

Abstract
PURPOSE
In National Wilms Tumor Study 5 (NWTS-5), tumor-specific combined loss of heterozygosity of chromosomes 1p and 16q (LOH1p/16q) was associated with adverse outcomes in patients with favorable histology Wilms tumor. The AREN0533/AREN0532 studies assessed whether augmenting therapy improved event-free survival (EFS) for these patients. Patients with stage I/II disease received regimen DD4A (vincristine, dactinomycin and doxorubicin) but no radiation therapy. Patients with stage III/IV disease received regimen M (vincristine, dactinomycin, and doxorubicin alternating with cyclophosphamide and etoposide) and radiation therapy.
METHODS
Patients were enrolled through the AREN03B2 Biology study between October 2006 and October 2013; all underwent central review of pathology, surgical reports, and imaging. Tumors were evaluated for LOH1p/16q by microsatellite testing. EFS and overall survival were compared using the log-rank test between NWTS-5 and current studies.
RESULTS
LOH1p/16q was detected in 49 of 1,147 evaluable patients with stage I/II disease (4.27%) enrolled in AREN03B2; 32 enrolled in AREN0532. LOH1p/16q was detected in 82 of 1,364 evaluable patients with stage III/IV disease (6.01%) in AREN03B2; 51 enrolled in AREN0533. Median follow-up for 83 eligible patients enrolled in AREN0532/0533 was 5.73 years (range, 2.84 to 9.63 years). The 4-year EFS for patients with stage I/II and stage III/IV disease with LOH1p/16 was 87.3% (95% CI, 75.1% to 99.5%) and 90.2% (95% CI, 81.8% to 98.6%), respectively. These results are improved compared with the NWTS-5 updated 4-year EFS of 68.8% for patients with stage I/II disease (P = .042), and 61.3% for patients with stage III/IV disease (P = .001), with trends toward improved 4-year overall survival. The most common grade 3 or higher nonhematologic toxicities with regimen M were febrile neutropenia (39.2%) and infections (21.6%).
CONCLUSION
Augmentation of therapy improved EFS for patients with favorable histology Wilms tumor and LOH1p/16q compared with the historical NWTS-5 comparison group, with an expected toxicity profile.
INTRODUCTION
Clinical trials for children with favorable histology Wilms tumor (FHWT) have used age, stage, and histology to tailor therapy according to risk of relapse, with the goal to optimize survival outcomes while minimizing toxicity.1 On the basis of this approach, event-free survival (EFS) and overall survival (OS) for children with FHWT have dramatically improved.2 Given previous extensive study of clinical parameters, additional advancement in outcomes on the basis of risk stratification is likely to involve molecular biologic prognostic markers.
Loss of heterozygosity (LOH) for polymorphic DNA markers seen on chromosomes 1p and 16q was suggested as a prognostic biomarker for Wilms tumor in several retrospective studies.3-5 National Wilms Tumor Study 5 (NWTS-5) prospectively evaluated LOH as a prognostic marker in more than 1,700 patients with FHWT. Results showed that LOH at either 1p or 16q was a predictor of increased relapse or death.6 The greatest effect was seen in patients with combined LOH of 1p and 16q (LOH1p/16q), found in approximately 6% of patients. For patients with stage I/II disease treated with regimen EE4A (vincristine [VCR] and dactinomycin [DACT]), 4-year relapse-free survival with and without LOH1p/16q was 71.9% and 91.2%, respectively (P = .001). For patients with stage III/IV disease treated with regimen DD4A (VCR, DACT, and doxorubicin [DOX]) and radiation therapy (RT), 4-year relapse-free survival with and without LOH1p/16q was 65.9% and 83%, respectively (P = .01). There was no association with the presence or absence of LOH1p/16q and the location of relapse; thus, augmentation with systemic therapy rather than local RT was considered an appropriate intensification strategy.
The AREN0532 and AREN0533 studies assessed whether augmentation of chemotherapy would improve EFS for patients with FHWT with LOH1p/16q. We report that prospectively augmenting therapy on the basis of this biomarker improved outcome.
METHODS
Patients
All patients were initially enrolled in the Children’s Oncology Group (COG) Biology and Classification Study AREN03B2. Real-time central review of pathology slides, chest computed tomography (CT) scans, and surgical reports were performed to confirm eligibility for the AREN0532 and AREN0533 therapeutic studies. All eligible patients were required to have newly diagnosed FHWT and to be younger than 30 years of age at diagnosis. Local staging criteria were as previously described for NWTS-5, except that patients previously designated as having stage II disease who underwent percutaneous needle biopsy before nephrectomy or had intraoperative tumor spillage were considered to have stage III disease. In addition, round, noncalcified lung nodules not in a fissure and visible on chest CT were considered stage IV, regardless of size, unless histologically proven not to be Wilms tumor.6,7 In NWTS-5, small lung nodules visible on CT scan but not on chest x-ray were staged according to preference of the local treating physician. Patients with stage I and II disease were enrolled in the AREN0532 study. Note that approximately 1 year after the study opened (October 2007), stage I and II FHWT without LOH1p/16q were no longer eligible to enroll in AREN0532. Patients with stage III FHWT were initially enrolled and treated in AREN0532 with regimen DD4A. They were eligible to be enrolled in AREN0533 no later than week 4 of induction therapy if LOH1p/16q was identified. All patients with stage IV disease were enrolled in study AREN0533. Patients with bilateral disease were not eligible. Adequate liver and cardiac function was required. No prior chemotherapy or RT was allowed except for patients with LOH1p/16q enrolling from AREN03B2 (stage I and II) and AREN0532 (stage III) or for those treated for emergent issues, as medically indicated.
Biologic specimens were obtained from the initial nephrectomy specimen or tumor biopsy, snap frozen, and shipped to the COG Biopathology Center, where testing of a single sample for tumor-specific LOH1p/16q was performed. Testing was conducted as previously described or by capillary electrophoresis of fluorescently detected short tandem repeat markers.6,8,9 Paraffin-fixed tissue could be used if fresh frozen was not available.
Before patient enrollment, research ethics board approval of both the AREN0532 and AREN0533 studies was obtained for all centers. Approval was obtained either through the National Institutes of Health Pediatric Central institutional review board or through local institutional review boards. Consent was obtained from parents or legal guardians of the patients. Age-appropriate consent or assent was obtained. This study was monitored by an independent Data Safety Monitoring Board.
Treatment
Chemotherapy.
All patients were treated based on an initial risk assignment and received standard, stage-dependent chemotherapy initiated within 14 days of primary nephrectomy or biopsy: patients with stage I and II disease received regimen EE4A, and patients with stage III and IV disease received regimen DD4A. On the basis of LOH testing, final risk assignments were issued within 3 weeks of initiation of treatment. Patients with stage I or II Wilms tumor (WT) with LOH1p/16q began regimen DD4A at week 4, for a total of 24 weeks of chemotherapy (cumulative DOX dose, 150 mg/m2; Fig 1; Table 1). Patients with stage III or IV WT with LOH1p/16q began regimen M at week 6 or on recovery from delayed nephrectomy. Regimen M was an augmented protocol including four cycles of cyclophosphamide and etoposide in addition to VCR, DACT, and DOX, for a total of 31 weeks (cumulative DOX dose, 195 mg/m2; cumulative cyclophosphamide dose, 8.8 g/m2; cumulative etoposide dose, 2000 mg/m2; Fig 1; Table 1).
FIG 1.

Treatment protocols used in the AREN0532 and AREN0533 studies. Regimen DD4A: vincristine, dactinomycin, and doxorubicin with no radiation therapy (RT); regimen M: vincristine, dactinomycin, and doxorubicin alternating with cyclophosphamide and etoposide with RT. A: dactinomycin 0.023 mg/kg/dose IV × 1 for infants < 1 year; 0.045 mg/kg/dose IV × 1 for children ≥ 1 year (maximum dose: 2.3 mg). C: cyclophosphamide 14.7 mg/kg/dose IV × 5 days for infants < 1 year; 440 mg/m2/dose IV × 5 days for children ≥ 1 year. D: doxorubicin 1.5 mg/kg/dose IV × 1 for infants < 1 year; 45 mg/m2/dose IV × 1 for children ≥ 1 year. D*: doxorubicin 1 mg/kg/dose IV × 1 for infants < 1 year; 30 mg/m2/dose IV × 1 for children ≥ 1 year. E: etoposide 3.3 mg/kg/dose IV × 5 days for infants < 1 year; 100 mg/m2/dose IV × 5 days for children ≥ 1 year. V: vincristine: 0.025 mg/kg/dose intravenously (IV) × 1 for infants < 1 year; 0.05 mg/kg/dose IV × 1 for children ≥ 1 year to 2.99 years; 1.5 mg/m2/dose IV × 1 for children ≥ 3 years (maximum dose: 2 mg). V*: vincristine: 0.034 mg/kg/dose IV × 1 for infants < 1 year; 0.067 mg/kg/dose IV × 1 for children ≥ 1 year to 2.99 years; 2 mg/m2/dose IV × 1 for children ≥ 3 years (maximum dose: 2 mg). RT: for local stage III tumors, 10.8 Gy flank radiation was used, with a 10.8 Gy boost for gross residual disease after surgery. Patients with preoperative tumor rupture, cytology-positive ascites, or diffuse peritoneal seeding were treated with whole-abdomen RT to a dose of 10.5 Gy. Patients with lung metastases received whole lung RT to a dose of 12 Gy in 1.5 Gy fractions (reduced to 10.5 Gy for patients < 12 months old). FHWT, favorable histology Wilms tumor; NWTS-5, National Wilms Tumor Study 5.
TABLE 1.
Stage-Specific Augmentation of Therapy

Radiation therapy.
All patients underwent abdominal local tumor staging. Patients with stage I and II disease did not receive RT. For local stage III tumors, 10.8 Gy flank RT was used, with a boost of 10.8 Gy only for patients with gross residual disease after surgery. Abdominal RT was delivered concurrently with induction chemotherapy for those with upfront nephrectomy or delayed until eventual nephrectomy at week 6 or 12. Patients with preoperative tumor rupture, large intraoperative spill outside the tumor bed, or diffuse peritoneal implants were treated with whole-abdomen irradiation to a dose of 10.5 Gy. Patients with stage IV disease with lung nodules received whole-lung RT to a dose of 12 Gy in 1.5 Gy fractions (reduced to 10.5 Gy for patients younger than 12 months old). Pulmonary RT was delivered regardless of pulmonary metastatic lesion response at week 6, because patients with LOH1p/16q were not eligible for omission of pulmonary RT by protocol. Flank, whole-abdomen, whole-lung, and extrapulmonary metastasis RT fields were designed to be administered in a manner similar to previous NWTS protocols.10,11
Surgery.
Radical nephrectomy with lymph node sampling was recommended according to previously reported guidelines.12 For tumors deemed by the treating institution to be unresectable at diagnosis, a minimum of a needle biopsy was required for initial histologic assessment, and delayed nephrectomy was performed after week 6 or week 12 on the basis of response to treatment.
Statistics
EFS was defined as the time from study enrollment to disease progression, disease recurrence, second malignant neoplasm, or death as a result of any cause, whichever occurred first. OS was defined as the time from study enrollment to death as result of any cause. EFS and OS were censored at the patient’s last contact date. EFS and OS rates were estimated using the Kaplan-Meier method, with CIs estimated by the Peto-Peto method,13 and were compared between groups using the log-rank test. Patient follow-up was current through the September 30, 2017 data freeze.
RESULTS
LOH1p/16q was detected in 49 of 1,147 evaluable patients (4.27%) with stage I/II disease enrolled in AREN03B2, 32 of whom enrolled in AREN0532 between October 2006 and October 2013. LOH1p/16q was detected in 82 of 1,364 (6.01%) evaluable patients with stage III/IV disease enrolled in AREN03B2, 51 of whom enrolled in AREN0533 between February 2007 and May 2013 (Fig 2). LOH1p/16q for each stage is listed in Table 2. Patient demographics are listed in Table 3. Median follow-up for 83 eligible patients enrolled in AREN0532/0533 was 5.73 years (range, 2.84 to 9.63 years; Table 4).
FIG 2.

Consort diagram of patients with combined loss of heterozygosity of chromosomes 1p and 16q (LOH1p/16q). FHWT, favorable histology Wilms tumor; LOH, loss of heterozygosity; NWTS-5, National Wilms Tumor Study 5; regimen DD4A, vincristine, dactinomycin, and doxorubicin with no radiation therapy; regimen EE4A, vincristine and dactinomycin; regimen M: vincristine, dactinomycin, and doxorubicin alternating with cyclophosphamide and etoposide with radiation therapy.
TABLE 2.
LOH1p/16q Positivity by Stage in All Patients Enrolled in AREN03B2 Between October 2006 and October 2013
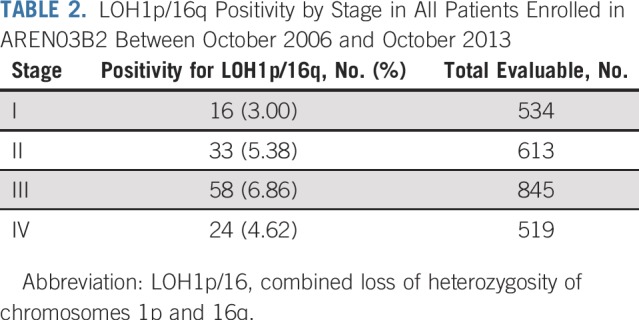
TABLE 3.
Demographics of Patients With LOH1p/16q Enrolled in AREN0532 and AREN0533
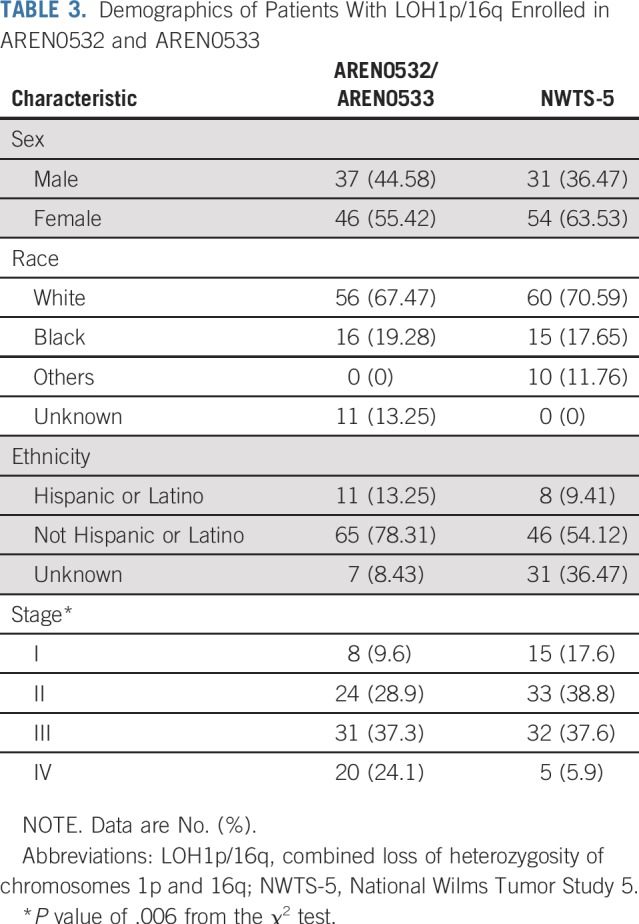
TABLE 4.
Comparison of EFS and OS Outcomes of Patients With LOH1p/16q

Patient Outcomes
At the data cutoff in September 2017, for 32 patients with stage I/II disease with LOH1p/16q who were treated with regimen DD4A, the number of events was four observed versus nine expected (on the basis of a 75% EFS for stage I/II with LOH1p/16q). Four-year EFS and OS estimates were 87.3% (95% CI, 75.1% to 99.5%) and 100%, respectively. Outcomes by individual stage are listed in Appendix Table A1 (online only). All four events were relapse or disease progression and occurred in patients with stage II disease. The sites of relapse were the lung (3), liver (1), and abdomen not including liver (1). The time to relapse from study entry was 0.6, 1.1, 1.3, and 3 years.
For 51 patients with stage III/IV disease with LOH1p/16q treated with regimen M and radiation to the lungs/flank/whole abdomen per study guidelines, six events were observed versus 18 expected (on the basis of a 65% EFS for stage III/IV with LOH), resulting in 4-year EFS and OS estimates of 90.2% (95% CI, 81.8% to 98.6%) and 96.1% (95% CI, 90.5% to 100%), respectively. Outcomes by individual stage are listed in Appendix Table A1. Four events were relapse or progression, and two patients had a second malignancy (anaplastic ependymoma of brain, T-cell lymphoma). The sites of relapse were the lung (2), liver (1), original tumor bed (1), left pleural cavity (1), and lymph nodes (1). The time to relapse from study entry was 0.8, 1.4, 2.6, and 4.7 years.
We assessed outcomes for patients with LOH at either 1p or 16q, although such patients did not automatically receive augmented therapy. Patients with stage I/II disease with LOH at either locus were not treated in the AREN0532 study after October 2007; therefore, reliable treatment and outcome data were not available. The outcomes of patients with stage III disease with LOH at either locus were previously reported.9 For patients with stage IV disease with complete lung nodule response (treated with regimen DD4A without lung RT), 4-year EFS estimates were 75.0% (95% CI, 49.0% to 100.0%) for LOH 1p alone, 66.7% (95% CI, 45.7% to 87.6%) for LOH 16q alone, and 83.4% (95% CI, 74.2% to 92.7%) for LOH at neither locus (P = 0.14; Appendix Fig A1, online only). For patients with stage IV disease with incomplete lung nodule response (treated with regimen M and lung RT), 4-year EFS estimates were 90.0% (95% CI, 68.9% to 100.0%) for LOH 1p alone, 75.0% (95% CI, 53.8% to 96.2%) for LOH 16q alone, and 92.4% (95% CI, 85.9% to 98.9%) for LOH at neither locus (P = .054; Appendix Fig A1). We also assessed for interactive effects on outcome between LOH and local lymph node involvement. In the context of the treatment regimens used in this study, patients with both LOH and lymph node involvement did not seem to have inferior outcomes compared with patients with none or just one of these features (Appendix Table A1).
Comparison With NWTS-5
We compared EFS and OS for the same combined stage groups of patients with LOH1p/16q in AREN0532/AREN0533 and NWTS-5 (Table 4). This comparison was based on the updated September 2017 data freeze analysis for the COG and NWTS-5 studies. For patients with stage I/II disease with LOH1p/16q, 4-year EFS estimates were 68.8% (95% CI, 55.2% to 82.3%) for NWTS-5 and 87.3% (95% CI, 75.1% to 99.5%) for AREN0532 (P = .042; Fig 3A). Four-year OS estimates for patients with stage I/II disease were 91.6% (95% CI, 83.6% to 99.6%) for NWTS-5 and 100% for AREN0532 (P = .096; Fig 3B). For patients with stage III/IV disease with LOH1p/16q, 4-year EFS estimates were 61.3% (95% CI, 44.9% to 77.6%) for NWTS-5 and 90.2% (95% CI, 81.7% to 98.6%) for AREN0533 (P = .001, Fig 4A). Four-year OS estimates for stage III/IV were 86.0% (95% CI, 74.5% to 97.5%) for NWTS-5 and 96.1% (95% CI, 90.5% to 100%) for AREN0533 (P = .087; Fig 4B).
FIG 3.

(A) Event-free survival for patients with stage I/II disease with combined loss of heterozygosity of chromosomes 1p and 16q treated in AREN0532 compared with National Wilms Tumor Study 5 (NWTS-5). (B) Overall survival for patients with stage I/II disease with combined loss of heterozygosity of chromosomes 1p and 16q treated in AREN0532 compared with NWTS-5.
FIG 4.

(A) Event-free survival for patients with stage III/IV disease with combined loss of heterozygosity of chromosomes 1p and 16q treated in AREN0533 compared with National Wilms Tumor Study 5 (NWTS-5). (B) Overall survival for patients with stage III/IV disease with combined loss of heterozygosity of chromosomes 1p and 16q treated in AREN0533 compared with NWTS-5.
We conducted additional analyses to control for potential effects of differences in staging definitions between NWTS-5 and AREN0532/0533. In AREN0532/0533, percutaneous needle biopsy and intraoperative spill confined to the flank were considered as criteria to upstage to stage III, whereas in NWTS-5, patients with these factors were considered to have stage II unless other factors warranting stage III designation were present. Only six of 31 patients in AREN0532/0533 underwent biopsies, five of which were open (inferior vena cava thrombus [4], tumor too large for resection [1]) and would have been considered stage III in NWTS-5. Twelve of 31 patients had intraoperative spill, 11 of whom had other reasons to be classified as stage III. Hence, only two patients with stage III disease would have been considered stage II in NWTS-5. When these two patients were excluded from the outcome analysis, the improved outcome in our study persisted (Table 4). Conversely, we performed an analysis excluding three patients with stage II in NWTS-5 with documented spill, and the improved outcome for the AREN0532 patients prevailed (Table 4).
The definition of stage IV disease also changed between NWTS-5 and the COG studies. To ensure that the excellent outcomes in patients with stage IV and LOH1p/16q were not due to preferential inclusion of patients with tiny lung nodules, we compared outcomes according to lung nodule size. Among 20 patients with stage IV disease and LOH1p/16q, EFS and OS estimates were similarly excellent between patients with a greatest lung nodule diameter of 1 cm or more versus less than 1 cm (Appendix Fig A2, online only). We also assessed how many NWTS-5 patients with stage I/II disease had nodules visible only on chest CT scan but not on chest x-ray to evaluate whether there were a significant number of low-stage patients who would have been considered to have stage IV disease in this study. Only three patients had CT-only nodules identified.
Toxicity and Adverse Events
For all patients with LOH1p/16q treated in either AREN0532 or AREN0533, no events were reported through the Adverse Event Expedited Reporting System; there were no unexpected toxicities and no deaths while receiving treatment (Appendix Table A2, online only). All reported grade III or higher toxicities occurred in patients receiving regimen M, the most common of which were febrile neutropenia (39.2%) and infections (21.6%). Note that the AREN0532 study only required reporting of grade IV and V toxicity for patients receiving regimen DD4A. The rate of individual nonhematologic grade III to V toxicities in regimen M was low (Appendix Table A3, online only). One patient receiving regimen M developed nonfatal sinusoidal obstructive syndrome.
DISCUSSION
The therapy augmentation strategies used in AREN0532 and AREN0533 resulted in EFS estimates that were superior to outcomes in NWTS-5 for patients with FHWT with LOH1p/16q. These improved outcomes were achieved for patients with stage I/II disease treated with regimen DD4A and for patients with stage III/IV disease treated with regimen M. A strong trend to improved OS was also observed. A limitation of LOH1p/16q as a significant prognostic marker is that it was found in only 6% of patients with FHWT enrolled in NWTS-5, a finding confirmed in our studies. Recent reports have identified that 1q gain, which is present in a much higher percentage of patients with FHWT (range, 14% to 39% by stage),14 is also a valuable adverse prognostic marker, particularly among those with advanced-stage disease.14-16 Analyses of the AREN0533 data suggest that regimen M is able to overcome the adverse prognostic effect of 1q gain in patients with stage IV FHWT with isolated pulmonary metastases.17 Similarly, patients with 1q gain are not good candidates for omission of lung RT in the setting of complete remission of lung nodules from initial chemotherapy.17 It should be noted that of 17 patients with stage IV disease with LOH1p/16q and 1q gain data, 15 were also found to have 1q gain. Given the considerable overlap, the role of each as an independent prognostic marker will be hard to discern. Additional prospective studies are planned using 1q gain as a stratification marker to examine the efficacy of novel therapeutic approaches using these biomarkers. Finally, intratumor genetic heterogeneity in WT is well described18 and may have affected detection of LOH in some patients.
There are a number of caveats to our conclusions. Of importance, this objective was not the primary statistical driver of AREN0532 and AREN0533. Neither study was statistically powered to assess differences in outcomes with augmented therapy for patients with LOH1p/16q because it was understood during study design that the anticipated low frequency of LOH1p/16q precluded a high level of power to detect these differences. Despite this, differences in the EFS were significant for patients at all stages when compared with the historical data. There were also improvements in the OS, but these did not reach the level of statistical significance. We acknowledge that potential pitfalls exist when comparing our study results with historical standards; newer standards of care, including improvements in supportive care over time and the potential for stage migration, can lead to differences resulting in bias. We have considered possible bias due to changes in staging between COG and NWTS-5 groups related to intraoperative spill, preoperative biopsy, and lung nodule size; yet, there was no suggestion that these factors played a role in the improved outcomes observed. Finally, although there were no unexpected acute toxicities in patients treated with regimen M, this protocol does introduce an increased risk for late effects, most notably for secondary leukemia due to cyclophosphamide and etoposide,19,20 and infertility related to the use of cyclophosphamide, particularly in boys.21 Recall that this late-effect impact is mitigated by the fact that approximately one third of these patients would have been exposed to at least these doses of alkylators as part of attempts at salvage post relapse. In our view, the marked improvement in EFS is sufficient to justify additional therapy up front. Although our study did not demonstrate a statistically significant improvement in OS associated with augmented therapy, the study was not powered to detect differences in OS, and there was a suggestion of improvement (P = .096 for stage I/II and P = .087 for stage III/IV). A nuanced discussion with the patient and/or family is required to explain the potential risks and benefits of augmented therapy. In our next therapeutic study we plan to test whether reduction of alkylator exposure in the context of a novel regimen for patients with advanced-stage disease would maintain the observed improvement in EFS while reducing the potential for late effects, especially second malignancy23 and infertility.
In summary, augmentation of therapy for FHWT with LOH1p/16q was associated with a statistically significant improvement in EFS. On the basis of these results, the COG Renal Tumor Committee now recommends LOH testing for all newly diagnosed FHWT patients. Our study has successfully demonstrated a proof of principle that augmentation of treatment can overcome a negative biomarker and provides strong encouragement in the evaluation of additional more sensitive and specific markers of outcome.
ACKNOWLEDGMENT
We thank the parents and children who enrolled in these studies and the many investigators of the Children’s Oncology Group, and the pathologists, surgeons, pediatricians, radiation oncologists, diagnostic imagers, statisticians, and other health professionals who treat the children entered in the Children’s Oncology Group renal tumor studies. We also thank the following research and protocol coordinators who worked on these studies over the years: Celeste Sabinske, Ellen Tsan, Tanya Wallas, Laetitia Goddet, Meera Raman, Teni Karimimian, and Mary Bancroft.
Appendix
FIG A1.

(A) Event-free survival for AREN0533 lung metastases only by size of nodule. (B) Overall survival for AREN0533 lung metastases only by size of nodule.
FIG A2.

(A) and (B) Event-free survival for AREN0533 lung metastases only, treated with DD4A and a complete response after induction, by loss of heterozygosity (LOH) of 1p and 16q. (C) and (D) Event-free survival for AREN0533 lung metastases only, treated with Regimen M and an incomplete response after induction, by loss of heterozygosity (LOH) of 1p and 16q.
TABLE A1.
AREN0532/AREN0533 Outcomes by Stage, LOH Status, and Lymph Node Status

TABLE A2.
Reportable Adverse Events in Patients With LOH1p/16q Treated in AREN0532 and AREN0533

TABLE A3.
Reportable Toxicities During Treatment With Regimen DD4A and Regimen M

Footnotes
Presented in part at the 2015 Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, May 29 to June 2, 2015.
Supported by Grants No. U10CA180886, U10CA180899, U10CA098543, U10CA098413, and U24CA114766 from the National Cancer Institute, National Institutes of Health, to support the Children’s Oncology Group. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Also supported by St Baldrick’s Foundation.
D.B.D. and C.V.F. are first coauthors.
ClinicalTrials.gov identifiers: NCT00352534 (AREN0532); NCT00379340 (AREN0533)
AUTHOR CONTRIBUTIONS
Conception and design: David B. Dix, Conrad V. Fernandez, Elizabeth A. Mullen, John A. Kalapurakal, Elizabeth J. Perlman, Nita L. Seibel, Marcio Malogolowkin, James Anderson, Paul E. Grundy, Jeffrey S. Dome
Provision of study materials or patients: Elizabeth A. Mullen, Eric J. Gratias, Marcio Malogolowkin, Paul E. Grundy, Jeffrey S. Dome
Collection and assembly of data: David B. Dix, Conrad V. Fernandez, Yueh-Yun Chi, Elizabeth A. Mullen, James I. Geller, Eric J. Gratias, Geetika Khanna, John A. Kalapurakal, Elizabeth J. Perlman, Nita L. Seibel, Peter F. Ehrlich, Marcio Malogolowkin, James Anderson, Julie Gastier-Foster, Robert C. Shamberger, Paul E. Grundy, Jeffrey S. Dome
Data analysis and interpretation: David B. Dix, Conrad V. Fernandez, Yueh-Yun Chi, Elizabeth A. Mullen, James I. Geller, John A. Kalapurakal, Elizabeth J. Perlman, Nita L. Seibel, Peter F. Ehrlich, James Anderson, Robert C. Shamberger, Yeonil Kim, Paul E. Grundy, Jeffrey S. Dome
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Augmentation of Therapy for Combined Loss of Heterozygosity 1p and 16q in Favorable Histology Wilms Tumor: A Children’s Oncology Group AREN0532 and AREN0533 Study Report
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Eric J. Gratias
Employment: MedSolutions, eviCore Healthcare
Stock and Other Ownership Interests: eviCore Healthcare, Express Scripts, Cigna
Travel, Accommodations, Expenses: eviCore Healthcare
James Anderson
Employment: Merck
Julie Gastier-Foster
Research Funding: Bristol-Myers Squibb (Inst), Incyte (Inst)
Jeffrey S. Dome
Patents, Royalties, Other Intellectual Property: Rockland Immunochemicals
No other potential conflicts of interest were reported.
REFERENCES
- 1.Dome JS, Fernandez CV, Mullen EA, et al. Children’s Oncology Group’s 2013 blueprint for research: Renal tumors. Pediatr Blood Cancer. 2013;60:994–1000. doi: 10.1002/pbc.24419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dome JS, Graf N, Geller JI, et al. Advances in Wilms tumor treatment and biology: Progress through international collaboration. J Clin Oncol. 2015;33:2999–3007. doi: 10.1200/JCO.2015.62.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grundy PE, Telzerow PE, Breslow N, et al. Loss of heterozygosity for chromosomes 16q and 1p in Wilms’ tumors predicts an adverse outcome. Cancer Res. 1994;54:2331–2333. [PubMed] [Google Scholar]
- 4.Grundy RG, Pritchard J, Scambler P, et al. Loss of heterozygosity on chromosome 16 in sporadic Wilms’ tumour. Br J Cancer. 1998;78:1181–1187. doi: 10.1038/bjc.1998.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skotnicka-Klonowicz G, Rieske P, Bartkowiak J, et al. 16q heterozygosity loss in Wilms’ tumour in children and its clinical importance. Eur J Surg Oncol. 2000;26:61–66. doi: 10.1053/ejso.1999.0742. [DOI] [PubMed] [Google Scholar]
- 6.Grundy PE, Breslow NE, Li S, et al. Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: A report from the National Wilms Tumor Study Group. J Clin Oncol. 2005;23:7312–7321. doi: 10.1200/JCO.2005.01.2799. [DOI] [PubMed] [Google Scholar]
- 7.Dome JS, Perlman EJ, Graf N. Risk stratification for Wilms tumor: Current approach and future directions. Am Soc Clin Oncol Educ Book. 2014:215–223. doi: 10.14694/EdBook_AM.2014.34.215. [DOI] [PubMed] [Google Scholar]
- 8.Dix DB, Seibel NL, Chi YY, et al. Treatment of stage IV favorable histology Wilms tumor with lung metastases: A report from the Children’s Oncology Group AREN0533 Study. J Clin Oncol. 2018;36:1564–1570. doi: 10.1200/JCO.2017.77.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandez CV, Mullen EA, Chi YY, et al. Outcome and prognostic factors in stage III favorable-histology Wilms tumor: A report from the Children’s Oncology Group Study AREN0532. J Clin Oncol. 2018;36:254–261. doi: 10.1200/JCO.2017.73.7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalapurakal JA, Dome JS, Perlman EJ, et al. Management of Wilms’ tumour: Current practice and future goals. Lancet Oncol. 2004;5:37–46. doi: 10.1016/s1470-2045(03)01322-6. [DOI] [PubMed] [Google Scholar]
- 11.Kalapurakal JA, Li SM, Breslow NE, et al. Influence of radiation therapy delay on abdominal tumor recurrence in patients with favorable histology Wilms’ tumor treated on NWTS-3 and NWTS-4: A report from the National Wilms’ Tumor Study Group. Int J Radiat Oncol Biol Phys. 2003;57:495–499. doi: 10.1016/s0360-3016(03)00598-4. [DOI] [PubMed] [Google Scholar]
- 12.Ehrlich PF, Hamilton TE, Gow K, et al. Surgical protocol violations in children with renal tumors provides an opportunity to improve pediatric cancer care: A report from the Children’s Oncology Group. Pediatr Blood Cancer. 2016;63:1905–1910. doi: 10.1002/pbc.26083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peto R, Peto J. Asymptotically efficient rank invariant test procedures. J R Stat Soc [Ser A] 1972;135:185–207. [Google Scholar]
- 14.Gratias EJ, Jennings LJ, Anderson JR, et al. Gain of 1q is associated with inferior event-free and overall survival in patients with favorable histology Wilms tumor: A report from the Children’s Oncology Group. Cancer. 2013;119:3887–3894. doi: 10.1002/cncr.28239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chagtai T, Zill C, Dainese L, et al. Gain of 1q as a prognostic biomarker in Wilms tumors (WTs) treated with preoperative chemotherapy in the International Society of Paediatric Oncology (SIOP) WT 2001 trial: A SIOP Renal Tumours Biology Consortium Study. J Clin Oncol. 2016;34:3195–3203. doi: 10.1200/JCO.2015.66.0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gratias EJ, Dome JS, Jennings LJ, et al. Association of chromosome 1q gain with inferior survival in favorable-histology Wilms tumor: A report from the Children’s Oncology Group. J Clin Oncol. 2016;34:3189–3194. doi: 10.1200/JCO.2015.66.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mullen E, Perlman E, Chi Y-Y, et al: Impact of 1q status on outcomes of stage IV favorable histology Wilms tumor (FHWT) patients treated on Children's Oncology Group (COG) study AREN0533: on behalf of the Renal Tumors Committee. Pediatr Blood Cancer 64:226772.S43, 2017.
- 18.Cresswell GD, Apps JR, Chagtai T, et al. Intra-tumor genetic heterogeneity in Wilms tumor: Clonal evolution and clinical implications. EBioMedicine. 2016;9:120–129. doi: 10.1016/j.ebiom.2016.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le Deley M-C, Leblanc T, Shamsaldin A, et al. Risk of secondary leukemia after a solid tumor in childhood according to the dose of epipodophyllotoxins and anthracyclines: A case-control study by the Société Française d’Oncologie Pédiatrique. J Clin Oncol. 2003;21:1074–1081. doi: 10.1200/JCO.2003.04.100. [DOI] [PubMed] [Google Scholar]
- 20.Smith DC, Esper P, Strawderman M, et al. Phase II trial of oral estramustine, oral etoposide, and intravenous paclitaxel in hormone-refractory prostate cancer. J Clin Oncol. 1999;17:1664–1671. doi: 10.1200/JCO.1999.17.6.1664. [DOI] [PubMed] [Google Scholar]
- 21.Green DM, Liu W, Kutteh WH, et al. Cumulative alkylating agent exposure and semen parameters in adult survivors of childhood cancer: A report from the St Jude Lifetime Cohort Study. Lancet Oncol. 2014;15:1215–1223. doi: 10.1016/S1470-2045(14)70408-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reference deleted. [Google Scholar]
- 23.Breslow NE, Takashima JR, Whitton JA, et al. Second malignant neoplasms following treatment for Wilm’s tumor: A report from the National Wilms’ Tumor Study Group. J Clin Oncol. 1995;13:1851–1859. doi: 10.1200/JCO.1995.13.8.1851. [DOI] [PubMed] [Google Scholar]