carbohydrate intake. In addition, GSD Ia is associated with hepatic steatosis, hyperlipidemia, hyperlactacidaemia, hepatocellular tumor formation and intestinal and renal impairments (1). Untreated GSD Ia patients display a protruding abdomen, hepatomegaly, wasted muscles, a bleeding tendency, truncal obesity, a rounded doll face and short stature. No cure is available yet and prevention of hypoglycemia and related metabolic dysfunctions are the main goals of dietary management and control of GSD Ia (2). In addition, GSD Ia has been associated with vitamin D deficiency. Suboptimal levels of serum 25-hydroxyvitamin-D (<30 ng/ml) were observed in most patients in a single center study of 20 patients, even if they were supplemented with vitamin D and calcium (3). Restrictive dietary plans, intestinal malabsorption, poor compliance to dietary plans and metabolic derangements may cause hypovitaminosis D in GSD Ia patients (3). Hypomagnesaemia, hypercalciuria and low tubular resorption of phosphate, along with vitamin D deficiency may reduce bone mineral content and matrix formation in GSD Ia and increase the risk of bone fractures and osteoporosis (4). Besides vitamin D, very limited information is available about other potential vitamins deficiencies in GSD Ia. Vitamin A may be particularly relevant for GSD Ia as these patients develop significant hepatic pathologies, such as hepatic steatosis, hyperlipidemia and adenomas that may affect the liver’s role in regulating vitamin A homeostasis.
Vitamin A is an essential fat-soluble vitamin and ~80% of the total vitamin A pool is stored as retinyl esters, mainly retinyl palmitate, in the liver. White adipose tissue (WAT) contains the second-largest pool of vitamin A (10–20%). Adequate hepatic storage is required to maintain plasma retinol levels around 2 μmol/L in healthy humans (1–1.5 μmol/L in mice) (5). Vitamin A plays important physiological roles in vision, reproduction, growth, development, immunity and metabolic programs (6). Impaired triglyceride and/or cholesterol metabolism often associates with impaired vitamin A metabolism and homeostasis (7, 8). Indeed, reduced serum retinol levels are associated with hepatic steatosis (9, 10), hypertriglyceridemia, glucose intolerance, insulin resistance and obesity (11, 12). On the other hand, excess of vitamin A metabolites may also cause hyperlipidemia by modulating hepatic triglyceride synthesis and very low-density lipoprotein (VLDL) production (13, 14). As steatosis and hypertriglyceridemia are prevalent metabolic symptoms in GSD Ia patients (1), it is relevant to determine whether this also associates with abnormal circulating vitamin A levels, as this may affect immune regulation, tissue differentiation and metabolic pathways in these patients. Thus, the aim of this study was to determine whether vitamin A metabolism is affected in GSD Ia patients and in liver-specific G6pc−/− knock-out mice.
Results
GSD Ia patients and L-G6pc−/− mice have elevated serum retinol levels
Twenty-two GSD Ia patients, all from different families, were included in this study and their general characteristics are presented in Table I. Mean serum triglyceride and cholesterol levels were above normal levels and all patients show hepatic steatosis, with mildly elevated serum AST and γGT levels. Seven (7) out of 22 patients showed osteopenia/osteoporosis. Proteinuria was observed in only 3 patients. Similarly, only 3 out of 22 patients showed insufficient serum 25(OH)D levels (<30 nmol/L). Serum retinol levels were determined in the 22 GSD Ia patients (mean age 26.1 ± 10.1) and 20 age- and sex-matched healthy controls (mean age 25.7 ± 10.2) (Fig. 1A). Circulating retinol levels were significantly increased in GSD Ia patients as compared to the healthy controls (2.96 ± 0.22 versus 2.31 ± 0.12 μM, respectively), with 10 out of 22 patients showing serum retinol levels above 2.7 μmol/L. No correlation was observed between serum retinol levels and sex or age of the patients and controls (Fig. 1B). We next aimed to analyze whether the elevated serum retinol levels are also observed in a mouse model of GSD Ia (L-G6pc−/− mice) (15), and if so, what molecular mechanisms may be involved. The induction of the GSD Ia phenotype in L-G6pc−/− mice was supported by: 1) almost complete absence of G6pc mRNA 10 days after tamoxifen treatment (Supplementary Fig. S3A), 2) increased liver weight (+47%); 3) elevated hepatic triglyceride levels (both concentration (+56%) and total pool (+120%), 4) minor effects on free cholesterol levels, but an increase in total hepatic pool of total cholesterol and 5) fasting hypoglycemia, when compared to control mice (Supplementary Fig. S1A). Excessive lipid accumulation in the L-G6pc−/− mice was also confirmed by H&E and ORO staining of the liver tissue (Supplementary Fig. S1B). Similar to GSD Ia patients, plasma retinol levels were significantly increased in L-G6pc−/− mice (2.33 ± 0.18 μM) as compared to control mice (1.23 ± 0.08 μM) (Fig. 2A).
Table I.
GSD 1a patient general characteristics
| Mean ± SD | |
|---|---|
| Gender (F/M) | 14/8 |
| Age (GSD 1a patients) | 26.1 ± 10.1 |
| Vitamin A (serum retinol; normal 1.2–2.7 μmol/L) | 3.0 ± 1.0 |
| Hypervitaminosis A: serum retinol > 2.7 μmol/L (no/yes) | 12/10 |
| Vitamin D (serum 25(OH)D; sufficient > 50 nmol/L) | 64.6 ± 27.4 |
| Hypovitaminosis D: serum 25(OH)D < 30 nmol/L (no/yes) | 19/3 |
| Hepatic steatosis (no/mild/severe/unknown) | 0/12/9/1 |
| Serum Triglycerides (normal < 1.7 mmol/L) | 12.7 ± 10.0 |
| Serum Total Cholesterol (normal < 5.2 mmol/L) | 7.5 ± 2.8 |
| Osteopenia/osteoporosis (no/yes) | 15/7 |
| Kidney disease/proteinuria (normal < 1–2 g/24 h) | 0.4 ± 0.7 |
| Proteinuria (no/yes) | 19/3 |
| AST (normal < 10–40 U/L) | 55.8 ± 34.1 |
| ALT (normal < 7–56 U/L) | 42.3 ± 25.8 |
| Total bilirubin (normal < 20.5 μmol/L) | 5.9 ± 1.0 |
| yGT (normal < 48 U/L) | 53.9 ± 33.1 |
Figure 1.
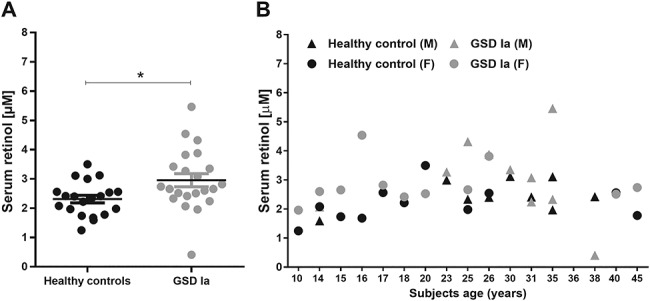
Serum retinol levels are increased in GSD Ia patients. Serum retinol levels were determined in GSD Ia patients (n = 22) and age- and sex-matched controls (n = 20) (A). No correlation was observed between age and sex (male: Δ or Female: O) in GSD Ia patients (grey symbols) nor in healthy controls (black symbols) (B).
Figure 2.
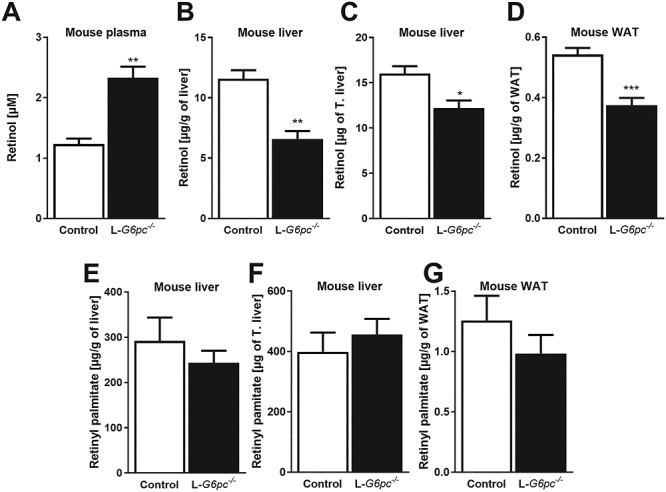
G6pc deficiency in mice increases plasma retinol and reduces hepatic retinol, while retinyl palmitate remains unchanged. Ten days after tamoxifen-induced deletion of the G6pc gene in hepatocytes, L-G6pc−/− and control mice (n = 6 each group) were sacrificed and analyzed for (A) plasma retinol levels, (B) liver retinol concentrations, (C) total liver retinol pool, (D) retinol concentrations in WAT, (E) liver retinyl palmitate concentrations, (F) total liver retinyl palmitate pool and (G) retinyl palmitate concentrations in WAT.
G6pc deficiency reduces hepatic retinol levels, while retinyl palmitate is unchanged
In contrast to plasma retinol, hepatic retinol concentrations (6.59 ± 0.66 versus 11.57 ± 0.72 μg/g liver) as well as the total hepatic pool of retinol were significantly lower in L-G6pc−/− mice compared to control mice (Fig. 2B and C). WAT is a second storage site of vitamin A and retinol levels were similarly reduced in WAT of L-G6pc−/− mice as compared to control mice (0.38 ± 0.02 versus 0.54 ± 0.02 μg/g WAT, respectively) (Fig. 2D).
On the other hand, hepatic retinyl palmitate levels were not different between L-G6pc−/− mice and control mice, not in concentration and not in the total pool (Fig. 2E and F) and the same was observed for retinyl palmitate concentrations in WAT (Fig. 2G). These results indicate that hepatic vitamin A metabolism is affected in the absence of G6pc, leading to increased levels of circulating retinol and reduced levels in liver and WAT, while hepatic vitamin A storage is maintained.
Normal expression of vitamin A-storing enzymes, together with increased expression of vitamin A-hydrolyzing enzymes in L-G6pc−/− fasted-mouse liver
Hepatic retinol and retinyl ester levels are a resultant of enzymes that catalyze the esterification of retinol (predominantly LRAT and to a lesser extent by DGAT1), 2) enzymes that hydrolyze retinyl esters (ATGL/PNPLA2 and PNPLA3) and 3) enzymes that convert retinol to retinoic acids (ADH and RALDH). Moreover, retinol promotes its own release from the liver to the circulation by binding to retinol binding protein 4 (RBP4) in hepatocytes. Hepatic mRNA levels of Lrat were significantly reduced in L-G6pc−/− mice as compared to control mice (Fig. 3A). However, LRAT protein levels appeared not different between L-G6pc−/− and control mice (Fig. 3B). Hepatic mRNA levels of alternative enzyme involved in retinol esterification and lipid droplet growth (Dgat1 and Dgat2) were elevated in L-G6pc−/− mice, but also for DGAT1 no clear increase in protein was detected (Fig. 3A,B). Specific antibodies against DGAT2 were not available. Hepatic mRNA levels of both Pnpla2 and Pnpla3 were strongly induced in L-G6pc−/− mice as compared to control mice, while Lipe mRNA levels were not changed (Fig. 3C). In line, ATGL protein levels sharply increased in L-G6pc−/− mice and total HSL protein levels were unchanged compared to control mice. Still, levels of active (phosphorylated) HSL (pHSL) were high in all L-G6pc−/− mice, while this was variable in the control mice (Fig. 3C). Hepatic mRNA levels of Rbp4 were similar in control and L-G6pc−/− mice (Fig. 3D). It is important to note, however, that hepatic and serum RBP4 protein levels are primarily regulated by the availability of retinol in the liver, where retinol binding promotes the secretion of RBP4 from hepatocytes (16). Conversely, the absence of retinol leads to strong hepatic accumulation of RBP4 even at stable Rbp4 mRNA levels, as observed in vitamin A-deficient mice (see Supplementary Fig. S2) and also observed in rat (17). Thus RBP4 protein levels were analyzed next. RBP4 protein levels in livers and WAT of L-G6pc−/− mice were similar to control mice (Fig. 4A and B, notably, hepatic RBP4 sometimes appears as a double band in western blot analyses, as observed by others (18–20), but with unknown cause). In contrast, serum RBP4 levels in L-G6pc−/− mice were clearly elevated ~3-fold compared to control mice (Fig. 4C). Sera of GSD Ia patients also contained significantly elevated levels of RBP4 compared to age- and sex-matched healthy control (Fig. 4D). Serum retinol levels were largely in line with serum RBP4 levels in healthy controls, while such association was less evident in GSD Ia patients (Fig. 4D).
Figure 3.
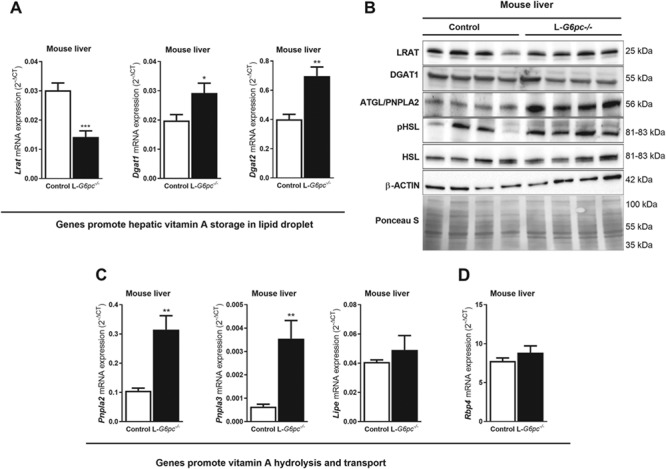
Hepatic expression of genes involved in vitamin A storage, hydrolysis and export in L-G6pc−/− fasted-mice. Ten days after tamoxifen-induced deletion of the G6pc gene in hepatocytes, L-G6pc−/− and control mice (n = 6 each group) were sacrificed and analyzed by Q-PCR (A, C and D) and Western blot analysis (n = 4 each group) (B) for hepatic expression of genes/proteins involved in (A) retinyl ester formation (Lrat, Dgat1 and Dagt2), (B) protein levels of LRAT, DGAT1, ATGL, pHSL, HSL, β-ACTIN and Ponceau S stainings (loading control), (C) retinol synthesis (Pnpla2/Atgl and Pnpla3) and (D) retinol export from the liver (Rbp4). Transcript analyses suggest that the balance between vitamin A storage/retinol synthesis in the liver shifts to retinol synthesis in L-G6pc−/− mice.
Figure 4.
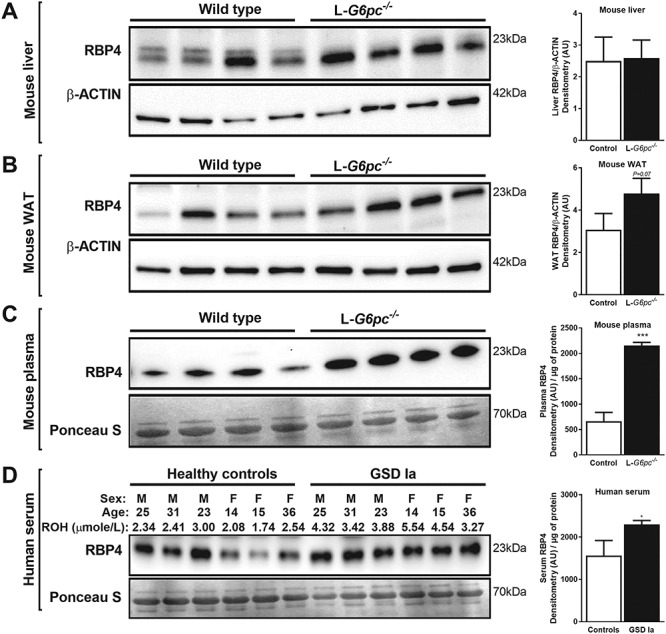
Serum RBP4 levels are elevated in L-G6pc−/− mice and GSD Ia patients. (A–C) Ten days after tamoxifen-induced deletion of the G6pc gene in hepatocytes, L-G6pc−/− and control mice (n = 4 each group) were sacrificed and analyzed by Western blotting for RBP4 protein levels in (A) liver, (B) WAT and (C) plasma. (D) Similarly, RBP4 protein levels were analyzed in sera of age- and sex-matched healthy controls and GSD Ia patients (n = 6 each group). β-ACTIN and Ponceau S stainings are included as loading controls. Protein signal intensities were quantified and are shown to the right.
G6pc deficiency suppresses the expression of retinoic acid-responsive Cyp26a1
Next, we aimed to analyze whether G6pc deficiency may affect the production of retinoic acids in the liver. Hepatic mRNA levels of Hsd17b13, a recently identified retinol dehydrogenase (21) and all 4 retinaldehyde dehydrogenases (Raldh1–4) were hardly affected by the absence of GSD Ia in mice (Fig. 5). Only a small significant increase in Raldh2 and decrease in Raldh4 were observed in G6pc deficient mice. However, mRNA levels of the highly retinoic acid-sensitive Cyp26a1 were strongly (89%) decreased (Fig. 5) compared to control mice. Unfortunately, specific antibodies against mouse Cyp26a1 were not available to confirm the effect of G6pc deficiency at the CYP26A1 protein level.
Figure 5.
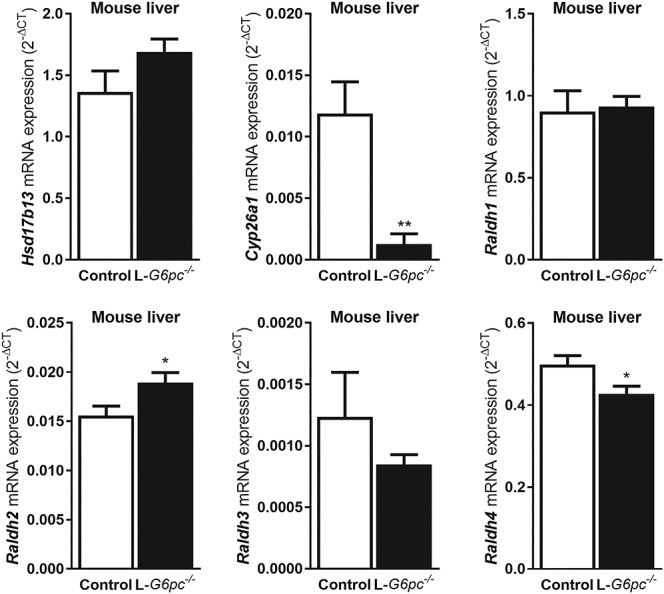
Gene expression of retinoic acid-responsive Cyp26a1 is strongly suppressed in the livers of L-G6pc−/− mice. Ten days after tamoxifen-induced deletion of the G6pc gene in hepatocytes, L-G6pc−/− and control mice (n = 6 each group) were sacrificed and analyzed by Q-PCR for hepatic mRNA levels of genes involved in the conversion of retinol to retinoic acids (Hsd1713, Raldh1, Raldh2, Raldh3, Raldh4) or catabolism of retinoic acids (Cyp26a1). Transcriptional regulation of Cyp26a1 is highly responsive to retinoic acids.
Liver-specific G6pc deficiency in mice does not cause hepatic inflammation, nor fibrosis
Finally, we analyzed whether abnormal vitamin A metabolism in L-G6pc−/− mice leads to hepatic inflammation and/or fibrosis. Hepatic mRNA levels of markers of inflammation, e.g. Cd68, Tnfα, Nos2, Ccl2, Il6 were reduced in L-G6pc−/− mice as compared to control mice (Fig. 6A). A similar suppression of hepatic mRNA levels of markers of fibrosis, e.g. Coll1a1, Acta2, Tgf-β and Timp1, was observed in L-G6pc−/− mice as compared to control mice (Fig. 6B).
Figure 6.
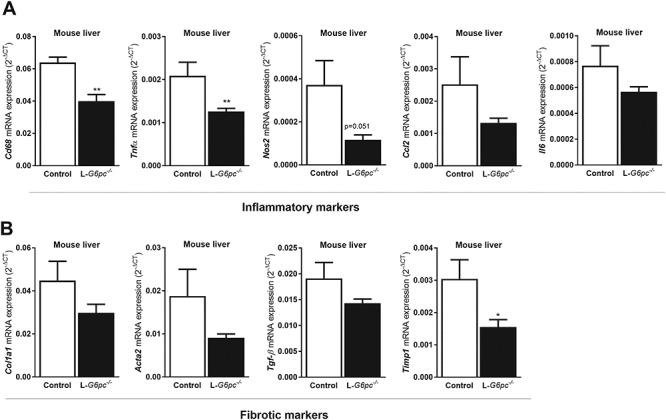
Hepatic G6pc-deficiency suppresses basal levels of inflammation and fibrosis in mice. Ten days after tamoxifen-induced deletion of the G6pc gene in hepatocytes, L-G6pc−/− and control mice (n = 6 each group) were sacrificed and analyzed by Q-PCR for hepatic mRNA levels of markers of (A) inflammation (Cd68, Tnfα, Nos2, Ccl2 and Il6) or (B) fibrosis (Col1a1, Acta2, Tgf-β and Timp1). Both markers of inflammatory and fibrosis were not increased in L-G6pc−/− livers. Instead, a significant reduction was observed for hepatic expression of Cd68, Tnfα and Timp1 in L-G6pc−/− mice compared to controls, while all other markers showed similar trends.
Taken together, our data show that G6pc deficiency leads to elevated serum retinol and RBP4 levels in humans and in mice. In contrast, hepatic retinol levels are reduced, most probably because of enhanced mobilization of retinol from retinyl ester stores and subsequent RBP4-mediated release from hepatocytes.
Discussion
This study shows for the first time that G6pc deficiency in human and mouse is associated with elevated levels of circulating retinol, concomitantly with an increase of circulating RBP4 levels. On the contrary, retinol levels are reduced in the liver and WAT. Tissue retinyl palmitate levels are not changed in liver-specific G6pc deficient mice, at least not within 10 days after ablation of the gene in hepatocytes. Hepatic expression profiling suggests that metabolism of retinyl ester to retinol that may promote the secretion of retinol-bound RBP4 to the circulation. A persistent increase in circulatory retinol may contribute to symptoms of GSD Ia patients and aggravate steatosis and osteoporosis.
Clinical management of GSD Ia is primarily aimed at maintaining steady circulating glucose levels by strictly controlled intake of dietary carbohydrates during day and night. Because of the impaired ability to produce glucose from glucose-6-phosphate (G6P), cellular glycogen content increases, in conjugation with elevated triglyceride storage leading to steatosis. GSD Ia is associated with hypovitaminosis D, which has been linked to the development of osteoporosis in these patients (3, 22). As fatty liver disease is associated with hypovitaminosis A (9, 10), we were interested whether GSD Ia patients may also show aberrant circulating vitamin A levels. To our surprise, we found that circulating retinol revels, as well as RBP4 levels, were significantly elevated, instead of being reduced, in GSD Ia patients and L-G6pc−/− mice. Apart from cases of excessive dietary vitamin A intake, elevated circulating retinol levels are a rare phenomenon. Serum retinol levels are, however, not a sensitive measure of sharp fluctuations in dietary intake of vitamin A as early work showed that daily retinyl palmitate supplementation in a range of 0 to 36 000 IU (= 0–12 times the RDA [Recommended Daily Allowance]) increased serum retinol levels by only 2% (= 0.04 μM) per 10 000 IU vitamin A (23). On average, we observed a 28% increase in serum retinol levels in GSD Ia patients compared to healthy controls, with 45% of the patients showing levels above the normal range. Thus, even though GSD Ia patients are often prescribed multivitamin supplements, it is unlikely to cause the increased circulating retinol levels in these patients. Notably, the incidence of hypervitaminosis A in GSD Ia patients in our study was higher than that of hypovitaminosis D. Impaired kidney function, a condition associated with GSD Ia (24), may also lead to elevated serum retinol levels (25, 26). However, in our study GSD Ia patients did not show severe kidney disease, protein ureia was elevated only in three GSD Ia patients. Moreover, the liver-specific G6pc−/− mice also showed markedly elevated retinol levels, while these animal do not develop any kidney abnormalities, not even 15 months after ablation of the gene in the liver (15). Thus, hepatic vitamin A metabolism likely contributes to the elevated serum levels of retinol and RBP4 in GSD Ia. In contrast to blood, tissue retinol levels in the liver and WAT were reduced in L-G6pc−/− mice compared to controls, while retinyl palmitate, the main storage form of vitamin A in the liver, was not changed. Indeed, protein levels of LRAT, the main hepatic enzyme catalyzing esterification of retinol, were normal in L-G6pc−/− mice. Remarkably, though, Lrat mRNA levels were significantly reduced in L-G6pc−/− mice. It remains to be determined why LRAT protein levels do not follow mRNA levels. One possibility is that LRAT is a stable protein and that it takes >10 days after G6pc gene deletion to observe a significant effect of LRAT protein. Expression profiling revealed an induction of retinyl ester-hydrolyzing activity and a reduction of retinoic acid catabolism. De novo lipogenesis is increased in G6PC deficiency and activation of the carbohydrate-response-element-binding protein (ChREBP) likely contributes to this phenomenon (27, 28). Hepatic Chrebp mRNA levels were indeed increased in L-G6pc−/− mice compared to controls (Supplementary Fig. S3). Notably, PNPLA3 expression is controlled by ChREBP (29) and NAFLD patients carrying the PNPLA3-I148M variant show reduced circulating retinol levels and enhanced hepatic retinyl palmitate contents (30, 31). Thus, ChREBP-mediated induction of PNPLA3, together with elevated levels of ATGL and possibly pHSL may contribute to the enhanced conversion of hepatic retinyl esters to retinol. The strong reduction in Cyp26a1 mRNA levels in L-G6pc−/− mice primarily hints to reduced production of retinoic acids, as they are potent inducers of Cyp26a1 transcription (5). Enhanced hepatic retinyl ester-hydrolysis and reduced retinoic acid catabolism are theoretically expected to lead to accumulation of retinol. However, hepatic retinol levels were actually reduced, while an increase was observed circulatory retinol and RBP4 in GDS Ia patients and L-G6pc−/− mice. Enhanced retinol production in the liver promotes its own release from hepatocytes, bound to RBP4, to the circulation and contributes to elevated plasma levels of retinol and RBP4 (32–34). Thus, we hypothesize that an enhanced production of retinol pushes itself out of the liver and contributes to the elevated serum levels of RBP4 and retinol found in GSD Ia patients and L-G6pc−/− mice. The reduced hepatic retinol levels may be an early response to the induced deletion of the G6pc gene in this mouse model (10 days gene deletion). Future studies may include L-G6pc mice after long-term gene deletion (15) to determine whether the low hepatic retinol levels persist.
Normal serum retinol levels range from 1.2 to 2.7 μmol/L in the Dutch population (35), thus the average level detected in GSD Ia patients is just above the higher end. This will not cause acute toxicity, but chronically elevated retinol in circulation may contribute to clinical symptoms associated with GSD Ia, especially in GSD Ia patients with levels above 4–5 μmol/L. Hypervitaminosis A promotes osteoclast formation, skeleton fragility and osteoporosis due to decreased cortical bone mass and bone formation (36). Hypervitaminosis A-associated osteoporosis may already occur at twice the recommended daily allowances (RDA) of vitamin A, which will not even lead to elevated serum retinol levels (36, 37). Osteoporosis is also observed in GSD Ia patients and typically linked to hypovitaminosis D, which is analyzed in routine surveillance (1, 3). Abnormal vitamin D and A levels may synergize in aberrant bone homeostasis and may need to be monitored both to prevent this complication in GSD Ia patients. In fact, there are quite a few additional commonalities in symptoms in GSD Ia and hypervitaminosis A, like impaired growth, dizziness and irritability (38–41). Though these symptoms likely primarily result of poorly controlled blood glucose levels, it could be that chronically elevated serum retinol levels may also contribute to such symptoms. Hypervitaminosis A causes hepatic steatosis in rats (42), while vitamin A deficiency reduces hepatic lipid accumulation (43).
Increased circulating retinol has been found to reduce the risk for hepatocellular carcinoma (HCC) (44, 45). GSD Ia patients are actually at risk for the development of hepatic adenomas that may progress to HCC. The effects on retinol and tumor development in GSD Ia therefore appear counterintuitive. However, hepatic retinol levels are reduced and may promote adenoma development specifically in the liver. Here also, it is of interest what the long-term effect is of the absence of hepatic G6PC activity on vitamin A metabolism in the liver (15). It may very well be that hepatic vitamin A stores get depleted in the long-term and predispose to liver tumor development in GSD Ia. One older patient indeed showed very low-circulating retinol levels (Fig. 1B), which suggests extremely low hepatic vitamin A stores.
The hepatic pathologies and disturbed vitamin A metabolism did not induce an inflammatory or fibrotic response in livers of L-G6pc−/− mice. In fact, all tested markers for hepatic inflammation and fibrosis were suppressed to greater or lesser extent in L-G6pc−/− mice. This may also be a result of changes in hepatic retinol metabolism as vitamin A metabolites are potent controllers of hepatic inflammation and fibrosis (46, 47).
Management of hypervitaminosis A is currently limited to controlling the dietary intake of vitamin A. Given the ‘metabolic origin’ of hypervitaminosis A in GSD Ia patients, it is important to monitor circulating retinol levels and refrain from vitamin A supplementation when plasma retinol levels are close to or above normal levels. Future studies need to establish the course of vitamin A levels in the absence of G6PC activity in patients and/or mice in order to determine the necessity of management of vitamin A levels in early and late stages of disease development.
Taken together, our study shows that vitamin A metabolism is disturbed in the absence of G6PC activity in mice and GSD Ia patients, resulting in elevated circulating retinol levels. This condition may contribute to various symptoms of GSD Ia, in particular, osteoporosis, which has been linked to hypovitaminosis D in these patients so far. Vitamin A is thus a second vitamin that needs attention in the management of GSD Ia.
Materials and Methods
Patients
The study was performed in accordance with the Declaration of Helsinki and the institutional rules for studying biological rest materials. Retinol analysis was performed in serum samples from 22 genetically confirmed GSD Ia patients (male n = 9 and female n = 13), who visited the Beatrix Children’ Hospital, UMCG. Samples were randomly obtained during the day. The controls included 20 healthy, age- and sex-matched control subjects (male n = 8 and female n = 12) aged between 10 and 45 years.
Animal model
The tamoxifen-inducible hepatocyte-specific G6pc-knock-out (L-G6pc−/−) mice were used in this study as a model of the liver-specific pathologies of GSD Ia (15). Briefly, G6pc recombinant mice with two loxP sites flank G6pc exon 3 (B.G6pclox/w) were crossed with transgenic mice expressing the tamoxifen-inducible recombinase (CREERT2) under control of the serum albumin promoter to confer hepatocyte-specific expression in B6.SAcreERT2/w mutant mice. Male B6.G6pcex3lox/ex3lox.SACreERT2/+ mice (8–12 weeks old) were injected intraperitoneally once daily with 100 μl tamoxifen (10 mg/ml, Sigma–Aldrich) for five consecutive days to obtain L-G6pc−/− mice. All mice were sacrificed 10 days after the last tamoxifen injection. Animal experiments were performed after approval of all procedures by the Institutional Animal Care and Use Committee, University of Groningen, the Netherlands. All animals (n = 6–7) were kept in an environment with alternating dark and light cycles (07:00 p.m.–07:00 a.m.), with controlled temperature (20–24°C) and relative humidity (55% ± 15%) and ad libitum access to food and water. Prior to sacrifice, the mice were fasted from 10:00 p.m. until 08:00 a.m. the next day. Tissue and plasma samples were collected for further analysis.
Standard protocols for histochemistry and quantification of serum/tissue cholesterol, triglyceride, retinol, retinyl esters, mRNA (quantitative reverse transcription-polymerase chain reaction [qRT-PCR]), protein (Western blotting) and the corresponding statistical analyses are presented in Supplementary Material and Methods.
Supplementary Material
Author contributions: Study concept and design: A.S., M.H.O., K.N.F.; acquisition of data: A.S., J.H., J.A.H., H.W., T.G.J.D., E.v.d.V., M.H.O.; analysis and interpretation of data: A.S., M.H.O., K.N.F.; drafting of the manuscript: A.S., T.G.J.D., G.M., F.R., M.H.O., K.N.F.; statistical analysis: A.S.; obtained funding: K.N.F.; technical assistance: J.H.; study supervision: M.H.O., K.N.F.
Acknowlegements
The authors are thankful to Trijnie Bos, Brenda Hijmans and Aycha Bleeker for providing the technical assistance in the execution of animal experiments in mice (L-G6pc−/−).
Conflict of Interest Statement: The authors certify that they have no affiliations with or involvement in any organization or entity with any financial or non-financial interest in the subject matter or materials described in this manuscript.
Funding
University of Groningen (Rosalind Franklin Fellowship to M.H.O.).
References
- 1. Bali D.S., Chen Y.-T., Austin S. and Goldstein J.L. (1993) Glycogen Storage Disease Type I In Pagon R.A., Adam M.P., Ardinger H.H., Wallace S.E., Amemiya A., Bean L.J., Bird T.D., Ledbetter N., Mefford H.C., Smith R.J. et al. (eds), GeneReviews(®). .University of Washington, Seattle, WA. [PubMed] [Google Scholar]
- 2. Ross K.M., Brown L.M., Corrado M.M., Chengsupanimit T., Curry L.M., Ferrecchia I.A., Porras L.Y., Mathew J.T. and Weinstein D.A. (2016) Safety and efficacy of chronic extended release cornstarch therapy for glycogen storage disease type I. JIMD Rep., 26, 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Banugaria S.G., Austin S.L., Boney A., Weber T.J. and Kishnani P.S. (2010) Hypovitaminosis D in glycogen storage disease type I. Mol. Genet. Metab., 99, 434–437. [DOI] [PubMed] [Google Scholar]
- 4. Cabrera-Abreu J., Crabtree N.J., Elias E., Fraser W., Cramb R. and Alger S. (2004) Bone mineral density and markers of bone turnover in patients with glycogen storage disease types I, III and IX. J. Inherit. Metab. Dis., 27, 1–9. [DOI] [PubMed] [Google Scholar]
- 5. Saeed A., Hoekstra M., Hoeke M.O., Heegsma J. and Faber K.N. (2017) The interrelationship between bile acid and vitamin A homeostasis. Biochim. Biophys. Acta, 1862, 496–512. [DOI] [PubMed] [Google Scholar]
- 6. Bar-El Dadon S. and Reifen R. (2017) Vitamin A and the epigenome. Crit. Rev. Food Sci. Nutr., 57, 2404–2411. [DOI] [PubMed] [Google Scholar]
- 7. Zhang M., Liu C., Hu M., Zhang J., Xu P., Li F., Zhong Z., Liu L. and Liu X. (2015) High-fat diet enhanced retinal dehydrogenase activity, but suppressed retinol dehydrogenase activity in liver of rats. J. Pharmacol. Sci., 127, 430–438. [DOI] [PubMed] [Google Scholar]
- 8. Shin M.-J., Kang S.-M., Jang Y., Lee J.H., Oh J., Chung J.H. and Chung N. (2007) Serum retinol binding protein 4 levels are associated with serum adiponectin levels in non-diabetic, non-obese subjects with hypercholesterolemia. Clin. Chim. Acta Int. J. Clin. Chem., 378, 227–229. [DOI] [PubMed] [Google Scholar]
- 9. Botella-Carretero J.I., Balsa J.A., Vázquez C., Peromingo R., Díaz-Enriquez M. and Escobar-Morreale H.F. (2010) Retinol and alpha-tocopherol in morbid obesity and nonalcoholic fatty liver disease. Obes. Surg., 20, 69–76. [DOI] [PubMed] [Google Scholar]
- 10. Villaça Chaves G., Pereira S.E., Saboya C.J. and Ramalho A. (2008) Non-alcoholic fatty liver disease and its relationship with the nutritional status of vitamin A in individuals with class III obesity. Obes. Surg., 18, 378–385. [DOI] [PubMed] [Google Scholar]
- 11. Liu Y., Chen H., Mu D., Fan J., Song J., Zhong Y., Li D. and Xia M. (2016) Circulating retinoic acid levels and the development of metabolic syndrome. J. Clin. Endocrinol. Metab., 101, 1686–1692. [DOI] [PubMed] [Google Scholar]
- 12. Godala M.M., Materek-Kuśmierkiewicz I., Moczulski D., Rutkowski M., Szatko F., Gaszyńska E., Tokarski S. and Kowalski J. (2017) The risk of plasma vitamin A, C, E and D deficiency in patients with metabolic syndrome: a case-control study. Adv. Clin. Exp. Med.. doi: 10.17219/acem/62453. [DOI] [PubMed] [Google Scholar]
- 13. Krupková M., Liška F., Šedová L., Křenová D., Křen V. and Šeda O. (2014) Pharmacogenomic analysis of retinoic-acid induced dyslipidemia in congenic rat model. Lipids Health Dis., 13, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vu-Dac N., Gervois P., Torra I.P., Fruchart J.C., Kosykh V., Kooistra T., Princen H.M., Dallongeville J. and Staels B. (1998) Retinoids increase human apo C-III expression at the transcriptional level via the retinoid X receptor. Contribution to the hypertriglyceridemic action of retinoids. J. Clin. Invest., 102, 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mutel E., Abdul-Wahed A., Ramamonjisoa N., Stefanutti A., Houberdon I., Cavassila S., Pilleul F., Beuf O., Gautier-Stein A., Penhoat A. et al. (2011) Targeted deletion of liver glucose-6 phosphatase mimics glycogen storage disease type 1a including development of multiple adenomas. J. Hepatol., 54, 529–537. [DOI] [PubMed] [Google Scholar]
- 16. Schina M., Koskinas J., Tiniakos D., Hadziyannis E., Savvas S., Karamanos B., Manesis E. and Archimandritis A. (2009) Circulating and liver tissue levels of retinol-binding protein-4 in non-alcoholic fatty liver disease. Hepatol. Res. Off. J. Jpn. Soc. Hepatol., 39, 972–978. [DOI] [PubMed] [Google Scholar]
- 17. Muto Y., Smith J.E., Milch P.O. and Goodman D.S. (1972) Regulation of retinol-binding protein metabolism by vitamin A status in the rat. J. Biol. Chem., 247, 2542–2550. [PubMed] [Google Scholar]
- 18. Thompson S.J., Sargsyan A., Lee S.-A., Yuen J.J., Cai J., Smalling R., Ghyselinck N., Mark M., Blaner W.S. and Graham T.E. (2017) Hepatocytes are the principal source of circulating RBP4 in mice. Diabetes, 66, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alapatt P., Guo F., Komanetsky S.M., Wang S., Cai J., Sargsyan A., Rodríguez Díaz E., Bacon B.T., Aryal P. and Graham T.E. (2013) Liver retinol transporter and receptor for serum retinol-binding protein (RBP4). J. Biol. Chem., 288, 1250–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhu C., Xiao Y., Liu X., Han J., Zhang J., Wei L. and Jia W. (2015) Pioglitazone lowers serum retinol binding protein 4 by suppressing its expression in adipose tissue of obese rats. Cell. Physiol. Biochem., 35, 778–788. [DOI] [PubMed] [Google Scholar]
- 21. Ma Y., Belyaeva O.V., Brown P.M., Fujita K., Valles K., Karki S., de Boer Y.S., Koh C., Chen Y., Du, X., et al. (2018) HSD17B13 is a hepatic retinol dehydrogenase associated with histological features of non-alcoholic fatty liver disease. Hepatol. Baltim. Md, 10.1002/hep.30350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Soejima K., Landing B.H., Roe T.F. and Swanson V.L. (1985) Pathologic studies of the osteoporosis of Von Gierke’s disease (glycogenosis 1a). Pediatr. Pathol., 3, 307–319. [DOI] [PubMed] [Google Scholar]
- 23. Wald N.J., Cuckle H.S., Barlow R.D., Thompson P., Nanchahal K., Blow R.J., Brown I., Harling C.C., McCulloch W.J. and Morgan J. (1985) The effect of vitamin A supplementation on serum retinol and retinol binding protein levels. Cancer Lett., 29, 203–213. [DOI] [PubMed] [Google Scholar]
- 24. Gjorgjieva M., Raffin M., Duchampt A., Perry A., Stefanutti A., Brevet M., Tortereau A., Dubourg L., Hubert-Buron A., Mabille M. et al. (2016) Progressive development of renal cysts in glycogen storage disease type I. Hum. Mol. Genet., 25, 3784–3797. [DOI] [PubMed] [Google Scholar]
- 25. Manickavasagar B., McArdle A.J., Yadav P., Shaw V., Dixon M., Blomhoff R., Connor G.O., Rees L., Ledermann S., Van’t Hoff W. et al. (2015) Hypervitaminosis A is prevalent in children with CKD and contributes to hypercalcemia. Pediatr. Nephrol. Berl. Ger., 30, 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vannucchi M.T., Vannucchi H. and Humphreys M. (1992) Serum levels of vitamin A and retinol binding protein in chronic renal patients treated by continuous ambulatorial peritoneal dialysis. Int. J. Vitam. Nutr. Res., 62, 107–112. [PubMed] [Google Scholar]
- 27. Bandsma R.H.J., Prinsen B.H., van Der Velden M., de S., Rake J.-P., Boer T., Smit G.P.A., Reijngoud D.-J. and Kuipers F. (2008) Increased de novo lipogenesis and delayed conversion of large VLDL into intermediate density lipoprotein particles contribute to hyperlipidemia in glycogen storage disease type 1a. Pediatr. Res., 63, 702–707. [DOI] [PubMed] [Google Scholar]
- 28. Grefhorst A., Schreurs M., Oosterveer M.H., Cortés V.A., Havinga R., Herling A.W., Reijngoud D.-J., Groen A.K. and Kuipers F. (2010) Carbohydrate-response-element-binding protein (ChREBP) and not the liver X receptor α (LXRα) mediates elevated hepatic lipogenic gene expression in a mouse model of glycogen storage disease type 1. Biochem. J., 432, 249–254. [DOI] [PubMed] [Google Scholar]
- 29. Perttilä J., Huaman-Samanez C., Caron S., Tanhuanpää K., Staels B., Yki-Järvinen H. and Olkkonen V.M. (2012) PNPLA3 is regulated by glucose in human hepatocytes, and its I148M mutant slows down triglyceride hydrolysis. Am. J. Physiol. Endocrinol. Metab., 302, E1063–E1069. [DOI] [PubMed] [Google Scholar]
- 30. Kovarova M., Königsrainer I., Königsrainer A., Machicao F., Häring H.-U., Schleicher E. and Peter A. (2015) The genetic variant I148M in PNPLA3 is associated with increased hepatic Retinyl-Palmitate storage in humans. J. Clin. Endocrinol. Metab., 100, E1568–E1574. [DOI] [PubMed] [Google Scholar]
- 31. Mondul A., Mancina R.M., Merlo A., Dongiovanni P., Rametta R., Montalcini T., Valenti L., Albanes D. and Romeo S. (2015) PNPLA3 I148M variant influences circulating retinol in adults with nonalcoholic fatty liver disease or obesity. J. Nutr., 145, 1687–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Suhara A., Kato M. and Kanai M. (1990) Ultrastructural localization of plasma retinol-binding protein in rat liver. J. Lipid Res., 31, 1669–1681. [PubMed] [Google Scholar]
- 33. Ronne H., Ocklind C., Wiman K., Rask L., Obrink B. and Peterson P.A. (1983) Ligand-dependent regulation of intracellular protein transport: effect of vitamin A on the secretion of the retinol-binding protein. J. Cell Biol., 96, 907–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dixon J.L. and Goodman D.S. (1987) Studies on the metabolism of retinol-binding protein by primary hepatocytes from retinol-deficient rats. J. Cell. Physiol., 130, 14–20. [DOI] [PubMed] [Google Scholar]
- 35. General overview of reference values NVKC 2019. [online] https://www.nvkc.nl/algemeen-overzicht-referentiewaarden.
- 36. Penniston K.L. and Tanumihardjo S.A. (2006) The acute and chronic toxic effects of vitamin A. Am. J. Clin. Nutr., 83, 191–201. [DOI] [PubMed] [Google Scholar]
- 37. Feskanich D., Singh V., Willett W.C. and Colditz G.A. (2002) Vitamin A intake and hip fractures among postmenopausal women. JAMA, 287, 47–54. [DOI] [PubMed] [Google Scholar]
- 38. Mahoney C.P., Margolis M.T., Knauss T.A. and Labbe R.F. (1980) Chronic vitamin A intoxication in infants fed chicken liver. Pediatrics, 65, 893–897. [PubMed] [Google Scholar]
- 39. Oliveira J.A., Silva-Netto C.R., Sala M.A., Lopes R.A. and Maia Campos G. (1990) Experimental hypervitaminosis A in the rat. 14. Morphological and morphometric study of changes in the esophageal epithelium. Rev. Odontol. Univ. Sao Paulo, 4, 200–205. [PubMed] [Google Scholar]
- 40. Özen H. (2007) Glycogen storage diseases: new perspectives. World J Gastroenterol: WJG, 13, 2541–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koeberl D.D., Kishnani P.S., Bali D. and Chen Y.-T. (2009) Emerging therapies for glycogen storage disease type I. Trends Endocrinol. Metab. TEM, 20, 252–258. [DOI] [PubMed] [Google Scholar]
- 42. Singh M. and Singh V.N. (1978) Fatty liver in hypervitaminosis A: synthesis and release of hepatic triglycerides. Am. J. Physiol., 234, E511–E514. [DOI] [PubMed] [Google Scholar]
- 43. Oliveros L.B., Domeniconi M.A., Vega V.A., Gatica L.V., Brigada A.M. and Gimenez M.S. (2007) Vitamin A deficiency modifies lipid metabolism in rat liver. Br. J. Nutr., 97, 263–272. [DOI] [PubMed] [Google Scholar]
- 44. Yuan J.-M., Gao Y.-T., Ong C.-N., Ross R.K. and Yu M.C. (2006) Prediagnostic level of serum retinol in relation to reduced risk of hepatocellular carcinoma. J. Natl. Cancer Inst., 98, 482–490. [DOI] [PubMed] [Google Scholar]
- 45. Clemente C., Elba S., Buongiorno G., Berloco P., Guerra V. and Di Leo A. (2002) Serum retinol and risk of hepatocellular carcinoma in patients with child-Pugh class a cirrhosis. Cancer Lett., 178, 123–129. [DOI] [PubMed] [Google Scholar]
- 46. Weiskirchen R. and Tacke F. (2014) Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg. Nutr., 3, 344–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhou T.-B., Drummen G.P.C. and Qin Y.-H. (2012) The controversial role of retinoic acid in fibrotic diseases: analysis of involved signaling pathways. Int. J. Mol. Sci., 14, 226–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.