

Abstract
5-Methylcytosine (5mC), generated through the covalent addition of a methyl group to the fifth carbon of cytosine, is the most prevalent DNA modification in humans and functions as a critical player in the regulation of tissue and cell-specific gene expression. 5mC can be oxidized to 5-hydroxymethylcytosine (5hmC) by ten–eleven translocation (TET) enzymes, which is enriched in brain. Alzheimer’s disease (AD) is the most common neurodegenerative disorder, and several studies using the samples collected from Caucasian cohorts have found that epigenetics, particularly cytosine methylation, could play a role in the etiological process of AD. However, little research has been conducted using the samples of other ethnic groups. Here we generated genome-wide profiles of both 5mC and 5hmC in human frontal cortex tissues from late-onset Chinese AD patients and cognitively normal controls. We identified both Chinese-specific and overlapping differentially hydroxymethylated regions (DhMRs) with Caucasian cohorts. Pathway analyses revealed specific pathways enriched among Chinese-specific DhMRs, as well as the shared DhMRs with Caucasian cohorts. Furthermore, two important transcription factor-binding motifs, hypoxia-inducible factor 2α (HIF2α) and hypoxia-inducible factor 1α (HIF1α), were enriched in the DhMRs. Our analyses provide the first genome-wide profiling of DNA hydroxymethylation of the frontal cortex of AD patients from China, emphasizing an important role of 5hmC in AD pathogenesis and highlighting both ethnicity-specific and overlapping changes of brain hydroxymethylome in AD.
Keywords: Alzheimerʼs disease, 5hmC, HIF-1α, HIF-2α
Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disorder (1). Although the etiology of AD remains mostly elusive, it is thought to involve complicated interactions between genetic and environmental factors and aging (2). Many studies have found that epigenetic regulations play a role in the etiological process of AD (3,4). DNA methylation is a reversible process that modifies the genome by adding a methyl group on the fifth carbon of cytosine to form 5-methylcytosine (5mC). Several large-scale methylome-wide studies using postmortem human brains have revealed important DNA methylation differences between AD and control (5–9). De Jager et al. found that the level of methylation at 71 loci was significantly associated with the burden of AD pathology using a DNA methylation array (6). Lunnon et al. performed an epigenome-wide association study (EWAS) of AD employing a sequential replication design across multiple tissues and identified a differentially methylated region (10). Similarly, Watson et al. identified 479 differentially methylated regions (DMRs) with a strong bias for hypermethylated changes in the superior temporal gyrus of patients with AD using a DNA methylation array (7).
5-Hydroxymethylcytosine (5hmC), an oxidized form of 5mC catalyzed by ten–eleven translocation (TET) proteins, was identified as part of DNA methylation dynamics (methylation/demethylation) in recent years (11,12). It has been shown that traditional bisulfite conversion-based sequencing or array analyses cannot distinguish 5hmC from 5mC. Because of this, the differences detected in previously published works could be based on the difference of either 5mC or 5hmC. Recent technological advances in mapping and tracing 5mC and 5hmC using antibodies allow further dissection of their functions. Studies show that DhMRs are significantly associated with AD in the brains of Caucasian patients with AD (5,13).
However, all of the genome-wide DNA methylation/hydroxymethylation studies in AD that have been performed to date are from Caucasian cohorts. Given that epigenetics varies dramatically across populations with different ethnic backgrounds (14,15), comparing human populations of different ethnic backgrounds will provide invaluable information and new insights into our understanding of epigenetic regulation of AD pathogenesis.
Here we collected human postmortem brain tissues from Chinese AD patients and age-matched unaffected controls and performed genome-wide profiling of both 5mC and 5hmC in the frontal cortex of these two groups of Chinese origin. We identified 16 165 DhMRs in the postmortem frontal cortex samples comparing AD and unaffected subjects. Surprisingly, no DMRs were found. DhMRs were primarily located in intragenic regions, especially CpG islands (CGI) within 500 bp (+/−) of transcription start sites (TSSs). We also observed that 6500 DhMRs overlapped those found in the Caucasian cohort. Pathway analyses revealed pathways enriched among Chinese-specific DhMRs, as well as the shared DhMRs with Caucasian cohorts. Furthermore, two important transcription factor binding motifs, hypoxia-inducible factor 2α (HIF2α) (P = 1.00e−33) and hypoxia-inducible factor 1α (HIF1α) (P = 1.00e−32), were enriched in the DhMRs. These findings provide the first genome-wide profiling of DNA hydroxymethylation of the frontal cortex of AD patients from China, emphasizing an important role of 5hmC in AD pathogenesis and highlighting both ethnicity-specific and overlapping changes of 5hmC in AD brains.
Results
Identification of differential methylated and hydroxymethylated regions in the brains of a cohort of Chinese Alzheimer’s disease patients
We employed hMe-Seal to profile the genome-wide distribution of 5hmC, and MeDIP-seq to profile the distribution of 5mC. In doing so, we identified DMRs/differentially hydroxymethylated regions (DhMRs) from five AD vs five control samples in the Chinese cohorts. Using an FDR of < 0.2, a total of 16 165 DhMRs were detected that were then annotated to 8149 genes (Fig. 1A and Supplementary Material, Table S1). Under FDR 0.1 and 0.05, there are 8404 and 4464 DhMRs, respectively. A threshold of 0.2 is preferred over 0.1 and 0.05 to include more top ranked genes while controlling for multiple testing issues when the sample size is relatively small. A threshold of 0.2 has been used in previous studies for similar reasons (16–19). Therefore, we continued with 16 165 DhMRs in our analyses. The genome-wide distribution of these bins with differential 5hmC signals are shown in Fig. 1A. Among these regions, 6600 DhMRs display hyper-hydroxymethylation in AD, while 9565 showed hypo-hydroxymethylation in AD; however, we did not identify any DMRs (FDR < 0.2). Although earlier works have identified DMRs in AD postmortem brain samples (7,8), the methylation arrays used in those studies were not able to distinguish 5hmC from 5mC (20,21). Thus, it is unclear whether the previously detected methylation differences resulted from 5hmC or 5mC changes (6–8). Our 5mC- and 5hmC-specific analyses suggest that 5hmC, rather than 5mC, is significantly altered in AD postmortem brain tissues. Based on the lack of DMRs, we focused our analyses on DhMRs.
Figure 1.
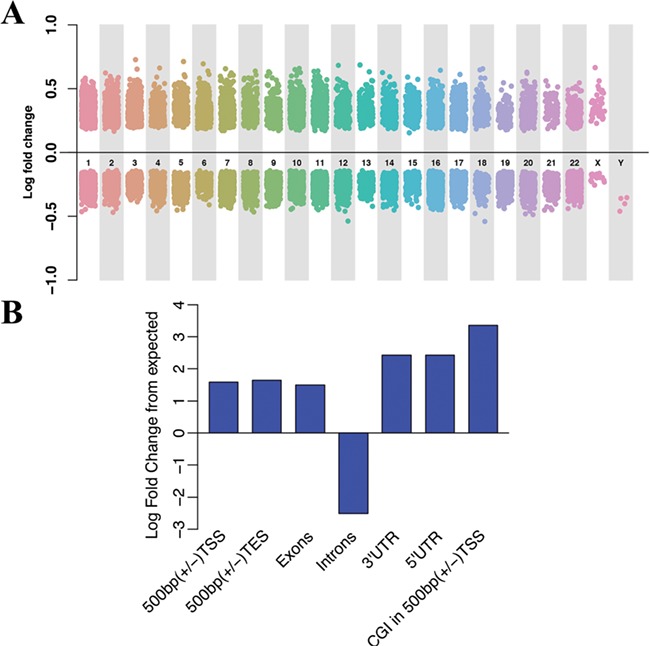
Genome-wide hydroxymethylation profiling in AD brain tissues. (A) Genome-wide hydroxymethylation profiling in AD brain tissues. The horizontal axis represents the human chromosomes, and the vertical axis indicates the levels of 5hmC enrichment. (B) Genomic distribution of DhMRs. Histogram of the log fold change from expected versus genomic locations. The height of the bar indicates the percentage of DhMRs located in the corresponding regions. TSS, transcription start site; TES, transcription end site; UTR, untranslated region; CGI, CpG island.
To better account for covariates including age, gender and postmortem interval in the differential analysis, we used DESeq2 to re-analyze the data. The significant regions identified by DSS are similar to DhMRs identified by DESeq2 adjusting for patient age, gender and postmortem interval. Around 90% of regions overlapped between the two results. For overlapping regions with Caucasian AD studies, more than 95% of regions are overlapped (Supplementary Material, Table S5). As the two results are almost identical, we focused on the regions identified from DSS for the following analysis.
We next characterized the DhMRs and evaluated the enrichment or depletion of their overlapped lengths with selective genomic features in contrast to the expected overlapped lengths in those corresponding regions. In general, in contrast to expected regions, DhMRs are enriched on all intragenic regions except introns, including 3′UTRs, 5′UTRs, exons, promoter regions (enrichment P value <1e−22) and CpG Islands (CGI) in 500 bp (+/−). The TSS was the most significant enriched region (P value < 2.2e−16, Fig. 1B).
Gene ontology and pathway analyses
To understand the potential biological roles underlying dynamic 5hmC changes in AD postmortem brains, we annotated the DhMRs to hg19 human RefSeq genes and further subjected these genes to gene ontology (GO) and MSigDB pathway analyses. Interestingly, phagocytosis (FDR = 4.63e−38), ventral spinal cord interneuron specification (FDR = 1.68e−25) and regulation of cellular response to insulin stimulus (FDR = 1.33e−24) were the most significantly enriched. The other enriched GO terms include oligodendrocyte development, positive regulation of apoptotic signaling pathway, positive regulation of gliogenesis and DNA methylation (Fig. 2A).
Figure 2.
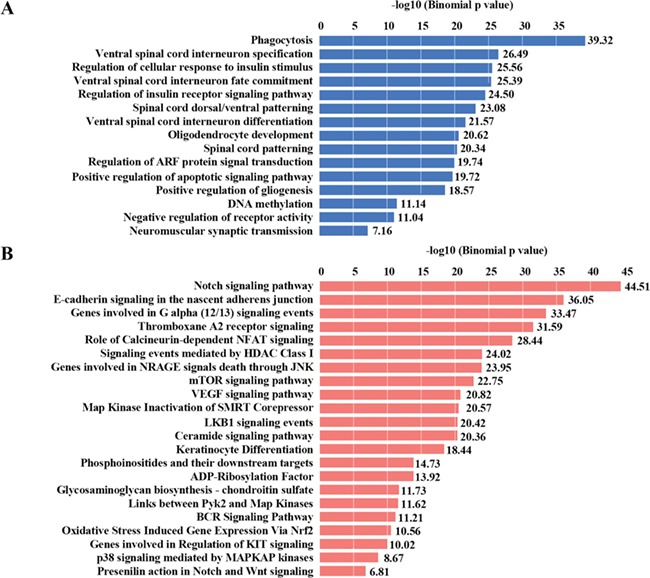
Specific biological pathways enriched among DhMRs. (A) GO analysis of DhMRs. (B) MSigDB pathway analysis of DhMRs. Significant GO categories and MSigDB pathway were identified by subjecting regions of DhMRs to GREAT (Genomic Regions Enrichment of Annotations Tool). Blue and red bars represent −log10 (binomial P value).
The top MSigDB pathways involved were the Notch signaling pathway (FDR = 4.09e−42), E-cadherin signaling in the nascent adherens junction (FDR = 3.88e−34) (22), thromboxane A2 receptor signaling (FDR = 2.54e−32) (23), calcineurin-dependent NFAT signaling (FDR = 4.03e−27) (24), signaling events mediated by HDAC Class I (FDR = 5.46e−23) (25), mTOR signaling pathway (FDR = 8.73e−22) (26), LKB1 signaling events (FDR = 3.79e−21) (27) and the VEGF signaling pathway (FDR = 6.61e−20) (28), which have all been shown to be involved in AD pathogenesis, such as Tau phosphorylation, excitotoxicity and Aβ synaptotoxicity (29,30). Further, other enriched MSigDB pathways, such as genes involved in G alpha (12/13) signaling events (FDR = 3.43e−34), NRAGE signals (FDR = 3.90e−29), Map kinase inactivation of SMRT Corepressor (FDR = 2.71e−21), BCR signaling pathway (FDR = 6.20e-12) and KIT signaling (FDR = 9.62e−11) were observed (Fig. 2B). However, their potential contribution to AD pathogenesis will need further study.
Unique and shared DhMRs in the postmortem brain tissues from Caucasian and Chinese AD patients
Previous work has assessed the methylation state in AD postmortem brain tissues of Caucasians, finding that the level of methylation at 71 loci was associated with the burden of AD pathology using Illumina Human Methylation 450, an array based on the bisulfite conversion method (6). To determine the impact of ethnicity on the epigenetic alterations in AD postmortem brain tissues, we first re-examined the 5hmC level at these 71 loci in Chinese samples. Interestingly, we observed 37 DhMRs significantly overlapped these loci (P value < 2.2e−16) (Table 1). However, the methylation array based on bisulfite conversion could not distinguish 5mC from 5hmC. The significant overlap that we observed here indicates that many of the previously identified differentially ‘methylated’ loci could be the result of 5hmC differences instead of 5mC changes.
Table 1.
Previously identified differentially ‘methylated’ loci overlap with DhMRs in this study
| Chr (DhMR) | Start (DhMR) | End (DhMR) | CpGa | Chr (71CpG)* | Position (71CpG)a | Gene |
|---|---|---|---|---|---|---|
| chr12 | 121890001 | 121900000 | 12 | 121890864 | cg11724984 | RNF34, KDM2B |
| chr10 | 73520001 | 73530000 | 10 | 73521631 | cg23968456 | CDH23, C10orf105, C10orf54 |
| chr1 | 43470001 | 43480000 | 1 | 43473840 | cg15821544 | SLC2A1, FLJ32224, U6 |
| chr17 | 74470001 | 74480000 | 17 | 74475294 | cg13076843 | UBE2O, AANAT, RHBDF2, AX747521, CYGB, PRCD |
| chr7 | 4780001 | 4790000 | 7 | 4786943 | cg25594100 | FOXK1, KIAA0415, MIR4656, RADIL |
| chr21 | 47850001 | 47860000 | 21 | 47855916 | cg00621289 | PCNT, DIP2A |
| chr17 | 1630001 | 1640000 | 17 | 1637391 | cg19803550 | PRPF8, TLCD2, MIR22HG, AF070569, MIR22, WDR81, SERPINF2, SERPINF1, SMYD4 |
| chr16 | 89590001 | 89600000 | 16 | 89598950 | cg03169557 | ANKRD11, SPG7, SNORD68, RPL13, CPNE7 |
| chr8 | 41510001 | 41520000 | 8 | 41519308 | cg05066959 | AGPAT6, NKX6-3, MIR486, JA429246, ANK1 |
| chr17 | 74470001 | 74480000 | 17 | 74475270 | cg05810363 | UBE2O, AANAT, RHBDF2, AX747521, CYGB, PRCD |
| chr17 | 80190001 | 80200000 | 17 | 80195180 | cg07012687 | CCDC57, SLC16A3, CSNK1D |
| chr8 | 41510001 | 41520000 | 8 | 41519399 | cg11823178 | AGPAT6, NKX6-3, MIR486, JA429246, ANK1 |
| chr17 | 74470001 | 74480000 | 17 | 74475355 | cg12163800 | UBE2O, AANAT, RHBDF2, AX747521, CYGB, PRCD |
| chr17 | 1370001 | 1380000 | 17 | 1373605 | cg05417607 | CRK, MYO1C, INPP5K, LOC100306951, PITPNA |
| chr19 | 44270001 | 44280000 | 19 | 44278628 | cg22904711 | SMG9, KCNN4, LYPD5, AK131520 |
| chr2 | 45170001 | 45180000 | 2 | 45178 474 | cg18556455 | SIX3 |
| chr10 | 105 420 001 | 105 430 000 | 10 | 105 420 501 | cg19007269 | SH3PXD2A, Y_RNA |
| chr1 | 55 240 001 | 55 250 000 | 1 | 55 247 356 | cg15645660 | HEATR8-TTC4, TTC4, PARS2, TTC22, C1orf177 |
| chr2 | 127 800 001 | 127 810 000 | 2 | 127 800 646 | cg22883290 | BIN1 |
| chr13 | 113 630 001 | 113 640 000 | 13 | 113 635 690 | cg09448088 | BC035340, MCF2L-AS1, MCF2L |
| chr19 | 1 070 001 | 1 080 000 | 19 | 1 071 176 | cg02308560 | CNN2, ABCA7, HMHA1, POLR2E, GPX4, SBNO2 |
| chr7 | 4 780 001 | 4 790 000 | 7 | 4 786 899 | cg07180538 | FOXK1, KIAA0415, MIR4656, RADIL |
| chr13 | 50 700 001 | 50 710 000 | 13 | 50 700 845 | cg20733077 | DLEU2, DLEU1, ST13P4 |
| chr19 | 49 960 001 | 49 970 000 | 19 | 49 962 324 | cg20618448 | CCDC155, PTH2, LOC100507003, SLC17A7, BC128433, PIH1D1, ALDH16A1,FLT3LG,RPL13A,SNORD32A,SNORD33,SNORD34,SNORD35A,RPS11,SNORD35B,MIR150 |
| chr7 | 65 740 001 | 65 750 000 | 7 | 65 746 852 | cg03193328 | - |
| chr13 | 113 630 001 | 113 640 000 | 13 | 113 634 042 | cg07883124 | BC035340, MCF2L-AS1, MCF2L |
| chr8 | 126 450 001 | 126 460 000 | 8 | 126 458 568 | cg10920329 | TRIB1, AK022787 |
| chr15 | 77 320 001 | 77 330 000 | 15 | 77 324 526 | cg11652496 | PSTPIP1, TSPAN3 |
| chr10 | 134 030 001 | 134 040 000 | 10 | 134 038 395 | cg25917732 | JAKMIP3, AL137551, DPYSL4, STK32C |
| chr8 | 41 510 001 | 41 520 000 | 8 | 41 514 039 | cg16140558 | AGPAT6, NKX6-3, MIR486, JA429246, ANK1 |
| chr20 | 39 310 001 | 39 320 000 | 20 | 39 314 091 | cg13579486 | MAFB |
| chr17 | 7 010 001 | 7 020 000 | 17 | 7 017 474 | cg18659586 | CLEC10A, ASGR2 |
| chr21 | 45 620 001 | 45 630 000 | 21 | 45 626 491 | cg15348679 | ICOSLG, DNMT3L |
| chr3 | 72 620 001 | 72 630 000 | 3 | 72 621 000 | cg16459281 | - |
| chr17 | 7 900 001 | 7 910 000 | 17 | 7 906 847 | cg04157161 | GUCY2D, ALOX15B |
| chr2 | 45 170 001 | 45 180 000 | 2 | 45 175 881 | cg22385702 | SIX3 |
| chr5 | 16 880 001 | 16 890 000 | 5 | 16 886 424 | cg06742628 | MYO10 |
aDe Jager et al. 2014.
To examine this further, we compared the 5hmC alterations between Chinese and Caucasian AD brains (5). Among the 16 165 DhMRs that we observed here, we identified 6500 DhMRs that overlap with the previously identified Caucasian DhMRs (among them, 4974 DhMRs displayed the same trends in changes between Chinese and Caucasian cohorts) which left 9665 DhMRs as Chinese-specific DhMRs. We then performed the GO and pathway analyses of these shared and Chinese-specific DhMRs. Using MSigDB pathway analysis of shared DhMRs, the top enriched pathways were E-cadherin signaling in the nascent adherens junction (FDR = 2.01e−19), genes involved in signaling by PDGF (FDR = 2.91e−19), regulation of RhoA activity (FDR = 1.84e−15), genes involved in signaling by Notch (FDR = 5.58e−15), genes involved in G alpha (12/13) signaling events (FDR = 1.95e−14) and genes involved in NCAM signaling for neurite out-growth (FDR = 5.42e−14) (Supplementary Material, Fig. S1B). Significant GO categories of shared DhMRs include phagocytosis (FDR = 1.90e−18), positive regulation of establishment of protein localization to the plasma membrane (FDR = 6.52e−16), spinal cord dorsal/ventral patterning (FDR = 1.58e−14) and positive regulation of gliogenesis (FDR = 1.73e−12) (Supplementary Material, Fig. S1A).
Among the Chinese-specific DhMRs, glucose and lipid metabolism-related pathways were significantly enriched, such as regulation of cellular response to insulin stimulus (FDR = 1.73e−20), regulation of insulin receptor signaling pathway (FDR = 3.39e−20), thromboxane A2 receptor signaling (FDR = 7.08e−28), genes involved in DAG and IP3 signaling (FDR = 6.5e−08), and the lissencephaly gene in neuronal migration and development (FDR = 3.65e−05) (Supplementary Material, Fig. S2A and B).
Selective transcription factor binding motifs are enriched within DhMRs
We then employed motif search algorithms (31) to comprehensively predict the potential transcription factor-binding sites within the Chinese 16 165 DhMRs, as well as Chinese-specific DhMRs and shared DhMRs. The top 10 binding motifs identified in every group are listed in Supplementary Material, Table S3. It is worth noting that among the top 10 binding motifs, the binding motifs of HIF1α and HIF2α, critical transcription factors responsible for gene regulation in response to normoxia and hypoxia, are enriched (Fig. 3). Other enriched binding motifs includes CLOCK, n-Myc, Max, bHLHE40, USF1, Arnt and Usf2.
Figure 3.
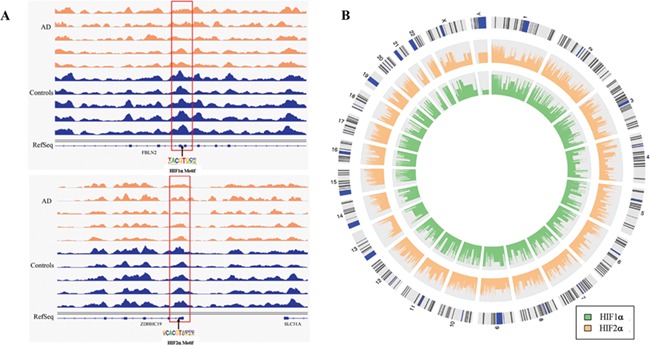
Characteristics of HIF motifs enriched among DhMRs. (A) HIF motif residing in the DhMR in AD (orange) and controls (blue), respectively. The upper half of A shows HIF-1α motif and lower half shows HIF-2α motif. (B) HIF-1α and HIF-2α binding motifs are enriched at DhMRs. The outer circle shows the human chromosomes. The inner three circles represent HIF-1α (green) and HIF-2α (orange) binding motif enrichment. The height of the histogram indicates the level of the log-odds score.
Further analysis revealed that 699 of the potential HIF1α binding sites (all overlapped or within 500 bp of the identified DhMR) were located at the gene promoter region and annotated to 367 genes. To further determine the impact of 5hmC alterations on gene expression, we performed RNA-sequencing using the same brain tissues and examined the expression of the genes close to DhMRs. Interestingly, 78 out of these 367 genes displayed altered gene expression at the cutoff of FDR < 0.2. Also, 831 of the potential HIF2α binding sites (all overlapping or within 500 bp of the identified DhMR) were located at the gene promoter region and annotated to 383 genes. Among these 383 genes, 90 showed differential expression (FDR < 0.2). Moreover, 55 genes overlapped between the 78 HIF1α and 90 HIF2α genes (Supplementary Material, Table S4). These results suggest that altered DNA hydroxymethylation status could potentially regulate the gene expression by altering the binding of transcriptional factors (such as HIF) and their target genes, which may contribute to the pathogenesis of ad.
Discussion
5hmC can be generated from 5mC being oxidized by Tet protein (11,12). 5hmC can be further oxidized by Tet proteins to produce 5-formylcytosine and 5-carboxylcytosine, which are quickly removed from the genome by thymine-DNA glycosylase (TDG) to initiate base excision repair (BER) (32–34). 5hmC not only acts as a transitory intermediate in DNA demethylation but also has been reported to be stably enriched in the central nervous system and bound by specific 5hmC-binding proteins (35–37). Therefore, 5hmC is now recognized as an important epigenetic mark and has been shown to play functional roles in a number of neurological disorders (38). Previous studies on DNA methylation of postmortem AD brain tissues have mostly focused on 5mC, both epigenome-wide studies (6–8) and gene specific studies (39–42), while 5hmC has been typically overlooked. More importantly, previous studies found that different ethnic backgrounds potentially modulate epigenetic profiles (14,15). For example, Fraser et al. found population-specific patterns of DNA methylation at over a third of all genes when they measured DNA methylation near the TSS of over 14 000 genes in 180 cell lines derived from one African and one European population (14). To better understand the role of DNA methylation (both 5mC and 5hmC) in AD pathogenesis, in this study we have generated the first methylation maps (both 5mC and 5hmC) of the human frontal cortex of AD patients from Chinese origin, which will provide valuable resources for future population-specific studies.
Intriguingly, unlike previous reports, we did not detect any loci with significant 5mC changes; however, we did identify significant differential 5hmC signals. We identified 16 165 DhMRs in AD patients from Chinese origin. DhMRs were primarily located in intragenic regions, including exons, UTRs and promoter regions but not introns, which was consistent with previous reports (5). GO and pathway analyses revealed multiple pathways with altered 5hmC, such as the Notch signaling pathway, HDAC CLASS I, mTOR signaling pathway and VEGF signaling pathway, many of which were previously linked to AD pathogenesis (26,43–45). If the change of 5hmC that we observed was based on the altered activity of DNA demethylation pathway, we should have observed the corresponding changes with 5mC. The lack of detectable 5mC changes in our analyses suggest that 5hmC might function as an independent epigenetic marker, rather than simply an intermediate of DNA demethylation, and contribute to AD pathogenesis.
Furthermore, significant differences in 5hmC changes between Chinese and Caucasian cohorts were observed: 9665 Chinese-specific DhMRs were identified. It is worth noting that some glucose and lipid metabolism-related pathways were significantly enriched among the Chinese-specific DhMRs, indicating that altered DNA hydroxymethylation of genes involved in glucose and lipid metabolism pathways could contribute to the pathogenesis of AD in China. Our results demonstrate the potential utility of Chinese-specific DNA methylation profiles, further supporting the population-specificity of epigenetic changes associated with AD.
HIF1α and HIF2α are two known critical regulators of the transcriptional response to hypoxic stress. HIF1α, a hypoxically inducible subunit of the HIF1 complex, is ubiquitously expressed in human and mouse tissues and regulates the expression of proteins involved in a myriad of processes, including angiogenesis, insulin growth factor signaling, iron metabolism, inflammation, erythropoiesis and apoptosis (46). Dysregulation of HIF1α and HIF2α can lead to impaired glucose uptake and metabolism, an invariable feature of AD brain (47,48). Consistent with this, we observed enrichment of glucose metabolism genes among the Chinese-specific DhMRs. Local hypoxia/ischemia augments the pathogenesis of ad by increasing the amyloidogenic processing of amyloid-β precursor protein (APP) in the brain, likely via HIF-1α-dependent transcriptional regulation of the β-secretase 1 (BACE1) gene through its binding to the hypoxia-response element in the BACE1 promoter and/or non-transcriptional activation of γ-secretases (49,50). These data taken together suggested that the 5hmC alteration could not only impact the transcripts associated with critical biological processes, but may also alter critical transcription factor binding dynamics, such as HIF1α and HIF2α, which could contribute to AD pathogenesis.
We acknowledge that our current analysis does not account for cellular composition variations across different samples. Although previous studies have reported potential impacts of changes in proportions of different cell types on the detection of differential signals from high-throughput data (9,51), in a separate analysis using DNA methylation 450K data generated from the ROS/MAP cohort, we did not observe any significant impact on detected signals with or without accounting for proportions of different cell types. Thus, the cellular composition is unlikely to alter the results significantly in this study.
In contrast with previous studies that used methods such as bisulfite pyrosequencing (39,52–56) and Illumina HumanMethylation450 beadset (6,7,9,10,40), the methods we have used are able to distinguish 5hmC from 5mC (37,57). In addition, we generated a genome-wide profile of DNA hydroxymethylation, providing a more comprehensive understanding for AD pathogenesis in contrast to single genes or only a subset of genes (39,53–55,58–60). We have also performed this genome-wide profiling of brain DNA methylation and DNA hydroxymethylation for the first time in AD patients of Chinese origin, which is of great significance. Nevertheless, we must acknowledge that there are some limits to our work. Based on the culture and traditions in China, organ donations after death are rare. Although we were able to identify significant differential 5hmC signals in this relatively small cohort, repetition in other larger groups of patients is warranted to validate our findings. Additionally, further functional studies are needed to better understand the relationship between the pathogenesis of AD and the cell-type-specific DNA hydroxymethylation changes.
In summary, we present the first genome-wide profiling of DNA hydroxymethylation in the frontal cortex of AD patients from China. Our analyses revealed Caucasian and Chinese-specific DhMRs as well as the shared DhMRs, which indicate a vital role of 5hmC in AD pathogenesis. Our results suggest that common pathways and ethnicity-specific pathways could be epigenetically mis-regulated and contribute to the pathogenesis of AD.
Materials and Methods
Subjects and brain samples
Tissue samples from the frontal cortex of patients with late-onset AD (LOAD) (n = 5) and neurologically normal controls (n = 5) were obtained from the Human Brain Bank, Chinese Academy of Medical Sciences & Peking Union Medical College, following relevant regulations and legislation as defined by China. All subjects were ethnic Han Chinese. Characteristics of AD patients and controls are listed in Supplementary Material, Table S1. Briefly, gender (% male, AD: 20% vs control: 40%), age at death (years, AD: 87.4 ± 7.9 vs control: 84.0 ± 3.5), postmortem interval (hours, AD: 4.6 ± 1.2 vs control: 5.7 ± 2.0) and years in storage (years, AD: 1.2 ± 0.4 vs control: 1.0 ± 0.4) were well matched (P value>0.05). Additional details regarding these subjects are shown in Supplementary Material, Table S2. Two controls suffered from diabetes, two controls had cancer, and two controls had coronary heart disease, but all were neurologically normal. Neuropathologic examination of every subject was recorded, and patients were diagnosed according to the National Institute on Aging/Alzheimer’s Association (NIA/AA) criteria (61). A single trained specialist dissected all samples, and the tissues used in this study were from the same sub-region of the frontal cortex from each subject. Only grey matter was included for this study. All of the AD patients had no familial history and were screened for APP, PSEN1 and PSEN2 to confirm the absence of any known mutations of these genes. The ethics committee of Central South University approved the study. Written consent for brain donations were obtained from all donors.
DNA and RNA isolation
Genomic DNA was extracted from ~ 100 mg of each frontal cortex using the standard phenol–chloroform extraction and ethanol precipitation method (62). The TRIzol solubilization and extraction method was used to isolate total RNA, as previously described (63). Concentrations and purity of DNA and RNA were evaluated with a NanoDrop spectrophotometer, and samples were stored at −20°C before further analysis.
DNA methylation/hydroxymethylation capture and sequencing
We employed a previously established chemical labeling and affinity purification method coupled with high-throughput sequencing (hMe-Seal) to profile the genome-wide distribution of 5hmC (37), and methylated DNA immunoprecipitation (MeDIP) coupled with high-throughput sequencing (MeDIP-seq) to profile 5mC (57). Briefly, genomic DNA was sonicated using a Covaris Ultrasonicator, yielding an average size of sonicated DNA fragments of ~ 200 bp. Four micrograms of fragmented DNA was used for the MeDIP/hMe-Seal assay. For the hMe-Seal assay, the T4 bacteriophage β-glucosyltransferase was used to transfer a chemically modified glucose, 6-N3-glucose, onto the hydroxyl group of 5hmC. After attaching a biotin tag to the 6-N3-glucose, the azide group was chemically modified for biotin detection, then 5hmC-containing DNA fragments were high-affinity captured for deep sequencing.
The captured DNA fragments containing 5hmC were used for library preparation using NEBNext® ChIP-Seq Library Prep Master Mix Set for Illumina (NEB, Cat# E6240L) per the manufacturer’s instructions with some modifications, i.e. no size selection and a 2.5-fold decrease in quantity of the final PCR primers (Universal primer and Index primer) for PCR-based final enrichment of the library.
For the MeDIP library preparation, we followed the protocol from Weber et al. with some modifications (64). Briefly, the genomic DNA was sonicated to 200 bp before starting end repair, dA tailing and adaptor ligation using the NEB kit NEBNext® DNA Library Prep Reagent Set for Illumina® (Cat#E6000S/L). DNA fragments with adaptors ligated were denatured at 98°C for 10 min followed immediately by incubation on ice for at least 2 min.
The denatured ssDNA fragments were used for immunoprecipitation without beads at 4°C overnight (O/N) with anti-5methylcytosine (5-mC) monoclonal antibody (Eurogentec, Cat# BI-MECY-0100) and mouse normal IgG (Millipore Cat# 632136) as IgG negative control. The following day, 30 μl of Dynabeads Protein G (Invitrogen, Cat# 10003D) were added to each of the O/N IP reaction tubes for 2 h with rotation. After IP with beads, six washes were conducted using 1× IP buffer followed by three washes with 1× IP buffer supplemented with 750 mm NaCl. The extensively washed beads were digested with proteinase K at 50°C for 2 h and then extracted with phenol:chloroform:isoamylalchohol = 25:24:1. The extracted aqueous phase was precipitated with 10% volume of 3 M NaAc, pH 5.2, 3 μl of glycogen at 5 mg/ml (Invitrogen, Cat# AM9510) and three volumes of absolute ethanol. Finally, the recovered ssDNA fragments were amplified using NEBNext Ultra II Q5 Master Mix (NEB, Cat# M0544S). The input DNA samples were used for library preparation in parallel with the captured DNA fragments containing the 5hmC or 5mC.
RNA-seq
RNA-seq library preparation and RNA-sequencing libraries were generated from 1000 ng of total RNA from triplicate samples using the TruSeq LT RNA Library Preparation Kit v2 (Illumina) following the manufacturer’s protocol. An Agilent 2100 Bioanalyzer and DNA1000 kit (Agilent) were used to quantify amplified cDNA and control for the quality of the libraries. A qPCR-based KAPA library quantification kit (KAPA Biosystems) was used to accurately quantify library concentration. Illumina MiSeq and NextSeq 500 were used to perform 75-cycle paired-end (PE) and single-read (SR) sequencing. Illumina HiSeq 2500 and NextSeq 500 were used to perform 100-cycle SR and 75-cycle PE sequencing. Image processing and sequence extraction occurred using the standard cloud-based Illumina pipeline in BaseSpace.
Bioinformatic analyses
The raw fastq files generated from the high-throughput sequencing were aligned to the human genome (hg19) using Bowtie. Uniquely aligned reads with no more than 2 mismatches in the first 25 bp were retained for downstream analyses (65). Aligned reads ranged from 15 to 20 million in number. The whole genome was cut into non-overlapping 10 kb bins. The genomic features and nearby genes for all bins were annotated with Homer (66). The mapped read counts in all bins were obtained for each sample. Bin counts were normalized by total read counts in each sample during data analysis. The DMR/DhMRs detection was performed by comparing the normalized bin-level read counts between AD and controls using DSS (67), which models the count data by negative binomial distribution and performs Wald tests for comparing two groups. After that, we corrected for multiple testing using a permutation-based method to calculate the empirical false discovery rate (FDR) for all bins (68). The permutation-based method to evaluate P values from high-throughput data analysis is widely used (69–74). The traditional multiple testing correction method (such as FDR) assumes that the P values follow a uniform distribution from null hypotheses. The assumption is often not satisfied in capture-sequencing data including ChIP-seq and hMe-Seal. This is because the statistical tests from capture sequencing data are often under-powered based on small sample sizes and/or low sequencing depths. From this perspective, a permutation test is a more appropriate procedure to correct for multiple testing in capture-sequencing data. In each permutation, we randomly shuffled the AD and control labels and repeated DSS on the permutated dataset to attain the null test statistics. The FDR can then be evaluated against the null distribution. All bins were ranked based on the FDR in increasing order, and bins lower than the FDR threshold (FDR < 0.2) were deemed DhMRs.
Gene Ontology (GO) and Molecular signature database (MSigDB) pathway analysis were conducted by GREAT (Genomic Regions Enrichment of Annotations Tool, http://bejerano.stanford.edu/great/public/html/ (75)). For the RNA-seq data, we first obtained the gene counts by recording the number of overlapped sequencing reads with gene locations. The raw counts were used as inputs to DSS to perform differential expression detections. We did not do extra data normalization because DSS automatically perform normalization to account for sequencing depth.
Statistical analysis
The DMRs/DhMRs detection was performed by comparing binned read counts in genomics windows between AD and unaffected control samples. The data are characterized by a negative binomial model, which captures the over-dispersion in the counts within the same group. Testing the significance of overlaps was performed by Fisher’s exact test on 2 × 2 contingency table. The Bioconductor package annotatr (76) was used for the genomic distribution analysis, and the binding motif enrichment analysis was conducted using Homer (66). The R programming language was used for all data analysis unless otherwise mentioned.
Supplementary Material
Funding
National Natural Science Foundation of China (81430023, 81301079, 81361120404 to B.T.); National Key Plan for Scientific Research and Development of China (2016YFC1306000, 2017YFC0909100 to B.T.); U.S.-China Program for Biomedical Collaborative Research of National Institute of Neurological Disorders and Stroke (NS083498 to X.Z.). The previously published Caucasian analyses were supported by NIH grants P30AG10161, R01AG17917 and U01AG61356.
Acknowledgements
We would like to thank Prof. Wenying Qiu from the Chinese Academy of Medical Sciences & Peking Union Medical College and Prof. Jiapei Dai from South Central University for Nationalities for their technical help. We thank all donors for their generous donation of tissue for this research. ROS/MAP resources can be requested at www.radc.rush.edu.
Conflict of Interest statement. None declared.
References
- 1. Scheltens P., Blennow K., Breteler M.M., de Strooper B., Frisoni G.B., Salloway S. and Van der W.M. (2016) Alzheimer’s disease. Lancet, 388, 505–517. [DOI] [PubMed] [Google Scholar]
- 2. Bettens K., Sleegers K. and Van Broeckhoven C. (2013) Genetic insights in Alzheimer’s disease. Lancet, 12, 92–104. [DOI] [PubMed] [Google Scholar]
- 3. Wen K.X., Milic J., El-Khodor B., Dhana K., Nano J., Pulido T., Kraja B., Zaciragic A., Bramer W.M. and Troup J. (2016) The role of DNA methylation and histone modifications in neurodegenerative diseases: a systematic review. PloS One, 11, e0167201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zuo L., Hemmelgarn B.T., Chuang C.C. and Best T.M. (2015) The role of oxidative stress-induced epigenetic alterations in amyloid-beta production in Alzheimer’s disease. Oxid Med Cell Longev, 2015, 604658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhao J., Zhu Y., Yang J., Li L., Wu H., De Jager P.L., Jin P. and Bennett D.A. (2017) A genome-wide profiling of brain DNA hydroxymethylation in Alzheimer’s disease. Alzheimers Dement, 13, 674–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. De Jager P.L., Srivastava G., Lunnon K., Burgess J., Schalkwyk L.C., Yu L., Eaton M.L., Keenan B.T., Ernst J. and McCabe C. (2014) Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nature Neurosci, 17, 1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Watson C.T., Roussos P., Garg P., Ho D.J., Azam N., Katsel P.L., Haroutunian V. and Sharp A.J. (2016) Genome-wide DNA methylation profiling in the superior temporal gyrus reveals epigenetic signatures associated with Alzheimer’s disease. Genome Med, 8, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bakulski K.M., Dolinoy D.C., Sartor M.A., Paulson H.L., Konen J.R., Lieberman A.P., Albin R.L., Hu H. and Rozek L.S. (2012) Genome-wide DNA methylation differences between late-onset Alzheimer’s disease and cognitively normal controls in human frontal cortex. J Alzheimers Dis, 29, 571–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gasparoni G., Bultmann S., Lutsik P., Kraus T.F.J., Sordon S., Vlcek J., Dietinger V., Steinmaurer M., Haider M. and Mulholland C.B. (2018) DNA methylation analysis on purified neurons and glia dissects age and Alzheimer’s disease-specific changes in the human cortex. Epigenetics hromatin, 11, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lunnon K., Smith R., Hannon E., De Jager P.L., Srivastava G., Volta M., Troakes C., Al-Sarraj S., Burrage J. and Macdonald R. (2014) Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nature Neurosci, 17, 1164–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tahiliani M., Koh K.P., Shen Y., Pastor W.A., Bandukwala H., Brudno Y., Agarwal S., Iyer L.M., Liu D.R. and Aravind L. (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science, 324, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kriaucionis S. and Heintz N. (2009) The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science, 324, 929–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ellison E.M., Bradley-Whitman M.A. and Lovell M.A. (2017) Single-Base resolution mapping of 5-Hydroxymethylcytosine modifications in hippocampus of Alzheimer’s disease subjects. J Mol Neurosci, 63, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fraser H.B., Lam L.L., Neumann S.M. and Kobor M.S. (2012) Population-specificity of human DNA methylation. Genome Biol, 13, R8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heyn H., Moran S., Hernando-Herraez I., Sayols S., Gomez A., Sandoval J., Monk D., Hata K., Marques-Bonet T. and Wang L. (2013) DNA methylation contributes to natural human variation. Genome Res, 23, 1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yamamoto S., Huang D., Du L., Korn R.L., Jamshidi N., Burnette B.L. and Kuo M.D. (2016) Radiogenomic analysis demonstrates associations between (18)F-fluoro-2-deoxyglucose PET, prognosis, and epithelial-mesenchymal transition in non-small cell lung cancer. Radiology, 280, 261–270. [DOI] [PubMed] [Google Scholar]
- 17. Penn A.M., Saly V., Trivedi A., Lesperance M.L., Votova K., Jackson A.M., Croteau N.S., Balshaw R.F., Bibok M.B. and Smith D.S. (2018) Differential proteomics for distinguishing ischemic stroke from controls: a pilot study of the SpecTRA project. Transl Stroke Res, 9, 590–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sadanandam A., Lyssiotis C.A., Homicsko K., Collisson E.A., Gibb W.J., Wullschleger S., Ostos L.C., Lannon W.A., Grotzinger C. and Del Rio M. (2013) A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nature Med, 19, 619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jang C., Lahens N.F., Hogenesch J.B. and Sehgal A. (2015) Ribosome profiling reveals an important role for translational control in circadian gene expression. Genome Res, 25, 1836–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nestor C., Ruzov A., Meehan R. and Dunican D. (2010) Enzymatic approaches and bisulfite sequencing cannot distinguish between 5-methylcytosine and 5-hydroxymethylcytosine in DNA. BioTechniques, 48, 317–319. [DOI] [PubMed] [Google Scholar]
- 21. Jin S.G., Kadam S. and Pfeifer G.P. (2010) Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res, 38, e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baki L., Marambaud P., Efthimiopoulos S., Georgakopoulos A., Wen P., Cui W., Shioi J., Koo E., Ozawa M., Friedrich V.L., Jr. (2001) Presenilin-1 binds cytoplasmic epithelial cadherin, inhibits cadherin/p120 association, and regulates stability and function of the cadherin/catenin adhesion complex. Proc Natl Acad Sci U S A, 98, 2381–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Soper J.H., Sugiyama S., Herbst-Robinson K., James M.J., Wang X., Trojanowski J.Q., Smith A.B. 3rd, Lee V.M., Ballatore C. and Brunden K.R. (2012) Brain-penetrant tetrahydronaphthalene thromboxane A2-prostanoid (TP) receptor antagonists as prototype therapeutics for Alzheimer’s disease. ACS Chem Neurosci, 3, 928–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sompol P., Furman J.L., Pleiss M.M., Kraner S.D., Artiushin I.A., Batten S.R., Quintero J.E., Simmerman L.A., Beckett T.L. and Lovell M.A. (2017) Calcineurin/NFAT signaling in activated astrocytes drives network hyperexcitability in abeta-bearing mice. J Neurosci, 37, 6132–6148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cuadrado-Tejedor M., Garcia-Barroso C., Sanchez-Arias J.A., Rabal O., Perez-Gonzalez M., Mederos S., Ugarte A., Franco R., Segura V. and Perea G. (2017) A first-in-class small-molecule that acts as a dual inhibitor of HDAC and PDE5 and that rescues hippocampal synaptic impairment in Alzheimer’s disease mice. Neuropsychopharmacology, 42, 524–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Norambuena A., Wallrabe H., McMahon L., Silva A., Swanson E., Khan S.S., Baerthlein D., Kodis E., Oddo S. and Mandell J.W. (2017) mTOR and neuronal cell cycle reentry: how impaired brain insulin signaling promotes Alzheimer’s disease. Alzheimers Dementia, 13, 152–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang J.W., Imai Y. and Lu B. (2007) Activation of PAR-1 kinase and stimulation of tau phosphorylation by diverse signals require the tumor suppressor protein LKB1. J Neuroscience, 27, 574–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Harris R., Miners J.S., Allen S. and Love S. (2018) VEGFR1 and VEGFR2 in Alzheimer’s disease. J Alzheimers Dis, 61, 741–752. [DOI] [PubMed] [Google Scholar]
- 29. Bjorklund G., Aaseth J., Dadar M. and Chirumbolo S. (2019) Molecular targets in Alzheimer’s disease. Mol Neurobiol, in press. [DOI] [PubMed] [Google Scholar]
- 30. Li H., Liu C.C., Zheng H. and Huang T.Y. (2018) Amyloid, tau, pathogen infection and antimicrobial protection in Alzheimer’s disease -conformist, nonconformist, and realistic prospects for AD pathogenesis. Transl Neurodegener, 7, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sharma D., Rajasekaran S. and Pathak S. (2014) An experimental comparison of PMSprune and other algorithms for motif search. Int J Bioinform Res Appl, 10, 559–573. [DOI] [PubMed] [Google Scholar]
- 32. He Y.F., Li B.Z., Li Z., Liu P., Wang Y., Tang Q., Ding J., Jia Y., Chen Z. and Li L. (2011) Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science, 333, 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ito S., Shen L., Dai Q., Wu S.C., Collins L.B., Swenberg J.A., He C. and Zhang Y. (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science, 333, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maiti A. and Drohat A.C. (2011) Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem, 286, 35334–35338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Szulwach K.E., Li X., Li Y., Song C.X., Wu H., Dai Q., Irier H., Upadhyay A.K., Gearing M. and Levey A.I. (2011) 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci, 14, 1607–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Spruijt C.G., Gnerlich F., Smits A.H., Pfaffeneder T., Jansen P.W., Bauer C., Munzel M., Wagner M., Muller M. and Khan F. (2013) Dynamic readers for 5-(hydroxy) methylcytosine and its oxidized derivatives. Cell, 152, 1146–1159. [DOI] [PubMed] [Google Scholar]
- 37. Song C.X., Szulwach K.E., Fu Y., Dai Q., Yi C., Li X., Li Y., Chen C.H., Zhang W. and Jian X. (2011) Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol, 29, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cheng Y., Bernstein A., Chen D. and Jin P. (2015) 5-Hydroxymethylcytosine: a new player in brain disorders? Exp Neurol, 268, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sanchez-Mut J.V., Aso E., Heyn H., Matsuda T., Bock C., Ferrer I. and Esteller M. (2014) Promoter hypermethylation of the phosphatase DUSP22 mediates PKA-dependent TAU phosphorylation and CREB activation in Alzheimer’s disease. Hippocampus, 24, 363–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chibnik L.B., Yu L., Eaton M.L., Srivastava G., Schneider J.A., Kellis M., Bennett D.A. and De Jager P.L. (2015) Alzheimer’s loci: epigenetic associations and interaction with genetic factors. Ann Clin Transl Neurol, 2, 636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu L., Chibnik L.B., Srivastava G.P., Pochet N., Yang J., Xu J., Kozubek J., Obholzer N., Leurgans S.E. and Schneider J.A. (2015) Association of Brain DNA methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with pathological diagnosis of Alzheimer disease. JAMA Neurol, 72, 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Foraker J., Millard S.P., Leong L., Thomson Z., Chen S., Keene C.D., Bekris L.M. and Yu C.E. (2015) The APOE gene is differentially methylated in Alzheimer’s disease. J Alzheimer’s Dis, 48, 745–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Francis R., McGrath G., Zhang J., Ruddy D.A., Sym M., Apfeld J., Nicoll M., Maxwell M., Hai B. and Ellis M.C. (2002) aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev Cell, 3, 85–97. [DOI] [PubMed] [Google Scholar]
- 44. Benito E., Urbanke H., Ramachandran B., Barth J., Halder R., Awasthi A., Jain G., Capece V., Burkhardt S. and Navarro-Sala M. (2015) HDAC inhibitor-dependent transcriptome and memory reinstatement in cognitive decline models. J Clin Invest, 125, 3572–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thomas T., Miners S. and Love S. (2015) Post-mortem assessment of hypoperfusion of cerebral cortex in Alzheimer’s disease and vascular dementia. Brain, 138, 1059–1069. [DOI] [PubMed] [Google Scholar]
- 46. Ke Q. and Costa M. (2006) Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol, 70, 1469–1480. [DOI] [PubMed] [Google Scholar]
- 47. Soucek T., Cumming R., Dargusch R., Maher P. and Schubert D. (2003) The regulation of glucose metabolism by HIF-1 mediates a neuroprotective response to amyloid beta peptide. Neuron, 39, 43–56. [DOI] [PubMed] [Google Scholar]
- 48. Liu Y., Liu F., Grundke-Iqbal I., Iqbal K. and Gong C.X. (2009) Brain glucose transporters, O-GlcNAcylation and phosphorylation of tau in diabetes and Alzheimer’s disease. J Neurochem, 111, 242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Correia S.C. and Moreira P.I. (2010) Hypoxia-inducible factor 1: a new hope to counteract neurodegeneration? J Neurochem, 112, 1–12. [DOI] [PubMed] [Google Scholar]
- 50. Sun X., He G., Qing H., Zhou W., Dobie F., Cai F., Staufenbiel M., Huang L.E. and Song W. (2006) Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci U S A, 103, 18727–18732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jaffe A.E. and Irizarry R.A. (2014) Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol, 15, R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Iwata A., Nagata K., Hatsuta H., Takuma H., Bundo M., Iwamoto K., Tamaoka A., Murayama S., Saido T. and Tsuji S. (2014) Altered CpG methylation in sporadic Alzheimer's disease is associated with APP and MAPT dysregulation. Hum Mol Genet, 23, 648–656. [DOI] [PubMed] [Google Scholar]
- 53. Smith A.R., Smith R.G., Condliffe D., Hannon E., Schalkwyk L., Mill J. and Lunnon K. (2016) Increased DNA methylation near TREM2 is consistently seen in the superior temporal gyrus in Alzheimer’s disease brain. Neurobiol Aging, 47, 35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nicolia V., Cavallaro R.A., Lopez-Gonzalez I., Maccarrone M., Scarpa S., Ferrer I. and Fuso A. (2017) DNA methylation profiles of selected pro-inflammatory cytokines in Alzheimer disease. J Neuropath Exp Neurol, 76, 27–31. [DOI] [PubMed] [Google Scholar]
- 55. Nicolia V., Ciraci V., Cavallaro R.A., Ferrer I., Scarpa S. and Fuso A. (2017) GSK3beta 5′-flanking DNA methylation and expression in Alzheimer’s disease patients. Curr Alzheimer Res, 14, 753–759. [DOI] [PubMed] [Google Scholar]
- 56. Mendioroz M., Celarain N., Altuna M., Sanchez-Ruiz de Gordoa J., Zelaya M.V., Roldan M., Rubio I., Larumbe R., Erro M.E. and Mendez I. (2016) CRTC1 gene is differentially methylated in the human hippocampus in Alzheimer’s disease. Alzheimers Res Ther, 8, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Weber M., Davies J.J., Wittig D., Oakeley E.J., Haase M., Lam W.L. and Schubeler D. (2005) Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet, 37, 853–862. [DOI] [PubMed] [Google Scholar]
- 58. Blanch M., Mosquera J.L., Ansoleaga B., Ferrer I. and Barrachina M. (2016) Altered mitochondrial DNA methylation pattern in Alzheimer disease-related pathology and in Parkinson disease. Am J Path, 186, 385–397. [DOI] [PubMed] [Google Scholar]
- 59. Cronin P., McCarthy M.J., Lim A.S.P., Salmon D.P., Galasko D., Masliah E., De Jager P.L., Bennett D.A. and Desplats P. (2017) Circadian alterations during early stages of Alzheimer's disease are associated with aberrant cycles of DNA methylation in BMAL1. Alzheimers Dementia, 13, 689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Villela D., Ramalho R.F., Silva A.R., Brentani H., Suemoto C.K., Pasqualucci C.A., Grinberg L.T., Krepischi A.C. and Rosenberg C. (2016) Differential DNA methylation of microRNA genes in temporal cortex from Alzheimer’s disease individuals. Neural Plast, 2016, 2584940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McKhann G.M., Knopman D.S., Chertkow H., Hyman B.T., Jack C.R.,.J., Kawas C.H., Klunk W.E., Koroshetz W.J., Manly J.J. and Mayeux R. (2011) The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dementia, 7, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Saldanha J., Gannicliffe A. and Itzhaki R.F. (1984) An improved method for preparing DNA from human brain. J Neurosci Methods, 11, 275–279. [DOI] [PubMed] [Google Scholar]
- 63. Rio D.C., Ares M., Jr., Hannon G.J. and Nilsen T.W. (2010) Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb Protoc, 2010, pdb.prot5439. [DOI] [PubMed] [Google Scholar]
- 64. Borgel J., Guibert S. and Weber M. (2012) Methylated DNA immunoprecipitation (MeDIP) from low amounts of cells. Methods Mol Biol, 925, 149–158. [DOI] [PubMed] [Google Scholar]
- 65. Langmead B., Trapnell C., Pop M. and Salzberg S.L. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol, 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H. and Glass C.K. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell, 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Feng H., Conneely K.N. and Wu H. (2014) A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res, 42, e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. McDaid A.F., Joshi P.K., Porcu E., Komljenovic A., Li H., Sorrentino V., Litovchenko M., Bevers R.P.J., Rueger S. and Reymond A. (2017) Bayesian association scan reveals loci associated with human lifespan and linked biomarkers. Nat Commun, 8, 15842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M. and Li W. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol, 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Muino J.M., Kaufmann K., Ham R.C., Angenent G.C. and Krajewski P. (2011) ChIP-seq analysis in R (CSAR): an R package for the statistical detection of protein-bound genomic regions. Plant Methods, 7, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kuan P.F., Chung D., Pan G., Thomson J.A., Stewart R. and Keles S. (2011) A statistical framework for the analysis of ChIP-Seq data. J Am Stat Assoc, 106, 891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yu G., Wang L.G. and He Q.Y. (2015) ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics, 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- 73. Gel B., Diez-Villanueva A., Serra E., Buschbeck M., Peinado M.A. and Malinverni R. (2016) regioneR: an R/Bioconductor package for the association analysis of genomic regions based on permutation tests. Bioinformatics, 32, 289–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shen-Orr S.S., Tibshirani R., Khatri P., Bodian D.L., Staedtler F., Perry N.M., Hastie T., Sarwal M.M., Davis M.M. and Butte A.J. (2010) Cell type-specific gene expression differences in complex tissues. Nat Methods, 7, 287–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M. and Bejerano G. (2010) GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol, 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cavalcante R.G. and Sartor M.A. (2017) annotatr: genomic regions in context. Bioinformatics (Oxford, England), 33, 2381–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.