

SUMMARY
Increased calpain activity is linked to neuroinflammation including a heritable retinal disease caused by hyper-activating mutations in the calcium-activated calpain-5 (CAPN5) protease. Although structures for classical calpains are known, the structure of CAPN5, a non-classical calpain, remains undetermined. Here we report the 2.8 Å crystal structure of the human CAPN5 protease core (CAPN5-PC). Compared to classical calpains, CAPN5-PC requires high calcium concentrations for maximal activity. Structure-based phylogenetic analysis and multiple sequence alignment reveal that CAPN5-PC contains three elongated flexible loops compared to its classical counterparts. The presence of a disease-causing mutation (c.799G>A, p.Gly267Ser) on the unique PC2L2 loop reveals a function in this region for regulating enzymatic activity. This mechanism could be transferred to distant calpains, using synthetic calpain hybrids, suggesting an evolutionary mechanism for fine-tuning calpain function by modifying flexible loops. Further, the open (inactive) conformation of CAPN5-PC provides structural insight into CAPN5-specific residues that can guide inhibitor design.
Graphical Abstract
In Brief
Velez et al. report the crystal structure of the calpain-5 protease core (CAPN5-PC). Sequence- and structure-based phylogenetic analysis reveals that CAPN5-PC contains three elongated loops compared to classical calpains. One loop contains a hyperactivating mutation that causes neovascular inflammatory vitreoretinopathy, revealing a function in this region for regulating proteolytic activity.
INTRODUCTION
Aberrant elevation of intracellular calcium levels leads to dysregulation of calpain-mediated proteolytic events, which can result in necrotic cell death and neuroinflammation (Artal-Sanz and Tavernarakis, 2005; Syntichaki et al., 2002). Excess calpain activity is implicated in a wide range of human neuroinflammatory diseases, including Parkinson’s disease, Alzheimer’s disease, stroke, multiple sclerosis, and retinal degeneration (Hoffmann et al., 2013; Hong et al., 1994; Samantaray et al., 2008; Shields et al., 1999; Trinchese et al., 2008). The only known inherited disease characterized by calpain hyperactivity is neovascular inflammatory vitreoretinopathy (NIV, OMIM: 193235), a dominantly inherited, severe inflammatory retinal disease caused by mutations in CAPN5 (Mahajan et al., 2012). The CAPN5 gene encodes for the Ca2+-activated cysteine protease, calpain-5 (CAPN5). Fifteen mammalian calpains comprise a family of Ca2+-dependent cysteine proteases that mediate limited proteolysis in the cell. These tightly regulated reactions make irreversible changes in their protein targets, facilitating a wide range of cellular processes, such as cell morphogenesis, cell signaling, membrane repair, and apoptosis (Campbell and Davies, 2012; Ono and Sorimachi, 2012; Singh et al., 2014).
Evolutionary gene duplication events have generated 15 human genes that encode a conserved calpain-like protease core domain. Calpains can be broadly divided into two subfamilies based on their domain architecture (Ono and Sorimachi, 2012). The “classical” calpains (e.g., CAPN1 and CAPN2) are comprised of a large and small subunit. The large subunit houses the protease core domain formed by two globular subdomains (PC1 and PC2) separated by a flexible linker, a calpain β sandwich domain (CBSW; DIII), and a Ca2+-binding penta-EF (PEF) hand domain (Figure 1A). The PEF-hand of the large subunit, PEF(L), serves as a dimerization module with the small subunit comprised of a glycine-rich region followed by a C-terminal PEF-hand, PEF(S). The PEF-hands are postulated to regulate enzymatic activity and form interactions with the endogenous calpain-inhibitor, calpastatin (CAST) (Campbell and Davies, 2012; Hanna et al., 2008). In contrast, the “non-classical” Capn5 and Capn6 genes were originally identified as orthologs of the tra-3 gene, a C. elegans gene encoding for the sex-determination factor TRA-3 (Matena et al., 1998). CAPN5 (also termed hTRA-3) and CAPN6 (a catalytically inactive calpain) are the most distant paralogs of the calpain family: they lack the PEF-hand domain and instead contain a C2 domain that is structurally similar to the CBSW domain (Figure 1B) (Rizo and Südhof, 1998). CAPN5 is broadly expressed, but displays high expression levels in select tissues, including the central nervous system (CNS), retina, and gastrointestinal tract (Dear et al., 1997; Dear and Boehm, 1999; Schaefer et al., 2016; Singh et al., 2014). Despite these differences, the CAPN5 protease core (termed CAPN5-PC) has conserved Ca2+-binding sites and is predicted to undergo activation in the presence of Ca2+ (Gakhar et al., 2016). It is unclear, however, what structural features distinguish CAPN5-PC from classical calpain core domains. Distinct structural features, such as surface-exposed flexible loops, may alter the enzyme’s activity independent of its Ca2+ requirements or non-catalytic domains.
Figure 1. Sequence-Based Phylogenetic Analysis of the Calpain Superfamily Reveals the Evolutionary Distance of CAPN5 from Classical Calpains.

(A) Schematic representation of classical calpain heterodimers and CAPN5. CAPN5 lacks the PEF-hand domains and instead contains a C2 domain following the CBSW domain.
(B) Cartoon representation of calpain domain architecture of classical calpains and CAPN5.
(C) Phylogenetic analysis workflow.
(D) The approximated calpain superfamily tree. The unrooted tree is represented with clades colored according to their representative nomenclature: classical (green), non-classical (gray), and CAPN5-like (red). The positions of human CAPN5 and rat CAPN1 are denoted by black dots for reference.
(E) The unrooted CAPN5 sub-tree is represented with clades colored by related organisms. Bootstrap values are projected onto the tree topology.
Studying human mutations is an effective way to probe the mechanisms regulating enzyme activity. Known CAPN5 mutations increase activity and cause NIV, while most other disease-causing calpain mutations result in a loss-of-function (e.g., CAPN1 mutations result in spastic paraplegia and CAPN3 mutations cause limb girdle muscular dystrophy type 2A) (Potz et al., 2016). Prior to blindness around the 5th decade of life, NIV progresses through a series of predictable pathological disease stages characterized by synaptic signaling defects (loss of b-wave on electroretinogram), inflammatory cell infiltration, neovascularization, and intraocular fibrosis. These 5 stages mimic common eye diseases that together account for a significant fraction of visual morbidity and blindness: uveitis, retinitis pigmentosa, proliferative diabetic retinopathy, and proliferative vitreoretinopathy (Velez et al., 2017). Thus, understanding how CAPN5 mutations lead to disease may provide insight into the mechanisms underlying common causes of blindness. To date, most NIV-causing mutations are located in the PC2 domain on gating loops near the active site. Three of the mutations are located on a flexible regulatory loop (termed the G1 gating loop) believed to gate access of substrates into the active site (Bassuk et al., 2015). NIV mutations are predicted to disrupt this regulatory function, leaving the active site always available to substrates and relieving the Ca2+ requirement for activation. Thus, NIV mutations are predicted to hyperactivate the protease. We recently identified a de novo CAPN5 mutation (p.R289W) in a pediatric patient with early-onset severe vitreoretinopathy, optic neuritis, and hearing loss. This mutation, which is located on a different loop, displayed higher enzymatic activity than the previously identified mutations, suggesting a correlation between the level of protease hyperactivity and disease severity (Velez et al., 2018).
The high levels of CAPN5 in the CNS, its genetic association with retinal disease, and successful oral delivery of calpain inhibitors to the retina imply CAPN5 inhibition is a promising strategy for NIV therapy (Oka et al., 2007). However, there are no high-resolution structures of CAPN5 to guide structure-based inhibitor design. There have been numerous efforts to inhibit calpains in disease, including Alzheimer disease, multiple sclerosis, spinal muscular atrophy, myocardial infarction, and cerebrovascular disease (Ono et al., 2016). The cysteine-active site and peptide sequence of CAST provides specificity for many inhibitors over other families of proteases (e.g., serine and aspartic proteases), but selectivity between cysteine proteases remains a challenge. Most calpain inhibitors are not specific and inhibit other cysteine proteases (Ono et al., 2016). Thus, most calpain inhibitors, to date, are too broad for clinical use. Here, we present a high-resolution structure of human CAPN5-PC along with detailed functional and biophysical analysis.
RESULTS
Broad Phylogenetic Analysis Reveals Conserved Residues that Are Unique to CAPN5
Calpains exhibit broad structural conservation of their protease core domains despite being highly divergent in their respective domain topologies. Nevertheless, broad sequence-based phylogenetic analysis could reveal unique features among calpain protease cores that have co-evolved to accommodate their respective non-catalytic domains (Masrati et al., 2018). We aligned a seed group of manually curated calpain sequences that share the protease core fold and produced a profile hidden Markov model (HMM) (Figure 1C) (Finn et al., 2011). The profile HMM was then used in a HMMER search to broaden the pool of calpain sequences. A fast approximation of the unrooted calpain superfamily tree (5,284 taxa) was constructed (Figure 1D) (Price et al., 2010). The resulting phylogenetic tree divided the calpain superfamily into clades based on their domain architecture (Figure S1). The CAPN5-like sequences were extracted from the approximated tree and aligned to construct a neighbor-joining maximum-likelihood tree (454 taxa) (Figure 1E). The CAPN5 sub-tree was divided into clades representing groups of related organisms. Despite the high diversity among the CAPN5 sequences, we identified 68 residues that were 100% conserved in chordates across evolution (Table S1). Of these 68 residues, 47 (69%) were localized to the PC domain, highlighting their functional importance in maintaining CAPN5 activity.
The Crystal Structure of CAPN5-PC Reveals an Inactive Conformation
We solved the crystal structure of the wild-type (WT) human CAPN5-PC in an inactive conformation. The crystals diffracted to 2.8 Å resolution and contained two molecules in the asymmetric unit (Table 1). No electron density was seen for the C-terminal 6x His tag of the construct, Ca2+ ions, or a peptide present in the crystallization solution (see STAR Methods). Alignment of the two molecules in the asymmetric unit yielded a root-mean-square deviation (RMSD) of 0.13 Å across 315 Cα atoms. The overall architecture (residues 4–349 in the construct) retained the conventional mixed α/β structure seen in other calpain protease cores: the catalytic subdomains (PC1 and PC2) were connected by a flexible linker (Figure 2A). There was one Ramachandran outlier present in the structure (Asp201 in chain B) (Table 1). This residue was located at the α8 helix, where the density map was slightly ambiguous. Nevertheless, the backbone continuity was sound near this residue. We anticipate that higher-resolution structures will resolve this outlier. ConSurf analysis was performed using the CAPN5-PC structure and the 454 CAPN5 sequences as input (Armon et al., 2001; Ashkenazy et al., 2016; Glaser et al., 2003). As expected, residues comprising CAPN5-PC’s hydrophobic core and active site groove displayed high evolutionary conservation scores and were among the 68 residues that were 100% conserved (Figure 2B; Table S1). The imidazole nitrogen (NE2) of His252 was located 3.7 Å away from the thiol sulfur (SG) of Cys81 (Figures 2C and S2). The orientation of the conserved tryptophan “wedge” residue (Trp286) in the active site groove precludes formation of the catalytic triad and forms π-π stacking interactions with His252 (Figure 2C). In active Ca2+-bound calpain protease core structures, the tryptophan wedge residue is displaced from its position between PC1 and PC2, allowing for formation of the fully competent catalytic triad (Moldoveanu et al., 2004b). Therefore, the unformed nature of the catalytic triad in our CAPN5-PC structure indicates it was in an inactive conformation (Otto and Schirmeister, 1997; Storer and Ménard, 1994).
Table 1.
Crystallographic Data Collection and Refinement Statistics
| Statistics | Human CAPN5-PC Wild-Type (Inactive) |
|---|---|
| Data Collection | |
| Beam line | ALS 4.2.2 |
| Wavelength (Å) | 1.0000 |
| Space group | P 1 21 1 |
| Unit cell dimensions (a, b, c, α, β, γ) | 84.0 Å, 51.6 Å, 110.6 Å, 90°, 110.4°, 90° |
| Resolution range (Å) | 51.8–2.8 |
| Total reflections | 43,742 (4,389) |
| Unique reflections | 22,239 (2,233) |
| Multiplicity | 2.0 (2.0) |
| Completeness (%) | 99.5 (99.1) |
| I/σ (I) | 10.8 (1.73) |
| Wilson B-factor (Å2) | 50.7 |
| Rmeas | 0.087 (0.623) |
| CC1/2 | 99.6 (80.4) |
| Refinement | |
| Resolution (Å) | 2.8 |
| No. of reflections used in refinement | 22,201 (2,226) |
| No. of reflections used for Rfree | 1,060 (119) |
| Rwork | 0.227 (0.339) |
| Rfree | 0.285 (0.442) |
| No. of non-hydrogen atoms | 5,595 |
| Protein | 5,595 |
| Water | 0 |
| B-factors (Å2) | 50.2 |
| RMSDa | |
| Bond length (Å) | 0.01 |
| Bond angle (°) | 1.12 |
| Ramachandran Plot (%) | |
| In preferred regions | 95.3 |
| In allowed regions | 4.6 |
| Outliers | 0.1 |
| PDB accession code | 6P3Q |
The numbers in parentheses are for the highest-resolution shell.
Root-mean-square deviation to ideal values.
Figure 2. Crystal Structure of CAPN5-PC.

(A) Ribbon tracing diagram representing domains PC1 and PC2 of CAPN5-PC. Catalytic residues and Trp286 are denoted by the cyan box. The G1 gating loop is denoted by the magenta box.
(B) Ribbon tracing diagram representing CAPN5-PC colored by the ConSurf evolutionary conservation scale. The orange dotted box denotes the active site groove.
(C) Active-site cleft of CAPN5-PC. Black dashes indicate the distance between the catalytic histidine and cysteine.
(D) Formed α7-region of CAPN5-PC.(E) Disordered α7-region of human CAPN1-PC (PDB: 2ARY).
To date, calpain inhibitor design has utilized the G1 gating loop as a conformational and sequential specificity determinant (Cuerrier et al., 2006; Moldoveanu et al., 2004a; Qian et al., 2008). We compared G1 loop conformations across known calpain structures (Figure S3A). Interestingly, the CAPN5-PC G1 loop adopted a conformation similar to that of certain active (Ca2+- and inhibitor-bound) calpain structures (e.g., leupeptin-, E-64-, and SNJ-1715-bound CAPN1) despite being in an inactive Ca2+-free state (Figure S3A). Because calpain activation requires a connection between the protease core subdomains, inhibition of this interaction through intercalation between PC1 and PC2 may be a viable strategy for designing CAPN5-specific inhibitors (Ono et al., 2016). These observations suggest that the inactive CAPN5-PC structure may serve as a usable template for rational inhibitor design. The CAPN5-PC structure had an inter-domain rotation angle of 19° with respect to its flexible linker when compared to the Ca2+-bound (active) human CAPN1-PC (PDB: 2ARY) (Figure S3B). This observed rotation angle corresponds to a more “open” conformation with respect to PC1 and PC2 compared to the “closed” conformations observed for active calpains. The relative domain rotation observed for CAPN5-PC was closed compared to conformations observed for inactive classical calpains (e.g., rat CAPN1-like, PDB: 1QXP) (Figure S3B), which contain rotation angles ranging from 23°C to 26°C. Previous small-angle X-ray scattering-guided (SAXS) modeling of CAPN5-PC in the absence of added Ca2+ by our group revealed a highly open conformation (Gakhar et al., 2016). It is unclear whether crystal packing influenced the conformation observed in our CAPN5-PC structure. We anticipate that Ca2+-bound or inhibitor-bound structures of CAPN5-PC will address this question.
Calpains contain an intrinsic reversible mechanism to inhibit the autolysis-generated protease core (Davis et al., 2007; Moldoveanu et al., 2003). This mechanism is mediated through instability of the α7 helix on PC1 in the absence of their non-catalytic domains (Moldoveanu et al., 2003). In classical calpains, instability of this α-helix in the vicinity of the active site is thought to cause a rearrangement of a nearby tryptophan side chain (Trp82) into the active site cleft, preventing formation of the catalytic triad. Calpain paralogs, containing a destabilizing glycine residue in the α7 helix, are predicted to be susceptible to this intrinsic silencing mechanism (e.g., human CAPN1, CAPN2, CAPN8, and CAPN11) (Moldoveanu et al., 2003). Conversely, alanine-containing protease cores, such as human CAPN5-PC and the highly catalytic rat CAPN1-PC, are expected to generate stable α7 helices, allowing them to be active in the absence of their respective non-catalytic domains (Moldoveanu et al., 2003). Indeed, we found that CAPN5-PC contained a well-formed α7 helix (Figure 2D) compared to the weakly catalytic human CAPN1-PC (PDB: 2ARY) (Figure 2E). This observation suggested that human CAPN5-PC could also be active in the absence of its CBSW and C2 domains.
The Calpain-5 Protease Core Undergoes Ca2+-Induced Conformational Changes
Thermostability was analyzed by recording differential scanning fluorimetry (DSF) data at different Ca2+ concentrations as CAPN5-PC was heated from 4°C to 95°C. With increasing amounts of Ca2+, the half-maximal change in melting temperature, Tmelt (DSF Kd), was observed at 2.5 ± 0.3 mM (Figure S4A), suggesting that CAPN5-PC displays Ca2+-stabilization in the millimolar range. Conformational changes of PC1-PC2 were measured in a standard calpain assay (Moldoveanu et al., 2002, 2003, 2004b). A sigmoidal increase in intrinsic tryptophan fluorescence (IWF) was observed in response to increasing Ca2+ concentrations. Half-maximal change in fluorescence occurred at 6.3 ± 1.0 mM Ca2+ (Figure 3A). The observed Ca2+ requirement for thermostability and increase in IWF signal indicated CAPN5-PC requires millimolar Ca2+ concentrations to achieve a conformational change. We performed SAXS analysis to gain better insight into CAPN5-PC’s behavior in solution. CAPN5-PC samples were thoroughly dialyzed against the SAXS buffer with either 5 mM EDTA (Ca2+-free) or 10 mM CaCl2 (Ca2+-bound). Our DSF and IWF data suggested that 10 mM CaCl2 would be sufficient to induce a conformational change that could be observed with SAXS. The merged scattering profiles for both samples displayed no signs of aggregation or radiation damage based on the Guinier plot at low q values (Figure 3B). Kratky plots adopted a bell-shaped curve for both samples, indicating that they were globular and well-folded (Figure S4B). The pairwise distance distribution P(r) function for both samples revealed distinct distribution profiles despite the subtle change in Dmax between Ca2+-free (Dmax = 69 Å) and Ca2+-bound CAPN5-PC (Dmax = 64 Å) (Figure 3C; Table S2). The Ca2+-free CAPN5-PC displayed a more extended shape in solution (Rg = 23.7 ± 0.3 Å) than the Ca2+-bound form (Rg = 21.9 ± 0.3 Å). Theoretical scattering patterns calculated from our crystal structure were in agreement with the experimental scattering data for Ca2+-free CAPN5-PC (χ2 = 1.55). The theoretical scattering pattern of our CAPN5-PC structure was a relatively poor fit to the experimental scattering profile for Ca2+-bound CAPN5-PC (χ2 = 8.58) (Figure 3D), further supporting that the crystal structure conformation was more consistent with an inactive state.
Figure 3. Ca2+-Dependent Conformational Changes and Activity of CAPN5-PC.

(A) IWF signal (I340) was monitored at 340 nm by exciting at 280 nm at varying Ca2+ concentrations. Data are fit to the Hill equation (R2 = 0.75) and are displayed as mean ± SEM (n = 3).
(B) Scaled buffer-subtracted scattering curves for CAPN5-PC in the presence of 5 mM EDTA or 10 mM Ca2+. The linear portion of the Guinier region is displayed in the inset (R2 = 0.95 and 0.93 for Ca2+-free and Ca2+-bound CAPN5-PC, respectively).
(C) Pairwise distance distribution function P(r) of the averaged data.
(D) Theoretical scattering patterns calculated from the CAPN5-PC structure showing agreement with the experimental data (χ2 = 1.55 and 8.58 for Ca2+-free and Ca2+-bound, respectively).
(E) Fluorescence tracing of sLY-AMC (0.4 mM) hydrolysis by 10 μM CAPN5-PC (WT or p.C81S) in the presence of 10 mM Ca2+.
(F) Michaelis-Menten analysis of sLY-AMC hydrolysis by 10 μM CAPN5-PC in the presence of 10 mM Ca2+ (R2 = 0.96). Data is displayed as mean ± SEM (n = 3).
(G) Effect of increasing concentrations of Ca2+ on Ac-LLY-AFC hydrolysis (0.1 mM) by 0.5 μM CAPN5-PC. Data are fit to the Substrate-Inhibition equation (R2 = 0.96). CAPN5-PC activity at μM Ca2+ levels (inset). Data are fit to the Hill equation (R2 = 0.97) and displayed as mean ± SEM (n = 3).
(H) Inhibition of Ac-LLY-AFC hydrolysis (0.2 mM) by 0.5 μM CAPN5-PC in the presence of 10 mM Ca2+ by E-64 or hCAST-B27. Data were fit to the Hill equation (R2 = 0.98 and 0.94 for E-64 and hCAST-B27, respectively) and displayed as mean ± SEM (n = 3).
The Calpain-5 Protease Core Requires High Calcium Concentrations for Maximal Enzymatic Activity
We probed CAPN5-PC activity using in vitro fluorescence resonance energy transfer (FRET) assays measuring hydrolysis of synthetic oligopeptides. When assayed with N-succinyl-Leu-Tyr-7-amido-4-methylcoumarin (sLY-AMC), CAPN5-PC was catalytically active in the presence of 10 mM Ca2+ and inactive in the presence of 5 mM EDTA (data not shown) (Sasaki et al., 1984). A catalytically inactive version of this enzyme, carrying a p.C81S mutation, displayed no observable activity (Figure 3E). The steady-state kinetic parameters of sLY-AMC hydrolysis by CAPN5-PC in the presence of 10 mM Ca2+ (kcat = 7.4 × 10−5 ± 3 × 10−6 s−1, Km = 131 ± 18 μM) fell in the range of values reported previously for classical calpain cores (Figure 3F; Table S3) (Davis et al., 2007; Moldoveanu et al., 2002, 2003, 2004b). We also assayed CAPN5-PC with another synthetic calpain substrate, Ac-Leu-Leu-Tyr-7-amino-4-trifluoromethylcoumarin (Ac-LLY-AFC), which displayed higher catalytic efficiency over sLY-AMC in the presence of 10 mM Ca2+ (kcat = 1.2 × 10−2 ± 8 × 10−4 s−1, Km = 153 × 102 ± 38 μM).
A Ca2+ titration in the presence of 0.1 mM Ac-LLY-AFC revealed that the half-maximal activity of CAPN5-PC occurred at 12 ± 4 mM Ca2+ (Figure 3G). This Ca2+ requirement is orders of magnitude higher than the reported [Ca2+] for half-maximal activity of the classical calpains CAPN1 and CAPN2 (3–50 μM and 200–1,000 μM, respectively) (Goll et al., 1992). CAPN5-PC displayed modest activity in the micromolar range (Figure 3G, inset). This high Ca2+ requirement could be an evolutionary adaptation for CAPN5’s expression in tissues with high basal Ca2+ levels. Interestingly, we observed a reduction in CAPN5-PC activity at Ca2+ concentrations >50 mM, suggesting that high Ca2+ levels may serve as an additional layer of regulation for CAPN5 activity. The Ca2+-requirement for CAPN5-PC activity in vitro is orders of magnitude higher than the nanomolar [Ca2+] observed in cells (Campbell and Davies, 2012), although the physiological relevance of the protease core alone is not known. It is hypothesized that localized increases in [Ca2+] result in transient calpain activation (i.e., influx at the site of a Ca2+-channel). Indeed, total intracellular [Ca2+] can increase up to three times the resting level during neuroinflammation (Hoffmann et al., 2003). CAPN5-PC activity was inhibited by E-64 (trans-epoxysuccinyl-L-leucylamido-(4-guanidino)-butane; IC50 = 1.2 ± 0.3 μM), a broad irreversible cysteine protease inhibitor, and the human CAST B27 peptide (residues 184–210; IC50 = 1.0 ± 0.2 μM) (Figure 3H) (Betts et al., 2003). Taken together, these results confirm that CAPN5-PC displays Ca2+-sensitive enzymatic activity characteristic of calpains but has varied proteolytic activity for different synthetic calpain substrates. Although CAPN5-PC activity can be inhibited by the B27 peptide in vitro, the full-length CAPN5 protein lacks the PEF-hand domains required to bind the full-length CAST (Hanna et al., 2008). Therefore, it is unlikely that CAST significantly inhibits CAPN5 activity in vivo, and other forms of regulation are important for this non-classical calpain.
Structure-Based Phylogenetic Analysis Reveals Unique Features in CAPN5-PC Are Absent in Classical Calpains
Although very divergent in their sequences and functions, calpains display remarkable similarity in the 3D structure of their protease cores (Campbell and Davies, 2012; Matena et al., 1998; Ono and Sorimachi, 2012). This structural similarity has presented challenges in the design of specific therapeutic inhibitors, because most calpain inhibitors are active-site directed (Ono et al., 2016). Thus, determining subtle structural similarities and differences between calpain protease cores would aid in the design of calpain-specific inhibitors. Earlier sequence-based phylogenetic studies of calpain paralogs highlighted that they have a long evolutionary history and are highly conserved across multiple species (Matena et al., 1998). However, the structural basis of their functional diversity is not completely understood, especially with regards to non-classical calpains. Primary sequence alignments and secondary structure predictions often miss key differences between structures due to the complexity of modeling non-local interactions between residues that are far away in their relative sequence position but are close in 3D space (Dill and MacCallum, 2012; Dill et al., 2008; Yang et al., 2018). Diversity in the length and composition of flexible loop regions can also produce large variations in sequence-based alignments (Carpentier and Chomilier, 2019). We therefore utilized a structure-based phylogenetic approach to examine the functional diversity among calpain protease cores (Balaji and Srinivasan, 2007). Twenty-six calpain protease core structures have been solved in the presence or absence of bound Ca2+ or an inhibitor (CAPN1, CAPN2, CAPN3, CAPN8, and CAPN9) (Davis et al., 2007; Moldoveanu et al., 2002; Ye et al., 2018). These structures were evaluated by their reported global validation metrics in PDB-REDO, including Rfree, Clashscore, Ramachandran outliers, side chain outliers, and normalized real-space R value outliers (Gore et al., 2017; Joosten et al., 2009). Structures meeting at least 3 out of the 5 metrics (20 structures total) were used in the analysis. Structures not meeting these criteria were excluded. Pairwise structural comparisons of PC1 and PC2 domains were used to calculate structural dissimilarity matrices (SDM) based on the RMSD between protein alpha carbon (Cα) atoms (Zhu et al., 2010). Structure-based phylogenetic trees were then generated from these SDM matrices using the unweighted pair group method with arithmetic mean (UPGMA) (Sneath and Sokal, 1973).
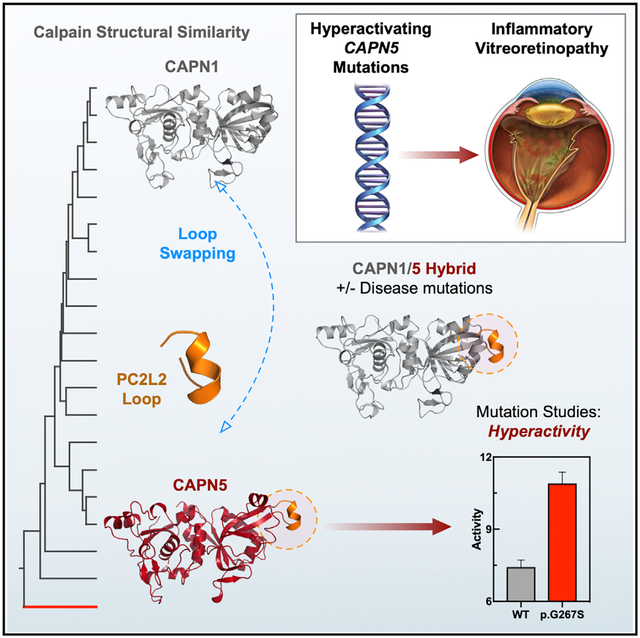
The structural phylogenetic tree for PC1 showed separation of calpain structures in two distinct clusters with a majority of structures grouped in the first cluster (Figure 4A). Interestingly, CAPN5 clustered separately from the other calpain PC1 structures. Superimposition of PC1 structures from this analysis revealed conformational differences with respect to their flexible loop regions (Figure 4B). Notably, CAPN5 PC1 contained a flexible, surface-exposed loop that was longer than corresponding loops on classical calpain structures. We termed this loop PC1L1, denoting its position on the PC1 domain and proximity to the N terminus (Figure 4B). Repeated analysis excluding the flexible loop regions showed similar separation of CAPN5 from classical calpain structures based on secondary structure elements alone (Figure S5). The structural phylogenetic tree for the PC2 domain similarly showed separation of calpain structures in two distinct clusters (Figures 4C and 4D). Interestingly, CAPN5’s PC2 domain contained two additional, elongated surface-exposed loops, which we termed PC2L1 and PC2L2 (denoting their proximity to the C terminus) (Figure 4D). Multiple sequence alignment of calpain isoforms revealed that these flexible loops correspond to “gaps” within the target-template alignment (Figures S6 and S7). Although the presence of these loops could be predicted from the primary sequence alignment, a detailed structure-based comparison provided more precise information on the secondary structure elements on these loops and their surrounding regions (Figure 4D). Additionally, CAPN5-PC contained an elongated α8 helix and several elongated β strands compared to classical PC2 domains (Figure 4D). Thus, without a crystal structure or accurate template, one could not reliably infer the presence of these motifs from the primary sequence alone. Interestingly, we observed variable evolutionary conservation scores among residues in the three elongated flexible loops on ConSurf analysis. Residues in the PC1L1 loop were highly conserved, while the PC2L1 loop displayed considerable variability (Figure 4E). The N-terminal region of the PC2L2 loop was highly conserved, while its α-helical residues were variable (Figure 4E). Taken together, these data reveal the presence of three elongated flexible loops on CAPN5-PC of unknown function that are sequentially conserved and structurally unique to CAPN5 compared to classical calpains.
Figure 4. Structural Phylogenetic Analysis Reveals Unique Features in CAPN5-PC.

(A) Structure-based phylogenetic tree of PC1 domains. The evolutionary distance was inferred using the UPGMA method. The tree is drawn to scale, with branch lengths corresponding to the Cα RMSD (Å) of the pairwise structural alignments. Branches are labeled according to the calpain paralog structures with respective PDB IDs in parentheses. The corresponding sequence-based tree is shown in the inset.
(B) Superimposition of PC1 structures used to construct the phylogenetic tree. Calpain structures are colored by their respective clustering in the phylogenetic tree.
(C) Structure-based phylogenetic tree of calpain PC2 domains.
(D) Superimposition of PC2 structures.
(E) Sequence logo diagrams showing the amino acid distribution in the PC1L1, PC2L1, and PC2L2 loops. The x axis lists the corresponding position in human CAPN5-PC. The height of each position is proportional to its conservation (among 454 CAPN5 sequences) and the height of the letter to its frequency at that position.
Disease-Causing Mutation in the Unique CAPN5 PC2L2 Loop Reveals a Regulation Mechanism for Calpain Activity
We next mapped known NIV mutations to the CAPN5-PC structure: p.R243L, p.L244P, p.K250N, p.G267S, and p.R289W (Figures 5A and 5B). Interrogation of our sequence-based phylogenetic data revealed the percent conservation (among chordates) of residues at NIV mutation sites: Arg243 (68.3%), Leu244 (73.3%), Lys250 (81.3%), Gly267 (81.3%), and Arg289 (53.1%). Interestingly, a previously identified patient mutation (CAPN5 c.799G>A, p.Gly267Ser) mapped to the PC2L2 loop on CAPN5-PC (Randazzo et al., 2019). This de novo heterozygous missense mutation was different than previously identified NIV mutations; it was not located on a gating loop near the active site (Figure 5B). The PC2L2 loop connects two β strands that contain the catalytic His252 and Asn284, suggesting that it may regulate CAPN5 activity through remote mechanisms (FigureS2). The patient harboring this mutation was a 44-year-old Caucasian male with a severe NIV phenotype and advanced disease (Figures 6A–6H). The patient displayed progressive visual problems from infancy with myopia, congenital nystagmus, and optic nerve myelination. By 19 years, the patient developed progressive, severe vitreoretinopathy, which resulted in sub-retinal exudative fluid and fibrosis leading to bilateral tractional retinal detachments. He later developed neovascular glaucoma in his fourth decade of life. The presence of this mutation on the PC2L2 loop suggested a functional role for this flexible region in regulating CAPN5 activity.
Figure 5. Disease-Associated Mutations in CAPN5-PC.

(A) Multiple sequence alignment of PC2 from human calpain paralogs showing the location of NIV-causing mutations and functional residues on the primary structure. The p.G267S mutation is located on the PC2L2 loop shared between CAPN5 and CAPN6.
(B) Ribbon tracing representation of CAPN5-PC (light gray) highlighting location of mutations implicated in NIV. Green ribbon, G1 gating loop; pink ribbon, G2 gating loop; orange ribbons, PC1L1, PC2L1, and PC2L2 loops.
Figure 6. The Unique CAPN5 PC2L2 Loop Remotely Regulates Calpain Activity.

Clinical features of a NIV patient harboring a p.G267S mutation.
(A) Fundus image of the normal retina.
(B) Fundus image of the affected patient showing severe vasoproliferative vitreoretinal pathology.
(C) Infrared image of the normal retina.
(D) Infrared image of the affected patient showing sub-retinal exudate.
(E) Fluorescein angiogram of the normal retina.
(F) Fluorescein angiography of the affected patient reveals central retinal degeneration.
(G) Optical coherence tomography (OCT) of the normal retina.
(H) OCT of the affected patient reveals gross disturbance of normal retinal morphology secondary to unusual retinal nerve fiber layer myelination and fibrosis.
(I) Michaelis-Menten analysis of sLY-AMC hydrolysis by 10 μM WT and p.G267S CAPN5-PC in the presence of 10 mM Ca2+ (R2 = 0.96 and 0.96 for WT and p.G267S, respectively). Data are displayed as mean ± SEM (n = 3).
(J) Schematic representation of hybrid calpain constructs. Alignment of the PC2L2 loop region between human CAPN5, rat CAPN1, and the rat CAPN1/5 hybrid is represented below. SS denotes secondary structure annotations corresponding to CAPN5-PC.
(K) Hydrolysis of 0.2 mM sLY-AMC by 10 μM MBP-rCAPN1/5 (WT and p.G267S) in the presence of 10 mM Ca2+.
We purified CAPN5-PC p.G267S and validated its correct folding by circular dichroism (CD) spectroscopy (Figure S8). The kinetic parameters for sLY-AMC hydrolysis by CAPN5-PC p.G267S (kcat = 1 × 10−4 ± 5 × 10−6 s−1, Km = 153 ± 18 μM) indicated it was hyperactive (Figure 6I; Table S3). Introduction of p.G267S mutation led to an increase in substrate turnover and catalytic efficiency, confirming that this PC2L2 loop mutation results in a hyperactivity. This subtle change in kcat is biologically meaningful due to the slow timescale of NIV disease progression. Patients are not born with the disease, and NIV often takes 20 or more years to develop (Bassuk et al., 2015; Mahajan et al., 2012; Velez et al., 2017, 2018; Wert et al., 2015). DSF and IWF results for CAPN5 p.G267S were indistinguishable from WT (data not shown) suggesting its hyperactivity was independent of Ca2+-induced conformational changes or stabilization. We therefore hypothesized that this loop’s length and dynamics may be evolutionarily designed to remotely regulate calpain activity. Using a synthetic biology approach, we introduced the CAPN5 PC2L2 loop (residues 260–275) into rat CAPN1-PC, a very distant calpain in terms of sequence and structural phylogeny (Figures 1D and 4). A recombinant CAPN1/5 hybrid molecule was created (Figure 6J) and purified with an N-terminal maltose binding protein (MBP) tag, which was more stable than the untagged enzyme (data not shown). The WT MBP-rCAPN1/5-PC hybrid molecule was catalytically active and hydrolyzed sLY-AMC. Introduction of the p.G267S mutation into the CAPN1/5 hybrid similarly led to an increase in activity, confirming that the PC2L2 loop can remotely regulate catalytic activity across ~43 Å (Figure 6K).
DISCUSSION
Calpain hyperactivity is implicated in a number of neurodegenerative diseases, and gain-of-activity mutations in CAPN5 cause NIV. However, the mechanisms underlying CAPN5’s role in the pathogenesis of NIV are poorly understood. The regulatory roles of classical calpains in mediating neuronal cell death and necrosis have been extensively studied, including the cleavage of JNK-interacting protein-1 and Bax, which trigger activation of cell death signaling pathways (Cheng et al., 2018; Wert and Mahajan, 2019). Aberrantly activated calpains (e.g., CAPN1 and CAPN2) also process a number of substrates in the CNS that promote cell necrosis, including vimentin, tubulins, and integrins (Azuma and Shearer, 2008; Cheng et al., 2018). These calpain-mediated events are thought to contribute to the pathogenesis of several neurodegenerative diseases, including retinal degeneration and hypoxia-induced glaucoma (Azuma and Shearer, 2008; Cheng et al., 2018; Huang et al., 2010). Detailed structural studies of classical calpains have revealed mechanistic insights into the substrate specificity between calpain isoforms (Moldoveanu et al., 2004a). The calpain active site is gated by two flexible loops on each side of the active site cleft. It is hypothesized that these loops mold around substrates and that the sequence variations in these loops confer specificity to calpain substrates and inhibitors (Moldoveanu et al., 2004a). We have previously demonstrated that activity of CAPN1 can be altered by the substitution of CAPN5 gating loops, with and without NIV disease mutations (Wert et al., 2015). It is unclear, however, if CAPN5 contributes to NIV pathogenesis through distinct or overlapping mechanisms and if it processes the same protein targets as CAPN1 and CAPN2 in the retina. Furthermore, the structural basis by which NIV mutations cause CAPN5 hyperactivity is poorly understood. These knowledge gaps highlight the need for detailed structural and functional studies of non-classical calpains, like CAPN5.
Here, we present the high-resolution structure of a non-classical calpain protease core. Most crystal structures of calpains that have been determined are activated forms (with bound Ca2+ ions and the catalytic triad in formation). It has been difficult to crystallize calpains without the active site cysteine-to-serine/alanine mutation (e.g., p.C81S). Here, we report the structure of the WT (no inactivating mutation) Ca2+-free CAPN5 protease core. We identified several unique features of CAPN5-PC: it contains elongated flexible loops (PC1L1, PC2L1, and PC2L2) and an elongated α8 helix compared to its classical counterparts (Figure 4). Although the functional significance of most of these regions remains unknown, the presence of the p.G267S mutation on the PC2L2 loop indicates a role for this region in regulating enzyme activity. This suggests that these features have co-evolved with CAPN5’s unique domain topology. Our ability to transfer this regulatory mechanism into rat CAPN1 by swapping in the CAPN5 PC2L2 loop supports the concept of an evolutionary mechanism for fine-tuning calpain function and activity by modifying the flexible loops. Furthermore, the identification of this unique structural motif on CAPN5-PC suggests that this region may be an attractive target for the design of specific allosteric inhibitors for CAPN5.
Previously identified NIV mutations in the PC2 domain are in close proximity to the active site groove and are predicted to disrupt regulatory motifs that would otherwise prevent aberrant CAPN5 activity. For example, the p.R243L, p.L244P, andp.K250N mutations are predicted to disrupt the gating function of the G1 loop, which may leave the active site groove accessible to substrates, even in the absence of Ca2+ (Figure 5B) (Bassuk et al., 2015; Mahajan et al., 2012; Velez et al., 2018). Thep.G267S mutation, however, is located far from the active site in a solvent-exposed region of the structure. The PC2L2 loop did not appear to make significant interactions with the rest of the PC2 domain nor the neighboring molecules. However, substitution from a glycine to a serine in this region may affect the dynamics or hydrogen-bonding network of this loop and could confer hyperactivity through remote effects. Interestingly, the phenotype of the p.G267S patient is distinct from originally identified NIV kindreds. In these original pedigrees, disease onset did not begin until the second decade of life. However, the p.G267S patient displayed pediatric retinal disease, suggesting a geno-type-phenotype correlation that may link enzymatic activity to disease severity. This correlation is supported by our previously published studies on the G1 loop mutations (e.g., p.R243L) (Wert et al., 2015) and the syndromic, pediatric NIV variant (p.R289W) (O’Keefe et al., 2019; Velez et al., 2018; Wang et al., 2018). More detailed kinetic studies of additional CAPN5 NIV mutants are required to determine if the level of hyperactivity correlates to disease onset and/or severity. Due to the location of the p.G267S mutation on a surface-exposed loop, it is possible that alterations in this region may affect protein-protein interactions. However, we observed a similar relative hyperactivity in the CAPN1/5 chimera in the absence of non-catalytic domains and binding partners. At this stage, we cannot formally exclude the possibility of the mutation affecting interactions with non-catalytic domains and/or other proteins, which would require future research on the full-length CAPN5 and identification of cognate interaction partners in the retina.
Our functional analyses addressed the activity and Ca2+ dependence of CAPN5-PC in vitro. IWF and SAXS revealed that CAPN5-PC undergoes a conformational change in the presence of Ca2+. A high-resolution structure of the Ca2+-bound CAPN5-PC would also reveal more detailed structural rearrangements that occur during this transition as well as elucidate the locations of its Ca2+-binding sites. We established that CAPN5-PC is active even in the absence of its CBSW and C2 domains and that it displays characteristic calpain activity when assayed with common calpain substrates. The observation that CAPN5-PC hydrolyzes similar peptide substrates suggests that there is some overlap in the protein targets of classical and non-classical calpains (Figure 3). Although we observe hyperactivity of CAPN5-PC p.G267S toward sLY-AMC, it is unclear if this would translate to hyperactivity toward the endogenous retinal substrates responsible for NIV development. Identification of the natural retinal substrates of CAPN5 will determine which signaling pathways are affected by hyperactivity. Our enzymatic studies also revealed the high Ca2+ requirements of CAPN5-PC activation in vitro. Interestingly, studies with CAPN2 showed that phosphorylation at Ser50 by extracellular signal related kinase (ERK) reduced the enzyme’s Ca2+requirement for activation (Glading et al., 2004). It is possible that post-translational modifications of CAPN5 may reduce its high Ca2+ requirement in vivo. The relatively low catalytic efficiency of CAPN5-PC in vitro may be attributed to the lack of structural support from its non-catalytic domains. This phenomenon has been observed for classical calpain cores (e.g., CAPN1), which display only 5%–10% of the activity of the whole enzyme (Campbell and Davies, 2012). Another possible explanation for the observed low kcat is that sLY-AMC and Ac-LLY-AFC are not natural or preferred substrates for CAPN5. These findings highlight the need to study enzymatic activity, Ca2+ dependence, and effects of disease-causing variants using the full-length CAPN5 protein and validated natural substrates.
While classical calpain regulation has been extensively studied, the mechanisms underlying CAPN5 regulation remain poorly understood (Campbell and Davies, 2012; Goll et al., 1992). CAST inhibits classical calpain activity by forming interactions with all five domains (Campbell and Davies, 2012; Hanna et al., 2008). Although our experiments show that CAPN5-PC activity can be inhibited by the CAST inhibitory region (B27 peptide) in vitro, the full-length CAPN5 protein does not contain the classical calpain domains required to bind full-length CAST. Therefore, it is likely that other forms of regulation are important. We anticipate that isolation of CAPN5-binding partners will further our understanding of its regulation at the cellular level. Our enzymatic studies revealed that CAPN5-PC requires millimolar concentrations of Ca2+ for maximal activity in vitro, similar to CAPN2. Because CAPN5 is highly expressed in cells with high resting concentrations of Ca2+, such as retinal photoreceptors (~300–700 nM) and intestinal epithelial cells (~140 nM), this high Ca2+ requirement may provide an additional safeguard to prevent aberrant CAPN5 activity in these cells (Brunet et al., 2000; Krizaj and Copenhagen, 2002; Schaefer et al., 2016, 2017). Our data provide important insight into the structural and functional mechanisms of CAPN5 activation and highlight unique characteristics of this disease-associated protein. Further structural and functional studies of CAPN5 will elucidate the mechanisms underlying NIV pathogenesis and guide the design of specific inhibitors to treat this blinding eye disease.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Vinit B. Mahajan (vinit.mahajan@stanford.edu). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Escherichia coli BL21 (DE3) cells were used for calpain expression and crystallization.
Patients
The patient described in the study was a 44-year-old Caucasian male with end-stage NIV disease harboring a CAPN5 c.799G > A, p.Gly267Ser mutation. The study adhered to the tenets of the Declaration of Helsinki. Informed consent was obtained from all patients for study participation as per standard NHS Trust policy.
METHOD DETAILS
Clinical testing
Peripheral blood samples were taken for next generation DNA sequencing as previously described (Randazzo et al., 2019). Clinical ophthalmic imaging (fundus photography, infrared imaging, fluorescein angiography, and spectral domain optical coherence tomography) was obtained with the Optos ultra-widefield camera and Heidelberg Spectralis Tracking Laser Tomography (v 6.6.2).
Sequence-based phylogenetic analysis
We first searched the UniProt database for reviewed entries denoted as calpains (containing a calpain protease core domain). This initial search yielded a total of 57 manually curated sequences. A seed multiple sequence alignment (MSA) was then constructed by using MAFFT v7 with default parameters (alignment strategy: FFT-NS-1) (Katoh and Standley, 2013). Using HMMER-3.1 and the seed alignment we produced an HMM profile and used it to broaden the search against the UniProt database (which was restricted to the phylum Chordata) (Finn et al., 2011). This search yielded a total of 8,113 proteins. We discarded fragmented sequences (< 300 amino acids) that appeared too short to truly represent the full protease core domain fold and redundant proteins were further filtered using CD-HIT v4 with a threshold of 100% sequence identity (Li and Godzik, 2006). This resulted in a pool of 5,284 proteins that were aligned using MAFFT v7 (alignment strategy FFT-NS-2) (Katoh and Standley, 2013). We used FastTree v2 to generate a fast approximation of the unrooted phylogenetic tree for the large pool of calpain sequences. Sequences representing the CAPN5 sub-tree (522 sequences) were extracted and realigned using MAFFT v7 (alignment strategy: FFT-NS-1) (Price et al., 2010). Taxa (sequences) producing many gaps in the alignment were removed using MaxAlign (Gouveia-Oliveira et al., 2007). Phylogenetic tree reconstruction of the CAPN5 family sub-tree (454 sequences) was performed using Geneious Tree Builder, Jukes-Cantor distance model, and Neighbor-Joining tree build method with 100 bootstrap replicas. Sequence logos were generated using Skylign (http://www.skylign.org).
Cloning and protein expression
Calpain protease core domain (PC1-PC2) sequences (residues 1–343) were cloned into a pUC57 vector with an Xho I restriction site, with an N-terminal thrombin cleavage site, and a C-terminal TEV protease cleavage site followed by a 6xHis tag, as previously described (Gakhar et al., 2016; Velez et al., 2018). Calpain constructs were transferred into the pMAL-C5X vector to obtain an N-terminal maltose binding protein (MBP) as a fusion partner that could be cleaved off with thrombin. Sequence of the calpain flanking regions was confirmed by sequencing of constructs. Plasmids were amplified and isolated from DH5α cells and then transformed into E. coli BL21 (DE3).
Protein expression and purification
For structural studies, samples of CAPN5-PC were prepared to a purity greater than 95%, as described below. E. coli BL21 (DE3) cells expressing CAPN5-PC were grown in 1 L shake-flask cultures, at 250 rpm in Terrific Broth (Invitrogen), at 37°C until an OD 600 of 1.0 and then induced with 0.2 mM IPTG. Cells were grown at 16°C for 19 hours, harvested, and centrifuged at 4,000 × g for 20 minutes and pellets (40 mL per Liter of cell culture) were re-suspended immediately or frozen at −20°C for later use. Cell pellets were re-suspended in 35 mL of lysis buffer (20 mM HEPES, 300 mM NaCl, 2 mM DTT, pH 7.5, one tablet of EDTA-free protease inhibitor (Roche), DNase (Roche)) and lysed using a sonic dismembrator (Fisher Model 705) and centrifuged for 1 hour at 18,000 × g at 4°C. Cell debris was discarded and the supernatant (30 mL) loaded onto a gravity-flow column packed with 15 mL (1CV) amylose resin (New England Biolabs). The column was washed with 5 to 10 CV of wash buffer (20 mM HEPES, 300 mM NaCl, pH 7.5) and eluted with 6 to 8 CV of elution buffer (20 mM HEPES, 300 mM NaCl, 10 mM maltose, pH 7.5). Eluted fractions were pooled and concentrated to 2 mL at 12 mg/mL (30 kDa NMWL spin concentrator; Millipore). MBP was removed with thrombin (1 units/mg of protein; thrombin from bovine plasma, T7201, Sigma-Aldrich) at 4°C for 16 hours and the reaction mix passed over Ni-NTA (QIAGEN) resin to remove cleaved-off MBP using 6 CV wash buffer (20 mM HEPES, 500 mM NaCl, 10 mM Imidazole, glycerol, pH 7.5). Cleaved and remaining un-cleaved CAPN5-PC was eluted with 5 CV of elution buffer (20 mM HEPES, 500 mM NaCl, 200 mM Imidazole, 5% glycerol, pH 7.5). The eluted protein was concentrated with a 10 kDa NMWL spin concentrator and passed over a Superdex 75 (GE) size-exclusion (SEC) column connected to an ÄTKA™ pure fast protein liquid chromatography (FPLC) system (GE Healthcare Inc.) to remove residual, un-cleaved fusion protein. The column was equilibrated with elution buffer (20 mM HEPES, 150 mM NaCl, 2 mM DTT, 5% glycerol, pH 7.5). For kinetic studies, samples of CAPN5-PC were prepared as described above, but with Tris-HCl instead of HEPES (Gakhar et al., 2016; Velez et al., 2018; Wert et al., 2015). Purity was assessed at each step on Coomassie-stained SDS-PAGE gels.
Crystallization and structure determination
Crystallization of the wild-type human CAPN5-PC was performed by the hanging drop vapor diffusion method at 18°C. Prior to addition to the drop, the final protein concentration was 10 mg/mL in 20 mM HEPES (pH 7.5), 150 mM NaCl, 2 mM DTT, and 5% glycerol. Crystals (soft, thin plates) initially grew overnight in 100 mM NaH2PO4 (pH 6.5) and 12% PEG 8000 (ProPlex HT-96 Screen, Molecular Dimensions). Optimization was carried out by screening around the initial hit condition with the addition of EDTA (from 0 to 75 mM) and/or CaCl2 (10 mM). Diffraction-quality crystals (large plates) were grown by mixing protein 1:1 with a reservoir solution containing 250 mM NaH2PO4 (pH 6.0), 12% PEG 8000, 36 mM EDTA, 10 mM CaCl2. Crystals grew thicker with the addition of 0.4 mM H-E(EDANS)-KIEIVRKKPIFKKATV-K(DABCYL)-OH peptide (Biopeptek) to the protein solution prior to mixing reservoir solution. Diffraction data were collected on flash-cooled crystals at the 4.2.2 beamline (Advanced Light Source, Lawrence Berkeley National Laboratory). Crystals belonged to the P 1 21 1 space group with two molecules per asymmetric unit. The datasets were indexed, integrated, and scaled using XDS (Kabsch, 2010). The data were truncated to 2.8 Å resolution, where I/σ (I) dropped to 1.73, to reduce noise in the electron density maps during refinement. Molecular replacement calculations were carried out with PHASER (McCoy et al., 2007) through the PHENIX software suite (Adams et al., 2011). Our previously published SAXS-guided homology model of CAPN5-PC was used as a search model (Gakhar et al., 2016). The structure was then refined with REFMAC5 (Murshudov et al., 2011) and PHENIX (Adams et al., 2011) using non-crystallographic symmetry (NCS; torsion-angle) restraints. All inspections and manual manipulations were completed with COOT (Emsley and Cowtan, 2004). Model geometry was improved using PDB-REDO (Joosten et al., 2009). Figures were generated in PyMOL (The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC). Crystallographic data collection and refinement statistics are summarized in Table 1. Atomic coordinates were deposited with the Protein Data Bank (PDB) under accession code 6P3Q.
ConSurf analysis
The CAPN5-PC crystal structure was used as input for analysis in the ConSurf server (Armon et al., 2001; Ashkenazy et al., 2016; Glaser et al., 2003). The 454 CAPN5 sequences from our sequence-based phylogenetic analysis underwent multiple sequence alignment using MAFFT and conservation scores were calculated using the Bayesian method option in ConSurf as previously described (Katoh and Standley, 2013; Wert et al., 2019). The ConSurf scores were mapped onto the B-value column of the CAPN5-PC crystal structure and visualized using PyMOL.
Circular dichroism analysis
Circular dichroism (CD) spectra were collected using a Jasco J-815 spectrometer in the 200–280 nm range with a 1 mm Spectrosil (Starna) quartz cuvette at 25°C, with a data interval of 0.1 nm and scan speed of 100 nm/min. Purified CAPN5-PC samples (wild-type and p.G267S) were dialyzed against 10 mM Na2PO4 (pH 7.5) and diluted to a concentration of 0.5 – 0.7 mg/mL. Ten measurements for each sample were averaged and normalized for total protein concentration after buffer subtraction.
Differential scanning fluorimetry
Purified CAPN5-PC samples (1 mg/mL) were analyzed by differential scanning fluorimetry (DSF) to test the ability of added calcium to bind and alter its thermal stability. Assays were performed on a Bio-Rad CFX96 Touch Thermal Cycler-CFX real-time-PCR system using the FRET channel and contained 300 mM NaCl, 20 mM HEPES (pH 7.5), 2 mM DTT, 5% glycerol, and 5x SYPRO-Orange dye (Invitrogen). Temperature was increased from 1°C /min from 4°C to 100°C in the presence of increasing concentrations of added CaCl2 (0.5 to 100 mM). As the temperature rose, the protein unfolded to expose hydrophobic residues to the SYPRO-Orange dye, leading to an increase in fluorescence signal. The temperature at which the protein starts to unfold is called the melting temperature (Tmelt). Fluorescence traces were analyzed with DMAN software (Wang et al., 2012).
Intrinsic tryptophan fluorescence measurements
Intrinsic tryptophan fluorescence measurements were performed on a fluorimetric plate reader (Tecan Spark, Männedorf Switzerland) at 22°C in a final volume of 100 μL, in 20 mM Tris-HCl (pH 7.5), 300 mM NaCl, 2 mM DTT, and 1 μM CAPN5-PC with 5 mM EDTA or CaCl2 (0.1 mM to 100 mM). Excitation and emission wavelengths were set to 280 nm and 340 nm, respectively. Fluorescence intensities (I340) were normalized and fit to the Hill equation.
SAXS data collection and analysis
Purified CAPN5-PC samples were thoroughly dialyzed against 20 mM Tris (pH 7.5), 150 mM NaCl, 5 mM DTT with either 5 mM EDTA or 10 mM CaCl2. SAXS data were collected on the 4–2 beamline at the Stanford Synchrotron Radiation Lightsource (SSRL), SLAC National Accelerator Laboratory. Scattering was measured by a Pilatus3 1M (Dectris) hybrid pixel array detector coaxial with the incident beam (Table S2). The SAXS data were collected (at 10°C) at three concentrations (0.3, 0.6, 1.2 mg/mL) in the order of lowest to highest concentration with 1 s exposures for 20 s resulting in 20 frames per concentration. Scattering data were reduced using SasTool and beamline scripts. The data from frames before radiation damage occurred were averaged. Scattering curves of the dialysate buffer were collected and subtracted from each protein scattering profile. To assess aggregation or interparticle interference, samples were assessed by analyzing the Guinier region (low-q region) of the scattering data using AutoRg and related programs from the ATSAS Program package (Konarev et al., 2006). Interparticle interference was observed at the highest concentration. Therefore, the curves from the lowest two concentrations (where Guinier regions were equivalent) were merged to give a single scattering profile for Ca2+-free and Ca2+-bound CAPN5-PC. The forward scattering I(0) and radius of gyration Rg were determined from Guinier approximation. The full scattering curves were estimated using the indirect Fourier transform method implemented in the program DATGNOM, along with the pair distribution function P(r), the maximum dimension of the macromolecule (Dmax), and the real space radius of gyration. The Porod volume was determined using the Porod invariant in PRIMUS and GNOM. Dimensionless Kratky analysis was used to determine the folded nature of CAPN5-PC samples in solution. Representative Kratky plots for partially-folded (unbound lysine riboswitch; 3LYSRR) and fully-unfolded protein (red; unfolded lysine riboswitch in Mg2+ free buffer; 2LYSRR) were used as reference (Garst et al., 2008). Simulated scattering curves were generated for the CAPN5-PC crystal structure using CRYSOL (Svergun et al., 1995).
Measurement of calpain activity
Calpain proteolytic activity was measured by hydrolysis of the synthetic calpain substrates, sLY-AMC (N-succinyl-Leu-Tyr-7-amido-4-methylcoumarin; Cayman) and Ac-LLY-AFC (Ac-Leu-Leu-Tyr-7-amino-4-trifluoromethylcoumarin; Fisher) (Wert et al., 2015). Briefly, 10 μM or 0.25 μM of purified calpain was added to a reaction buffer containing 20 mM Tris-HCl (pH 7.5), 300 mM NaCl, 2 mM DTT, and 0.1 to 100 mM CaCl2 or 5 mM EDTA and incubated on ice prior to reaction in 96-well plates (100 μL per reaction). Varying concentrations of substrate (10 μM to 1 mM) were added and the reaction was incubated at 37°C for 1 hour on a fluorimetric plate reader (Tecan Spark, Männedorf Switzerland). All experiments were performed in triplicate. The amount of AMC or AFC released during the experiment was calculated based on a standard curve of AMC or AFC concentrations (from 2 nM to 5 μM) and kinetic parameters were calculated by direct fitting to the Michaelis-Menten equation in GraphPad Prism 8. Inhibition experiments were carried out in similar manner with either E-64 (trans-epoxysuccinyl-L-leucylamido-(4-guanidino)-butane (Sigma) or the human CAST B27 domain (residues 184–210; Calbiochem) in the presence of 0.2 mM Ac-LLY-AFC. Reaction rates at various inhibitor concentrations (10 nM to 10 μM) were normalized and fit to the Hill equation in GraphPad Prism 8.
Structure-based phylogenetic analysis of calpain protease core domains
Calpain protease core structures were evaluated by their reported global validation metrics in PDB-REDO (Rfree, Clashscore, Ramachandran outliers, Sidechain outliers, and RSRZ outliers) (Gore et al., 2017; Joosten et al., 2009). Structures meeting at least 3 out of the 5 metrics, with percentile scores greater than X-ray structures of a similar resolution, were further evaluated. Structures not meeting these criteria were excluded (PDB: 1DF0, 1QXP; 1KFX, 1KFU, and 1U5I). Structures of individual calpain domains (PC1 and PC2) were superimposed using PyMOL to calculate the pairwise RMSD between protein alpha carbon atoms (Cα; The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC). A structural dissimilarity matrix (SDM) was constructed using the Cα RMSD values in order to generate a phylogenetic tree. The phylogenetic tree was constructed using the UPGMA (Unweighted Pair Group Method with Arithmetic Mean) method in MEGA7 (Molecular Evolutionary Genetics Analysis) software (Kumar et al., 2016; Sneath and Sokal, 1973).
QUANTIFICATION AND STATISTICAL ANALYSIS
Figure 3A: Intrinsic fluorescence intensity at varying Ca2+ concentrations. Data were fit to the Hill equation in GraphPad Prism 8 (R2 = 0.75) and displayed as mean ± SEM (n = 3; number of reactions).
Figure 3B: The linear portion of the Guinier region of the SAXS data (such that q × Rg < 1.3) is displayed in the inset. Data were fit to a linear regression in GraphPad Prism 8 (R2 = 0.95 and 0.93 for Ca2+-free and Ca2+-bound CAPN5-PC, respectively) and represent merged scattering curves from the lowest two concentrations (where Guinier regions were equivalent).
Figure 3D: Fit of the theoretical scattering patterns (calculated from the CAPN5-PC structure) to the experimental scattering data. The goodness of fit (χ2) was calculated in CRYSOL. χ2 = 1.55 and 8.58 for Ca2+-free and Ca2+-bound CAPN5-PC, respectively.
Figure 3F: Kinetic analysis of sLY-AMC hydrolysis by wild-type CAPN5-PC. Data were fit to the Michaelis-Menten equation in GraphPad Prism 8 (R2 = 0.96) and displayed as mean ± SEM (n = 3; number of reactions).
Figure 3G: Kinetic analysis of CAPN5-PC as a function of increasing Ca2+ concentrations. Data were fit to the Substrate-Inhibition equation in GraphPad Prism 8 (R2 = 0.96) and displayed as mean ± SEM (n = 3; number of reactions). The inset represents CAPN5-PC activity at micromolar Ca2+ levels. Data were fit to the Hill equation (R2 = 0.97) and displayed as mean ± SEM (n = 3; number of reactions).
Figure 3H: Inhibition of CAPN5-PC activity by E-64 and hCAST-B27. Reaction rates were normalized to fractional activity and fit to the Hill equation in GraphPad Prism 8 (R2 = 0.98 and 0.94 for E-64 and hCAST-B27, respectively). Data are displayed as mean ± SEM (n = 3; number of reactions).
Figure 6I: Kinetic analysis of sLY-AMC hydrolysis by wild-type and p.G267S CAPN5-PC. Data were fit to the Michaelis-Menten equation in GraphPad Prism 8 (R2 = 0.96 and 0.96 for wild-type and p.G267S, respectively) and displayed as mean ± SEM (n = 3; number of reactions).
Figure S4B: The melting temperature (Tmelt) of CAPN5-PC as a function of increasing Ca2+ concentrations. Data were fit to the Hill equation GraphPad Prism 8 (R2 = 0.96) and are displayed as mean ± SD (n = 3; number of reactions).
DATA AND CODE AVAILABILITY
Atomic coordinates for the reported structure have been deposited with the Protein Data Bank (PDB) under accession code 6P3Q.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. coli BL21 (DE3) cells | Invitrogen | Cat. # C600003 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ampicillin | Fisher Scientific | Cat. # BP1760–5 |
| Isopropyl β-D-Thiogalactoside (IPTG) | Sigma-Aldrich | Cat. # I6758 |
| Thrombin from bovine plasma | Millipore | Cat. # T7201 |
| SYPRO-Orange | Invitrogen | Cat. # S6651 |
| Amylose resin | New England Biolabs | Cat. # E8021L |
| Ni-NTA resin | QIAGEN | Cat. # 30210 |
| Superdex 75 Increase 10/300 GL | GE Healthcare | Cat. # 29148721 |
| N-succinyl-Leu-Tyr-7-amido-4-methylcoumarin (sLY-AMC) | Cayman Chemical | Cat. # 10008120 |
| Ac-Leu-Leu-Tyr-7-amino-4-trifluoromethylcoumarin (Ac-LLY-AFC) | Fisher Scientific | Cat. # MP3AFC11610 |
| Trans-epoxysuccinyl-L-leucylamido-(4-guanidino)-butane (E64) | Sigma-Aldrich | Cat. # E3132 |
| Human calpastatin B27 domain | Calbiochem | Cat. # 208902 |
| ProPlex HT-96 Screen | Molecular Dimensions | Cat. # MD1–42 |
| H-E(EDANS)-KIEIVRKKPIFKKATV-K(DABCYL)-OH | Biopeptek | Order #. BPT18360101–3 |
| Deposited Data | ||
| hCAPN5-PC | This work | PDB: 6P3Q |
| rCAPN1-PC complexed with ZLAK-3001 | Qian et al., 2008 | PDB: 2R9C |
| rCAPN1-PC complexed with ZLAK-3002 | Qian et al., 2008 | PDB: 2R9F |
| rCAPN1-PC complexed with SNJ-1945 | Cuerrier et al., 2006 | PDB: 2G8J |
| rCAPN1-PC complexed with leupeptin | Moldoveanu et al., 2004a | PDB: 1TL9 |
| rCAPN1-PC complexed with E64 | Moldoveanu et al., 2004a | PDB: 1TLO |
| rCAPN1-PC complexed with SNJ-1715 | Cuerrier et al., 2006 | PDB: 2G8E |
| hCAPN1-PC | Davis et al., 2007 | PDB: 2ARY |
| hCAPN1-PC complexed with ZLLYCH2F | Li et al., 2006 | PDB: 1ZCM |
| hCAPN9-PC complexed with leupeptin | Davis et al., 2007 | PDB: 2P0R |
| hCAPN9-PC | Davis et al., 2007 | PDB: 1ZIV |
| rCAPN2-PC | Moldoveanu et al., 2004a | PDB: 1MDW |
| rCAPN2 complexed with calpastatin | Hanna et al., 2008 | PDB: 3BOW |
| rCAPN2-PC complexed with calpastatin | Moldoveanu et al., 2004a | PDB: 3DF0 |
| hCAPN3-PC complexed with leupeptin | Yeetal., 2018 | PDB: 6BKJ |
| hCAPN3-PC complexed with E64 | Yeetal., 2018 | PDB: 6BJD |
| hCAPN3-PC C129S mutant | Yeetal., 2018 | PDB: 6BDT |
| hCAPN3-PC C129A mutant | Yeetal., 2018 | PDB: 6BGP |
| hCAPN8-PC | Davis et al., 2006 | PDB: 2NQA |
| Recombinant DNA | ||
| pMAL-C5X-hCAPN5-PC | This work | N/A |
| pMAL-C5X-hCAPN5-PC mutants | This work | N/A |
| pMAL-C5X-rCAPN1/5-PCDC2-loop | This work | N/A |
| pMAL-C5X-rCAPN1/5-PCDC2-loop mutants | This work | N/A |
| Software and Algorithms | ||
| XDS | Kabsch, 2010 | http://xds.mpimf-heidelberg.mpg.de/ |
| PHASER | McCoy et al., 2007 | http://www.ccp4.ac.uk/html/phaser.html |
| PHENIX | Adams et al., 2011 | http://www.phenix-online.org/ |
| REFMAC | Murshudov et al., 2011 | http://www.ccp4.ac.uk/html/refmac5.html |
| COOT | Emsley and Cowtan, 2004 | https://www2.mrc-lmb.cam.ac.uk/Personal/pemsley/coot/ |
| SasTool | 4–2 Beamline, SSRL | https://www-ssrl.slac.stanford.edu/smb-saxs/content/documentation/sastool |
| ATSAS | Konarev et al., 2006 | https://www.embl-hamburg.de/biosaxs/software.html |
| ScAtter | SIBLYS Beamline, ALS | https://bl1231.als.lbl.gov/scatter/ |
| CRYSOL | Svergun et al., 1995 | https://www.embl-hamburg.de/biosaxs/crysol.html |
| DMAN | Wang et al., 2012 | http://www.structuralchemistry.org/pcsb/dman.php |
| MODELLER | Webb and Sali, 2017 | https://salilab.org/modeller/ |
| PDB-REDO | Gore et al., 2017; Joosten et al., 2009 | https://pdb-redo.eu/ |
| PyMOL | Schrödinger, LLC | https://pymol.org/2/ |
| HMMER 3.1 | Finn et al., 2011 | http://hmmer.org/ |
| MAFFT v7 | Katoh and Standley, 2013 | https://mafft.cbrc.jp/alignment/software/ |
| CD-HIT | Li and Godzik, 2006 | http://weizhongli-lab.org/cd-hit/ |
| MaxAlign 1.1 | Gouveia-Oliveira et al., 2007 | http://www.cbs.dtu.dk/services/MaxAlign/ |
| FastTree 2 | Price et al., 2010 | http://www.microbesonline.org/fasttree/ |
| Geneious Tree Builder | Geneious, LLC | https://www.geneious.com/ |
| ConSurf | Armon et al., 2001; Ashkenazy et al., 2016 | https://consurf.tau.ac.il/ |
| MEGA7 | Kumar et al., 2016 | https://www.megasoftware.net/ |
| EsPript | Patrice Gouet and Xavier Robert | http://espript.ibcp.fr/ |
| GraphPad Prism | GraphPad Software, Inc. | https://www.graphpad.com/ |
Highlights.
The structure of the human calpain-5 protease core (CAPN5-PC) is determined
CAPN5-PC contains three elongated loops compared to its classical counterparts
One loop contains a mutation identified in a patient with NIV (p.Gly267Ser)
The p.Gly267Ser mutation causes hyperactivity in CAPN5-PC and CAPN1/5-PC hybrids
ACKNOWLEDGMENTS
We would like to acknowledge use of resources at the Carver College of Medicine’s Protein and Crystallography Facility at the University of Iowa. Part of this work was conducted at the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory and the Stanford Synchrotron Radiation Laboratory (SSRL), Stanford Linear Accelerator Center (SLAC), supported by DOE Office of Biological and Environmental Research (BER). We thank Jay Nix and staff at the ALS 4.2.2 beamline. We thank Tsutomu Matsui and the staff at the SSRL 4-2 beamline. Part of this work was performed at the Stanford Macromolecular Structure Knowledge Center. We thank Amnon Kohen and Zhen Xu for critical review, and Kellie A. Schaefer, Hanna J. Koster, and Irini Petros for technical assistance. This work was supported by NIH (R01EY026682, R01EY024665, R01EY025225, R01EY024698, R21AG050437, and P30EY026877 to V.B.M. and A.G.B. and F30EYE027986 and T32GM007337 to G.V.). R.E.M. is funded by the NIHR Oxford Biomedical Research Centre. S.W. is supported by the DOE BER and by the NIH (including P41GM103393 and R01HD084422); Stanford PRECOURT Institute, Stanford Child Health Research Institute; Pediatric Cancer Research Foundation; and NSF Major Research Instrumentation (DBI-1625906). Use of SSRL is supported by the DOE Office of Basic Energy Sciences (DE-AC02-76SF00515). The SSRL Structural Molecular Biology Program is supported by the DOE BER and by NIGMS (including P41GM103393). This research used resources of ALS, which is a DOE Office of Science User Facility under contract DE-AC02-05CH11231. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the sponsor.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.12.077.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Echols N, Headd JJ, Hung LW, Jain S, Kapral GJ, Grosse Kunstleve RW, et al. (2011). The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armon A, Graur D, and Ben-Tal N (2001). ConSurf: an algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J. Mol. Biol 307, 447–463. [DOI] [PubMed] [Google Scholar]
- Artal-Sanz M, and Tavernarakis N (2005). Proteolytic mechanisms in necrotic cell death and neurodegeneration. FEBS Lett. 579, 3287–3296. [DOI] [PubMed] [Google Scholar]
- Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, and Ben-Tal N (2016). ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44 (W1), W344–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma M, and Shearer TR (2008). The role of calcium-activated protease calpain in experimental retinal pathology. Surv. Ophthalmol 53, 150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaji S, and Srinivasan N (2007). Comparison of sequence-based and structure-based phylogenetic trees of homologous proteins: Inferences on protein evolution. J. Biosci 32, 83–96. [DOI] [PubMed] [Google Scholar]
- Bassuk AG, Yeh S, Wu S, Martin DF, Tsang SH, Gakhar L, and Mahajan VB (2015). Structural modeling of a novel CAPN5 mutation that causes uveitis and neovascular retinal detachment. PLoS ONE 10, e0122352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts R, Weinsheimer S, Blouse GE, and Anagli J (2003). Structural determinants of the calpain inhibitory activity of calpastatin peptide B27-WT. J. Biol. Chem 278, 7800–7809. [DOI] [PubMed] [Google Scholar]
- Brunet JP, Cotte-Laffitte J, Linxe C, Quero AM, Géniteau-Legendre M, and Servin A (2000). Rotavirus infection induces an increase in intracellular calcium concentration in human intestinal epithelial cells: role in microvillar actin alteration. J. Virol 74, 2323–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell RL, and Davies PL (2012). Structure-function relationships in calpains. Biochem. J 447, 335–351. [DOI] [PubMed] [Google Scholar]
- Carpentier M, and Chomilier J (2019). Protein multiple alignments: sequence-based versus structure-based programs. Bioinformatics 35, 3970–3980. [DOI] [PubMed] [Google Scholar]
- Cheng SY, Wang SC, Lei M, Wang Z, and Xiong K (2018). Regulatory role of calpain in neuronal death. Neural Regen. Res 13, 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuerrier D, Moldoveanu T, Inoue J, Davies PL, and Campbell RL (2006). Calpain inhibition by alpha-ketoamide and cyclic hemiacetal inhibitors revealed by X-ray crystallography. Biochemistry 45, 7446–7452. [DOI] [PubMed] [Google Scholar]
- Davis TL, Paramanathan R, Butler-Cole C, Finerty PJ Jr., Weigelt J, Sundstrom M, Arrowsmith CH, Edwards AM, Bochkarev A, Dhe-Paganon S, Structural Genomics Consortium (SGC). (2006). Catalytic Domain of Human Calpain 8 PDB: 2NQA. [Google Scholar]
- Davis TL, Walker JR, Finerty PJ Jr., Mackenzie F, Newman EM, and Dhe-Paganon S (2007). The crystal structures of human calpains 1 and 9 imply diverse mechanisms of action and auto-inhibition. J. Mol. Biol 366, 216–229. [DOI] [PubMed] [Google Scholar]
- Dear TN, and Boehm T (1999). Diverse mRNA expression patterns of the mouse calpain genes Capn5, Capn6 and Capn11 during development. Mech. Dev 89, 201–209. [DOI] [PubMed] [Google Scholar]
- Dear N, Matena K, Vingron M, and Boehm T (1997). A new subfamily of vertebrate calpains lacking a calmodulin-like domain: implications for calpain regulation and evolution. Genomics 45, 175–184. [DOI] [PubMed] [Google Scholar]
- Dill KA, and MacCallum JL (2012). The protein-folding problem, 50 years on. Science 338, 1042–1046. [DOI] [PubMed] [Google Scholar]
- Dill KA, Ozkan SB, Shell MS, and Weikl TR (2008). The protein folding problem. Annu. Rev. Biophys 37, 289–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Finn RD, Clements J, and Eddy SR (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39, W29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gakhar L, Bassuk AG, Velez G, Khan S, Yang J, Tsang SH, and Mahajan VB (2016). Small-angle X-ray scattering of calpain-5 reveals a highly open conformation among calpains. J. Struct. Biol 196, 309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garst AD, Héroux A, Rambo RP, and Batey RT (2008). Crystal structure of the lysine riboswitch regulatory mRNA element. J. Biol. Chem 283, 22347–22351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glading A, Bodnar RJ, Reynolds IJ, Shiraha H, Satish L, Potter DA, Blair HC, and Wells A (2004). Epidermal growth factor activates m-calpain (calpain II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol. Cell. Biol 24, 2499–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser F, Pupko T, Paz I, Bell RE, Bechor-Shental D, Martz E, and Ben-Tal N (2003). ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19, 163–164. [DOI] [PubMed] [Google Scholar]
- Goll DE, Thompson VF, Taylor RG, and Zalewska T (1992). Is calpain activity regulated by membranes and autolysis or by calcium and calpastatin? BioEssays 14, 549–556. [DOI] [PubMed] [Google Scholar]
- Gore S, Sanz García E, Hendrickx PMS, Gutmanas A, Westbrook JD, Yang H, Feng Z, Baskaran K, Berrisford JM, Hudson BP, et al. (2017). Validation of Structures in the Protein Data Bank. Structure 25, 1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouveia-Oliveira R, Sackett PW, and Pedersen AG (2007). MaxAlign: maximizing usable data in an alignment. BMC Bioinformatics 8, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna RA, Campbell RL, and Davies PL (2008). Calcium-bound structure of calpain and its mechanism of inhibition by calpastatin. Nature 456, 409–412. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Kann O, Ohlemeyer C, Hanisch UK, and Kettenmann H (2003). Elevation of basal intracellular calcium as a central element in the activation of brain macrophages (microglia): suppression of receptor-evoked calcium signaling and control of release function. J. Neurosci 23, 4410–4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann DB, Williams SK, Bojcevski J, Müller A, Stadelmann C, Naidoo V, Bahr BA, Diem R, and Fairless R (2013). Calcium influx and calpain activation mediate preclinical retinal neurodegeneration in autoimmune optic neuritis. J. Neuropathol. Exp. Neurol 72, 745–757. [DOI] [PubMed] [Google Scholar]
- Hong SC, Goto Y, Lanzino G, Soleau S, Kassell NF, and Lee KS (1994). Neuroprotection with a calpain inhibitor in a model of focal cerebral ischemia. Stroke 25, 663–669. [DOI] [PubMed] [Google Scholar]
- Huang W, Fileta J, Rawe I, Qu J, and Grosskreutz CL (2010). Calpain activation in experimental glaucoma. Invest. Ophthalmol. Vis. Sci 51, 3049–3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosten RP, Salzemann J, Bloch V, Stockinger H, Berglund AC, Blanchet C, Bongcam-Rudloff E, Combet C, Da Costa AL, Deleage G, et al. (2009). PDB_REDO: automated re-refinement of X-ray structure models in the PDB. J. Appl. Cryst 42, 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010). Xds. Acta Crystallogr. D Biol. Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, and Standley DM (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev PV, Petoukhov MV, Volkov VV, and Svergun DI (2006). ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Cryst 39, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizaj D, and Copenhagen DR (2002). Calcium regulation in photoreceptors. Front. Biosci 7, d2023–d2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, and Tamura K (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol 33, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, and Godzik A (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. [DOI] [PubMed] [Google Scholar]
- Li Q, Hanzlik RP, Weaver RF, and Schönbrunn E (2006). Molecular Mode of Action of a Covalently Inhibiting Peptidomimetic on the Human Calpain Protease Core. Biochemistry 45, 701–708. [DOI] [PubMed] [Google Scholar]
- Mahajan VB, Skeie JM, Bassuk AG, Fingert JH, Braun TA, Daggett HT, Folk JC, Sheffield VC, and Stone EM (2012). Calpain-5 mutations cause autoimmune uveitis, retinal neovascularization, and photoreceptor degeneration. PLoS Genet. 8, e1003001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masrati G, Dwivedi M, Rimon A, Gluck-Margolin Y, Kessel A, Ashkenazy H, Mayrose I, Padan E, and Ben-Tal N (2018). Broad phylogenetic analysis of cation/proton antiporters reveals transport determinants. Nat. Commun 9, 4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matena K, Boehm T, and Dear N (1998). Genomic organization of mouse Capn5 and Capn6 genes confirms that they are a distinct calpain subfamily. Genomics 48, 117–120. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Cryst 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldoveanu T, Hosfield CM, Lim D, Elce JS, Jia Z, and Davies PL (2002). A Ca(2+) switch aligns the active site of calpain. Cell 108, 649–660. [DOI] [PubMed] [Google Scholar]
- Moldoveanu T, Hosfield CM, Lim D, Jia Z, and Davies PL (2003). Calpain silencing by a reversible intrinsic mechanism. Nat. Struct. Biol 10, 371–378. [DOI] [PubMed] [Google Scholar]
- Moldoveanu T, Campbell RL, Cuerrier D, and Davies PL (2004a). Crystal structures of calpain-E64 and -leupeptin inhibitor complexes reveal mobile loops gating the active site. J. Mol. Biol 343, 1313–1326. [DOI] [PubMed] [Google Scholar]
- Moldoveanu T, Jia Z, and Davies PL (2004b). Calpain activation by cooperative Ca2+ binding at two non-EF-hand sites. J. Biol. Chem 279, 6106–6114. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, and Vagin AA (2011). REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Keefe G, Hanif AM, Mahajan VB, and Jain N (2019). Early Onset Neovascular Inflammatory Vitreoretinopathy Due to a De Novo CAPN5 Mutation: Report of a Case. Ocul. Immunol. Inflamm 27, 706–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Nakajima T, Tamada Y, Shearer TR, and Azuma M (2007). Contribution of calpains to photoreceptor cell death in N-methyl-N-nitrosourea-treated rats. Exp. Neurol 204, 39–48. [DOI] [PubMed] [Google Scholar]
- Ono Y, and Sorimachi H (2012). Calpains: an elaborate proteolytic system. Biochim. Biophys. Acta 1824, 224–236. [DOI] [PubMed] [Google Scholar]
- Ono Y, Saido TC, and Sorimachi H (2016). Calpain research for drug discovery: challenges and potential. Nat. Rev. Drug Discov 15, 854–876. [DOI] [PubMed] [Google Scholar]
- Otto HH, and Schirmeister T (1997). Cysteine Proteases and Their Inhibitors. Chem. Rev 97, 133–172. [DOI] [PubMed] [Google Scholar]
- Potz BA, Abid MR, and Sellke FW (2016). Role of Calpain in Pathogenesis of Human Disease Processes. J. Nat. Sci 2, e218. [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, and Arkin AP (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Cuerrier D, Davies PL, Li Z, Powers JC, and Campbell RL (2008). Cocrystal structures of primed side-extending alpha-ketoamide inhibitors reveal novel calpain-inhibitor aromatic interactions. J. Med. Chem 51, 5264–5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randazzo NM, Shanks ME, Clouston P, and MacLaren RE (2019). Two Novel CAPN5 Variants Associated with Mild and Severe Autosomal Dominant Neovascular Inflammatory Vitreoretinopathy Phenotypes. Ocul. Immunol. Inflamm 27, 693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J, and Südhof TC (1998). C2-domains, structure and function of a universal Ca2+-binding domain. J. Biol. Chem 273, 15879–15882. [DOI] [PubMed] [Google Scholar]
- Samantaray S, Ray SK, and Banik NL (2008). Calpain as a potential therapeutic target in Parkinson’s disease. CNS Neurol. Disord. Drug Targets 7, 305–312. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Kikuchi T, Yumoto N, Yoshimura N, and Murachi T (1984). Comparative specificity and kinetic studies on porcine calpain I and calpain II with naturally occurring peptides and synthetic fluorogenic substrates. J. Biol. Chem 259, 12489–12494. [PubMed] [Google Scholar]
- Schaefer KA, Toral MA, Velez G, Cox AJ, Baker SA, Borcherding NC, Colgan DF, Bondada V, Mashburn CB, Yu CG, et al. (2016). Calpain-5 Expression in the Retina Localizes to Photoreceptor Synapses. Invest. Ophthalmol. Vis. Sci 57, 2509–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer K, Mahajan M, Gore A, Tsang SH, Bassuk AG, and Mahajan VB (2017). Calpain-5 gene expression in the mouse eye and brain. BMC Res. Notes 10, 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields DC, Schaecher KE, Saido TC, and Banik NL (1999). A putative mechanism of demyelination in multiple sclerosis by a proteolytic enzyme, calpain. Proc. Natl. Acad. Sci. USA 96, 11486–11491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Brewer MK, Mashburn CB, Lou D, Bondada V, Graham B, and Geddes JW (2014). Calpain 5 is highly expressed in the central nervous system (CNS), carries dual nuclear localization signals, and is associated with nuclear promyelocytic leukemia protein bodies. J. Biol. Chem 289, 19383–19394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneath PHA, and Sokal RR (1973). Unweighted pair group method with arithmetic mean In Numerical Taxonomy: The Principles and Practice of Numerical Classification (W.H. Freeman and Company; ), pp. 230–234. [Google Scholar]
- Storer AC, and Ménard R (1994). Catalytic mechanism in papain family of cysteine peptidases. Methods Enzymol 244, 486–500. [DOI] [PubMed] [Google Scholar]
- Svergun DI, Barberato C, and Koch MHJ (1995). CRYSOL - a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. J. Appl. Cryst 28, 768–773. [Google Scholar]
- Syntichaki P, Xu K, Driscoll M, and Tavernarakis N (2002). Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature 419, 939–944. [DOI] [PubMed] [Google Scholar]
- Trinchese F, Fa’ M, Liu S, Zhang H, Hidalgo A, Schmidt SD, Yamaguchi H, Yoshii N, Mathews PM, Nixon RA, and Arancio O (2008). Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J. Clin. Invest 118, 2796–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velez G, Bassuk AG, Colgan D, Tsang SH, and Mahajan VB (2017). Therapeutic drug repositioning using personalized proteomics of liquid biopsies. JCI Insight 2, 97818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velez G, Bassuk AG, Schaefer KA, Brooks B, Gakhar L, Mahajan M, Kahn P, Tsang SH, Ferguson PJ, and Mahajan VB (2018). A novel de novo CAPN5 mutation in a patient with inflammatory vitreoretinopathy, hearing loss, and developmental delay. Cold Spring Harb. Mol. Case Stud 4, a002519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CK, Weeratunga SK, Pacheco CM, and Hofmann A (2012). DMAN: a Java tool for analysis of multi-well differential scanning fluorimetry experiments. Bioinformatics 28, 439–440. [DOI] [PubMed] [Google Scholar]
- Wang Y, Li H, Zang S, Li F, Chen Y, Zhang X, Song Z, Peng Q, and Gu F (2018). Photoreceptor Cell-Derived CAPN5 Regulates Retinal Pigment Epithelium Cell Proliferation Through Direct Regulation of SLIT2 Cleavage. Invest. Ophthalmol. Vis. Sci 59, 1810–1821. [DOI] [PubMed] [Google Scholar]
- Webb B, and Sali A (2017). Protein Structure Modeling with MODELLER. Methods Mol Biol 1654, 39–54. [DOI] [PubMed] [Google Scholar]
- Wert KJ, and Mahajan VB (2019). In Vivo Expression of Mutant Calpains in the Eye Using Lentivirus. Methods Mol. Biol 1915, 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wert KJ, Bassuk AG, Wu WH, Gakhar L, Coglan D, Mahajan M, Wu S, Yang J, Lin CS, Tsang SH, and Mahajan VB (2015). CAPN5 mutation in hereditary uveitis: the R243L mutation increases calpain catalytic activity and triggers intraocular inflammation in a mouse model. Hum. Mol. Genet 24, 4584–4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wert KJ, Koch SF, Velez G, Hsu CW, Mahajan M, Bassuk AG, Tsang SH, and Mahajan VB (2019). CAPN5 genetic inactivation phenotype supports therapeutic inhibition trials. Hum. Mutat 40, 2377–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gao J, Wang J, Heffernan R, Hanson J, Paliwal K, and Zhou Y (2018). Sixty-five years of the long march in protein secondary structure prediction: the final stretch? Brief. Bioinform 19, 482–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Campbell RL, and Davies PL (2018). Structures of human calpain-3 protease core with and without bound inhibitor reveal mechanisms of calpain activation. J. Biol. Chem 293, 4056–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Liao B, and Li R (2010). A Method for Constructing Phylogenetic Tree Based on a Dissimilarity Matrix . MATCH Commun. Math. Comput. Chem 63, 483–492. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates for the reported structure have been deposited with the Protein Data Bank (PDB) under accession code 6P3Q.