

Abstract
PURPOSE
In men with metastatic germ cell tumors (GCTs), risk-directed treatment is determined, in part, by a distinction between seminoma and nonseminomatous GCT (NSGCT). The importance of NSGCT cell type is uncertain. We evaluated the long-term impact of teratoma on survival in patients with NSGCT.
METHODS
Prechemotherapy, primary tumors from patients who received platinum-based chemotherapy were studied, and the histology was confirmed by a genitourinary pathologist. The cumulative incidence of disease-related death (CIDD) was the primary end point, and a competing-risk analysis was performed.
RESULTS
Tumors were available from 232 patients, including 193 with NSGCT. An element of teratoma was present in 82 NSGCT primary tumors (42%). With a median follow-up of 17 years (range, 0.3 to 35 years), 58 patients with NSGCT died, 47 as a result of GCT and 11 as a result of other causes. Most GCT deaths occurred within the first 5 years and were associated with pretreatment risk status (P < .001). Death as a result of other causes rose steadily after 15 years and was not associated with risk status (P = .66). A higher CIDD was observed in patients who had NSGCT with teratoma than those with NSGCT without teratoma and seminoma (5-year CIDD rate, 27.4%, 17.4%, and 10.3%, respectively; P = .03). A higher CIDD was observed in patients who had NSGCT with mature teratoma compared with those with either NSGCT with immature teratoma or NSGCT without teratoma (5-year CIDD rate, 38.1%, 19.9%, and 17.4%, respectively; P = .01).
CONCLUSION
The presence of teratoma, particularly mature teratoma, in an NSGCT primary tumor is associated with a higher CIDD, consistent with the hypothesis that differentiation is associated with adverse outcomes. Death as a result of non-GCT causes is not associated with risk status and must be separated from GCT death when evaluating long-term survival.
INTRODUCTION
Two unique features characterize germ cell tumors (GCTs), the most common solid malignancy in men 15 to 40 years of age.1,2 First, platinum-based chemotherapy (PC) cures a majority of patients with advanced GCT.3,4 Second, GCTs display a multitude of undifferentiated to terminally differentiated cell types.
GCT originates in the primordial germ cell, and its cell types parallel those observed during embryogenesis. Seminoma and embryonal carcinoma (EC) are both undifferentiated. However, unlike seminoma, EC is pluripotent, expresses an embryonic stem-cell transcription factor signature, and is the origin of the diverse cell types seen in nonseminomatous GCT (NSGCT).5 Extraembryonic differentiation into yolk sac and trophoblastic lineages is represented by the production of α-fetoprotein (AFP) or human chorionic gonadotropin (HCG), respectively, with or without histologic evidence of yolk sac tumor (YST) or trophoblastic elements (choriocarcinoma [CC] or syncytiotrophoblasts). Differentiation into embryonic ectoderm, mesoderm, and endoderm gives rise to teratoma and its characteristic mixture of somatic cell types.
Because of GCT responsiveness to PC, the identification of patients with metastatic GCT who are more or less likely to be cured is important. The International Germ Cell Cancer Collaborative Group (IGCCCG) developed a validated classification on the basis of 5-year progression-free and overall survival. Both anatomy (primary site, sites of metastasis) and tumor biology (serum tumor markers, seminoma v NSGCT) are represented.6 Approximately 10%, 20% to 30%, and 50% to 60% of good-, intermediate-, and poor-risk patients, respectively, will succumb to progressive GCT.
The tumor biology elements in the IGCCCG classification imply that differentiation is involved in the responsiveness of GCTs to PC. Primary pure seminoma, which displays a minimal ability to differentiate, has a higher cure rate than NSGCT.6,7 The chemotherapy sensitivity of extraembryonic differentiation (YST, CC, syncytiotrophoblasts) varies from very high to very low, which parallels the serum levels of AFP or HCG. EC may produce either marker without histologic evidence of YST, CC, or syncytiotrophoblasts per se. Teratoma is resistant to chemotherapy, a consequence of terminal differentiation (eg, mature glands, muscle). Because teratoma does not secrete a serum marker, the impact of somatic differentiation on outcome is uncertain. To study the impact of teratoma (representing somatic differentiation) on treatment outcome, we evaluated whether the presence of teratoma in the pretreatment primary tumor is associated with treatment resistance and survival in patients with metastatic NSGCT who received PC.
METHODS
Patient Eligibility
To maximize long-term follow-up, a retrospective review of primary tumor specimens obtained between April 1975 and May 1996 from patients with GCT who received modern-dose, first-line PC at Memorial Sloan Kettering Cancer Center (MSKCC) was conducted. All specimens and data were obtained according to an institutional review board–approved protocol.
Tumor Specimen and Clinical Characteristics
Primary tumors from 232 patients with metastatic GCT were identified; 193 had NSGCT. The primary tumor slides were reviewed by experienced genitourinary pathologists at MSKCC (S.K.T. and V.E.R.). All cell types, including germ cell neoplasia in situ, seminoma, EC, CC, YST, immature teratoma, mature teratoma, and syncytiotrophoblasts, were recorded. Demographic variables, PC treatment regimens, serum tumor markers (AFP, HCG, lactate dehydrogenase), and IGCCCG risk classification were obtained through chart review. Details on the modern-dose PC regimens that the patients received have been published previously.8-15
Statistical Analysis
The date of last follow-up or death and the cause of death were recorded for all patients. Survival was defined as the time from diagnosis to death or last contact. Overall survival was estimated using the Kaplan-Meier method. Because of possible confounding from deaths as a result of other causes (DOC), a competing-risk approach was used to evaluate cumulative incidence of disease-related death (CIDD). The associations between factors of interest and CIDD or DOC were evaluated using the Fine and Gray test.16,17 NSGCT primary tumors were divided into those with and those without teratoma, and teratoma as either mature or immature. The relationship between CIDD and the presence of mature teratoma was tested in a proportional subdistribution hazards model.16,17 To account for changes in chemotherapy regimen over time, a chronologic analysis evaluated outcomes in the 1970s, 1980s, and 1990s. Patient clinical characteristics were compared using the χ2 test for categorical variables and the two-sample t test for continuous variables. All analyses were performed using SAS 9.4 (SAS Institute, Cary, NC) and R 3.1.1 statistical software.
RESULTS
Survival and Cumulative Incidence of Death in All Patients
The overall survival of all 232 patients, by IGCCCG risk and major histologic group, are shown in Figs 1A to 1C. The median follow-up for survivors was 17 years (range, 0.30 to 35 years; interquartile range, 10.3 years). Because of the long follow-up during which age-related cardiovascular and a second non-GCT cancer may appear and the increased risk of these events in patients with GCT treated with PC, we determined the causes of death.
FIG 1.

Overall survival for all patients (n = 232). (A) All patients. (B) All patients by International Germ Cell Cancer Collaborative Group (IGCCCG risk). (*) one patient with seminoma who did not have any markers was classified as having good risk. (C) All patients by histologic subtype. NSGCT, nonseminomatous germ cell tumor.
The CIDD and the cumulative incidence of DOC (CIDOC) distributions show that death as a result of disease occurred most frequently in the 5 years after diagnosis, whereas DOC began to rise approximately 15 years after diagnosis, and more sharply after 20 years, for all patients and when stratified by IGCCCG risk status (Figs 2A and 2B). The overall and risk status CIDD and CIDOC distributions for patients with NSGCT did not differ from that of the entire cohort (Figs 2C and 2D). The CIDD was correlated with the IGCCCG risk stratum both in all patients (5-year CIDD rate: good, 6.5%; intermediate, 33.4%; poor, 56.4%; P < .001; Fig 2B) and in those with only NSGCT (5-year CIDD rate: good, 5.8%; intermediate, 34.0%; poor, 56.4%; P < .001; Fig 2D), whereas the CIDOC was not (5-year CIDOC rate: good, 1.4%; intermediate, 0%; poor, 0% [P = .93; Fig 2B] v good, 0%; intermediate, 0%; poor, 0% [P = 0.66; Fig 2D], respectively).
FIG 2.

Cumulative incidence of death by cause. (A) All patients. (B) All patients by International Germ Cell Cancer Collaborative Group (IGCCCG risk) (*) one patient with seminoma who did not have any markers was classified as having good risk. Five-year cumulative incidence of disease-related death rate: good, 6.5%; intermediate, 33.4%; poor, 56.4%; P < .001; 5-year cumulative incidence of death as a result of other causes rate: good, 1.4%; intermediate, 0%; poor, 0%; P = .93. (C) Patients with nonseminomatous germ cell tumor. (D) Patients with nonseminomatous germ cell tumor by International Germ Cell Cancer Collaborative Group (5-year cumulative incidence of disease-related death rate: good, 5.8%; intermediate, 34.0%; poor, 56.4%; P < .001; 5-year cumulative incidence of death as a result of other causes rate: good, 0%; intermediate, 0%; and poor, 0%; P = .66).
A statistically significant difference in CIDD was observed when teratoma was present in the primary tumor compared with when it was not (5-year CIDD rate: 27.4% NSGCT with teratoma v 17.4% NSGCT without teratoma v 10.3% in seminoma; P = .03; Fig 3). Because pure seminoma only exceptionally displays differentiation and has a better prognosis than NSGCT (only five patients with seminoma died as a result of disease),6,7 subsequent analyses were limited to the NSGCT cohort.
FIG 3.

Cumulative incidence of disease-related death by histology. NSGCT, nonseminomatous germ cell tumor.
Cumulative Incidence of Death in Patients With NSGCT
Of the 58 NSGCT deaths, 47 patients died as a result of disease and 11 as a result of other causes. NSGCT cancer deaths were analyzed further. Deaths as a result of disease in the teratoma-positive cohort were almost invariably associated with treatment-resistant nonteratoma GCT and one or more sites of nonretroperitoneal visceral metastases (24 of 26; 92%; Appendix Table A1, online only). Deaths as a result of disease that occurred after a late relapse (more than 2 years from completion of PC) occurred in 10 (12.2%) of 82 patients who had teratoma in the primary tumor compared with seven (6.3%) of 111 patients whose primary tumor was negative for teratoma. (Appendix Table A1). GCT deaths as a result of teratoma with secondary somatic-type malignancy (malignant transformation) occurred in three patients with teratoma-positive primary tumors and one with a teratoma-negative tumor (Appendix Table A1).
Because the cohort spans from 1975 to 1996, we assessed for chronologic changes. Consistent with other studies,18,19 outcomes improved over time (Appendix Fig A1, online only). Treatment regimens more closely resembled the frontline etoposide- or ifosfamide-containing PC regimens used today (Appendix Table A2, online only). Of note, the impact of teratoma on CIDD became stronger with each passing decade (Appendix Fig A1), which supports the relevance of our findings to the modern-day treatment of advanced GCTs.
Examination of teratoma subtype in patients whose primary tumors had mature teratoma showed a higher CIDD than in those with NSGCT with immature teratoma and NSGCT without teratoma (5-year CIDD rate, 38.1%, 19.9%, 17.4%, respectively; P = .01; Fig 4). In multivariable analyses, mature teratoma was independently associated with a higher CIDD after adjusting for IGCCCG risk status and age (hazard ratio, 1.91; 95% CI, 1.02 to 3.6; P = .04; Appendix Table A3, online only). Mature teratoma remained independently associated with a higher CIDD in a model that included nonpulmonary visceral metastases, AFP and HCG as continuous variables, and age (hazard ratio, 2.16; 95% CI, 1.1 to 4.1; P = .02; data not shown).
FIG 4.

Cumulative incidence of disease-related death among patients with nonseminomatous germ cell tumor (NSGCT) according to type of teratoma. Ten patients with both immature and mature teratoma and one patient with teratoma with secondary somatic-type malignancy were excluded from this analysis.
Patient and Primary Tumor Characteristics of NSGCT
The characteristics of 193 patients with NSGCT are listed in Appendix Table A4 (online only). The distribution of IGCCCG risk class and outcomes of each closely approximated that reported by the IGCCCG6 (Appendix Table A5, online only). Pathologic review identified 438 distinct histologies within the primary tumors of the 193 patients with NSGCT (Appendix Table A6, online only). Most primary NSGCT tumors (166; 86%) had two or more histologies. Somatic malignant transformation of teratoma was identified in one primary tumor. A sensitivity analysis with and without this tumor yielded similar results. Six patients had mediastinal primary NSGCT; a component of teratoma was present in three. Analysis with and without the six patients with mediastinal primary NSGCT did not influence the findings with regard to the presence or absence of teratoma.
Analysis of Teratoma Within NSGCT
At least one component of teratoma (mature, immature, or malignant transformation of teratoma) was identified in the primary tumors of 82 patients (42%); 111 patients had NSGCT without teratoma (Table 1). The teratoma-positive and -negative groups were well-balanced for the IGCCCG risk group and the distribution and degree of serum tumor marker elevations (Table 1).
TABLE 1.
Clinical Characteristics of Patients with Teratoma-Positive and Teratoma-Negative NSGCT

Both teratoma-positive and teratoma-negative NSGCTs displayed an array of histologic subtypes (Table 1). No differences in the frequencies of EC, CC, and seminoma were found between the two cohorts. YST and syncytiotrophoblasts were more frequently present within teratoma-positive tumors than in the teratoma-negative tumors (P < .01 for both comparisons). We fit multivariable competing-risk models with the presence of syncytiotrophoblast, presence of yolk sac, and histology (teratoma v all other NSGCT v seminoma). Neither YST nor syncytiotrophoblast histology were statistically significant, and the relationship between teratoma and CIDD persisted (data not shown). The disparity in the frequency of YST and syncytiotrophoblasts between the two groups is likely due to greater association of differentiation with EC compared with YST. Of 33 tumors with YST and teratoma, 26 (79%) also had EC, whereas only 10 (43%) of 23 tumors with YST without teratoma had EC. In addition, of the 36 tumors with EC and YST (with or without teratoma), 16 (44%) also had syncytiotrophoblasts compared with only one (5%) of 20 tumors comprising YST without EC.
DISCUSSION
This series provides new information on the relationship between cause of death and histologic subtype in patients with metastatic GCT. Three findings are important. First, an accounting for DOC after treatment of GCT is critically important when evaluating prognostic factors and long-term outcomes in a curable disease such as GCT. Second, after accounting for DOC, the presence of teratoma in the primary tumor was associated with treatment-resistant metastatic disease and worse CIDD. Third, mature and not immature teratoma may be powering this association, which raises some concern about the recent elimination of this distinction from the WHO classification.
Studies of risk factors for death as a result of GCT or any highly curable cancer must account for DOC when follow-up is long. As patients age after successful treatment, DOC will increase. In addition, cardiovascular disease and second malignant tumors are well-known consequences of treatment in survivorship studies of patients who received PC for metastatic GCT.20,21 An accounting for DOC allows for an accurate determination of disease-specific survival and an evaluation of factors that might modify it.
After accounting for DOC, teratoma in the primary tumor was a pretreatment prognostic factor for CIDD in patients with metastatic NSGCT. The higher CIDD was not due to teratoma per se; 92% of teratoma deaths were associated with treatment-resistant, nonteratomatous GCT (eg, CC, YST, EC) at nonretroperitoneal metastatic sites (Appendix Table A1). Because normal somatic tissues (eg, muscle) generally are resistant to the effects of chemotherapy, we hypothesize that the genetic mechanisms responsible for somatic differentiation to teratoma and its treatment resistance also are present in the nonteratoma malignant GCT elements. This hypothesis is supported by the IGCCCG’s incorporation of differentiation into risk stratification and experimental evidence.3,6,22-27 In the IGCCCG classification, seminoma, which displays minimal differentiation and better survival, and NSGCT are classified separately. NSGCT, which reflects the aforementioned relationship of embryogenesis to GCT tumorigenesis, may display one or multiple cell types and reflects the capacity of EC to differentiate into extraembryonic elements (YST, syncytiotrophoblasts, CC), somatic elements (teratoma), both, or neither. Although an increased serum level of AFP and/or HCG usually is accompanied by pathologic evidence of YST, syncytiotrophoblast, and CC elements, pathologic evidence may be absent, which implies that the genetic programming for AFP and/or HCG production exists in EC itself. Because the pretreatment level of AFP and HCG determines risk status in nonmediastinal NSGCT, extraembryonic differentiation is associated with outcome. Similarly, the worse CIDD in the teratoma cohort implies that the genetic machinery for somatic differentiation, and therefore treatment resistance, exists in pluripotent EC. Experimentally, in vitro silencing of NANOG and OCT3/4 led to lineage specification and loss of pluripotency in both GCTs and embryonic stem cells, and loss of pluripotency-specific surface markers (including OCT3/4) was followed by differentiation and emergence of cisplatin resistance.22-24 Genomic profiling revealed loss of OCT3/4 expression in resistant metastatic tumor,25 and in a separate study, a differentiation gene signature was associated with worse survival, independent of IGCCCG risk.26 Thus, differentiation and cisplatin resistance in GCT are linked.27
Our study of CIDD and its long-term follow-up is unique in that most prior studies that evaluated the prognostic significance of teratoma have focused on overall survival in patients who underwent retroperitoneal lymph node dissection. Some studies suggested that only residual disease of less than 1 cm after PC should be considered, although debate exists.28-30 In the study with the longest median follow-up (15.5 years), only 26% of patients had teratoma in the primary tumor, and 77% of the patients had good risk, the risk group in which the least effect would be expected.28,30 A second study had 84% good-risk patients and a median follow-up of less than 5 years.29,30 In our study, the distribution of risk strata and teratoma were similar to that reported by the IGCCCG. The 17-year median follow-up and end point of CIDD allowed for observations that were not possible in other studies.
A higher likelihood of teratoma exists at metastatic sites when teratoma is present within the primary tumor.31-34 Late relapse is a recognized clinical GCT entity characterized by relapse more than 2 years after completing primary PC; an increased serum tumor marker (usually AFP), which reflects the presence of nonteratoma GCT elements; and frequent occurrence of teratoma and somatic malignant transformation.35 In our series, death as a result of late relapse and teratoma with secondary somatic-type malignancy were uncommon but more frequent in the teratoma cohort. Hence, our series is consistent with others that reported late recurrences of GCT with viable nonteratomatous GCT or teratoma with secondary somatic-type malignancy, distinct clinical entities associated with a dismal prognosis without surgery, which underscores the drug resistance of these nonteratoma elements.35-39 Our findings support the notion that teratoma in the primary tumor is associated with both early and delayed disease-related mortality. Complete resection of teratoma should be an important consideration.
Although the most recent WHO classification of GCT eliminated the distinction between mature and immature teratoma, our study was started when these two cell types were recognized as distinct. In a secondary analysis, we found that NSGCT with mature teratoma was associated with a higher CIDD than NSGCT without teratoma; immature teratoma was not associated with a higher CIDD. Mature teratoma was also an independent predictor of worse CIDD in a multivariable analysis that adjusted for IGCCCG prognostic classification. This finding suggests that teratoma subtype may be powering the observed association between teratoma and worse CIDD. A reason for this finding is not clear. Unlike extraembryonal differentiation, teratoma produces no tumor marker. In an attempt to identify immunohistochemical markers of outcome, tumors with a staining pattern of lower Ki67 expression, high apoptosis, and higher p53 expression were associated with a worse prognosis in patients with EC.40 Although both immature and mature teratoma generally showed low Ki67 expression by immunohistochemistry, mature teratoma and not immature teratoma exhibited high expression of apoptosis.41 We speculate that the difference in CIDD between immature and mature teratoma may be due to the delayed recurrence of the more terminally differentiated mature teratoma, but additional investigation is warranted.
There are limitations to this study. First, it is a retrospective, single-center study. Second, the patient population represents a cohort early in the modern-dose platinum era. However, the time interval was chosen to maximize long-term follow-up; all patients received modern-dose PC regimens; and the strongest association between teratoma in the primary tumor and worse CIDD was observed during the 1990s, the most recent time period assessed. Third, the patients may have presented with more-advanced bulky disease. However, IGCCCG risk stratification in our series was not different from that reported, and mature teratoma was an independent predictor in multivariable analysis.6 Fourth, surgical approaches may be different, although all operations were performed according to Whitmore’s MSKCC standards of the time.42 Fifth, experienced genitourinary pathologists reviewed every case to eliminate the problem of temporal changes in histologic classification. We did not wish to rely on old pathology reports. Although teratoma should be easily recognizable, the distinction between mature and immature teratoma is more nuanced and subject to interobserver variability, which may limit the application to the larger oncologic community. Finally, our study did not include a molecular analysis. Our group has shown that TP53 and MDM2 alterations in patients with primary mediastinal NSGCT are associated with inferior outcomes.43 However, mediastinal primary site did not influence the results of our analysis.
Despite these limitations, the large sample size, long median follow-up, and the competing-risk method that separated non-GCT causes of death from GCT-specific mortality enabled the observation that teratoma in the primary tumor, particularly mature teratoma, is associated with a worse outcome. The biology and natural history of teratoma is complex. Teratoma is not inert; its genomic instability is at least as great as EC.44 Modern molecular or genetic analysis may be useful if it can be applied rapidly at diagnosis. To untangle the relationship between teratoma and long-term outcome and to clarify the role of teratoma in treatment planning, additional long-term follow-up studies are needed.
APPENDIX
FIG A1.

Chronologic changes in cumulative incidence of disease-related death (CIDD). (A) All patients. (B) The 1970s CIDD by histology. (C) The 1980s CIDD by histology. (D) The 1990s CIDD by histology. NSGCT, nonseminomatous germ cell tumors.
TABLE A1.
Defining Deaths as a Result of Disease in Patients With Nonseminomatous Germ Cell Tumors

TABLE A2.

TABLE A3.
Multivariable Analysis That Evaluated the Impact of Mature Teratoma, IGCCCG Risk Classification, and Age on Cumulative Incidence of Disease-Related Death
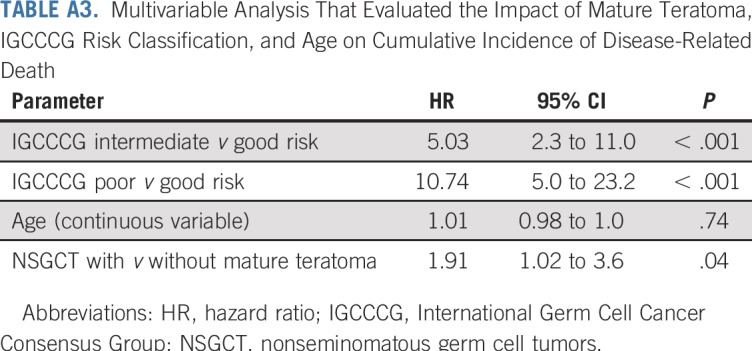
TABLE A4.
Patient Characteristics of Patients With Nonseminomatous Germ Cell Tumor
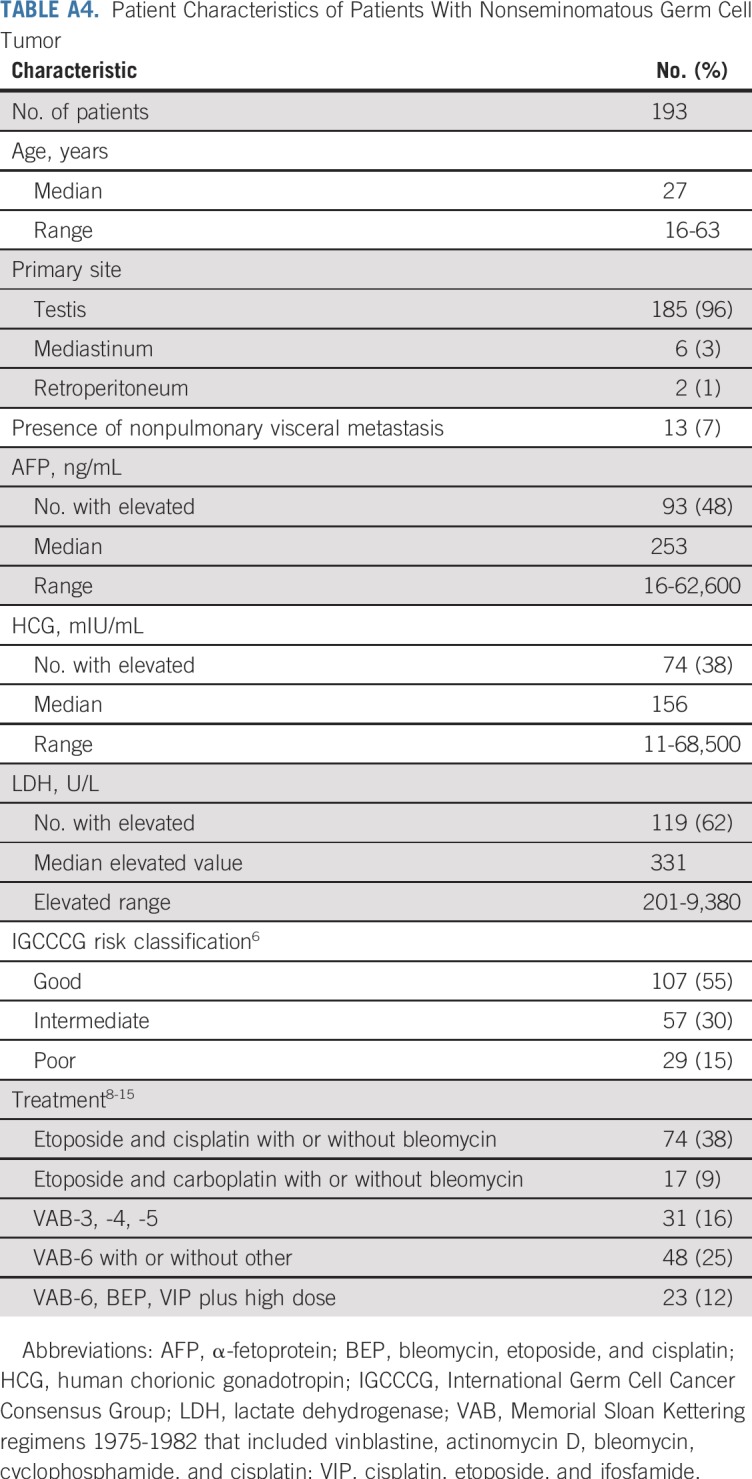
TABLE A5.
IGCCCG Risk Distribution and Outcome Comparison
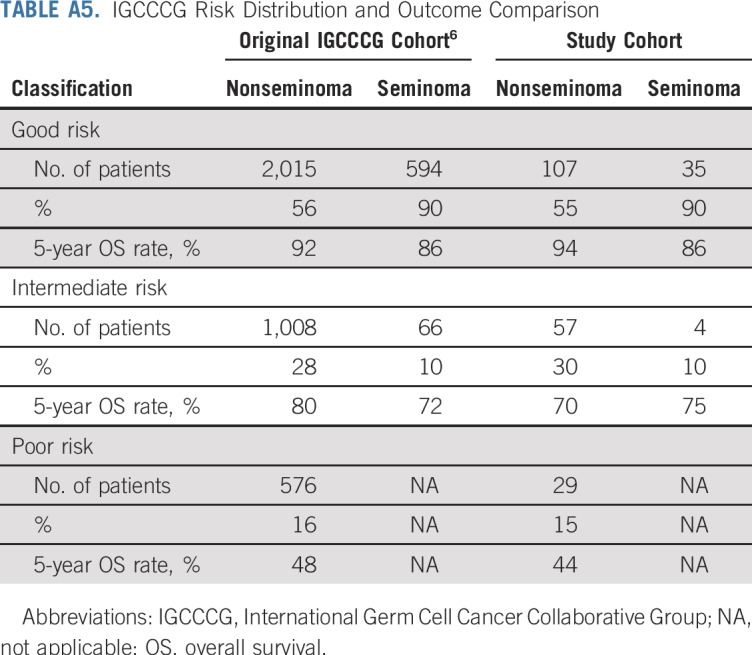
TABLE A6.
Histologies Present in the Nonseminomatous Germ Cell Tumor Cohort (n = 193)
Footnotes
Presented at the 2017 American Society of Clinical Oncology Annual Meeting, Chicago, IL, June 2-6, 2017.
Supported by National Cancer Institute Cancer Center Support grant P30 CA008748 and the Byrne Fund.
See accompanying Editorial on page 2314
AUTHOR CONTRIBUTIONS
Conception and design: All authors
Administrative support: George J. Bosl
Provision of study material or patients: Robert J. Motzer, Dean F. Bajorin, George J. Bosl
Collection and assembly of data: Samuel A. Funt, Sujata Patil, Darren R. Feldman, Robert J. Motzer, George J. Bosl
Data analysis and interpretation: Samuel A. Funt, Sujata Patil, Darren R. Feldman, Robert J. Motzer, Dean F. Bajorin, Victor E. Reuter, George J. Bosl
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Impact of Teratoma on the Cumulative Incidence of Disease-Related Death in Patients With Advanced Germ Cell Tumors
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Samuel A. Funt
Stock and Other Ownership Interests: Kite Pharma, UroGen Pharma (I), Hubble (I), Second Science, Allogene Therapeutics, Neogene Therapeutics (I), Kronos Bio (I), Vida Ventures (I)
Consulting or Advisory Role: AstraZeneca (Inst), MedImmune (Inst), Decibel Therapeutics (Inst)
Research Funding: Genentech (Inst), Roche (Inst), AstraZeneca (Inst), Decibel Therapeutics (Inst)
Travel, Accommodations, Expenses: Bristol-Myers Squibb, AstraZeneca, MedImmune
Darren R. Feldman
Research Funding: Novartis, Seattle Genetics, Decibel Therapeutics (Inst)
Other Relationship: UpToDate
Robert J. Motzer
Consulting or Advisory Role: Pfizer, Novartis, Eisai, Exelixis, Merck, Genentech, Roche, Incyte, Eli Lilly
Research Funding: Pfizer (Inst), Bristol-Myers Squibb (Inst), Eisai (Inst), Novartis (Inst), Genentech (Inst), Roche (Inst)
Dean F. Bajorin
Honoraria: Merck Sharp & Dohme
Consulting or Advisory Role: Bristol-Myers Squibb, Novartis, Roche, Genentech, Merck, Eli Lilly, Fidia Farmaceutici, UroGen Pharma, Pfizer, EMD Serono
Research Funding: Novartis (Inst), Genentech (Inst), Roche (Inst), Merck (Inst), Bristol-Myers Squibb (Inst), AstraZeneca (Inst), Astellas Pharma (Inst), Seattle Genetics (Inst)
Travel, Accommodations, Expenses: Roche, Genentech, Merck, Bristol-Myers Squibb, Eli Lilly, UroGen Pharma
Victor E. Reuter
Consulting or Advisory Role: Cepheid
No other potential conflicts of interest were reported.
REFERENCES
- 1.Nigam M, Aschebrook-Kilfoy B, Shikanov S, et al. Increasing incidence of testicular cancer in the United States and Europe between 1992 and 2009. World J Urol. 2015;33:623–631. doi: 10.1007/s00345-014-1361-y. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 3. Funt SA, Feldman DR, Bosl GJ: The Management of advanced germ cell tumors in 2016: The Memorial Sloan Kettering approach. Oncology (Williston Park) 30:653-664, 2016. [PubMed]
- 4.Hanna N, Einhorn LH. Testicular cancer: A reflection on 50 years of discovery. J Clin Oncol. 2014;32:3085–3092. doi: 10.1200/JCO.2014.56.0896. [DOI] [PubMed] [Google Scholar]
- 5.Santagata S, Ligon KL, Hornick JL. Embryonic stem cell transcription factor signatures in the diagnosis of primary and metastatic germ cell tumors. Am J Surg Pathol. 2007;31:836–845. doi: 10.1097/PAS.0b013e31802e708a. [DOI] [PubMed] [Google Scholar]
- 6.International Germ Cell Cancer Collaborative Group International Germ Cell Consensus Classification: A prognostic factor-based staging system for metastatic germ cell cancers. J Clin Oncol. 1997;15:594–603. doi: 10.1200/JCO.1997.15.2.594. [DOI] [PubMed] [Google Scholar]
- 7.Bajorin DF, Mazumdar M, Meyers M, et al. Metastatic germ cell tumors: Modeling for response to chemotherapy. J Clin Oncol. 1998;16:707–715. doi: 10.1200/JCO.1998.16.2.707. [DOI] [PubMed] [Google Scholar]
- 8.Reynolds TF, Vugrin D, Cvitkovic E, et al. VAB-3 combination chemotherapy of metastatic testicular cancer. Cancer. 1981;48:888–898. doi: 10.1002/1097-0142(19810815)48:4<888::aid-cncr2820480405>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 9.Vugrin D, Cvitkovic E, Whitmore WF, Jr, et al. VAB-4 combination chemotherapy in the treatment of metastatic testis tumors. Cancer. 1981;47:833–839. doi: 10.1002/1097-0142(19810301)47:5<833::aid-cncr2820470502>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 10.Vugrin D, Whitmore WF, Jr, Golbey RB. VAB-5 combination chemotherapy in prognostically poor risk patients with germ cell tumors. Cancer. 1983;51:1072–1075. doi: 10.1002/1097-0142(19830315)51:6<1072::aid-cncr2820510616>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 11.Bosl GJ, Geller NL, Vogelzang NJ, et al. Alternating cycles of etoposide plus cisplatin and VAB-6 in the treatment of poor-risk patients with germ cell tumors. J Clin Oncol. 1987;5:436–440. doi: 10.1200/JCO.1987.5.3.436. [DOI] [PubMed] [Google Scholar]
- 12.Motzer RJ, Mazumdar M, Gulati SC, et al. Phase II trial of high-dose carboplatin and etoposide with autologous bone marrow transplantation in first-line therapy for patients with poor-risk germ cell tumors. J Natl Cancer Inst. 1993;85:1828–1835. doi: 10.1093/jnci/85.22.1828. [DOI] [PubMed] [Google Scholar]
- 13.Motzer RJ, Mazumdar M, Bajorin DF, et al. High-dose carboplatin, etoposide, and cyclophosphamide with autologous bone marrow transplantation in first-line therapy for patients with poor-risk germ cell tumors. J Clin Oncol. 1997;15:2546–2552. doi: 10.1200/JCO.1997.15.7.2546. [DOI] [PubMed] [Google Scholar]
- 14.Bosl GJ, Gluckman R, Geller NL, et al. VAB-6: An effective chemotherapy regimen for patients with germ-cell tumors. J Clin Oncol. 1986;4:1493–1499. doi: 10.1200/JCO.1986.4.10.1493. [DOI] [PubMed] [Google Scholar]
- 15.Bosl GJ, Geller NL, Bajorin D, et al. A randomized trial of etoposide + cisplatin versus vinblastine + bleomycin + cisplatin + cyclophosphamide + dactinomycin in patients with good-prognosis germ cell tumors. J Clin Oncol. 1988;6:1231–1238. doi: 10.1200/JCO.1988.6.8.1231. [DOI] [PubMed] [Google Scholar]
- 16.Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;94:496–509. [Google Scholar]
- 17.Gray RJ. A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann Stat. 1988;16:1141–1154. [Google Scholar]
- 18.Aareleid T, Sant M, Hédelin G. Improved survival for patients with testicular cancer in Europe since 1978. Eur J Cancer. 1998;34:2236–2240. doi: 10.1016/s0959-8049(98)00313-x. [DOI] [PubMed] [Google Scholar]
- 19.Sonneveld DJ, Hoekstra HJ, van der Graaf WT, et al. Improved long term survival of patients with metastatic nonseminomatous testicular germ cell carcinoma in relation to prognostic classification systems during the cisplatin era. Cancer. 2001;91:1304–1315. [PubMed] [Google Scholar]
- 20.van den Belt-Dusebout AW, de Wit R, Gietema JA, et al. Treatment-specific risks of second malignancies and cardiovascular disease in 5-year survivors of testicular cancer. J Clin Oncol. 2007;25:4370–4378. doi: 10.1200/JCO.2006.10.5296. [DOI] [PubMed] [Google Scholar]
- 21.Haugnes HS, Bosl GJ, Boer H, et al. Long-term and late effects of germ cell testicular cancer treatment and implications for follow-up. J Clin Oncol. 2012;30:3752–3763. doi: 10.1200/JCO.2012.43.4431. [DOI] [PubMed] [Google Scholar]
- 22.Chadalavada RSV, Korkola JE, Houldsworth J, et al. Constitutive gene expression predisposes morphogen-mediated cell fate responses of NT2/D1 and 27X-1 human embryonal carcinoma cells. Stem Cells. 2007;25:771–778. doi: 10.1634/stemcells.2006-0271. [DOI] [PubMed] [Google Scholar]
- 23.Mueller T, Mueller LP, Luetzkendorf J, et al. Loss of Oct-3/4 expression in embryonal carcinoma cells is associated with induction of cisplatin resistance. Tumour Biol. 2006;27:71–83. doi: 10.1159/000092324. [DOI] [PubMed] [Google Scholar]
- 24.Kushwaha R, Jagadish N, Kustagi M, et al. Interrogation of a context-specific transcription factor network identifies novel regulators of pluripotency. Stem Cells. 2015;33:367–377. doi: 10.1002/stem.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor-Weiner A, Zack T, O’Donnell E, et al. Genomic evolution and chemoresistance in germ-cell tumours. Nature. 2016;540:114–118. doi: 10.1038/nature20596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korkola JE, Houldsworth J, Feldman DR, et al. Identification and validation of a gene expression signature that predicts outcome in adult men with germ cell tumors. J Clin Oncol. 2009;27:5240–5247. doi: 10.1200/JCO.2008.20.0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korkola JE, Houldsworth J, Bosl GJ, et al. Molecular events in germ cell tumours: Linking chromosome-12 gain, acquisition of pluripotency and response to cisplatin. BJU Int. 2009;104:1334–1338. doi: 10.1111/j.1464-410X.2009.08855.x. [DOI] [PubMed] [Google Scholar]
- 28.Ehrlich Y, Brames MJ, Beck SDW, et al. Long-term follow-up of cisplatin combination chemotherapy in patients with disseminated nonseminomatous germ cell tumors: Is a postchemotherapy retroperitoneal lymph node dissection needed after complete remission? J Clin Oncol. 2010;28:531–536. doi: 10.1200/JCO.2009.23.0714. [DOI] [PubMed] [Google Scholar]
- 29.Kollmannsberger C, Daneshmand S, So A, et al. Management of disseminated nonseminomatous germ cell tumors with risk-based chemotherapy followed by response-guided postchemotherapy surgery. J Clin Oncol. 2010;28:537–542. doi: 10.1200/JCO.2009.23.0755. [DOI] [PubMed] [Google Scholar]
- 30.Bosl GJ, Motzer RJ. Weighing risks and benefits of postchemotherapy retroperitoneal lymph node dissection: Not so easy. J Clin Oncol. 2010;28:519–521. doi: 10.1200/JCO.2009.25.0738. [DOI] [PubMed] [Google Scholar]
- 31.Donohue JP, Rowland RG, Kopecky K, et al. Correlation of computerized tomographic changes and histological findings in 80 patients having radical retroperitoneal lymph node dissection after chemotherapy for testis cancer. J Urol. 1987;137:1176–1179. doi: 10.1016/s0022-5347(17)44439-9. [DOI] [PubMed] [Google Scholar]
- 32.Toner GC, Panicek DM, Heelan RT, et al. Adjunctive surgery after chemotherapy for nonseminomatous germ cell tumors: Recommendations for patient selection. J Clin Oncol. 1990;8:1683–1694. doi: 10.1200/JCO.1990.8.10.1683. [DOI] [PubMed] [Google Scholar]
- 33.Steyerberg EW, Keizer HJ, Fosså SD, et al. Prediction of residual retroperitoneal mass histology after chemotherapy for metastatic nonseminomatous germ cell tumor: Multivariate analysis of individual patient data from six study groups. J Clin Oncol. 1995;13:1177–1187. doi: 10.1200/JCO.1995.13.5.1177. [DOI] [PubMed] [Google Scholar]
- 34.Beck SDW, Foster RS, Bihrle R, et al. Teratoma in the orchiectomy specimen and volume of metastasis are predictors of retroperitoneal teratoma in post-chemotherapy nonseminomatous testis cancer. J Urol. 2002;168:1402–1404. doi: 10.1016/S0022-5347(05)64458-8. [DOI] [PubMed] [Google Scholar]
- 35.Dieckmann K-P, Albers P, Classen J, et al. Late relapse of testicular germ cell neoplasms: A descriptive analysis of 122 cases. J Urol. 2005;173:824–829. doi: 10.1097/01.ju.0000154013.96349.36. [DOI] [PubMed] [Google Scholar]
- 36.Loehrer PJ, Sr, Hui S, Clark S, et al. Teratoma following cisplatin-based combination chemotherapy for nonseminomatous germ cell tumors: A clinicopathological correlation. J Urol. 1986;135:1183–1189. doi: 10.1016/s0022-5347(17)46031-9. [DOI] [PubMed] [Google Scholar]
- 37.Carver BS, Shayegan B, Serio A, et al. Long-term clinical outcome after postchemotherapy retroperitoneal lymph node dissection in men with residual teratoma. J Clin Oncol. 2007;25:1033–1037. doi: 10.1200/JCO.2005.05.4791. [DOI] [PubMed] [Google Scholar]
- 38.Svatek RS, Spiess PE, Sundi D, et al. Long-term outcome for men with teratoma found at postchemotherapy retroperitoneal lymph node dissection. Cancer. 2009;115:1310–1317. doi: 10.1002/cncr.24145. [DOI] [PubMed] [Google Scholar]
- 39.Donadio AC, Motzer RJ, Bajorin DF, et al. Chemotherapy for teratoma with malignant transformation. J Clin Oncol. 2003;21:4285–4291. doi: 10.1200/JCO.2003.01.019. [DOI] [PubMed] [Google Scholar]
- 40.Mazumdar M, Bacik J, Tickoo SK, et al. Cluster analysis of p53 and Ki67 expression, apoptosis, alpha-fetoprotein, and human chorionic gonadotrophin indicates a favorable prognostic subgroup within the embryonal carcinoma germ cell tumor. J Clin Oncol. 2003;21:2679–2688. doi: 10.1200/JCO.2003.03.136. [DOI] [PubMed] [Google Scholar]
- 41. Bosl G, Reuter V, Mazumdar M, et al: Significant differences in Ki67, p53 expression and apoptosis characterize germ cell tumor (GCT) differentiation and progression. J Clin Oncol 19, 2000 (suppl; abstr 2602) [Google Scholar]
- 42.Ray B, Hajdu SI, Whitmore WF., Jr Proceedings: Distribution of retroperitoneal lymph node metastases in testicular germinal tumors. Cancer. 1974;33:340–348. doi: 10.1002/1097-0142(197402)33:2<340::aid-cncr2820330207>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 43.Bagrodia A, Lee BH, Lee W, et al. Genetic determinants of cisplatin resistance in patients with advanced germ cell tumors. J Clin Oncol. 2016;34:4000–4007. doi: 10.1200/JCO.2016.68.7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodriguez E, Mathew S, Reuter V, et al. Cytogenetic analysis of 124 prospectively ascertained male germ cell tumors. Cancer Res. 1992;52:2285–2291. [PubMed] [Google Scholar]