

Abstract
The incidence of stroke and dementia are diverging across the world, rising for those in low- and middle-income countries and falling in those in high-income countries. This suggests that whatever factors cause these trends are potentially modifiable. At the population level, neurological disorders as a group account for the largest proportion of disability-adjusted life years globally (10%). Among neurological disorders, stroke (42%) and dementia (10%) dominate. Stroke and dementia confer risks for each other and share some of the same, largely modifiable, risk and protective factors. In principle, 90% of strokes and 35% of dementias have been estimated to be preventable. Because a stroke doubles the chance of developing dementia and stroke is more common than dementia, more than a third of dementias could be prevented by preventing stroke. Developments at the pathological, pathophysiological, and clinical level also point to new directions. Growing understanding of brain pathophysiology has unveiled the reciprocal interaction of cerebrovascular disease and neurodegeneration identifying new therapeutic targets to include protection of the endothelium, the blood-brain barrier, and other components of the neurovascular unit. In addition, targeting amyloid angiopathy aspects of inflammation and genetic manipulation hold new testable promise. In the meantime, accumulating evidence suggests that whole populations experiencing improved education, and lower vascular risk factor profiles (e.g., reduced prevalence of smoking) and vascular disease, including stroke, have better cognitive function and lower dementia rates. At the individual levels, trials have demonstrated that anticoagulation of atrial fibrillation can reduce the risk of dementia by 48% and that systolic blood pressure lower than 140 mmHg may be better for the brain. Based on these considerations, the World Stroke Organization has issued a proclamation, endorsed by all the major international organizations focused on global brain and cardiovascular health, calling for the joint prevention of stroke and dementia. This article summarizes the evidence for translation into action.
Keywords: Stroke, Dementia, Prevention, Risk factor reduction, Policy, Cognitive impairment, Alzheimer’s disease, Neurovascular unit, Treatment, Resilience
1. Background
Worldwide, neurological disorders represent the leading causes of disability-adjusted life years (DALYs) accounting for 10% of the global burden of disease. Nearly half of DALYs from neurological disorders arise from stroke (42%) and 10% from dementia [1]. Because stroke doubles the chances of developing dementia [2] and 90% of strokes are preventable [3,4], mitigating stroke risk at the population level, and for those at increased individual risk, provides the most immediate and promising opportunity to reduce the rates of both stroke and dementia through the same international and national policies. However, if we are to succeed, radical new approaches are needed moving well beyond current paradigms [5]. The most pervasive is our categorical, binary “you have it or you don’t” approach to the dementia syndrome, often described wrongly as “a disease”. Neuro-pathological studies point to multiple and interactive pathologies to account for most cases of dementia [6,7], of which the vascular component remains the only treatable and preventable one. All major dementias have a vascular component, ranging from 61% in frontotemporal dementia to 80% in Alzheimer’s disease (AD), the presence of a vascular component doubling the likelihood that the neurodegenerative pathology will have manifested as dementia in life [8].
To speed progress, we need to apply multidimensional and dynamic approaches to understand pathophysiological interactions against the changing background of aging. This article documents the relationships between stroke and dementia, with an emphasis on the current or potentially treatable and preventable components.
1.1. The bases for action
Stroke and dementia often co-occur and pose risks for each other [9]. Vascular and neurodegenerative pathologies interact additionally and also synergistically, even in asymptomatic individuals [10]. There is circumstantial evidence of substantial drops of dementia (approximately 25% in age-specific prevalence) in high-income countries in which stroke incidence and mortality has declined substantially [11].
Epidemiological evidence suggests that preventing stroke could also prevent some dementias [12,13]. Treating atrial fibrillation with anticoagulants not only prevents stroke but is also associated with substantial reduction in dementia risk compared with no anticoagulant treatment [14]. A multifactorial data-driven analysis using the Alzheimer’s Disease Neuroimaging Initiative data suggested that the first step that leads to late-onset AD is vascular dysregulation [15].
The World Health Summit in 2017 and 2018 has offered a unique platform to highlight the opportunities to prevent stroke together. The Summit attracts academics, policymakers, heads of pharmaceutical and device industries, and entrepreneurs. The World Health Summit’s hallmark is the interdisciplinary and the multistakeholder participation and interaction it provides, including representation from low- and middle-income countries and a focus on the UN’s sustainable developmental goals under the leadership of the World Health Organization.
Preparing for an aging population is vital to the achievement of the integrated 2030 Agenda, with aging cutting across the goals on poverty eradication, good health, gender equality, economic growth, and decent work, reduced inequalities and sustainable cities. This underlines the importance of the Berlin Manifesto by the World Stroke Organization (WSO) and endorsing organizations, which sets forth a new paradigm for research and evidence in the area of stroke and dementia, and therefore a landmark for a life-course approach to healthy aging.
Before the 2018 Summit, a number of individuals working on understanding the interaction of stroke and dementia and trying to prevent them together were able to gather in Berlin on October 13-14, 2018 to highlight what is known and what needs to be done next. The others, who could not attend, have also contributed to this article by providing summaries of their research.
In principle, 90% of strokes and approximately 35% of dementias are preventable [3,4,16]. However, stroke and dementia statistics are typically considered in isolation. The fact that stroke doubles the chance of developing dementia [2] has not usually been considered in calculating the percentage of potentially preventable dementias, suggesting that their actual proportion could be higher than the estimated one third [16].
The growing evidence has led the WSO to update a proclamation, now calling for the joint prevention of stroke and dementia and endorsed by 23 international, regional, and national organizations (Fig. 1) [17].
Fig. 1.

Logos of the endorsing organizations [17].
Four of the endorsing organizations, the WSO, World Heart Federation, World Hypertension League, and the European Society of Hypertension have formed a further alliance for vascular health as a step toward implementing the Proclamation [18].
2. Epidemiology
2.1. Diverging trends in dementia
Recent epidemiological trends in global burden of dementia suggest that the prevalence and incidence of dementia in the developed world is stable and even declining. This change has been attributed to improved risk factor control, a declining incidence of stroke, and improvements in societal conditions, education, and health care. However, the scenario in the Global South is quite different. It is increasingly evident that populations in low- and middle-income countries (as defined by the World Bank [19] using gross national income per capita [in US dollars] as those countries with a gross national income of $12,055 or less) are witnessing a rising burden of dementia [1]. Societies in low- and middle-income countries are in a state of transition, with a shift in demography and urbanization. Developing societies also substantially differ from Western countries in their attitudes toward dementia, and their risk factor profiles, marked by lower education and socioeconomic status, a higher cardiovascular disease burden, and greater genetic variability. Care provision also differs markedly in these resource-limited countries. Insights from understanding diverging trends among various countries provide a unique opportunity to reduce the global burden of dementia. Vascular contribution to dementia is significantly higher in developing countries than in the West because of the increasing incidence of vascular risk factors, such as diabetes mellitus, smoking, metabolic syndrome, and hypertension [20]. Preventing stroke and reducing vascular risk factor burden will therefore be an effective strategy to reduce the dementia burden in developing countries. Furthermore, emerging evidence emphasizes the interactions between different risk and protective life-course factors in diverse genetic, environmental, and cultural contexts, contributing to the complexity of dementia risk and resilience. While education is a very important protective factor, it’s interaction with variables such as linguistic diversity and occupation/employment influence its effect [21,22]. Therefore, the investigation of dementia in diverse settings, including a more global perspective, is crucial for a comprehensive understanding of the condition and the identification of novel solutions [23].
2.2. Population studies and their contribution to understanding of the role of vascular health in brain aging
Population studies conducted over the last 40 years have contributed to our understanding of the clinical dementia syndrome and cognitive decline. This includes both traditional descriptive epidemiological measurement within older people over time and investigating risk and relationships to the natural history of the syndrome with vascular phenotypes. Population studies have varied in their intensity of vascular biomarker examination including the range of markers that have been measured and whether these are able to differentiate so-called ‘normal’ aging and pathological aging related to dementia in later life. It is clear that dementia can change in populations with a reduction observed in many high-income countries in the rates of either or both incidence and prevalence [24]. This is in the context of quite dramatic changes in these populations in the rates of stroke and vascular disease, estimated to be due to half primary prevention and half secondary prevention. Such findings suggest that societies should urgently capitalize on knowledge about how to improve health profiles for the brain at all ages in all global contexts and invest in new types of research for populations (particularly in low- and middle-income countries) and dementia risk reduction. Indeed, reduction in the number and severity of people with cognitive impairment and dementia will have personal, societal, and economic benefits (Fig. 2) [11,25–28].
Fig. 2.
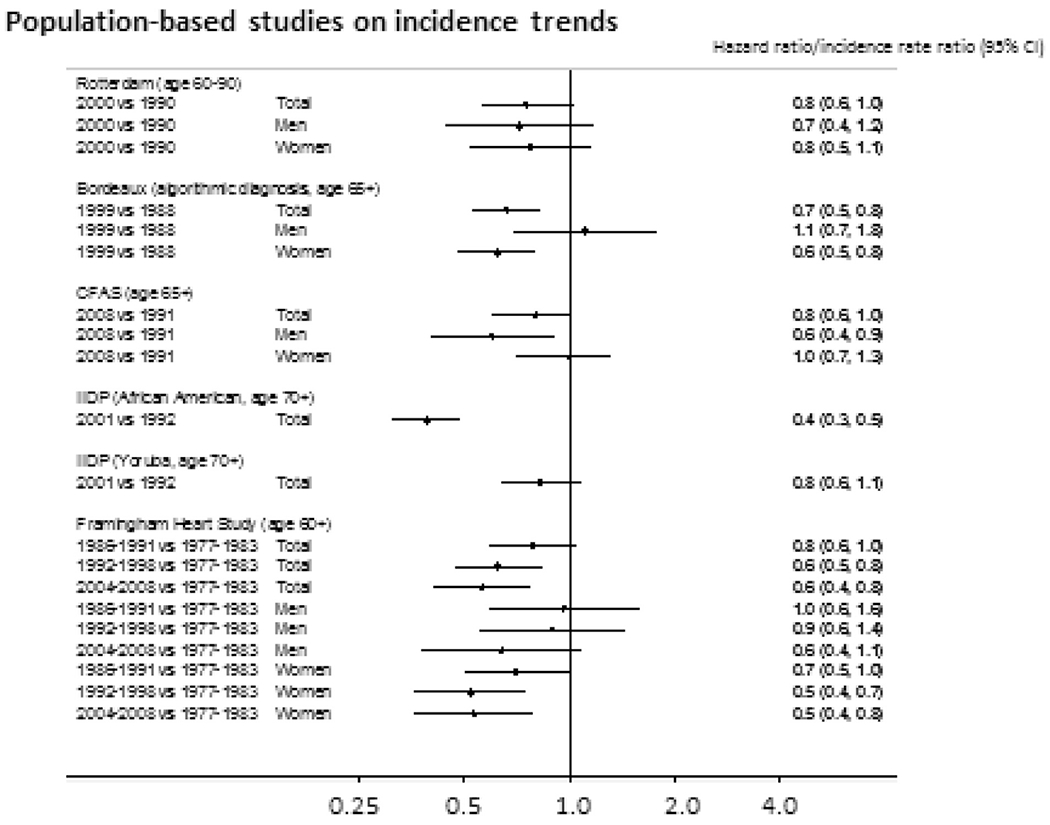
Results from population-based studies on incidence trends: Hazard ratio and incidence rate ratio from five different studies from across the world [11].
3. Pathophysiology
Strokes can lead to (or aggravate preexisting) cognitive impairment and even lead to poststroke dementia. In addition, covert stroke and silent brain ischemia contributes to cognitive impairment and dementia. In addition to poststroke and vascular dementia, however, it is clearly recognized that vascular mechanisms also contribute to neurodegeneration, dementia and AD: established risk factors for vascular disease and stroke are also associated with Alzheimer’s dementia, and vascular dysfunction is an early pathophysiological event in AD. In the future, the concept of the neurovascular unit will help to disentangle the joint mechanisms of microvascular dysfunction and neurodegeneration as a common pathway to stroke and dementia.
3.1. Blood-brain barrier: structure, function, and role in neurodegenerative disorders and AD
Blood vessels in the brain are organized with impressive precision, patterned in parallel with the major brain circuits tasked with sensation, memory, and motion. This tight interrelationship may reflect key functional roles of the vasculature in normal function of neurons in the healthy brain, during aging and in neurodegenerative disorders such as AD [29–31]. Recent studies suggest that changes in the blood-brain barrier (BBB) integrity and cerebral blood flow deficits may occur early in the AD continuum and therefore may precede changes in amyloid-β (Aβ) and tau biomarkers, neurodegeneration, and cognitive impairment [29]. (see Fig. 3, revised Jack model of Alzheimer’s disease biomarkers to include role of brain vasculature) [31] In addition, the link between BBB breakdown and neurodegeneration in humans and animal models has been well documented in rare human monogenic neurological disorders with the primary genetic defect in nonneuronal cells of the BBB [32].
Fig. 3.
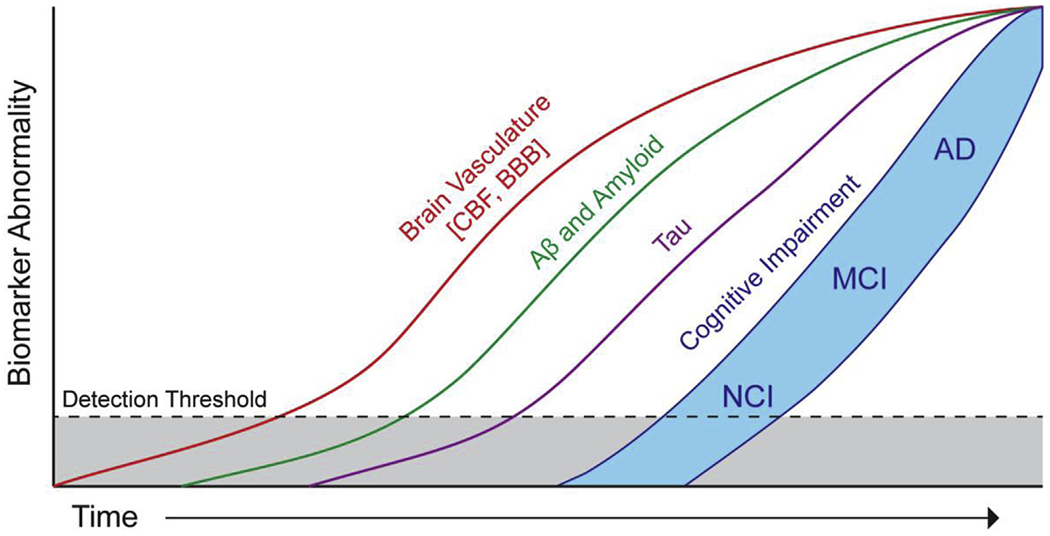
Hypothetical updated Jack model of Alzheimer’s disease biomarkers to include the role of brain vasculature. Hypothetical model of AD biomarker changes illustrating that early cerebral blood flow and blood-brain barrier biomarkers and vascular dysfunction may contribute to initial stages of AD pathophysiological progression from NCI to MCI to AD, which is followed by cerebrospinal fluid and brain changes in Aβ and amyloid, and tau biomarkers. All biomarker curves converge at the top right-hand corner of the plot, that is, the point of maximum abnormality. The horizontal axis of disease progression is expressed as time. Cognitive response is illustrated as a zone (blue filled area) with low- and high-risk borders. Subjects with high risk of AD-related cognitive impairment are shown with a cognitive response curve that is shifted to the left. In contrast, the cognitive response curve is shifted to the right in subjects with a protective genetic profile, high cognitive reserve, and the absence of comorbid brain pathologies [31]. Abbreviations: AD, Alzheimer’s disease; CBF, cerebral blood flow; BBB, blood-brain barrier; MCI, mild cognitive impairment; NCI, no cognitive impairment.
In addition, genes underlying inheritance or increased susceptibility of familial forms of AD, and some other neurodegenerative disorders, such as amyotrophic lateral sclerosis, Huntington’s and Parkinson’s disease, are associated with BBB breakdown and neurovascular dysfunction [29,31].
BBB breakdown is an independent early biomarker of human cognitive dysfunction [33]. Using two independent biomarkers of BBB breakdown—(1) dynamic imaging analysis of regional BBB permeability in the living human brain and (2) evaluation of brain capillary damage using a novel cerebrospinal fluid (CSF) biomarker of BBB-associated mural cells—a recent study has shown that individuals with clinical dementia rating score of 0.5 (vs. cognitive controls) have increased BBB permeability to gadolinium-based contrast agent in the hippocampus but not in other brain regions [33]. This is consistent with previous findings showing that BBB breakdown during normal aging and in individuals with mild cognitive impairment starts in the hippocampus [34]. Interestingly, individuals with a clinical dementia ratingscore of 0.5 compared with controls, exhibited regional BBB hippocampal and parahippocampal breakdown regardless of whether they were positive or negative for CSF Aβ1-42 and pTau, that is, classical AD biomarkers [33]. These findings were replicated when cognitive dysfunction was assessed in different domains including memory, attention/executive function, and language. This study also found increased CSF biomarkers suggesting progressive damage of pericytes with more advanced cognitive impairment regardless of CSF Aβ1-42 or pTau status [33]. Collectively, these findings suggest that BBB breakdown is an early biomarker of human cognitive dysfunction independent of Alzheimer’s Aβ and tau biomarkers [33].
Findings from animal studies suggest that a number of mechanisms may link neuronal injury to the presence of BBB breakdown as we recently reviewed [32] and as shown in Fig. 4 (BBB-mediated neurodegeneration) [32]. In this article, some examples of how BBB breakdown can lead to vascular-mediated neuronal degeneration, injury, and loss are illustrated (1): one possible pathway involves red blood cell-derived neurotoxic hemoglobin and free iron (Fe2+) and both can lead to production of reactive oxygen species causing oxidant neuronal injury (2); blood-derived proteins such as fibrinogen and thrombin could be directly or indirectly neurotoxic by activating microglia (see point #3, below), whereas plasminogen when converted into plasmin in the brain tissue leads to degradation of extracellular matrix causing detachment of neurons and cell death (3); fibrinogen activates microglia, which promotes neuroinflammation and demyelination (4); albumin leads to the development of vasogenic edema, capillary hypoperfusion, and low oxygen tissue levels (5); BBB breakdown could be associated with a loss of immune privilege, resulting in development of anti-brain antibodies against different cell components in neurons; and (6) finally, loss of BBB, endothelial and mural cells may lead to a loss of important neurotrophic support. An additional or alternative potential mechanism, not shown in Fig. 4 [32], is the calcium ion signaling path (aka “Calcium Hypothesis”), which is a putative cascade of molecular events that may account for more direct causal relationship between early cerebral blood flow deficits and subsequent decrements in neural functioning and connectivity and behavioral problems or symptoms.
Fig. 4.
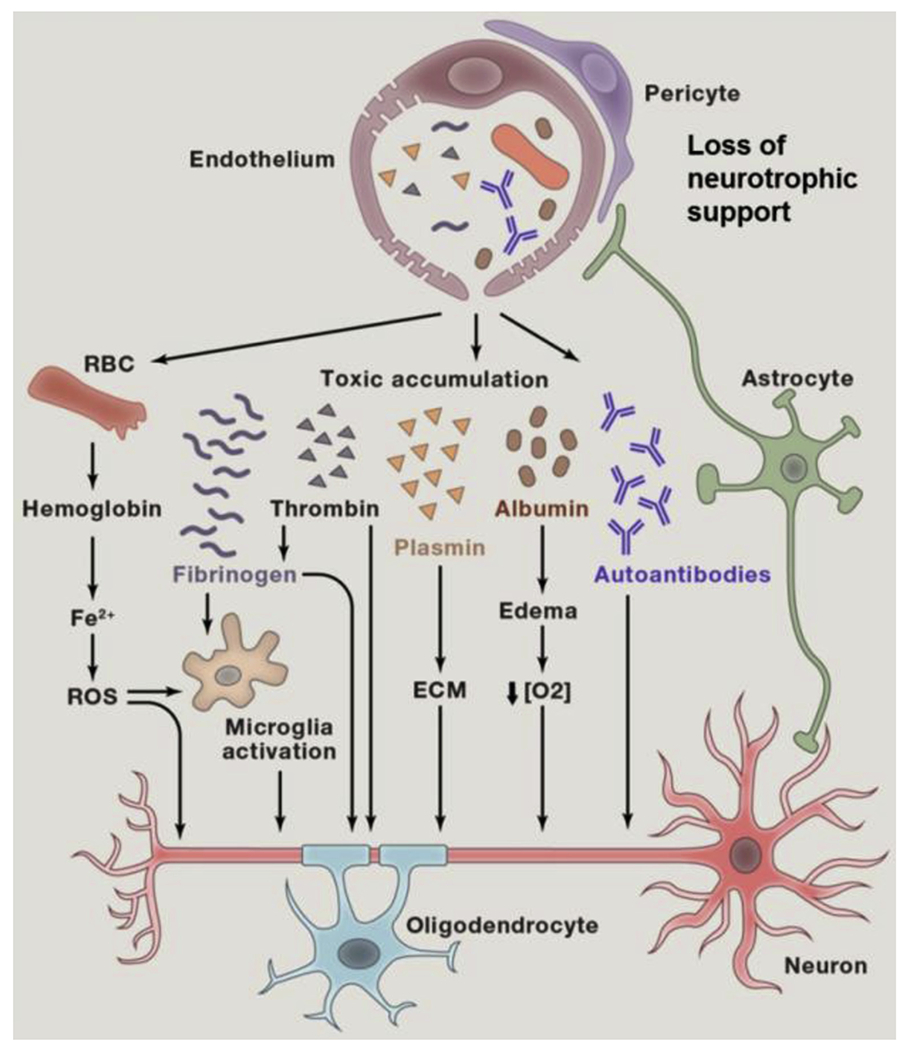
Blood-brain barrier–mediated neurodegeneration [32].
Recent studies in a new model of small vessel disease (SVD) using pericyte-deficient mice [35] have shown that BBB breakdown affects early white matter preceding changes in cognitive function and neuronal loss [35]. Particularly, blood-derived fibrinogen and its polymerized form fibrin, which is converted in tissue from fibrinogen by thrombin, both lead to autophagy of oligodendrocytes and pericytes, resulting in a progressive loss of both cell types and loss of myelin and axons [35]. In short, these studies provide compelling evidence for the hypothesis that functional changes in brain vasculature play an important contributory role in the pathogenesis of neurodegenerative disorders. Thus, there is the promising potential for developing novel therapeutic strategies based on the idea that chronic or sustain hypoglycemic conditions play a crucial role in the etiologies of neurodegenerative disorders, dementia, and AD.
However, the hypothesis that early deficits in cerebral blood flow (due to small vessel pathologies or breakdown in the integrity of BBB), which trigger the cascade of molecular signals that subsequently may mediate cognitive impairment has not been tested in prospective longitudinal studies. The prospects of developing future interventions, based on the putative hypothesis that a causal relationship exists between early brain metabolic compromise (e.g., vascular damage) and subsequent downstream cognitive impairment, will require a more detailed account of the signaling paths. For example, how cascade of molecular mechanisms initiated the breakdown in the integrity BBB affects the functioning of a neuron or how these early upstream events lead to the selectivity-specificity in the injury of neurons or neural nets.
3.2. Ischemia as an accelerated form of neurodegeneration
The proposition that brain vascular changes might play a vital role in the etiology of neurodegeneration is one of the earliest theories on the origins of this condition. The circumstantial evidence for some form of mechanistic links between cognition/dementia and aberrant changes in the structure-function of cerebral microvasculature, particularly those that adversely affect the transport of glucose-oxygen or otherwise create chronic deficiencies in perfusion, has been steadily gaining strength. Increasing numbers of epidemiological findings are providing further corroborating support for the importance of a possible mechanistic relationship. However, the exact mechanistic links are not clear, and the notion that early brain vascular changes might be an important preventable target has not been validated in large-scale prospective population-based longitudinal studies.
The possibility of neurophysiological links between early vascular changes (i.e., regional hypoglycemic events) and decrements in neuronal functions (e.g., loss of synapse/dendrite pruning) has been outlined as a key testable postulate for the “Calcium Hypothesis of Brain Aging and Dementia” [36–38]. The hypothesis proposes thata transient large increases of Ca2+ (e.g., as in stroke or traumatic brain injury) produce equivalent neuronal damage as a small but prolonged dishomeostasis in (Ca2+). The essence of this postulate’s claim (prediction) is that the actual neurodegenerative process due to stroke is the same as those due to other triggers that produce similar disruptive process. This postulate, yet to be validated, recognizes that both small sustained and large rapid-transient increases of Ca2+ can each generate pathogenic conditions that contribute to aging-related deficits and pathologies associated with neurodegeneration. Each condition may recruit different compensatory mechanisms and homeostatic responses, but the net effect is a brain more vulnerable to dementia or some form of chronic brain disorder.
The original formulation of the “Calcium Hypothesis of Alzheimer’s disease and Brain Aging” in the late 1970s was influenced by (1) studies showing age-related changes in calcium ion signaling (e.g., prolonged hyperpolarization, changes in LTP) and (2) concepts derived from “ischemic cascade” about vital role of calcium ion signaling in apoptosis. At the time, the strategic intent was to propose a unifying hypothesis to account for both age-related changes and disease-related decrements in neural functioning. As an integral part of the early struggles to develop an extramural research program on the neurobiology of aging and dementia/AD at the National Institute on Aging and National Institutes of Health, the other rationale for the Calcium Hypothesis was to promote a shift in the focus of National Institute on Aging-funded research away from the nearly exclusive domain of descriptive studies toward molecular mechanism of brain aging. Since the 1980s, the hypothesis has been updated several times [37,38]. The key postulates of the Calcium Hypothesis propose the following:
Sustained dyshomeostasis of cytosol calcium ion concentration [Ca2+]I (beyond normal modulations that occur during the typical depolarization-repolarization cycles of a healthy neuron) represents the pivotal precursor for neuronal dysfunctions (e.g., synapse loss, dendrite pruning, loss of network connectivity or apoptosis) associated with dementia, AD, and aging.
The breakdown of Ca2+ ion signaling system within a neuron is the common mechanistic thread for a number of neurodegenerative disorders. Prolonged bouts of failure in an array of cellular components/mechanism to maintain calcium ion concentration [Ca2+]I at 10-9 M represent the final common path for malfunctioning of a neuron and catastrophic failure of a neural network.
The concept of the final common path suggest the existence of several potential upstream antecedent conditions or precursors that could trigger the deterioration in Ca2+ ion signaling system or chronic dyshomeostasis [Ca2+]I. For example, small vessel pathology/hypoglycemia/metabolic or energy crisis/ischemic cascade; free radical oxidative damage; excessive repetitive stimulation (e.g., epilepsy/glutamate excitotoxicity); exogenous toxins (e.g., amyloid proteins; mitochondrial defect); membrane alteration that influence the functioning of transmembrane proteins (e.g., receptors, ion channels, amyloid precursor protein [APP] etc).
The optimal functioning of a neuron requires tight regulation of a complex energy-dependent system to maintain the cytosol concentration of [Ca2+]I at 10-9 M, which requires energy consuming work against a steep gradient of 1000 fold difference between the extracellular Ca2+ concentration of 10-3M and 10-9 M within the cell. Some of the main components of the intricate cellular machinery to sustain the homeostasis of cytosol [Ca2+]I include Ca2+ permeable voltage or ligand-gated ion channels, Na+/Ca2+ exchanger, Ca2+ buffers, calmodulin–CaM, Calbindin, endoplasmic reticulum, mitochondria, buffers, adenosine triphosphate (ATP)-dependent ion pumps, ATPase or other regulatory mechanisms. The early precursor event signals, such as those mentioned previously, ischemic cascade or metabolic oxidative or proteotoxic stressor, set into motion the complex interactions of intercellular components leading to dyshomeostasis cytosol [Ca2+]I. The multifaceted nature of Ca2+ signal regulation requires the use of system approach both to account for the interrelationships among key components and for developing novel paradigms for discovery of therapeutic targets.
The downstream consequences of dyshomeostasis cytosol [Ca2+]I (i.e., deterioration of Ca2+ handling systems of a neuron) include changes in internal effector signals from second messengers (e.g., CaM, A-Kinase/C-Kinase/G-Kinase TnC etc.) and cellular responses mediated by third messengers or transcription factors (e.g., c-fos/c-jun/RNA/Telomerase/Protein synthesis), which promote either regeneration (growth, protein synthesis, synapse formation, memory) or degeneration (synapse loss, pruning dendrite arbor, apoptosis)
The downstream cellular responses-mechanisms prompted dyshomeostasis cytosol [Ca2+]I represent a continuum of molecular processes (a common thread) associated with the developing nervous system, aging-related changes, and neurodegeneration. The speculative claim of the hypothesis, which needs to be tested, asserts that the plasticity of neuroarchitecture is regulated by a functional equilibrium between Ca2+-mediated molecular mechanisms promoting growth/regeneration and those Ca2+-mediated processes that control regression/degeneration; thus, degeneration merely reflects a shift in equilibrium.
A key claim, which also needs to be validated, asserts that the same or similar molecular events that encompass the ischemic cascade could also be involved over an extended time scale and account for the sequence of cellular mechanisms that underlie a number of neurodegenerative disorders, dementia, and AD. The similarity or the differences between the two processes mediating neural injury or dysfunction is explained on the basis of the relationship between the amount of the perturbation in the Δ [Ca2+] I and the duration (Δ T) of the deregulation in the calcium homeostasis is a constant:
This relationship suggests (Fig. 5) that large change in Δ [Ca2+] I over a short period (ΔT) (e.g., stroke) will result in similar cellular damage as will a small change in Δ [Ca2+] I that is sustained over a prolonged period (Δ T) (e.g., dementia):
Fig. 5.
The crucial requirement for optimal functioning, in fact the viability, of a neuron is the requirement to a constant supply of energy. Any deficiencies in perfusion that deprives the adequate supply of oxygen and glucose to the neuron for more than 60–90 seconds will initiate an ischemic cascade leading to impaired functioning. Deprivation that persists beyond three hours will most likely result in irreversible injury or possibly apoptosis [39,40].
One of the fundamental premises of the “Calcium Hypothesis” is that an unstable energy supply or impaired system of energy metabolism may underlie several neurodegenerative diseases. The hypothesis accounts for several explanations for diminution of glucose supply to the neuron. For example, breakdown of BBB, atherosclerosis may compromise blood flow supply by narrowing the lumen of blood vessels leading to a reduction in glucose supply of blood flow. Another possible reason might be deficiencies in glucose transporter protein (e.g., GLUT3).
Regardless of the precursor, whether due to blocked blood vessels or some other mechanism for hindering the supply of glucose, low energy levels in the neuron, results in a shift to anaerobic metabolism; thus, the release of lactic acid, which disrupts the normal acid-base balance within the neuron. Low energy levels also lead to failure in the production of high energy phosphate compounds such as ATP leading to a cascade of further downstream failure of energy-dependent processes. The sequence of molecular mechanism that is detrimental to the optimal function of neuron may include the following:
Failure of ion-transport pumps which lead to depolarization that allows Ca2+ ions to flow into the neuron;
Failure of energy-dependent Ca2+ ion pumps to transport calcium out of the neuron leads to high level intracellular Ca2+ concentration;
The Ca2+ triggers the release of neurotransmitter glutamate, which stimulates AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors and Ca2+-permeable NMDA (N-methyl-D-aspartate receptor) receptors, thus allowing the entry of more Ca2+ into the neuron;
Excessive Ca2+ concentration in the neuron triggers the release of more glutamate and the process of excitotoxicity, which leads to the generation of harmful chemicals (e.g., free radicals, reactive O2 species, and calcium-dependent enzymes such as calpain, endonucleases, ATPases, and phospholipases);
As phospholipase breakdown the membrane, the neuron becomes more permeable to ions and harmful chemicals that flow into the cell;
Mitochondria break down releasing toxins and apoptotic factors into the cell;
The caspase-dependent apoptosis cascade is initiated causing cells to “commit suicide”;
If the cell dies through necrosis, it releases glutamate and toxic chemicals into the environment around it. Toxins poison nearby neurons, and glutamate can overexcite them;
If and when the brain is reperfused, a number of factors lead to reperfusion injury;
An inflammatory response is mounted, and phagocytic cells engulf damaged but still viable tissue;
A parsimonious strategy for comparing, contrasting, or integrating various models or putative mechanisms for linking early upstream events (e.g., breakdown of BBB with subsequent downstream clinical sequels) will require each alternative theory to account for a precise cascade of events (or mechanisms) that may influence the performance of a neuron/a network/a system.
The aforementioned sequence of interdependent molecular events describes the steps that lead to a catastrophic failure of a neuron. Slight variations or modifications of the time scale could account for the prolonged period of degenerative process in chronic brain disorders. In addition, cascade of molecular mechanism between vascular damage and neuronal dysfunction provide the rationale for the Berlin Manifesto for treating dementia by treating and preventing stroke.
The progressive decline in the optimal performance of a neural net is the vital feature of chronic brain disorders associated with neurodegeneration. Although various ideas on the origins of neurodegeneration may start with different assumptions, virtually all models of pathogenesis invoke some form of deficit in functional connectivity of a neural net. Persistent waning of synaptic transmission, continued pruning of dendritic arbors, loss of synapses, and decrements in repair and restoration are the most proximal events or abnormalities that underlie the expression of clinical features of virtually all neurodegenerative disorders.
The proposed mechanistic explanation provides the rationale for considering interventions developed for stroke and other cardiovascular disorders as potential promising interventions to prevent dementia.
3.3. Vascular dysregulation as a key triggering factor of late-onset AD
Big data-driven analysis for identifying multifactorial mechanisms in late-onset AD showed an early dysregulation of cerebral blood flow, which anteceded other brain alterations, including amyloid deposition, glucose hypometabolism, and neuronal hyperactivity. In a multimodal imaging study [41], it was observed that cerebrovascular dysregulations are not only concurrent with AD progression, but, importantly, they are also the most likely events triggering this neurodegenerative condition. In comparison, other biological alterations (e.g., amyloid deposition, hypometabolism) could not explain by themselves the multifactorial spatiotemporal alterations observed in late-onset AD. However, this study also highlighted the critical importance of considering multiple biological factors and their direct interactions (instead of only one factor, e.g., amyloid/vascular/metabolic) when studying this complex disorder.
Having identified causal drivers of neurodegenerative progression, the same group proceeded to identify effective interventions at the individual level. A novel concept of personalized therapeutic intervention fingerprint was proposed [42], predicting the effectiveness of potential interventions (e.g., vascular improvement, physical exercise, antiamyloid, antitau, and neurogenesis) for controlling a patient’s neurodegenerative evolution. The personalized therapeutic intervention fingerprint vastly outperformed cognitive/clinical evaluations when predicting individual blood gene expression profiles, revealing a strong relationship between predicted therapeutic needs and individual molecular profiles. Interestingly, this recent data-driven study showed that patients primarily needing a vascular-focused intervention have a small number of altered genes and molecular pathways in the blood while patients primarily needing amyloid-, tau-, or structural-focused interventions have an increasingly higher number of altered genes and molecular pathways. This finding may be reflective of the characteristic ordering on pathophysiological events associated with neurodegenerative progression. Vascular dysregulation may be starting when the patient’s molecular integrity damage is still at an early stage. Contrariwise, tau spreading and structural atrophy may start when a vast number of molecular functions are affected.
3.4. The neurovascular unit and the neurovascular biology of cognitive impairment
The brain lacks energy reserves and is vitally dependent on a continuous and well-regulated delivery of oxygen and glucose through the cerebral blood supply. Structural and functional alterations of cerebral blood vessels have emerged as a key correlate of conditions associated with cognitive impairment. The concept of “neurovascular unit” (NVU) was introduced in 2001 to highlight the close developmental, structural, and functional interactions between brain cells and cerebral blood vessels, and their coordinated reaction to injury [43]. Blood vessels in the brain are organized with surprising precision, patterned in parallel with the major brain circuits tasked with sensation, memory, and motion. This tight interrelationship reflects key functional roles of vasculature in normal brain function. Comprised of neurons, glia, perivascular cells, for example, perivascular macrophages and vascular cells (endothelium, smooth muscle cells, and pericytes), the NVU is responsible for matching the delivery of blood to the brain with local energy needs dictated by brain activity. The NVU is also involved in regulating the molecular exchange between BBB, clearance of metabolic byproducts through perivascular, paravascular, and transvascular routes, trafficking of immune cells, and trophic support to brain cells (matrix, growth factors, etc.) [43]. NVU dysfunction, affecting not just blood flow regulation but also other NVU functions, is central to the pathobiology of dementia and alters the homeostasis of the brain microenvironment in regions involved in cognition leading to cognitive impairment. Thus, neurovascular dysfunction is observed not only in vascular cognitive impairment, but also in AD, attesting to the significant overlap between these conditions [44]. In addition, major risk factors for cognitive impairment, such as hypertension, obesity, apolipoprotein 4 (ApoE4) genotype, and high salt intake, are also associated with neurovascular dysfunction [44]. Although activation of innate immunity, vascular oxidative stress, and inflammation are major pathogenic factors, the underlying molecular mechanisms and their link to cognitive impairment remain poorly understood and are a fruitful area of research with major diagnostic and therapeutic implications for both vascular and neurodegenerative dementias.
3.5. Amyloid pathology, neurovascular dysfunction, and cognitive impairment
Studies over the past decade have shown that Aβ has profound effects on all aspects of neurovascular function. Using transgenic mice that overexpress mutated forms of the APP it was shown that Aβ has profound cerebrovascular effects resulting in vasoconstriction, suppression of the coupling between neural activity and CBF, and impairment of endothelium-dependent vasodilation [45–47]. Importantly, Aβ alters the relationship between arterial pressure and CBF (cerebrovascular autoregulation), such that the brain becomes unable to maintain CBF during reduction in intravascular pressure [48]. As a result, APP mice have increased susceptibility to ischemic brain injury because of neurovascular dysfunction and inability to develop an effective compensatory collateral circulation [49]. Subsequent studies demonstrated that the cerebrovascular dysfunction is mediated by the interaction of Aβ with the innate immunity receptor CD36 leading to NOX2-dependent vascular oxidative stress [50,51]. The major cellular sources of radicals are perivascular macrophages, innate immune cells nestled around penetrating cerebral blood vessels including resistance arterioles. These cells express CD36 and, when engaged by Aβ, produce vascular oxidative stress via NOX2 [52].
The cellular mechanisms by which Aβ induces oxidative stress and alters endothelial function are related to peroxynitrite production leading to endothelial DNA damage with consequent activation of the DNA repair enzyme polyADP ribose polymerase. PolyADP ribose is cleaved into ADP ribose, which in turn activates transient receptor potential melastatin-2 channels, leading to endothelial calcium overload and endothelial dysfunction [53]. These observations have firmly established that Aβ, in addition to its well-known deleterious effects on neurons, also impairs cerebral perfusion, providing an added insult to the brain and amplifying its overall damaging effects on cognitive function [54].
The deleterious cerebrovascular effects of Aβ have been validated by recent clinical-pathological and imaging studies demonstrating that neurovascular dysfunction, ischemic lesions, and AD pathology most often coexist in the same brain, making “mixed dementia” the most common cause of cognitive impairment in the elderly [6]. Furthermore, as shown in the section on human studies, the early role of neurovascular dysfunction in the pathobiology of AD predicted a decade earlier by animal studies [54], was recently confirmed in patients with AD (Fig. 3) [31].
3.6. Inflammation mediates interaction between stroke and AD in preclinical models
Using complementary preclinical comorbid AD/stroke models, it has been demonstrated that in a transgenic mouse model of AD (APP23 mice), small striatal infarcts induced with the vasoconstrictor endothelin-1 resulted in enhanced levels of inflammatory cytokines and AD-like pathology [15]. Using a nontransgenic comorbid AD/stroke rat model, we demonstrated that the combination of AD-like pathology induced by intracerebroventricular injections of the toxic 25-35 fragment of Aβ peptide (Aβ25-35) and stroke (unilateral endothelin-1 injections in the striatum) in the rat also resulted in enhanced neuroinflammatory responses and AD-like pathology in the neocortex and hippocampus and were correlated with memory deficits detected using the Barnes circular platform test [55–59]. In addition, selective inhibition of the inflammatory transcription factor, NF-κB blocked pathological and memory deficits associated with the rat AD model [60]. This rat model of AD/stroke demonstrated progressive longer-term deterioration of cognitive function. In parallel, there was a progressive increase in the size of the stroke-induced infarct and the neuroinflammatory response with time in the combined models of stroke and AD [57]. Intriguingly, it has also been shown that producing a subcortical small infarct leads to extensive inflammation of the white matter in the rat [61]. Findings using these rat models of AD and stroke were the first documented experimental evidence demonstrating the reciprocal relationships between Aβ, stroke, and neuroinflammation.
More recent findings using a transgenic APP21 rat, with human Swedish/Indiana mutations of the APP [62], have revealed age-dependent increases in activated microglia, diffusely distributed within the white matter tracts [63] that correlate with executive dysfunction [64], both of which are exacerbated after a subcortical endothelin-1-induced stroke [65]. Using a positron emission tomography (PET) tracer for activated microglia, we also observed persistent expression of activated microglia in the comorbid AD/stroke transgenic rats [66]. Overall, preclinical findings demonstrate that interactions between Aβ, stroke and neuroinflammation, predominantly within the white matter tracts could be occurring within the brains of some patients after a stroke that may explain why they go on to develop dementia. Therefore, preventing stroke should prevent persistent white matter inflammation that could lead to enhanced degeneration and cognitive impairment.
3.7. Hypertension, dietary salt, neurovascular dysfunction, and dementia
Although well known to alter cerebral endothelial-dependent relaxation and autoregulation, it remains unclear how hypertension, a major cause of vascular cognitive impairment, influences brain function leading to cognitive dysfunction. Recent research demonstrated that hypertension disrupts the neurovascular unit resulting in a mismatch between the energy demands imposed by neural activity and the delivery of oxygen and glucose through blood flow. Using mouse models of hypertension, it has been demonstrated that hypertension blunts the increase in CBF induced by somatosensory stimulation (functional hyperemia) [67]. The effect is not related to suppression of the neural activity driving the vascular response but to a selective disruption of the ability of cerebral resistance vessels to dilate in response to neural stimuli. Subsequent investigations determined that the neurovascular coupling deficit was mediated by NOX2-derived reactive oxygen species [68], which in conjunction with endothelial nitric oxide, led to formation of peroxynitrite and suppression of neurovascular function [69]. The effect is sexually dimorphic, not being observed in young females, and exhibits a previously unrecognized dependence on the phase of the estrous cycle [70]. Remarkably, in a model of slowly developing hypertension produced by AngII, the suppression in functional hyperemia is observed before the elevation of blood pressure, unveiling a unique sensitivity of the neurovascular unit to the neurohumoral factors underlying hypertension [71]. Furthermore, a contribution of microglial cells in AngII-mediated vascular oxidative stress was discovered, highlighting the contribution of other cells in the neurovascular unit to the effects of this peptide on neurovascular regulation [72]. More recently, it was found that perivascular macrophages are the major source of cerebrovascular oxidative stress in mouse models of life-long hypertension [73]. Remarkably, blocking the production of radicals selectively in these cells prevented the neurovascular dysfunction and cognitive impairment produced by hypertension [73].
Dietary salt has emerged as a major independent risk factor for stroke and dementia. Recent experimental studies indicate that the gut-brain immune axis may be responsible for the link between high dietary salt and cognitive impairment [74]. A salt-rich diet in mice leads to a Th17 lymphocyte adaptive response in the gut which increases circulating levels of IL17. IL17 in turn acts on the cerebral endothelium to induce Rho kinase-mediated inhibitory phosphorylation of endothelial nitric oxide (NO) synthase and reduced NO production. Loss of endothelial NO in turn results in reduced resting cerebral perfusion, endothelial vasomotor dysfunction, and cognitive impairment [74]. Consistent with epidemiological data indicating that excessive dietary salt is an independent risk factor for cerebrovascular injury leading to stroke and dementia, these effects are independent of salt-induced elevations of arterial pressure. These findings unveiled novel gut-brain axes whereby adaptive immune responses generated in the gut by the microbiota or by dietary salt are able to influence the susceptibility of the brain to ischemic injury or induce endothelial dysfunction and cognitive impairment.
These observations, collectively, highlight the distinct role that the cells associated with the NVU play in the neurovascular dysfunction induced by vascular risk factors or Aβ. While in hypertension and amyloid pathology, perivascular macrophages are critical for the neurovascular and cognitive dysfunction, in high-salt diet, endothelial dysfunction is the major pathogenic factor. Furthermore, recent evidence implicates transient plugging of neocortical capillaries by circulating leukocytes in the oligemia described in mouse models of amyloid accumulation and AD.
Cerebral blood flow deficits of ~ 30% have been noted in patients with AD, and similar deficits are replicated in mouse models of Aβ overexpression [75,76]. These brain blood flow decreases appear early in the development of AD, likely contribute to the cognitive symptoms of the disease, and may accelerate disease progression by decreasing the clearance of Aβ, impairing neural function, and driving neuroinflammation. The mechanisms underlying this decrease in brain blood flow, however, remain poorly understood. In recent work [77], in vivo nonlinear microscopy was used to explicitly examine the cellular origin of the blood flow deficit in the APP/PS1 and 5xFAD mouse models of Aβ overexpression. While obstructions in arterioles or venules were not seen in these models, an elevation in the number of nonflowing capillaries in AD mice (~2%) as compared with those in wildtype controls (~0.4%) was observed. Most of these stalled capillaries had a neutrophil firmly adhered to the capillary wall, implicating vascular inflammation, caused by exposure to Aβ oligomers, as a likely cause. A high dose of antibodies against the neutrophil surface protein, Ly6G, led to an immediate release of the adhered neutrophils, which was accompanied by a rapid ~20% increase in cortical blood flow. This blood flow increase in turn was associated with an improvement in performance on spatial and working memory tasks in the AD mice, measured just a few hours after antibody administration. Isotype control antibodies did not release stalls, increase brain blood flow, or improve cognitive performance. This work establishes neutrophil adhesion in brain capillaries as a significant contributor to the cortical blood flow deficits in AD mouse models and suggests a more hopeful view of the cognitive dysfunction in AD, where an acute increase in cortical blood flow can improve cognitive function.
3.8. Vascular dysfunction may promote neurodegenerative pathology
Experimental studies have revealed that vascular risk factors, such as hypertension, promote amyloid pathology in models of Aβ accumulation (Fig. 6). [78] Hypertension stimulates the secretase pathways in the brain to produce Aβ [79,80], which is toxic not only to neurons and their synapses but also to the endothelium and smooth muscle, which in turn further exacerbates reduced blood flow and ischemia. Clinical studies support these observations by demonstrating increased amyloid accumulation in patients with vascular risk factors [81,82]. In addition to enzyme breakdown and microglial processing, amyloid is also thought to be cleared along the perivascular spaces especially during deep sleep [83]. Clearance may decrease with aging, more so in ApoE e4 carriers. Vascular pathology and sleep disruption can impair amyloid removal and both can deteriorate with aging.
Fig. 6.
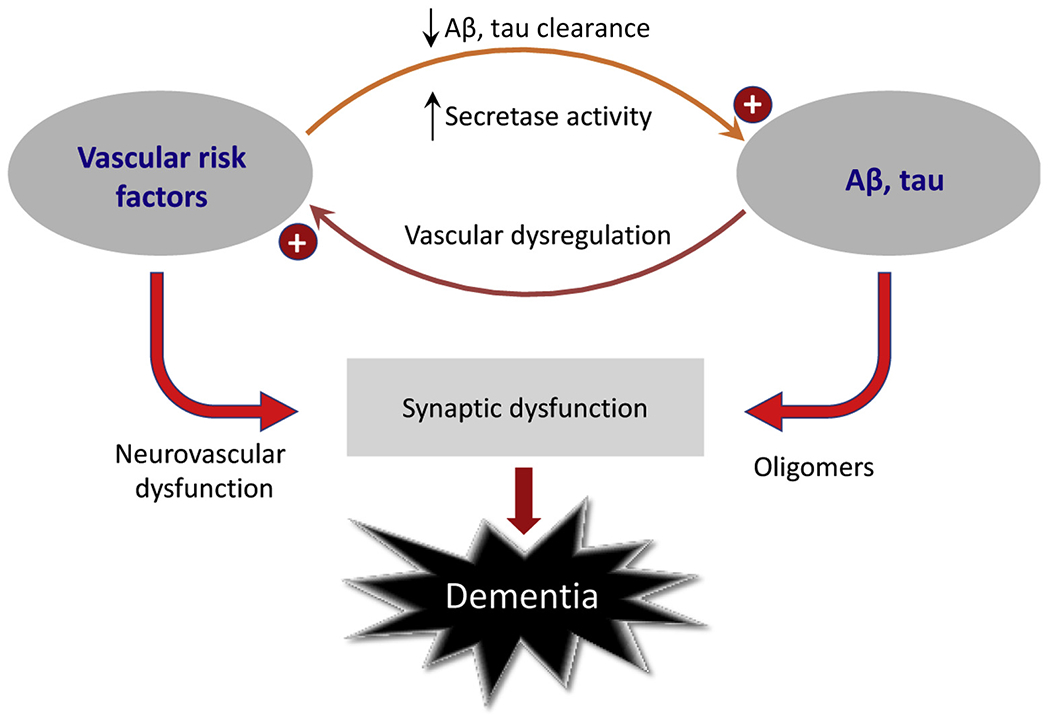
Synergistic interaction between vascular risk factors and AD pathology: Vascular risk factors promote Aβ and tau accumulation by enhancing their production and reducing vascular clearance. In turn, AD pathology induces neurovascular dysregulation which aggravates the deleterious impact of vascular risk factors. Abbreviation: AD, Alzheimer’s disease.
A key role of ApoE4 genotype on the integrity of the white matter, a frequent target of hypertensive microangiopathy and AD, was unveiled by a recent study in which 3-photon microscopy was used to examine the microcirculation of the white matter of the corpus callosum in the normal state and during hypoperfusion [84]. An increased susceptibility of the white matter to hypoperfusion and damage and to cognitive impairment was found, providing the putative mechanistic bases of the increased susceptibility of ApoE4-positive individuals not only to AD but also to hypertensive SVD and white matter damage.
Therefore, the destiny of the neurovascular unit, which incorporates the vascular and parenchymal cellular components, and of toxic proteins such as Aβ and tau, are intertwined and must be better understood to prevent and combat the inevitable erosion of memory, thinking, and autonomy that eventually ensues in the dementia syndromes.
3.9. Protein misfolding meets neurovascular disease: A dual threat to the brain aging gracefully
The intriguing and complex relationships between cerebrovascular disease and the misfolded proteins implicated in neurodegenerative disorders are becoming increasingly apparent. Rapid advancements in experimental science and in biochemical and neuroimaging techniques now enable in vivo detection of fluid and imaging biomarkers in both clinically symptomatic and presymptomatic stages of dementing illnesses. The interrelationships can be causal bidirectionally, additive, interactive, and synergistic, influenced by genetic and environmental factors. They also vary by stage of development or senescence. For example, aerobic glycolysis may be occurring in the default mode regions of the human brain only in the third and fourth decades [85]. Advances in computational prowess, data science and analytics, such as deep learning techniques, are rendering these complex relationships and individual heterogeneity more decipherable [86], as we can now study how these relationships play out in real time in animal models and in human disease in the emerging era of precision brain theragnostics.
We can see patterns of amyloid 40-42 and tau, as well as other misfolded protein deposits in neurodegenerative disorders in relation to different degrees and type of vasculopathy, not only at autopsy [87] but now also in vivo. We are beginning to understand the complex “go-no go” signaling pathways between different cells within the neuroglial-vascular unit. These include pericytes, smooth muscle, and endothelial cells, astrocytes and microglia, which enable or disrupt functional neural networks and can reach a threshold that impairs cognition, mood, and behavior [31].
Specifically, we can now quantify in CSF and on PET the key Alzheimer proteinopathies—amyloid and tau (Aβ 40-42 and hyperphosphorylated tau and total tau), but not yet synuclein and TDP43, which are also common with aging [88]. We can map their topographical patterns at baseline [89] and over time, accounting for genetic risk particularly APoE ε4 carrier status [90]. We can also monitor changes in response to interventions such as antiamyloid antibody infusions [91]. We can quantify brain tissue atrophy, stroke, (overt and covert), focal and diffuse SVD [92], more quickly and reliably than ever before using computational methods [93,94], including differential microbleed patterns related to AD [95]. We can measure cerebral blood flow and cerebrovascular reactivity, as well as BBB leakage, which may provide early warning signals before atrophy and molecular deposits appear [29]. Regional vulnerabilities and changes in structural integrity using diffusion tensor imaging and water fraction can also be tracked [96]. Functional network connectivity using computational methods allow us to map the multiple separate trajectories of these biomarkers on brain images, informed by genetics and biochemistry of CSF, and perhaps soon peripheral blood [97]. These imaging techniques are already providing new insights into interrelationships among these various surrogates of brain pathology at different stages of clinical manifestation as measured by clinical, cognitive, and functional measures. In particular, vascular dysregulation appears to be one of the first signs in the temporal evolution of AD [15], which gradually unfolds in the brain over one or two decades before the deposits of amyloid and tau start to become visible with molecular PET imaging [98].
For example, in an amyloid PET-magnetic resonnance imaging (MRI) study of patients with severe white matter disease, cortical thickness patterns differed in the amyloid positive versus negative cases; namely there was more bilateral perisylvian, and right medial frontal and orbital thinning and relative sparing of the medial temporal lobe and precuneus in the amyloid burden cases [99]. Furthermore, a high burden of vascular risk factors interacts with amyloid burden to increase tau deposition in the inferior temporal region [100]. Tau PET imaging in amyloid negative patients with subcortical vascular cognitive impairment shows relatively more uptake in the temporal regions compared with normal controls suggesting amyloid-independent pathways in vascular disease [101]. A case study of subacute stroke showed tau uptake in the infarct, further hinting at tauischemia relationships hitherto unsuspected [102].
Ischemia stimulates the gamma and beta secretase pathway in the brain to produce Aβ 40-42 which is toxic to neurons and their synapses, and also vascular endothelium and smooth muscle, which can in turn further exacerbate reduced blood flow and ischemia. In addition to enzyme breakdown and microglial processing, amyloid is cleared along the perivascular spaces especially during deep sleep. Clearance decreases with aging, especially in APoE ε4 carriers. Vascular pathology and sleep disruption can impair amyloid removal and both can deteriorate with aging. Tau phosphorylation, and its deposition and spread can also be driven by ischemia. Therefore, the destiny of the neurovascular unit, which incorporates the vascular and parenchymal cellular components, and toxic proteins such as Aβ and tau, are intertwined and must be understood to prevent and combat the inevitable erosion of memory, thinking, and autonomy that eventually ensues in the dementia syndromes.
3.10. Role of capillary dysfunction in stroke and dementia
In the context of cerebrovascular disease, it is generally assumed that limited blood supply and ischemia are the primary source of hypoxic tissue injury and neurological symptoms. The extraction of oxygen from blood, however, is contingent on the uniform distribution of blood across tissue capillaries, and deteriorating capillary morphology and function may therefore reduce tissue oxygenation by causing oxygenated blood to be “shunted” through the microcirculation [103]. Such capillary flow changes, termed capillary dysfunction, may cause hypoxic tissue injury and neurological symptoms, even if blood supply and vessel patency remain inconspicuous as assessed by current diagnostic methods [104,105]. Microvascular flow disturbances have now been demonstrated in AD [106] (Fig. 7) and mild cognitive impairment [107], and estimates of the accompanying degree of tissue hypoxia correlate with the extent of cognitive decline in AD [108]. Capillary dysfunction may therefore contribute to disease changes not traditionally ascribed to the vascular system and to the complex disease mechanism in cerebral SVD [109].
Fig. 7.
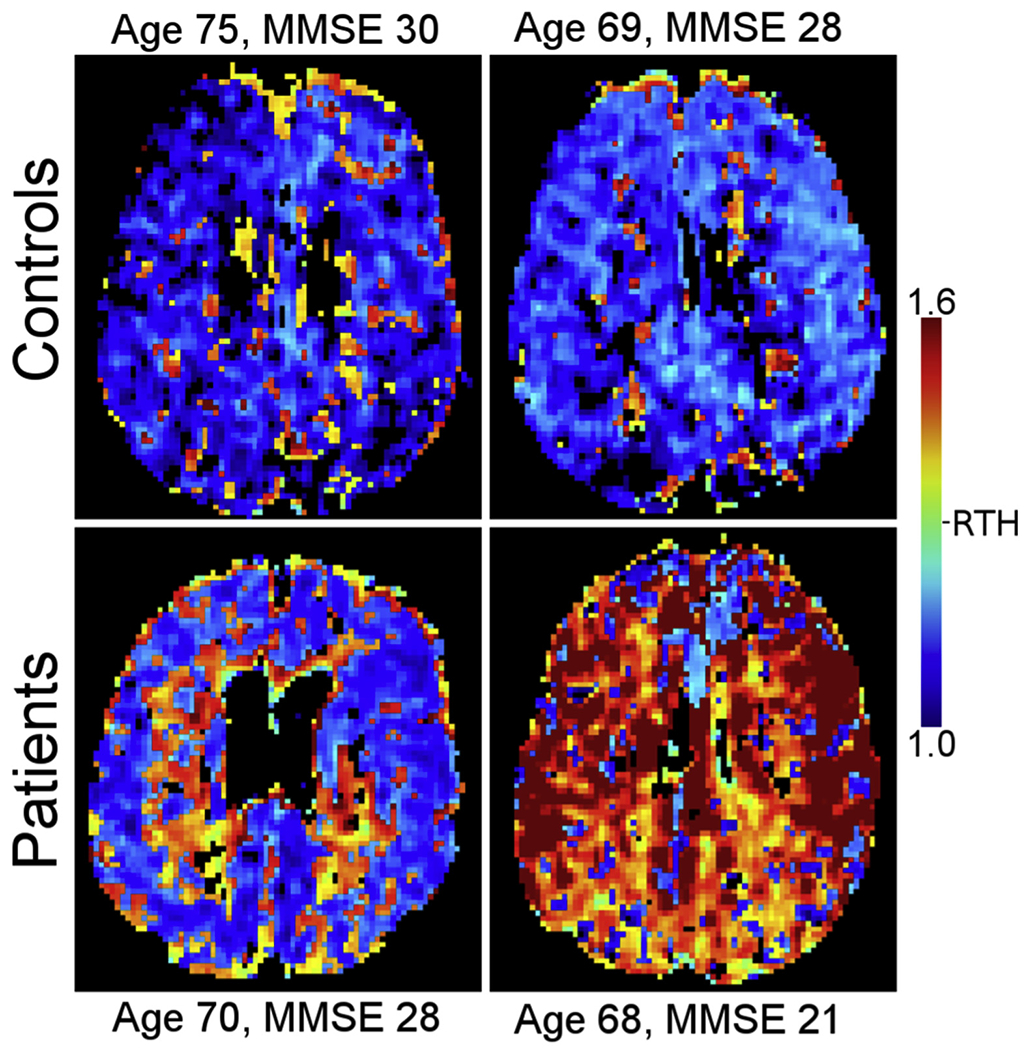
Capillary function can be estimated by dynamic susceptibility contrast MRI. Blood mean transit time, a traditional marker of cerebral perfusion, is supplemented with the standard deviation of blood transit times within each image voxel. The ratio of the two, dubbed relative transit-time heterogeneity is taken as an index of the microvasculature’s ability to maintain uniform perfusion. The figure shows parametric RTH maps in patients with AD and healthy controls of similar age. Note how RTH tends to be elevated in AD patients compared with controls, suggesting that capillary flow homogenization is impaired in these patients [107]. Abbreviations: AD, Alzheimer’s disease; RTH, relative transit-time heterogeneity; MMSE, Mini Mental State Examination.
4. Novel targets for prevention and therapy
Dysfunction of the neurovascular unit is increasingly recognized as a crucial joint mechanism for both stroke and dementia, and the breakdown of the BBB is an early event in cognitive dysfunction in AD. BBB breakdown, cerebral SVD along with amyloid angiopathy and perivascular inflammation all emerge as attractive new therapeutic agents for stroke and dementia prevention alike. In addition, targeting “classic” stroke risk factors such as hypertension or atrial fibrillation will not only reduce stroke risk but also cut down dementias.
4.1. Cerebral SVD: Novel targets for prevention
Cerebral SVD causes 45-65% of dementias, 20-25% of strokes, and worsens long-term outcome after acute brain insults [110]. SVD is due to an intrinsic disorder of the perforating arterioles, capillaries, and probably venules, in which, in the sporadic form, involves most components of the neuro-glio-vascular unit. There is endothelial including pericyte dysfunction which consists of subtle BBB leakage, impaired vasoreactivity, increased pulsatility due to vessel stiffness and mismatch of blood supply to demand which results in impaired perivascular space function, interstitial fluid and waste drainage (including Aβ), and net interstitial fluid increase [111]; defective oligodendrocyte precursor cell maturation impairs myelination, myelin repair, and facilitates axon disruption [112]. Perivascular inflammation which occurs early in the disease development although remains poorly understood; damage to matrix proteins and astrocyte dysfunction which likely impairs fluid transport and neuronal energy supply (although is currently less understood). This means that the mechanisms of brain damage leading to dementia, that are typically labeled as ischemic, are in fact much more complex than simply falling blood flow. The dysregulation of dynamic vascular function with altered BBB function, impaired vasoreactivity, failure to match blood supply to demand, and increased vessel pulsatility appear increasingly critical to creating a situation of failed brain fluid management that results in failed fluid and waste clearance, protein deposition, myelin, astrocyte and neuronal damage. These dysfunctions may account for ischemic damage, which is secondary and likely occurs late. Furthermore, there is increasing evidence that the perivascular space dysfunction influences the ability to clear Aβ from the brain and that alterations in the production of Aβ maybe also be related to the inflammation seen pathologically in SVD, providing a mechanistic link between the pathological processes described in vascular disease and AD. While these dysfunctions in the main may arise through risk factor exposures, the heritability of SVD features suggests premorbid vulnerability, supported by recent model [112] and gene [113] analyses.
This constellation of intertwined cellular defects offers multiple intervention targets [114]. Examples include endothelial stabilizing, improved nitric oxide bioavailability and BBB integrity, antiinflammatory approaches, reduced vascular smooth muscle proliferation and increase relaxation, improved oligodendrocyte precursor cell maturation, and improved neuronal energy transfer. Existing drugs with relevant pleiotropic effects include cilostazol (a phosphodiesterase 3’ inhibitor), nitric oxide donors, the family of phosphodiesterase 5 inhibitors, statins (for their antiinflammatory effects), and several classes of antihypertensive drugs. Risk factor exposures that worsen vascular dysfunction are the same as for stroke, dementia, and for intermediary indicators of vascular damage (white matter hyperintensities, lacunes, cortical thinning, and ventricular enlargement) and include the obvious candidates of hypertension, hypercholesterolemia, and diabetes. However, much more attention should be paid to smoking cessation, reduction in air pollution, and reduction in dietary salt and dietary changes including increasing vegetables (a source of nitrates) and increased regular exercise as important ways to maintain good cerebrovascular function. Furthermore, the association between lower educational attainment and increased risk of stroke [115], vascular brain damage, and dementia indicates that improvements in education should be regarded as a population-level life-course intervention to prevent, or at least mitigate, the burden of stroke and dementia.
4.2. Poststroke dementia: Mechanisms and opportunities for prevention
Stroke defines a population at high risk of developing dementia. This is true for both prestroke dementia (pooled prevalence in population-based studies about 10%) and poststroke dementia. Reported figures for poststroke (<1 year) dementia vary depending on case-mix, reaching up to 40% in hospital-based studies of recurrent stroke that do not exclude prestroke dementia [116,117]. Independent predictors for poststroke dementia include older age, low educational attainment, vascular risk factors (in particular diabetes and atrial fibrillation), severe stroke (higher National Institutes of Health stroke scale score), previous cognitive decline, multiple/recurrent strokes, and leukoaraiosis/white matter hyperintensities on brain imaging [116,117]. These findings emphasize the opportunity of dementia prevention through stroke prevention while also offering criteria for risk stratification and identifying patients that might benefit from close monitoring. Early cognitive testing (<7 days) by the Montreal Cognitive Assessment predicts long-term cognitive outcome, functional outcome, and mortality after stroke and should thus become part of clinical routine [118]. Recent studies using circulating biomarkers and longitudinal MRI have emphasized a role of secondary neurodegeneration after stroke as a source of ongoing neuroaxonal injury, which may extend many months and possibly years after stroke [119,120]. Whether secondary neurodegeneration contributes to cognitive decline after stroke is unknown and subject of current investigation. In the absence of efficient preventive treatments for neurodegenerative dementia including AD, preventing stroke and vascular brain injury currently seems the most efficient strategy to reduce the burden of dementia worldwide.
4.3. Cerebral amyloid angiopathy as a target for dementia prevention
Cerebral amyloid angiopathy (CAA), representing deposition of the Aβ peptide in small arteries and capillaries of the cerebral cortex and leptomeninges, is best recognized as a common cause of spontaneous intracerebral hemorrhage. A range of clinical-radiologic and clinical-pathologic studies, however, have also implicated CAA as an important contributor to age-related cognitive impairment. In the epidemiologic Religious Orders Study/Memory and Aging Project sample, moderate-severe CAA was common (i.e., 35.8% of 1079 autopsied brains) and when present, accounted for a mean 20.6% of the premortem cognitive decline in those individuals [121].
Elucidating the mechanisms by which advanced CAA causes cognitive impairment would be a critical step toward developing rational preventive therapies. CAA is associated with several types of brain injuries that are potential mediating mechanisms for cognitive impairment. Among these are white matter T2 hyperintensities, alterations in structural brain connectivity, cerebral microinfarcts, and loss of tissue in the cortical gray matter, white matter, and basal ganglia [121]. Among these candidate lesions, structural connectivity appears to have the strongest link to cognitive performance, whereas microinfarcts appear to be the most prevalent individual lesion type, numbering in the thousands in brains with advanced CAA. These observations suggest a model by which CAA-related cognitive impairment occurs because of progressive structural disconnection, driven by the cumulative effects of numerous small, spatially distributed brain lesions.
4.4. Less dementia with oral anticoagulation in atrial fibrillation
The association between atrial fibrillation and dementia is well documented; however, it was not clear if oral anticoagulant treatment offers protection from dementia. A retrospective registry study was conducted in Sweden with the aim to compare the incidence of new dementia in patients with atrial fibrillation treated with and without oral anticoagulants, and to explore if there was a difference between novel anticoagulants and warfarin. All patients with hospital diagnosis of atrial fibrillation between 2006 and 2014 were identified from the national health care registries. Patients with a previous diagnosis of dementia were excluded. Propensity score matching, falsification endpoints, and analyses according to intention to treat and treatment principles were performed. The study included 444,106 patients and over 1.5 million years at risk. Patients using oral anticoagulants at baseline had 29% lower risk of receiving a diagnosis of dementia during follow-up than patients without anticoagulant treatment (HR: 0.71, 95% CI: 0.68–0.74). Analyzed on treatment, that is, according to oral anticoagulant exposure during follow-up, showed that the risk was almost half as high with oral anticoagulants (HR: 0.52, 95% CI: 0.50–0.55). Direct comparison between new oral anticoagulants and warfarin showed no difference (HR: 0.97, 95% CI: 0.67–1.40) (Fig. 8) [14].
Fig. 8.
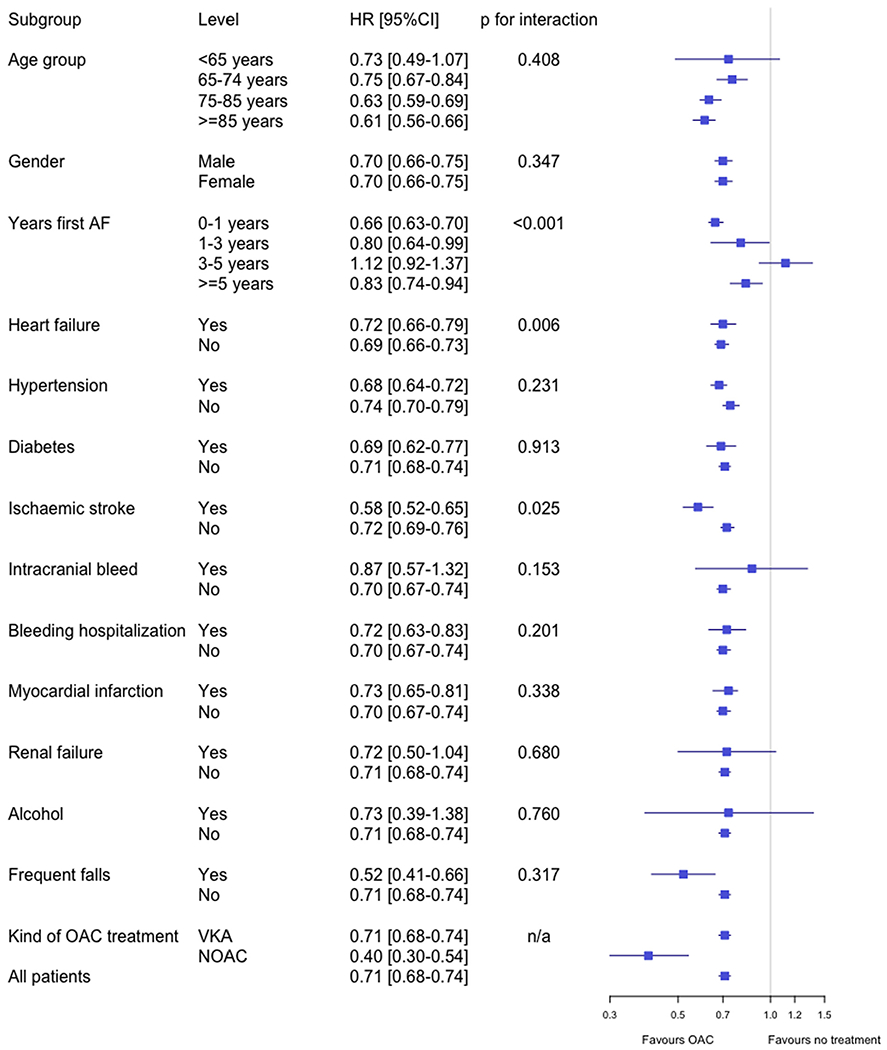
Risk of dementia with and without oral anticoagulation treatment at baseline (“intention to treat”). Multivariable Cox regression on propensity score matched cohorts [14]. Abbreviations: AF, atrial fibrillation; CI, confidence interval; HR, hazard ratio; NOAC, non-Vitamin K oral anticoagulant; VKA, Vitamin K antagonist.
5. Call to action
The greatest potential gains in dementia prevention could come from preventing stroke and cerebrovascular disease.
The following initiatives are offered as examples of different possible approaches: No value judgment is implied compared with other uncited approaches.
5.1. Action plan for stroke in Europe
Several actions to reduce the burden of noncommunicable diseases are in progress from the United Nations and the World Health Organization; however, there is no detailed action plan specifically for stroke. In Europe, two previous pan-European consensus meetings, the 1995 and 2006 Helsingborg meetings [122,123] were convened to review the scientific evidence and the state of current services, and to set targets for the development of stroke care for the decade to follow. The European Stroke Organization has now prepared a European Stroke Action Plan (ESAP) for the years 2018 to 2030, in cooperation with the Stroke Alliance for Europe. The ESAP adheres to the format of the Helsingborg Declarations, presenting a review of the “state of the art,” the state of current services, research and development priorities, and targets for a series of domains in stroke care (organization of stroke services, management of acute stroke, prevention, rehabilitation, evaluation of stroke outcome and quality assessment). The ESAP includes two additional domains, on primary prevention and life after stroke, along with research and development priorities for translational stroke research. The ESAP provides 32 specific targets for seven domains. Four overarching targets were identified (Table 1).
Table 1.
Overarching targets for 2030 in the ESAP
| For each domain of the 2018 to 2030 ESAP, specific targets are being set. |
| Beyond these targets, four overarching targets for 2030 have been identified: |
| 1. To reduce the absolute number of strokes in Europe by 10%. |
| 2. To treat 90% or more of all patients with stroke in Europe in a dedicated stroke unit as the first level of care. |
| 3. To have national plans for stroke encompassing the entire chain of care from primary prevention to life after stroke. |
| 4. To fully implement national strategies for multisector public health interventions to promote and facilitate a healthy lifestyle, and reduce environmental (including air pollution), socioeconomic, and educational factors that increase the risk of stroke. |
Abbreviation: ESAP, European Stroke Action Plan.
The ESAP was launched at the European Stroke Organization Congress in Gothenburg in May 2018 and published in the European Stroke Journal [124]. An ESAP implementation plan will be prepared by European Stroke Organization after having assessed an updated epidemiological report on stroke incidence, prevalence, and mortality and receiving detailed and reliable data from quality registries from national stroke and patient societies. Progress toward the targets and research and development priorities laid out in the ESAP will be reviewed in 2021 and 2024, with a midterm review scheduled for 2024. The extent to which the targets have been achieved will be reviewed in 2030.
It is hoped that the ESAP will have a major impact in reducing the burden of stroke in Europe, with important spin-off effects also in the prevention of dementia on the European population.
5.2. Canadian initiatives on stroke and dementia
Dementia is a growing health concern in Canada [125] (Fig. 9), and vascular health is one important risk factor. Focus on the prevention and management of circulatory diseases has the potential to reduce the risk of dementia [126]. Canada faces many unique challenges with the delivery of health care. Mandated by the Canada Health Act, health care is delivered by 10 provinces and 3 territorial ministries; however, the federally supported Canadian Institutes of Health Research (CIHR) represents the main funding agency for health research.
Fig. 9.
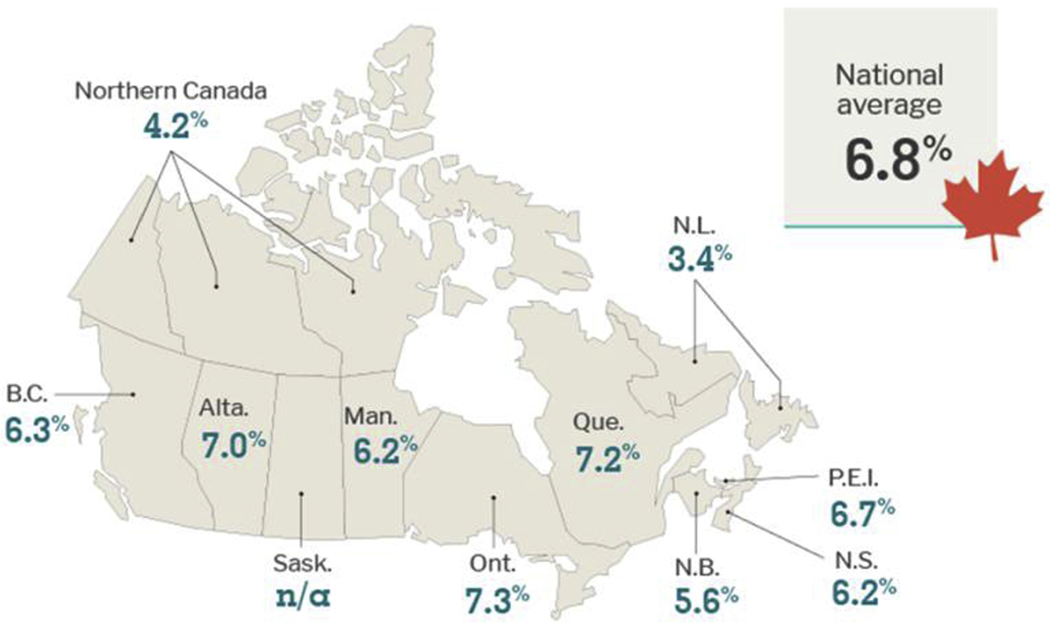
The age-standardized prevalence rate of dementia in Canada is 6.8%; however, there is variation across provincial and territorial boundaries across Canada [125]. Abbreviations: BC, British Columbia; Alta, Alberta; Sask, Saskatchewan; Man, Manitoba; Ont, Ontario; Que, Quebec; NB, New Brunswick; NS, Nova Scotia; PEI, Prince Edward Island; NL, Newfoundland.
Three CIHR Institutes, including the Institutes of Aging, Circulatory and Respiratory Health, and Neurosciences, Mental Health and Addiction, share mandates regarding vascular health and dementia. Stroke and dementia researchers have excelled in open competitions; however, strategic research investments have also been used to focus attention of specific issues. The Canadian Consortium on Neurodegeneration and Aging ($25 M over 5 years) investment includes one platform that focuses on vascular dementia. In addition, CIHR has funded the Cardiovascular Research Network ($4.3 M over 5 years). These networks provide additional support for training, pilot project funding, and network coordination to enhance investigator and team grant success in the field.
Overall, Canada’s scientific community in vascular health is mobilized to address the dementia challenge by focusing both on multi-stroke vascular dementias, and on the contribution of vascular health to the cascade of events involved in neurodegenerative diseases causing dementia.
5.3. Turning knowledge into action: Lessons from stroke
Stroke, ischemic heart disease, vascular kidney disease, and dementia share many common risk factors, including hypertension, diabetes, high blood lipids, physical inactivity, obesity, and others. For stroke and dementia, cardiac conditions with high risk for brain and systemic emboli (e.g., atrial fibrillation/flutter, heart failure, etc.) are also well-known risk factors. The presence of vascular risk factors not only affects the occurrence of vascular cognitive decline and dementia but also is predictive for onset and progress of “degenerative” dementias (AD spectrum) [127].
Chronic heart failure, chronic kidney disease, diabetes, and hypertension can be accompanied by cognitive decline and dementia, which again may be treatable, and in some cases, with treatment of the underlying condition (e.g., kidney transplantation) even reversible. In this context, diabetes and hypertension play a special role because they are on one hand important risk factors for the development of stroke, heart failure, and chronic kidney disease and on the other hand, can be linked to cognitive impairment without having caused detectable stroke yet [128,129].
Stroke and dementia statistics are usually studied in isolation, and the fact that stroke doubles the chances of developing dementia is frequently ignored when calculating the percentage of potentially preventable or even reversible dementias.
It seems straight forward, that by influencing these risk factors both vascular and degenerative dementias would be preventable. Unfortunately, the development of dementia takes longer than the occurrence of recurrent stroke, and most trials that showed efficacy in preventing stroke had follow-ups too short to show effects on dementia, and most did not try to look at it specifically. Recently, two studies showed a decrease in the incidence of dementia by intervening with multiple risk factors [130] and by anticoagulation for atrial fibrillation [14].
Given the central roles of hypertension, diabetes, and hypercholesterinemia for both stroke and dementia incidence, it may be expected that rigid prevention of stroke would also lead to a reduction of vascular dementia and a later onset of degenerative dementias.
6. Conclusions
Neurological disorders account for the largest number of DALYs worldwide [1]. More than half result from stroke and dementia. Both conditions arise from similar treatable risk and protective factors, and growing evidence suggests that preventing stroke can also prevent some dementias.
The neurovascular unit represents a core feature of organizations of the brain with its elements in dynamic equilibrium. Disruption of one of its components has consequences for all the others. On the positive side, each malfunctioning element represents a potential new therapeutic target. Vascular mechanisms and neurodegeneration meet at the neurovascular unit. Multiple mechanisms imply multiple interactions. While technological advances allow us to pry increasingly more deeply into the micro level of structure and function with promises of future new therapeutic approaches, much can be done at the macro level right now.
Initiatives in Europe, Canada, and worldwide show the power of systematic approaches in reducing the incidence of stroke and the potential for beginning to also do this for dementia. All major world organizations dealing with the brain and the heart have committed to the joint prevention of stroke and dementia [17]. These conditions represent such huge problems that even modest progress would have great effects. Because the risk factors for stroke and dementia are the same, coordinating efforts among disciplines alone could have a major impact not only on stroke and dementia but also on heart disease and vascular kidney and retinal disease. The evidence for doing so is incontestable; the time to act is now.
7. Potential recommendations for an action plan
These recommendations did not arise from a formal process but are offered to encourage discussion and action and largely correspond to the more explicit and detailed aims outlined in the editorial accompanying this article.
Specific Aims:
Aim # 1 - Establish an International Consortium for Longitudinal Studies on the links between aging-stroke/cerebrovascular and cognitive disorders-dementia. The rationale for such a consortium is mandated by the need for very large population-based, genetically diverse, cohorts as essential research and development resources. The key objectives of this project will require the following:
Accurate detection (reliable identification) of asymptomatic people (in the general population) at elevated risk for cognitive decline and dementia;
Discovery-development-validation technologies/protocols for accurate identification of asymptomatic people (preclinical stages) at elevated risk for these conditions at similar levels of risk factors but different susceptibility or effects of yet undetermined risk factors for dementia; and,
Consensus on study design for large-scale multisite/international prospective prevention study(ies) or trial(s).
Aim # 2 - Conduct a comprehensive review-evaluation of ongoing major longitudinal studies/cohorts/important-relevant data series that can be incorporated. Identify key partners and centers that should be invited to contribute or join the consortium.
Aim # 3 – Establish an “Executive Committee” and “Project Management Team” with mandates to develop drafts for
International agreement for “public-private partnerships” and standards regarding best practices for data deposit, management, access, and sharing. Set up coordinating mechanism which will enable public-private enterprises to accelerate innovation on new technologies for early detection and interventions of cerebrovascular disease, stroke, traumatic brain injury, dementia, and other chronic brain diseases.
International oversight and governance mechanism for administering such an undertaking must include policies, rules, and a system for decisions that govern data sharing and/or access to these resources.
Consider the prospects of a two-year study to demonstrate the feasibility of the project, which will focus on designing the collaborative multisite study and evaluating alternative models for the governance and financing of the proposed study.
Aim # 4 – Prepare a proposal for the initial two-year pilot phase of what would eventually become a decade long multinational enterprise. This document should outline: 1) long-term and short-term aims, 2) study design, 3) scientific expertize of key investigators, 4) resources and technical capabilities available to the project (e.g., data management), 5) description of available cohorts and characteristics of prospective subject, 6) time line and deliverables, and 7) budget.
The aim of the two-year proposal is to establish a new type of research resource, an international research and development infrastructure that can provide a comprehensive database on well-characterized cohorts. Current resources, established cohorts, and ongoing longitudinal studies are fragmented and inadequate, because of (1) relatively small sample sizes and, (2) lack of guarantee for long-term funding required to underwrite the longevity of cohorts.
The creation of a unique shared-resource, being proposed, will be based on formal agreements between governments, agencies, and/or research institutions from several world regions. The reasoning is that no single country, agency, institution, company, or industry has all the assets to pursue this type of research independently. Although existing population-based cohorts and/or studies could offer valuable data, they do not individually fulfill all the specific needs for prospective prevention validation studies. For example, the development of new computational models to establish probability profiles for predicting the relative risks for memory disorders/dementia in asymptomatic populations will require large databases from varied sources and domains of measurements (e.g., biomarkers, genetic, behavioral, imaging, and other information on comorbid conditions).
Brain vascular disease can be identified in vivo. Risk factors for brain vascular disease are mostly known. Effective low cost interventions for some aspects of brain vascular disease, especially for stroke, are available. The big unknown remains as to what extent prevention of brain vascular disease will substantially reduce dementia or AD.
RESEARCH IN CONTEXT.
Systematic review: All co-authors are leaders in their fields. Each were asked to limit their references to a key few.
Interpretation: Each co-author was asked to link expertise to the larger context provided by the Berlin Manifesto. This resulted in particularly animated and fruitful integration of important aspects of the basic pathophysiology of cognitive disorders.
Future directions: There needs to be: a greater emphasis on common vascular and neurodegenerative pathophysiological mechanisms; funding for population and clinical interventions, studies and trials; a symposium integrating what is known, identifying opportunities for treatment and prevention; and formation of a small leadership group to act as guiding integrators and synergizers of the initiatives of this major field of therapeutic and preventable opportunities.
Acknowledgments
The authors wish to acknowledge:
All those who made it possible for the Satellite to take place. The World Health Summit Satellite Symposium “Dementia Prevention by Stroke Prevention” was organized by Martin Dichgans, Matthias Endres, Vladimir Hachinski, Pierluigi Nicotera (DZNE, Bonn, Germany), and Arno Villringer (Max-Planck-Institute, Leipzig, Germany). It was supported by the Center for Stroke Research Berlin, CompetenceNet Stroke, Institute for Stroke and Dementia Research (ISD), Munich, German Center for Neurodegenerative Diseases (DZNE), Munich Cluster of Excellence SyNergy, and Cluster of Excellence NeuroCure Berlin.
Ulrich Dirnagl and Arno Villringer who served as chairs during the symposium.
Dr. Jörg Heldmann, Managing Director World Health Summit, was very supportive to accommodate us during the WHS.
Dr. Ulrike Lachmann, Center for Stroke Research Berlin and Competence Net Stroke who got everything organized. Rebecca Clarke, assistant to VH who had the challenging task of reconciling the growing and changing contributions and references.
J.W. received support from European Union Horizon 2020, PHC-03-15, project No 666881, ‘SVDs@Target’, Fondation Leducq Transatlantic Network of Excellence for the Study of Perivascular Spaces in Small Vessel Disease, ref no. 16 CVD 05, and the UK Dementia Research Institute which receives its funding from DRI Ltd, funded by the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK.
B.Z. is supported by the National Institutes of Health grants 5P01AG052350 and 5P50AG005142, in addition to the Alzheimer’s Association strategic 509279 grant and the Foundation Leducq Transatlantic Network of Excellence for the Study of Perivascular Spaces in Small Vessel Disease reference no. 16 CVD 05.
M.E. has received funding from the DFG, BMBF, DZNE, DZHK, EU, Corona Foundation, and Fondation Leducq. M.D. received funding from the EU (the European Union’s Horizon 2020 research and innovation programme under grant agreement No 666881, SVDs@target and grant agreement No 601456, CVgenes@target, DFG, BMBF, DZNE, Fondation Leducq, and the Corona Foundation.
References
- [1].Corporate, Author(s) GBD 2016 Dementia Collaborators. Global, regional, and national burden of Alzheimer’s disease and other dementias. Lancet Neurol 2019;18:88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kuzma E, Lourida I, Moore SF, Levine DA, Ukoumunne OC, Llewellyn DJ. Stroke and dementia risk: A systematic and meta-analysis. Alzheimers Dement 2018;14:1416–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].O’Donnell MJ, Xavier D, Liu L, Zhang H, Chin SL, Rao-Melacini P, et al. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): A case-control study. Lancet 2010;376:112–23. [DOI] [PubMed] [Google Scholar]
- [4].Feigin VL. Global burden of stroke and risk factors in 188 countries, during 1990-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet Neurol 2016;15:913–24. [DOI] [PubMed] [Google Scholar]
- [5].Hachinski V. Dementia: Paradigm shifting into high gear. Alzheimers Dement 2019;15:985–94. [DOI] [PubMed] [Google Scholar]
- [6].Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007;69:2197–204. [DOI] [PubMed] [Google Scholar]
- [7].Azarpazhooh MRAA, Cipriano LE, Munoz DG, Sposato LA, Hachinski V. Concomitant vascular and neurodegenerative pathologies double the risk of dementia. Alzheimers Dement 2018;14:148–56. [DOI] [PubMed] [Google Scholar]
- [8].Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain 2013;136:2697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jin Y, Di Legge S, Ostbye T, Feightner JW, Hachinski V. The reciprocal risks of stroke and cognitive impairment in an elderly population. Alzheimers Dement 2006;2:171–8. [DOI] [PubMed] [Google Scholar]
- [10].Rabin JS, Schultz AP, Hedden T, Viswanathan A, Marshall GA, Klein KE, et al. Interactive associations of vascular risk and beta-amyloid burden with cognitive decline in clinically normal elderly individuals. Findings from the Harvard Aging Brain Study. JAMA Neurol 2018;75:1124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wu Y-T, Beiser AS, Breteler MMB, Fratiglioni L, Helmer C, Hendrie HC, et al. The changing prevalence and incidence of dementia over time - current evidence. Nat Rev Neurol 2017;13:1081–8. [DOI] [PubMed] [Google Scholar]
- [12].Sposato LA, Kapral MK, Wu J, Gill SS, Hackam DG, Cipriano LE, et al. Declining incidence of stroke and dementia: Coincidence or prevention opportunity? JAMA Neurol 2015;72:1529–31. [DOI] [PubMed] [Google Scholar]
- [13].Cerasuolo JO, Cipriano LE, Sposato LA, Kapral MK, Fang JM, Gil SS, et al. Population-based stroke and dementia incidence trends: Age and sex variations. Alzheimers Dement 2017;13:1081–8. [DOI] [PubMed] [Google Scholar]
- [14].Friberg L, Rosenqvist M. Less dementia with oral anticoagulation in atrial fibrillation. Eur Heart J 2018;39:453–60. [DOI] [PubMed] [Google Scholar]
- [15].Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Pérez JM, Evans AC, ADNI. Early role of vascular dysregulation on late-onset Alzheimer’s disease progression: evidence from a multi-factorial, data-driven analysis. Nat Commun 2016;7:11934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Livingston G, Sommerlad A, Vasiliki O, Costafreda SG, Huntley J, Ames D, et al. Dementia prevention intervention and care. Lancet 2017;390:2673–734. [DOI] [PubMed] [Google Scholar]
- [17].Hachinski V. World Stroke Organization. Stroke and potentially preventable dementias proclamation. Stroke 2015;46:3039–40. [DOI] [PubMed] [Google Scholar]
- [18].Hachinski V, Ganten D, Lackland D, Kreutz R, Tsioufis C, Hacke W, WSO The World Heart Federation, The World Hypertension League and the European Society of Hypertension. Implementing the proclamation of stroke and potentially preventable dementias. Int J Stroke 2018;13:780–6. [DOI] [PubMed] [Google Scholar]
- [19].The World Bank. World Bank Country and Lending Groups, https://datahelpdesk.worldbank.org/knowledgebase/articles/906519-worldbank-country-and-lending-groups Accessed June 26, 2019.
- [20].Kalaria RN, Maestre GE, Arizaga R, Friedland RP, Galasko D, Hall K, et al. Alzheimer’s disease and vascular dementia in developing countries: Prevalence, management, and risk factors. Lancet Neurol 2008;7:812–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Alladi S, Bak TH, Duggirala V. Bilingualism delays age at onset of dementia, independent of education and immigration status. Neurology 2013;81:1938–44. [DOI] [PubMed] [Google Scholar]
- [22].Iyer GK, Alladi S, Bak TH, Shailaja M, Mamidipudi A, Rajan A, et al. Dementia in developing countries: Does education play the same role in India as in the West? Dement Neuropsychol 2014;8:132–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Alladi S, Hachinski V. World dementia: One approach does not fit all. Neurology 2018;91:264–70. [DOI] [PubMed] [Google Scholar]
- [24].Stephan BCM, Birdi R, Tang EYH, Cosco TD, Donini LM, Licher S, et al. Secular trends in dementia prevalence and incidence worldwide: A systematic review. J Alzheimers Dis 2018;66:653–80. [DOI] [PubMed] [Google Scholar]
- [25].Matthews FE, Brayne C, Lowe J, McKeith I, Wharton SB, Ince P. Epidemiological pathology of dementia: Attributable-risks at death in the Medical Research Council Cognitive Function and Ageing Study. Plos Med 2009;6:e1000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Matthews FE, Arthur A, Barnes LE, Bond J, Jagger C, Robinson L, et al. A two-decade comparison of prevalence of dementia in individuals aged 65 years and older from three geographical areas of England: Results of the Cognitive Function and Ageing Study I and II 2013;382:1405–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Norton S, Matthews FE, Brayne C. A commentary on studies presenting projections of the future prevalence of dementia. BMC Public Health 2013;13:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Matthews FE, Stephan BCM, Robinson L, Jagger C, Barnes LE, Arthur A, et al. A two decade dementia incidence comparison from the Cognitive Function and Ageing Studies I and II. Nat Commun 2016;7:11398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-Brain Barrier: From physiology to disease and back. Physiol Rev 2019;99:21–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sweeney MD, Montagne A, Sagare AP, Nation DA, Schneider LS, Chui HC, et al. Vascular dysfunction: The disregarded partner of Alzheimer’s disease. Alzheimers Dement 2019;5:158–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sweeney MD, Kisler K, Montagne A, Toga A, Zlokovic BV. The role of brain vasculature in neurodegenerative disorders. Nat Neurosci 2018;21:1318–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhao Z, Nelson A, Betsholtz C, Zlokovic BV. Establishment and dysfunction of blood-brain barrier. Cell 2015;163:1064–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nation DA, Sweeney MD, Montagne A, Sagare AP, D’Orazio LM, Pachicano M, et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med 2019;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015;85:296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Montagne A, Nikolakopoulou AM, Zhao Z, Sagare AP, Si G, Lazic D, et al. Pericyte degeneration causes white matter dysfunction in the mouse CNS. Nat Med 2018;24:326–37. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [36].Khachaturian ZS. The role of calcium regulation in brain aging: Reexamination of a hypothesis. Aging 1989;1:17–34. [DOI] [PubMed] [Google Scholar]
- [37].Khachaturian ZS. Calcium hypothesis of Alzheimer’s disease and brain aging. Ann NY Acad Sci 1994;747:1–11. JF D, Willem HG, Treber J, Khachaturian ZS, editors. [DOI] [PubMed] [Google Scholar]
- [38].AACH Workgroup. Calcium Hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement 2017;13:178–82. [DOI] [PubMed] [Google Scholar]
- [39].Xing C, Arai K, Lo EH, Hommel M. Pathophysiologic cascade in ischemic stroke. Int J Stroke 2012;7:378–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wikipedia c. Ischemic Cascade: Wikipedia, The Free Encyclopedia. Available from: https://en.wikipedia.org/ischemice_cascade; 2018. Accessed December 10, 2018.
- [41].Iturria-Medina Y, Carbonell F, Sotero RC, Chouinard-Decorte F, Evans ACADNI. Multifactorial causal model of brain (dis)organization and therapeutic intervention: Application to Alzheimer’s disease. Neuroimage 2017;152:60–77. [DOI] [PubMed] [Google Scholar]
- [42].Iturria-Medina Y, Carbonell FM, Evans AC, ADNI. Multimodal imaging-based therapeutic fingerprints for optimizing personalized interventions: Application to neurodegeneration. Neuroimage 2018;179:40–50. [DOI] [PubMed] [Google Scholar]
- [43].Iadecola C. The neurovascular unit coming of age: A journey through meurovascular coupling in health and disease. Neuron 2017;96:17–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Iadecola C. The pathobiology of vascular dementia. Neuron 2013;80:844–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Iadecola C, Zhang F, Niwa K, Eckman C, Turner SK, Fischer E, et al. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci 1999;2:157–61. [DOI] [PubMed] [Google Scholar]
- [46].Niwa K, Porter VA, Kazama K, Cornfield D, Carlson GA, Iadecola C. A beta-peptides enhance vasoconstriction in cerebral circulation. Am J Physiol 2001;281:H2417–24. [DOI] [PubMed] [Google Scholar]
- [47].Niwa K, Younkin L, Ebeling C, Turner SK, Westaway D, Younkin S, et al. Abeta 1-40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc Natl Acad Sci USA 2000;97:9735–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Niwa K, Kazama K, Younkin L, Younkin SG, Carlson GA, Iadecola C. Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am J Physiol 2002;283:H315–23. [DOI] [PubMed] [Google Scholar]
- [49].Zhang F, Eckman C, Younkin S, Hsiao KK, Iadecola C. Increased susceptibility to ischemic brain damage in transgenic mice overexpressing the amyloid precursor protein. J of Neurosci 1997;17:7655–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Park L, Zhou J, Zhou P, Pistick R, Jamal El S, Younkin L, et al. Innate immunity receptor CD36 promotes cerebral amyloid angiopathy. Proc Natl Acad Sci USA 2013;110:3089–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, et al. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci USA 2008;105:1347–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Park L, Uekawa K, Garcia-Bonilla L, Koizumi K, Murphy M, Pitstick R, et al. Brain perivascular nacrophages initiate the neurovascular dysfunction of Alzheimer Aβ peptides. Circ Res 2017;121:258–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Park L, Wang G, Moore J, Girouard H, Zhou P, Anrather J, et al. The key role of transient receptor potential melastatin-2 channels in amyloid-β–induced neurovascular dysfunction. Nat Commun 2014;5:5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Iadecola C Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci 2004;5:347–60. [DOI] [PubMed] [Google Scholar]
- [55].Whitehead S, Cheng G, Hachinski V, Cechetto D. Interaction between a rat model of cerebral ischemia and beta-amyloid toxicity: II. Effects of triflusal. Stroke 2005;36:1782–9. [DOI] [PubMed] [Google Scholar]
- [56].Whitehead S, Hachinski V, Cechetto D. Interaction between a rat model of cerebral ishemia and beta-amyloid toxicity: Inflammatory response. Stroke 2005;36:107–12. [DOI] [PubMed] [Google Scholar]
- [57].Whitehead SN, Cheng G, Hachinski VC, Cechetto DF. Progressive increase in infarct size, neuroinflammation, and cognitive deficits in the presence of high levels of amyloid. Stroke 2007;38. [DOI] [PubMed] [Google Scholar]
- [58].Amtul Z, Whitehead SN, Keeley RJ, Bechberger J, Fisher AL, McDonald RJ, et al. Comorbid rat model of ischemia and beta-amyloid toxicity: Striatal and cortical degeneration. Brain Pathol 2015;25:24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Weishaupt N, Caughlin S, Yeung K, Whitehead S. Differential anatomical expression of ganglioside GM1 species containing d18:1 or d20:1 Sphingosine detected by MALDI imaging mass spectrometry in mature rat brain. Front Neuronat 2015;9:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Cheng G, Whitehead S, Hachinski V, Cechetto D. Effects of pyrrolidine dithiocarbamate on beta-amyoid (25-35) induced inflammatory responses and memory deficits in the rat. Neurobiol Dis 2006;23:140–51. [DOI] [PubMed] [Google Scholar]
- [61].Weishaupt N, Riccio P, Dobbs T, Hachinski VC, Whitehead SN. Characterization of behaviour and remote degeneration following thalamic stroke in the rat. Int J Mol Sci 2015;16:13921–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Agca C, Fritz JJ, Walker LC, Levey AI, Chan AW, Lah JJ, et al. Development of transgenic rats reducing human beta-amyloid precursor protein as a model for Alzheimer’s disease: Transgene and endogenous APP genes are regulated tissue-specifically. BMC Neurosci 2008;9:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Weishaupt N, Liu Q, Shin S, Singh R, Agca Y, Agca C, et al. APP21 transgenic rat develop age-dependent cognitive impairment and microglia accumulation within white matter tracts. J Neuroinflammation 2018:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Levit A, Regis AM, Gibson A, Hough OH, Maheshwari S, Agca Y, et al. Impaired behavioural flexibility related to white matter microgliosis in the TgAPP21 rat model of Alzheimer disease. Brain Behav Immun 2019. February 15 [Epub ahead of print]. 10.1016/j.bbi.2019.02.013. [DOI] [PubMed] [Google Scholar]
- [65].Levit ARA, Garabon JR, Oh SH, Rajakumar N, Hachinski V, Agca Y, et al. Behavioural inflexibility in a comorbid rat model of striatal ischemic injury and mutant haPP overexpression. Behav Brain Res 2017;333:267–75. [DOI] [PubMed] [Google Scholar]
- [66].Levit A, Fox MS, Qi Q, Hachinski V, Thiessen JD, Whitehead SN. Multimodal imaging of white matter inflammation in a rat model of striatal ischemic stroke. Neurology 2018;90:2–226. [Google Scholar]
- [67].Kazama K, Wang G, Frys K, Anrather J, Iadecola C. Angiotensin II attenuates functional hyperemia in the mouse somatosensory cortex. Am J Physiol 2003;285:H1890–9. [DOI] [PubMed] [Google Scholar]
- [68].Kazama K, Anrather J, Zhou P, Girouard H, Frys K, Milner TA, et al. Angiotensin II impairs neurovascular coupling in neocortex through NADPH oxidase-derived radicals. Circ Res 2004;95:1019–26. [DOI] [PubMed] [Google Scholar]
- [69].Girouard H, Park L, Anrather J, Zhou P, Iadecola C. Cerebrovascular nitrosative stress mediates neurovascular and endothelial dysfunction induced by angiotensin II. Arterioscler Thromb Vasc Biol 2007;27:303–9. [DOI] [PubMed] [Google Scholar]
- [70].Capone C, Anrather J, Milner TA, Iadecola C. Estrous cycle-dependent neurovascular dysfunction induced by angiotensin II in the mouse neocortex. Hypertension 2009;54:302–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Capone C, Faraco G, Park L, Cao X, Davisson RL, Iadecola C. The cerebrovascular dysfunction induced by slow pressor doses of angiotensin II precedes the development of hypertension. Am J Physiol 2011;300:H397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Capone C, Faraco G, Anrather J, Zhou P, Iadecola C. Cyclooxygenase 1-derived prostaglandin E2 and EP1 receptors are required for the cerebrovascular dysfunction induced by angiotensin II. Hypertension 2010;55:911–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Faraco G, Sugiyama Y, Lane D, Garcia-Bonilla L, Chang H, Santisteban MM, et al. Perivascular macrophages mediate the neurovascular and cognitive dysfunction associated with hypertension. J Clin Invest 2016;126:4674–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Faraco G, Brea D, Garcia-Bonilla L, Wang G, Racchumi G, Chang H, et al. Dietary salt promotes neurovascular and cognitive dysfunction through a gut-initiated TH17 response. Nat Neurosci 2018;21:240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Niwa K, Kazama K, Younkin SG, Carlson GA, Iadecola C. Alterations in cerebral blood flow and glucose utilization in mice overexpressing the amyloid precursor protein. Neurobiol Dis 2002;9:61–8. [DOI] [PubMed] [Google Scholar]
- [76].Montagne A, Nation DA, Pa J, Sweeney MD, Toga AW, Zlokovic BV. Brain imaging of neurovascular dysfunction in Alzheimer’s disease. Acta Neuropathol 2016;131:687–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Hernandez Cruz JC, Bracko Oliver O, Kersbergen CJ, Muse V, Haft-Javaherian M, Berg M, et al. Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nat Neurosci 2019;22:413–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Iadecola C. The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol 2010;120:287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Faraco G, Park L, Zhou P, Luo W, Paul SM, Anrather J, et al. Hypertension enhances Aβ-induced neurovascular dysfunction, promotes β-secretase activity, and leads to amyloidogenic processing of APP. J Cereb Blood Flow Metab 2016;36:241–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Carnevale D, Mascio G, D’Andrea I, Fardella V, Bell RD, Branchi I, et al. Hypertension induces brain β-amyloid accumulation, cognitive impairment, and memory deterioration through activation of receptor for advanced glycation end products in brain vasculature. Hypertension 2012;60:188–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Gottesman RF, Schneider ALC, Zhou Y, Coresh J, Green E, Gupta N, et al. Association between midlife vascular risk factors and estimated brain amyloid deposition. JAMA 2017;317:1443–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Rodrigue KM, Rieck JR, Kennedy KM, Devous MD, Diaz-Arrastia R, Park DC. Risk factors for β-amyloid deposition in healthy aging: vascular and genetic effects. JAMA Neurol 2013;70:600–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol 2015;11:457–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Koizumi K, Hattori Y, Ahn SJ, Buendia I, Ciacciarelli A, Uekawa K, et al. ApoE4 disrupts neurovascular regulation and undermines white matter integrity and cognitive function. Nat Commun 2018;9:3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Goyal MS, Vlassenko AG, Blazey TM, Su Y, Couture LE, Durbin TJ, et al. Loss of brain aerobic glycolysis in normal human aging. Cell Metab 2017;26:353–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Nestor SM, Misic B, Ramirez J, Zhao J, Graham SJ, Verhoeff NPLG, et al. Small vessel disease is linked to disrupted structural network covariance in Alzheimer’s disease. Alzheimers Dement 2017;134:171–86. [DOI] [PubMed] [Google Scholar]
- [87].Kapasi A, DeCarli C, Schneider JA. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol 2017;134:171–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].McAleese KE, Walker L, Erskine D, Thomas AJ, McKeith IG, Attems J. TDP-43 pathology in Alzheimer’s disease, dementia with Lewy bodies and ageing. Brain Pathol 2017;27:472–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Sepulcre J, Grothe MJ, Sabuncu M, Chhatwal J, Schultz AP, Hansweeuw B, et al. Hierarchical organization of tau and amyloid deposits in the cerebral cortex. JAMA Neurol 2017;74:813–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Sepulcre J, Grothe MJ, d’Oleire Uquillas F, Ortiz-Teran L, Diez I, Yang HS, et al. Neurogenetic contributions to amyloid beta and tau spreading in the human cortex. Nat Med 2018;24:1910–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016;537:50–6. [DOI] [PubMed] [Google Scholar]
- [92].Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R, et al. Standards for reporting vascular changes on nEuroimaging (STRIVE v1). Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 2013;12:822–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Ramirez J, Gibson E, Quddus A, Lobaugh NJ, Feinstein A, Levine B, et al. Lesion Explorer: a comprehensive segmentation and parcellation package to obtain regional volumetrics for subcortical hyperintensities and intracranial tissue. Neuroimage 2011;54:963–73. [DOI] [PubMed] [Google Scholar]
- [94].Ramirez J, McNeely AA, Scott CJ, Stuss DT, Black SE. Subcortical hyperintensity volumetrics in Alzheimer’s disease and normal elderly in the Sunnybrook Dementia Study: Correlations with atrophy, executive function, mental processing speed, and verbal memory. Alzheimers Res Ther 2014;6:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Pettersen JA, Aviv RI, Black SE, Fox AJ,Lim A, Murray BJ. Global Hemispheric CT hypoperfusion may differentiate headache with associated neurological deficits and lymphocytosis from acute stroke. Stroke 2008;39:492–3. [DOI] [PubMed] [Google Scholar]
- [96].Heiss WD, Rosenberg GA, Thiel A, Berlot R, de Reuck J. Neuroimaging in vascular cognitive impairment: a state-of-the-art review. BMC Med 2016;14:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Palmqvist S, Scholl M, Strandberg O, Mattsson N, Stomrud E, Zetterberg H, et al. Earliest accumulation of β-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nat Commun 2017;8:1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Montagne A, Zhao Z, Zlokovic BV. Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J Exp Med 2017;217:3151–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Kim CH, Seo SW, Kim GH, Shin JS, Cho H, Noh Y, et al. Cortical thinning in subcortical vascular dementia with negative 11C-PiB PET. J Alzheimers Dis 2012;31:315–23. [DOI] [PubMed] [Google Scholar]
- [100].Rabin JS, Yang HS, Schultz AP, Hanseeuw BJ, Hedden T, Viswanathan A, et al. Vascular risk and β-amyloid are synergistically associated with cortical tau. Ann Neurol 2019;85:272–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Kim HJ, Park S, Cho H, Jang YK, San Lee J, Jang H, et al. Assessment of extent and role of tau in subcortical vascular cognitive impairment using 18F-AV1451 positron emission tomography imaging. JAMA Neurol 2018;75:999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Cho SH, Cho H, Park S, Ryu YH, Choi JY, Lyoo CH, et al. Increased uptake of AV-1451 in a subacute infarction lesion. Yonsei Med J 2018;59:563–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Jespersen SN, Østergaard L. The roles of cerebral blood flow, capillary transit time heterogeneity and oxygen tension in brain oxygenation and metabolism. J Cereb Blood Flow Metab 2012;32:264–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Østergaard L, Jespersen SN, Mouridsen K, Mikkelsen IK, Jonsdottir KY, Tietze A, et al. The role of the cerebral capillaries in acute ischemic stroke: The extended penumbra model. J Cereb Blood Flow Metab 2013;33:635–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Østergaard L, Jespersen SN, Engedahl T, Gutierrez Jimenez E, Ashkanian M, Hansen MB, et al. Capillary dysfunction: its detection and causative role in dementias and stroke. Curr Neurol Neurosci Rep 2015;15:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Eskildsen SF, Gyldensted L, Nagenthiraja K, Nielsen RB, Hansen MB, Dalby RB, et al. Increased cortical capillary transit time heterogeneity in Alzheimer’s disease: A DSC-MRI perfusion study. Neurobiol Aging 2017;50:107–18. [DOI] [PubMed] [Google Scholar]
- [107].Nielsen RB, Parbo P, Ismail R, Dalby RB, Tietze A, Brærndgaard H, et al. Impaired perfusion and capillary function in mild cognitive impairment: Relation to oxygenation and amyloidosis. (In review, Alzheimers Dement) 2018.
- [108].Nielsen RB, Egefjord L, Angleys H, Mouridsen K, Gejl M, Moller A et al. Capillary dysfunction is associated with symptom severity and neurodegeneration in Alzheimer’s disease. Alzheimers Demen 2017;13:1143–53. [DOI] [PubMed] [Google Scholar]
- [109].Østergaard L, Engedal TS, Moreton F, Hansen MB, Wardlaw JM, Dalkara T, et al. Cerebral small vessel disease: Capillary pathways to stroke and cognitive decline. J Cereb Blood Flow Metab 2016;36:302–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Wardlaw JM, Smith C, Dichgans M. Small vessel disease: Mechanisms and clinical implications. Lancet Neurol 2019;18:684–96. [DOI] [PubMed] [Google Scholar]
- [111].Wardlaw JM, Makin SJ, Valdés Hernández MC, Armitage PA, Heye AK, Chappell FM, et al. Blood-brain barrier failure as a core mechanism in cerebral small vessel disease and dementia: evidence from a cohort study. Alzheimers Dement 2017;13:634–43. [Google Scholar]
- [112].Rajani RM, Quick S, Ruigrok SR, Graham D, Harris SE, Verhaaren BFJ, et al. Reversal of endothelial dysfunction reduces white matter vulnerability in cerebral small vessel disease in rats. Sci Transl Med 2018;10. [DOI] [PubMed] [Google Scholar]
- [113].Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A, et al. Multiancestry genome-wide association study of 520, 000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet 2018;50:524–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Bath PM, Wardlaw JM. Pharmacological treatment and prevention of cerebral small vessel disease: a review of potential interventions. Int J Stroke 2015;10:469–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Pendlebury ST, Rothwell PM. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: A systematic review and meta-analysis. Lancet Neurol 2009;8:1006–18. [DOI] [PubMed] [Google Scholar]
- [116].Dichgans M, Leys D. Vascular cognitive impairment. Circ Res 2017;3:573–91. [DOI] [PubMed] [Google Scholar]
- [117].Zietemann V, Georgakis MK, Dondaine T, Müller C, Mendyk AM, Kopczak A, et al. Early MoCA predicts long-term cognitive and functional outcome and mortality after stroke. Neurology 2018;20:e1838–50. [DOI] [PubMed] [Google Scholar]
- [118].Tiedt S, Duering M, Barro C, Kaya AG, Boeck J, Bode FJ, et al. Serum neurofilament light: A biomarker of neuroaxonal injury after ischemic stroke. Neurology 2018;14:e1338–47. [DOI] [PubMed] [Google Scholar]
- [119].Duering M, Righart R, Wollenweber FA, Zietemann V, Gesierich B, Dichgans M. Acute infarcts cause focal thinning in remote cortex via degeneration of connecting fiber tracts. Neurology 2015;84:1685–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Boyle PA,Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol 2018;83:74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Greenberg SM, Al-Shahi Salman R, Biessels GJ, van Buchem M, Cordonnier C, Lee JM, et al. Outcome markers for clinical trials in cerebral amyloid angiopathy. Lancet Neurol 2014;13:419–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Aboderin I, Venables G, Asplund K. Stroke management in Europe. J Intern Med 1996;240:173–80. [DOI] [PubMed] [Google Scholar]
- [123].Kjellström T, Norrving B, Shatchkute A. Helsingborg Declaration 2006 on European Stroke Strategies. Cerebrovasc Dis 2007;23:229–41. [DOI] [PubMed] [Google Scholar]
- [124].Norrving B, Barrick J, Davalos A, et al. Action plan for stroke in Europe 2018-2030. Eur Stroke J 2018;3:309–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].CIHI. Dementia in Canada - How dementia impacts Canadians (digital report). Available at: https://www.cihi.ca/en/dementia-in-Canada2018 Accessed July 6, 2018.
- [126].InterAcademy Partnership for Health T, Italy. IAP for Health: A call for action to tackle the growing burden of dementia. 2018; (July 6). Available from: http://www.interacademies.org Accessed July 6, 2018.
- [127].Gorelick PB. Risk factors for vascular dementia and Alzheimer disease. Stroke 2004;35:2620–2. [DOI] [PubMed] [Google Scholar]
- [128].Kennelly SPLB, Kenny RA. Blood pressure and dementia - A comprehensive review. Ther Adv Neurol Disord 2009;2:241–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Deckers KCI, vanBoxtel MP, Verhey FR, Irving K, Brayne C, Kivipelto M, et al. Dementia risk in renal dysfunction: A systematic review and meta-analysies of prospective studies. Neurology 2017;88:198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Ngandu T, Lethisalo I, Solomon A, Levälahti E, Ahtiluoto S, Antikainen R, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): A randomized controlled trial. Lancet 2015;385:2255–63. [DOI] [PubMed] [Google Scholar]