

The quinoline moiety of the title quinoline carboxamide derivative is not planar as a result of a slight puckering of the pyridine ring. The secondary amine has a slightly pyramidal geometry.
Keywords: crystal structure, hydrogen bonding, quinoline, carboxamide, amine
Abstract
The structure of the title quinoline carboxamide derivative, C26H25N3O, is described. The quinoline moiety is not planar as a result of a slight puckering of the pyridine ring. The secondary amine has a slightly pyramidal geometry, certainly not planar. Both intra- and intermolecular hydrogen bonds are present. Hirshfeld surface analysis and lattice energies were used to investigate the intermolecular interactions.
Chemical context
Quinoline (1-aza-naphthalene or benzo[b]pyridine) is a natural heterocyclic building block often used as a template for derivatization and generation of drug-like libraries for the discovery of novel bioactive ligands (Mugnaini et al., 2009 ▸; Musiol, 2017 ▸). Quinoline-based compounds are well known for their antimalarial activity (Antony & Parija, 2016 ▸), although a large spectrum of other biological activities, such as anticancer, antimicrobial, anti-inflammatory, antioxidant, antihypertensive and against neurodegenerative diseases, have also been ascribed to these types of heterocyclic compounds (Nainwal et al., 2019 ▸).
This work is a continuation of our investigation into the preparation, structural analysis and pharmacological properties of substituted heterocyclics including, for example, new insights in the discovery of novel h-MAO-B inhibitors obtained by the structural characterization of a series of N-phenyl-4-oxo-4H-chromene-3-carboxamide derivatives (Gomes et al., 2015a
▸). Other chromone and coumarin carboxamides are discussed in Gomes et al. (2015b
▸, 2016 ▸).
Here we report the synthesis and structural characterization of a quinoline-3-carboxamide derivative, 4-(3,4-dimethylanilino)-N-(3,4-dimethylphenyl)quinoline-3-carboxamide, 1.
Structural commentary
An ellipsoid plot for compound 1 is shown in Fig. 1 ▸. The quinoline ring system is not planar, with atoms C2 and C4 deviating from the mean plane of the quinoline ring by −0.110 (3) and 0.125 (3) Å, respectively, and C6 lying −0.100 (3) Å below the mean plane. The pyridine ring is slightly puckered with a screw-boat conformation, Q = 0.087 (3)Å, θ = 106 (2)° and φ = 25 (2)°. The mean plane of this ring makes a dihedral angle of 7.49 (13)° with the mean plane of the benzene ring of the quinoline moiety. The angles between the mean planes of the quinoline ring and the benzene rings with pivot atoms C321 and C411 are 28.99 (11) and 59.16 (11)° respectively. The dihedral angle between the mean plane of these benzene rings is 64.71 (14)°.
Figure 1.

A view of the asymmetric unit of 1 with the atom-numbering scheme. Displacement ellipsoids are drawn at the 50% probability level.
The amide group attached to C3 is coplanar with the quinoline ring system. The C—N rotamer of the amide has an anti conformation placing the quinoline ring trans in relation to the ring with pivot atom C321. The amide group atoms are essentially coplanar with the quinoline ring with deviations of −0.034 (3), (C31), −0.009 (2) (O31), 0.009 (2), (N32) and 0.145 (3) Å (C321). The geometric arrangement of the amide permits the formation of an intramolecular hydrogen bond between the amine hydrogen atom and the carboxyl group of the amide, N41—H41⋯O31; geometric parameters are given in Table 1 ▸. A further intramolecular hydrogen bond, C326—H326⋯O31, occurs.
Table 1. Hydrogen-bond geometry (Å, °).
Cg is the centroid of the N1/C2–C4/C4A/C8A ring.
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N41—H41⋯O31 | 0.84 (4) | 1.93 (3) | 2.635 (3) | 142 (3) |
| C326—H326⋯O31 | 0.95 | 2.40 | 2.887 (3) | 112 |
| N32—H32⋯N1i | 0.90 (4) | 2.07 (4) | 2.891 (3) | 150 (3) |
| C2—H2⋯N1i | 0.96 (3) | 2.55 (3) | 3.477 (4) | 163 (2) |
| C416—H416⋯O31ii | 0.95 | 2.39 | 3.252 (4) | 150 |
| C326—H326⋯Cg iii | 0.95 | 2.82 | 3.398 (3) | 120 |
Symmetry codes: (i) 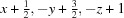 ; (ii)
; (ii) 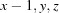 ; (iii)
; (iii) 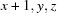 .
.
The secondary amine has a slightly pyramidal geometry, certainly not planar. The angles C411—N41—C4, C41—N41—H41 and C411—N41—H41 are 125.7 (2), 112 (2) and 115 (2)°, respectively, the sum of which (352.7°) is less than 360°; in addition, atom H41 lies 0.41 (3) Å out of the C4/N41/C411 mean plane, confirming the sp 3 hybridization of N41. An inspection of the amine bond lengths shows that there is a slight asymmetry of the electronic distribution around it: C4—N41 = 1.364 (3) Å while N41—C411 = 1.437 (4) Å, suggesting there is higher density between the nitrogen and the carbon atom of the quinoline ring system. However, these bonds and angles are typical for a Cquinoline–NH–C–R group, see the Database Survey below. As a consequence of the screw-boat pucker of the pyridine ring, the C4—N41 bond is displaced from the pyridine mean plane with a deviation of 0.159 (2) Å for N41; atom C411 is displaced by 0.965 (3) Å and consequently, the N41—C411 bond lies further from the mean plane.
Supramolecular features
In the crystal, the molecules are linked by N32—H32⋯N1(x + 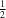 , −y +
, −y +  , −z + 1), hydrogen bonds, forming C6 chains which run parallel to the a-axis formed by the action of the 21 screw axis at (, 0,
, −z + 1), hydrogen bonds, forming C6 chains which run parallel to the a-axis formed by the action of the 21 screw axis at (, 0, 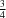 ). This is supplemented by the weak C2—H2⋯N1(x + , −y + , −z + 1) hydrogen bond, Table 1 ▸ and Fig. 2 ▸. The other weak hydrogen bonds, C416—H416⋯O31 and C418—H41B⋯O31, both involve atom O31 as an acceptor and link the chains described above to form a sheet which extends along the b-axis direction.
). This is supplemented by the weak C2—H2⋯N1(x + , −y + , −z + 1) hydrogen bond, Table 1 ▸ and Fig. 2 ▸. The other weak hydrogen bonds, C416—H416⋯O31 and C418—H41B⋯O31, both involve atom O31 as an acceptor and link the chains described above to form a sheet which extends along the b-axis direction.
Figure 2.

A view of the N32—H32⋯N1 C6 chain running along the a axis with the supplementary C2—H2⋯N1 bond. Hydrogen atoms not involved in the hydrogen bonding are omitted for clarity.
No π–π interactions occur but there is a possible C—H⋯π interaction, C326—H326⋯Cg, involving the pyridine ring (Table 1 ▸), which is discussed more fully below.
Hirshfeld surface analysis and lattice energies
Hirshfeld surfaces (McKinnon et al., 2004 ▸) and two-dimensional fingerprint (FP) plots provide complementary information concerning the intermolecular interactions discussed above. The analyses were generated using Crystal Explorer 3.1 (Wolff et al., 2012 ▸). The lattice energies for 1 were analysed after performing calculations as implemented in the PIXEL program (Gavezzotti, 2003 ▸, 2008 ▸). The total stabilization energy of the crystal packing, E tot is −207.0 kJ mol−1, distributed as Coulombic, (E coul = −112.9 kJ mol−1), polarization (E pol = −52.8 kJ mol−1), dispersion (E disp = −251.6 kJ mol−1) and repulsion (E rep = 210.4 kJ mol−1). The dispersive energy contributes the most to the total stabilization energy of the lattice, in addition to the C—H⋯O hydrogen bonds, and to the C—H⋯π interaction. The stabilization energy comes from six sub-structural motifs made by the molecule pairs I to VI that are shown in Figs. 3 ▸ to 8, together with the symmetry codes as well as the respective energies. They contribute a total energy of −369.4 kJ mol−1 for the lattice, half of it, −184.7 kJ mol−1 attributed to the (x, y, z) molecule. That energy corresponds approximately to 88% of the total stabilization energy of the network.
Figure 3.

Molecule pairs Ia/Ib: x − 1, y, z (top) and x + 1, y, z (bottom). Values of energies by pair: E tot = −55.9 kJ mol−1, E coul = −21.4 kJ mol−1, E pol = −10.0 kJ mol−1, E disp = −79.5 kJ mol−1 and E rep = 55.0 kJ mol−1. Interaction energies were calculated using PIXEL3.1 (Gavezzotti, 2003 ▸, 2008 ▸) based on densities computed with G09 using the mp2/6–31** level of theory.
The percentages of atom⋯atom close contacts taken from the FP plot (McKinnon et al., 2004 ▸) for 1 shows that, apart from the H⋯H contacts (58.4%), there are high percentages of C⋯H/H⋯C close contacts (27.0%) and of N⋯H/H⋯N close contacts (6.5%), see Table 2 ▸.
Table 2. Percentages for atom⋯atom close contacts.
| Compound | H⋯H | H⋯O/O⋯H | H⋯C/C⋯H | C⋯C | O⋯C/C⋯O | N⋯N | H⋯N/N⋯H | C⋯N/N⋯C |
|---|---|---|---|---|---|---|---|---|
| 1 | 58.4 | 4.3 | 27.0 | 2.5 | 0.6 | 0.2 | 6.5 | 0.5 |
Apart from the intramolecular hydrogen bond with N41, the carboxyl oxygen atom O31 involves its lone pairs in another two intermolecular C—H⋯O interactions, O31⋯H416—C416 and O31⋯H41B—C418. The first interaction creates chains running along the a-axis direction that are further stabilized by C—H⋯π interactions (C326—H326⋯Cg pyridine), as can be identified by the red spots in the Hirshfeld Surface (McKinnon et al., 2004 ▸) for the molecule, Fig. 9 ▸, and they form two molecule pairs, identified as sub-structures Ia/Ib in Fig. 3 ▸. Each of those pairs contribute −55.9 kJ mol−1 to the stabilization of the lattice, mainly dispersion energy. The second interaction, O31⋯H41B—C418, makes another two molecule pairs, IIIa/IIIb, Fig. 5 ▸. In this substructure the Coulombic energy is higher than the dispersive energy, which is indicative of the minor importance of the interactions involving the aromatic rings. These hydrogen bonds can also be identified as red spots in the HS, Fig. 9 ▸.
Figure 9.

Several faces of the HS plotted over d norm for 1 showing the red spots that indicate close contacts between atoms, which are identified in the figures.
Figure 5.

Molecule pairs IIIa/IIIb: (x + , −y + , z + 1 (top) and x − , −y + , z + 1 (bottom). Values of energies by pair: E
tot = −30.0 kJ mol−1, E
coul = −11.3 kJ mol−1, E
pol = −4.5 kJ mol−1, E
disp = −36.0 kJ mol−1, E
rep = 21.8 kJ mol−1. Interaction energies were calculated using PIXEL3.1 (Gavezzotti, 2003 ▸, 2008 ▸) based on densities computed with G09 using the mp2/6–31** level of theory.
The nitrogen atom N32 acts as a donor for N1 (N32—H32⋯N1). N1 also acts as an acceptor for C6, making a C6—H6⋯N1 hydrogen bond, seen as a red spot in Fig. 9 ▸. Those interactions give sub structural motifs IIa/IIb, Fig. 4 ▸. The molecules are linked by N32—H32⋯N1(x + , −y + , −z + 1) hydrogen bonds, forming C6 chains which run parallel to the a-axis direction, formed by the action of the 21 screw axis at (, 0, ). This is supplemented by the weak C2—H2⋯N1(x + , −y + , −z + 1) hydrogen bond, Figs. 3 ▸ and 4 ▸.
Figure 4.

Molecule pairs IIa/IIb: x − , –y + , −z + 1 (top) and x − , −y + , −z + , –y + , −z + 1 (bottom). Values of energies by pair: E
tot = −52.3 0 kJ mol−1, E
coul = −59.10 kJ mol−1, E
pol = −26.9 0 kJ mol−1, E
disp = −41.5 0 kJ mol−1 and E
rep = 75.2 0 kJ mol−1. Interaction energies were calculated using PIXEL3.1 (Gavezzotti, 2003 ▸, 2008 ▸) based on densities computed with G09 using the mp2/6–31** level of theory.
In addition, the C—H⋯π interaction can also be identified in the HS of the molecule, Fig. 9 ▸. The interaction connects the molecules in zigzag chains running along the c-axis direction, as a result of the propagation of the molecule pairs IVa/IVb depicted in Fig. 6 ▸.
Figure 6.

Molecule pairs IVa/IVb: −x + , −y + 1, z + (left) and −x + , −y + 1, z − (right). Values of energies by pair: E
tot = −20.7 kJ mol−1, E
coul = −7.4 kJ mol−1, E
pol = −4.4 kJ mol−1, E
disp = −31.3 kJ mol−1, E
rep = 22.5 kJ mol−1. Interaction energies were calculated using PIXEL3.1 (Gavezzotti, 2003 ▸, 2008 ▸) based on densities computed with G09 using the mp2/6–31** level of theory.
Apart from the sub-structural motifs described, there are two extra molecule pairs, identified as Va/Vb and VIa/VIb, which are also illustrated in Figs. 7 ▸ and 8 ▸: the two molecules involved are at x, y, z (green-coloured molecule) and −x + , −y + 1, z − /−x + , −y + 1, z + (black-coloured molecule) for Va/Vb and x − , −y + , −z + 1/x − , −y + , −z + 1 for VIa/VIb. Although these molecules do not exhibit atom⋯atom close contacts, each pair provides a significant contribution to the overall lattice stabilization energy of −14.5 and −11.3 kJ mol−1, respectively for V and VI. The grey molecules drawn in this figure indicate a possible pathway for electronic delocalization within the network of molecules.
Figure 7.

Molecule pairs Va/Vb: −x + , y + 1, z − (left) and −x + , y + 1, z + (right). Values of energies by pair: E
tot = −14.5 kJ mol−1, E
coul = −5.0 kJ mol−1, E
pol = −2.5 kJ mol−1, E
disp = − 23.4 kJ mol−1, E
rep = 16.4 kJ mol−1. Interaction energies were calculated using PIXEL3.1 (Gavezzotti, 2003 ▸, 2008 ▸) based on densities computed with G09 using the mp2/6–31** level of theory.
Figure 8.

Molecule pairs VIa/VIb, (x − , −y + , −z + 1 (left) and x − , −y + , −z + 1(right). Values of energies by pair: E
tot = −11.3 kJ mol−1, E
coul = −3.3 kJ mol−1, E
pol = −2.2 kJ mol−1, E
disp = −15.5 kJ mol−1, E
rep = 9.6 kJ mol−1. Interaction energies were calculated using PIXEL3.1 (Gavezzotti, 2003 ▸, 2008 ▸) based on densities computed with G09 using the mp2/6–31** level of theory.
Database survey
A search of the Cambridge Structural Database (CSD, Version 5.40, November 2019 update; Groom et al., 2016 ▸) for 3,4-disubstituted quinoline with an N—H unit attached to C4 revealed two compounds: SEZJIR (3-acetyl-4-aminoquinoline; Lokaj et al., 2007 ▸) with a carbonyl group attached to C3 and an amino group attached to C4 and PABPUD {4-[3-(N,N-dimethylamino)propylamino]-3-nitroquinoline; Boyd et al., 1992 ▸} with an amino group attached to C4 and a nitro group attached to C3. In both of these compounds, there is no puckering of the pyridine ring and the quinoline ring system is essentially planar. In both cases, a hydrogen atom forms an intramolecular hydrogen bond between an amino hydrogen and the carbonyl oxygen in both independent molecules of the asymmetric unit (SEZJIR) or between the amino hydrogen and a nitro group oxygen atom (PABPUD). In both structures, the C(pyridine)⋯N(amino) distances are significantly shorter than those in 1, viz. 1.325 and 1.335 Å for the two molecules in the asymmetric unit of SEZJIR and 1.320 Å in PABPUD. The corresponding value in 1 is 1.364 (3) Å.
A survey of quinoline compounds, with an R factor of 10% or less with a Cquinoline–NH–Caryl/sp3 unit attached to C4 of the quinoline moiety gave 56 hits for 63 individual molecules, including 1. The Cquinoline—N distances lie in the range 1.319 to 1.438 Å with an average value of 1.360 Å.
The situation is more complex for the N—Caryl/sp3 bond and for the Cquinoline—N—Caryl/sp3 angle. A scatterplot of these revealed two populations, one in which the N atom is attached to a benzene ring and the other in which the connection is to an sp 3 carbon. UNIKUZ [6-(t-butylsulfonyl)-N-(5-fluoro-1H-indazol-3-yl)quinolin-4-amine methanol solvate; Haile et al., 2016 ▸) is included in the first group. The Caryl—N distances lie in the range 1.396 to 1.438 Å with an average value of 1.418 Å and an average Cquinoline—N—Caryl/sp3 angle of 126.105°. In the second case, the Caryl/sp3—N distances lie in the range 1.439 to 1.478 Å with an average value of 1.458 Å, with an average Cquinoline—N—Caryl/sp3 angle of 123.98°.
As noted above, the conformation around the amino N atom is slightly pyramidal. In their paper on bond lengths in organic compounds, Allen et al. (2006 ▸) discuss the planarity and pyramidality of amino compounds. They state that for planar N atoms, the mean valence angle is greater than 117.6° while for pyramidal N atoms the mean valence angle lies in the range 108 to 114°. The value for 1 is 117.56°. There are three other structures in this survey which have average valence angles close to but less than 117°. The valence angles are 116.57° in DAMIOT {2,3-bis[(2,6-dimethylphenyl)sulfanyl]-N-phenylquinolin-4-amine; Florke & Egold, 2016 ▸}, 117.41° in MEQKEY (2,4-dianilino-3-ethylquinoline; Katritzky et al., 2000 ▸) and 117.04° in OTAMOM {2-(4-methoxyphenyl)-N-[2-(2-phenylvinyl)phenyl]quinolin-4-amine; Mphahlele & Mphahlele, 2011 ▸}. These four compounds are thus neither strictly planar nor pyramidal.
There are two compounds in the database which have an amide group attached to C3, GICGIL [2-chloro-N-(4-fluorophenyl)-6-methylquinoline-3-carboxamide; Govender et al., 2018 ▸] and SUZHEB (N-isopropyl-6-methyl-2-phenylquinoline-3-carboxamide; Benzerka et al., 2010 ▸). In both these compounds, the amide group is inclined to the quinoline moiety, unlike in molecule 1.
Synthesis and crystallization
The title quinolone derivative 1 was synthesized by a one-pot reaction between 4-oxo-1,4-dihydroquinoline-3-carboxylic acid and 3,4-dimethylaniline in the presence of POCl3 following a procedure described previously (Cagide et al., 2015 ▸). The title compound was obtained in 70% yield and characterized by NMR. It was re-crystallized from dichloromethane to yield crystals suitable for X-ray diffraction, m.p. 489–493 K.
NMR data were acquired on a Bruker AMX 400 spectrometer, recorded at room temperature in 5 mm outer-diameter tubes. The samples were prepared in deuterated dimethylsulfoxide (DMSO) with tetramethylsilane (TMS) as internal reference. Chemical shifts are expressed as δ (ppm) values relative to TMS; coupling constants (J) are given in Hz. Atoms are labelled with their numerical designation as per Fig. 1 ▸. See Supporting Information for spectra.
4-(3,4-Dimethylanilino)- N -(3,4-dimethylphenyl)quinoline-3-carboxamide
1H NMR (400 MHz, DMSO): 10.16 (1H, s, CONH), 9.43 (1H, s, NH), 8.82 (1H, s, H-2), 8.14 (1H, dd, J = 1.0, 8.5 Hz, H-8), 7.95 (1H, dd, J = 0.84, 8.4 Hz, H-5), 7.73 (1H, ddd, J = 1.0, 6.9, 8.4 Hz, H-6), 7.46 (1H, ddd, J = 1.0, 6.9, 8.5 Hz, H-7), 7.18 (1H, d, J = 2.0 Hz, H-412), 7.12 (1H, dd, J = 2.1, 8.0 Hz H-326), 7.00 (1H, d, J = 8.0 Hz, H-325), 6.93 (1H, d, J = 8.0 Hz, H-415), 6.84 (1H, d, J = 2.1 Hz, H-322), 6.72 (1H, dd, J = 2.0, 8.0 Hz, H-416), 2.01 (3H, s, CH3), 2.07 (3H, s, CH3), 2.16 (6H, s, 2 × CH3).
13C NMR (100 MHz, DMSO): 165.4 (CONH), 149.9 (C-2), 149.1 (C-8A), 146.6 (C-4), 140.3 (C-411), 136.5 (C-414), 136.3 (C-321), 135.6 (C-324), 131.2 (C-413), 130.5 (C-323), 130.2 (C-6), 129.6 (C-415), 129.2 (C-5), 129.0 (C-325), 125.0 (C-7), 124.2 (C-8), 121.6 (C-412), 121.5 (C-322), 120.8 (C-4A), 117.8 (C-325), 117.7 (C-416), 114.4 (C-3), 19.5 (CH3), 19.3 (CH3), 18.7 (CH3), 18.5 (CH3).
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 3 ▸. The H atoms were included in idealized positions and treated as riding atoms: C—H = 0.95–0.98 Å with U iso(H) = 1.2U eq(C) or 1.5U eq(C) for methyl H atoms. Those attached to N and C2 [C—H = 0.96 (3) Å] were refined. The latter was refined since it is involved in a short contact with H32, which is attached to N32. Although in the riding model for H2 the H-atom position is within the highest contour on the difference map, it is not at the centre. In the refined model it is. The H⋯H distances are 1.87 and 1.93 Å for the riding and refined models, respectively. The angles around C2 are N1—C2—C3 = 125.9 (3) and 125.9°(3); N—C2—H2 = 117 and 111.9 (17)° and C3—C2—H2 = 117 and 122.2 (17)° for riding and refined H atoms, respectively. In the case of H32, the N32—H32 distance changes from 0.89 (3) to 0.90 (4) Å and the angle C31—N32—H32 changes from 120 (2) to 119 (2)° for riding to refined, respectively, which are really insignificant shifts. Hence, in this case the short contact does induce a shift in the angular position of H2 from its calculated position.
Table 3. Experimental details.
| Crystal data | |
| Chemical formula | C26H25N3O |
| M r | 395.49 |
| Crystal system, space group | Orthorhombic, P212121 |
| Temperature (K) | 100 |
| a, b, c (Å) | 6.2502 (3), 15.7915 (6), 20.7395 (9) |
| V (Å3) | 2046.99 (15) |
| Z | 4 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.08 |
| Crystal size (mm) | 0.30 × 0.05 × 0.01 |
| Data collection | |
| Diffractometer | Rigaku FRE+ equipped with VHF Varimax confocal mirrors and an AFC12 goniometer and HyPix 6000 detector |
| Absorption correction | Multi-scan (CrysAlis PRO; Rigaku OD, 2018 ▸) |
| T min, T max | 0.487, 1.000 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 28997, 3754, 3390 |
| R int | 0.089 |
| (sin θ/λ)max (Å−1) | 0.602 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.044, 0.094, 1.08 |
| No. of reflections | 3754 |
| No. of parameters | 287 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.22, −0.20 |
| Absolute structure | Flack x determined using 1238 quotients [(I +)−(I −)]/[(I +)+(I −)] (Parsons et al., 2013 ▸) |
| Absolute structure parameter | 0.2 (10) |
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S2056989020000298/zl2767sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989020000298/zl2767Isup2.hkl
Spectra. DOI: 10.1107/S2056989020000298/zl2767sup3.tif
CCDC reference: 1879928
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
The authors thank the staff at the National Crystallographic Service, University of Southampton, for the data collection, help and advice (Coles & Gale, 2012 ▸).
supplementary crystallographic information
Crystal data
| C26H25N3O | Dx = 1.283 Mg m−3 |
| Mr = 395.49 | Mo Kα radiation, λ = 0.71075 Å |
| Orthorhombic, P212121 | Cell parameters from 5133 reflections |
| a = 6.2502 (3) Å | θ = 1.6–27.0° |
| b = 15.7915 (6) Å | µ = 0.08 mm−1 |
| c = 20.7395 (9) Å | T = 100 K |
| V = 2046.99 (15) Å3 | Needle, yellow |
| Z = 4 | 0.30 × 0.05 × 0.01 mm |
| F(000) = 840 |
Data collection
| Rigaku FRE+ equipped with VHF Varimax confocal mirrors and an AFC12 goniometer and HyPix 6000 detector diffractometer | 3754 independent reflections |
| Radiation source: Rotating Anode, Rigaku FRE+ | 3390 reflections with I > 2σ(I) |
| Confocal mirrors, VHF Varimax monochromator | Rint = 0.089 |
| Detector resolution: 10 pixels mm-1 | θmax = 25.4°, θmin = 1.6° |
| profile data from ω–scans | h = −7→7 |
| Absorption correction: multi-scan (CrysAlis PRO; Rigaku OD, 2018) | k = −19→19 |
| Tmin = 0.487, Tmax = 1.000 | l = −24→24 |
| 28997 measured reflections |
Refinement
| Refinement on F2 | Secondary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Hydrogen site location: mixed |
| R[F2 > 2σ(F2)] = 0.044 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.094 | w = 1/[σ2(Fo2) + (0.0402P)2 + 0.5091P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.08 | (Δ/σ)max < 0.001 |
| 3754 reflections | Δρmax = 0.22 e Å−3 |
| 287 parameters | Δρmin = −0.20 e Å−3 |
| 0 restraints | Absolute structure: Flack x determined using 1238 quotients [(I+)-(I-)]/[(I+)+(I-)] (Parsons et al., 2013) |
| Primary atom site location: structure-invariant direct methods | Absolute structure parameter: 0.2 (10) |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O31 | 0.5597 (3) | 0.49964 (12) | 0.49759 (10) | 0.0221 (5) | |
| N1 | −0.0092 (4) | 0.69133 (14) | 0.44267 (11) | 0.0174 (5) | |
| N32 | 0.5344 (4) | 0.62696 (15) | 0.54659 (11) | 0.0162 (5) | |
| H32 | 0.470 (5) | 0.678 (2) | 0.5477 (15) | 0.028 (9)* | |
| N41 | 0.2497 (4) | 0.44714 (15) | 0.42129 (13) | 0.0195 (5) | |
| H41 | 0.368 (6) | 0.441 (2) | 0.4400 (16) | 0.026 (9)* | |
| C2 | 0.1722 (5) | 0.66921 (17) | 0.47005 (13) | 0.0161 (6) | |
| H2 | 0.233 (5) | 0.7147 (17) | 0.4948 (14) | 0.014 (7)* | |
| C3 | 0.2727 (4) | 0.58912 (17) | 0.46478 (13) | 0.0154 (6) | |
| C4 | 0.1727 (5) | 0.52774 (16) | 0.42608 (13) | 0.0158 (6) | |
| C5 | −0.1008 (5) | 0.50493 (18) | 0.33862 (13) | 0.0196 (6) | |
| H5 | −0.037379 | 0.452194 | 0.327658 | 0.024* | |
| C4A | −0.0119 (4) | 0.55400 (17) | 0.38887 (13) | 0.0161 (6) | |
| C6 | −0.2769 (5) | 0.53228 (19) | 0.30555 (15) | 0.0237 (7) | |
| H6 | −0.332691 | 0.499128 | 0.271220 | 0.028* | |
| C7 | −0.3757 (5) | 0.60896 (19) | 0.32201 (15) | 0.0241 (7) | |
| H7 | −0.504081 | 0.625506 | 0.300995 | 0.029* | |
| C8 | −0.2880 (5) | 0.65997 (19) | 0.36813 (14) | 0.0209 (6) | |
| H8 | −0.352964 | 0.712802 | 0.377996 | 0.025* | |
| C8A | −0.1007 (5) | 0.63448 (17) | 0.40134 (13) | 0.0160 (6) | |
| C31 | 0.4680 (4) | 0.56876 (16) | 0.50328 (13) | 0.0158 (6) | |
| C321 | 0.7023 (4) | 0.61744 (16) | 0.59179 (13) | 0.0146 (6) | |
| C322 | 0.6840 (5) | 0.66241 (17) | 0.64919 (14) | 0.0173 (6) | |
| H322 | 0.559581 | 0.695626 | 0.656408 | 0.021* | |
| C323 | 0.8416 (4) | 0.66032 (18) | 0.69622 (14) | 0.0184 (6) | |
| C324 | 1.0277 (5) | 0.61192 (18) | 0.68531 (13) | 0.0181 (6) | |
| C325 | 1.0432 (5) | 0.56727 (18) | 0.62795 (14) | 0.0191 (6) | |
| H325 | 1.167921 | 0.534397 | 0.620254 | 0.023* | |
| C326 | 0.8845 (4) | 0.56865 (17) | 0.58145 (14) | 0.0168 (6) | |
| H326 | 0.899709 | 0.536669 | 0.542929 | 0.020* | |
| C327 | 0.8115 (5) | 0.70814 (19) | 0.75834 (15) | 0.0250 (7) | |
| H32A | 0.934273 | 0.745612 | 0.765419 | 0.037* | |
| H32B | 0.800307 | 0.667948 | 0.794181 | 0.037* | |
| H32C | 0.680368 | 0.741992 | 0.755852 | 0.037* | |
| C328 | 1.2038 (5) | 0.6077 (2) | 0.73465 (15) | 0.0254 (7) | |
| H32D | 1.253251 | 0.665128 | 0.744633 | 0.038* | |
| H32E | 1.323221 | 0.574413 | 0.717458 | 0.038* | |
| H32F | 1.149786 | 0.580841 | 0.774012 | 0.038* | |
| C411 | 0.1222 (5) | 0.37148 (17) | 0.41833 (14) | 0.0194 (6) | |
| C412 | 0.2064 (5) | 0.30036 (17) | 0.38929 (13) | 0.0202 (7) | |
| H412 | 0.346385 | 0.302560 | 0.371508 | 0.024* | |
| C413 | 0.0886 (5) | 0.22452 (18) | 0.38552 (13) | 0.0192 (6) | |
| C414 | −0.1149 (5) | 0.22182 (18) | 0.41275 (14) | 0.0208 (7) | |
| C415 | −0.1955 (5) | 0.29255 (18) | 0.44308 (14) | 0.0228 (7) | |
| H415 | −0.333552 | 0.290071 | 0.462158 | 0.027* | |
| C416 | −0.0793 (5) | 0.36751 (18) | 0.44637 (14) | 0.0200 (6) | |
| H416 | −0.137324 | 0.415606 | 0.467629 | 0.024* | |
| C417 | 0.1826 (5) | 0.14893 (18) | 0.35187 (16) | 0.0275 (7) | |
| H41D | 0.090414 | 0.132683 | 0.315709 | 0.041* | |
| H41E | 0.193326 | 0.101625 | 0.382290 | 0.041* | |
| H41F | 0.325447 | 0.163163 | 0.335672 | 0.041* | |
| C418 | −0.2461 (5) | 0.14148 (19) | 0.40803 (16) | 0.0281 (7) | |
| H41A | −0.384750 | 0.150125 | 0.429087 | 0.042* | |
| H41B | −0.169739 | 0.095098 | 0.429390 | 0.042* | |
| H41C | −0.268722 | 0.127195 | 0.362554 | 0.042* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O31 | 0.0180 (10) | 0.0128 (9) | 0.0356 (12) | 0.0031 (8) | −0.0017 (9) | −0.0058 (9) |
| N1 | 0.0155 (12) | 0.0137 (11) | 0.0229 (13) | −0.0001 (10) | 0.0022 (11) | −0.0008 (10) |
| N32 | 0.0136 (12) | 0.0114 (11) | 0.0236 (13) | 0.0014 (10) | −0.0003 (10) | −0.0012 (10) |
| N41 | 0.0152 (13) | 0.0120 (12) | 0.0314 (14) | 0.0002 (10) | −0.0025 (12) | −0.0058 (10) |
| C2 | 0.0173 (14) | 0.0107 (13) | 0.0202 (15) | −0.0031 (12) | 0.0028 (12) | −0.0018 (11) |
| C3 | 0.0133 (14) | 0.0137 (13) | 0.0194 (14) | −0.0012 (12) | 0.0052 (12) | −0.0011 (11) |
| C4 | 0.0152 (14) | 0.0139 (13) | 0.0184 (15) | −0.0010 (11) | 0.0042 (12) | −0.0010 (11) |
| C5 | 0.0203 (15) | 0.0159 (14) | 0.0226 (15) | −0.0033 (13) | 0.0013 (13) | −0.0032 (12) |
| C4A | 0.0146 (14) | 0.0146 (14) | 0.0193 (14) | −0.0032 (12) | 0.0040 (12) | 0.0013 (11) |
| C6 | 0.0273 (17) | 0.0212 (15) | 0.0224 (15) | −0.0067 (14) | −0.0068 (14) | −0.0013 (12) |
| C7 | 0.0203 (16) | 0.0232 (16) | 0.0287 (17) | −0.0014 (13) | −0.0065 (13) | 0.0028 (13) |
| C8 | 0.0195 (15) | 0.0171 (14) | 0.0260 (16) | 0.0008 (13) | 0.0010 (13) | 0.0024 (12) |
| C8A | 0.0164 (14) | 0.0138 (13) | 0.0179 (14) | −0.0031 (12) | 0.0048 (12) | −0.0003 (11) |
| C31 | 0.0145 (14) | 0.0123 (12) | 0.0208 (15) | −0.0020 (12) | 0.0057 (12) | 0.0000 (12) |
| C321 | 0.0136 (14) | 0.0103 (13) | 0.0200 (14) | −0.0020 (11) | 0.0009 (12) | 0.0035 (11) |
| C322 | 0.0163 (14) | 0.0118 (13) | 0.0238 (16) | −0.0002 (12) | 0.0036 (12) | 0.0004 (11) |
| C323 | 0.0171 (14) | 0.0180 (14) | 0.0202 (15) | −0.0030 (13) | 0.0036 (12) | 0.0041 (11) |
| C324 | 0.0159 (15) | 0.0152 (14) | 0.0232 (15) | −0.0035 (12) | 0.0031 (12) | 0.0057 (12) |
| C325 | 0.0138 (14) | 0.0168 (14) | 0.0267 (16) | 0.0018 (12) | 0.0048 (12) | 0.0032 (12) |
| C326 | 0.0151 (14) | 0.0148 (13) | 0.0205 (15) | −0.0018 (11) | 0.0034 (12) | −0.0011 (12) |
| C327 | 0.0246 (16) | 0.0234 (16) | 0.0269 (17) | 0.0001 (14) | 0.0022 (14) | −0.0033 (13) |
| C328 | 0.0186 (16) | 0.0316 (17) | 0.0261 (16) | −0.0015 (14) | 0.0003 (13) | 0.0025 (13) |
| C411 | 0.0241 (16) | 0.0132 (13) | 0.0208 (15) | −0.0013 (12) | −0.0066 (13) | −0.0002 (12) |
| C412 | 0.0225 (16) | 0.0174 (14) | 0.0209 (16) | 0.0005 (13) | −0.0013 (13) | 0.0001 (12) |
| C413 | 0.0279 (17) | 0.0128 (14) | 0.0171 (15) | 0.0022 (12) | −0.0057 (13) | −0.0007 (11) |
| C414 | 0.0223 (16) | 0.0205 (15) | 0.0197 (16) | −0.0021 (12) | −0.0065 (13) | 0.0028 (12) |
| C415 | 0.0198 (15) | 0.0252 (16) | 0.0235 (16) | −0.0019 (13) | −0.0022 (13) | 0.0035 (12) |
| C416 | 0.0206 (15) | 0.0150 (14) | 0.0245 (16) | 0.0005 (12) | −0.0025 (13) | −0.0018 (12) |
| C417 | 0.0307 (17) | 0.0171 (15) | 0.0347 (18) | −0.0001 (14) | 0.0016 (15) | −0.0046 (13) |
| C418 | 0.0298 (18) | 0.0213 (16) | 0.0333 (18) | −0.0062 (14) | −0.0042 (15) | 0.0036 (13) |
Geometric parameters (Å, º)
| O31—C31 | 1.238 (3) | C323—C327 | 1.505 (4) |
| N1—C2 | 1.315 (4) | C324—C325 | 1.386 (4) |
| N1—C8A | 1.367 (4) | C324—C328 | 1.505 (4) |
| N32—C31 | 1.350 (3) | C325—C326 | 1.384 (4) |
| N32—C321 | 1.416 (4) | C325—H325 | 0.9500 |
| N32—H32 | 0.90 (4) | C326—H326 | 0.9500 |
| N41—C4 | 1.364 (3) | C327—H32A | 0.9800 |
| N41—C411 | 1.437 (4) | C327—H32B | 0.9800 |
| N41—H41 | 0.84 (4) | C327—H32C | 0.9800 |
| C2—C3 | 1.416 (4) | C328—H32D | 0.9800 |
| C2—H2 | 0.96 (3) | C328—H32E | 0.9800 |
| C3—C4 | 1.405 (4) | C328—H32F | 0.9800 |
| C3—C31 | 1.494 (4) | C411—C412 | 1.379 (4) |
| C4—C4A | 1.449 (4) | C411—C416 | 1.389 (4) |
| C5—C6 | 1.367 (4) | C412—C413 | 1.408 (4) |
| C5—C4A | 1.413 (4) | C412—H412 | 0.9500 |
| C5—H5 | 0.9500 | C413—C414 | 1.392 (4) |
| C4A—C8A | 1.411 (4) | C413—C417 | 1.503 (4) |
| C6—C7 | 1.401 (4) | C414—C415 | 1.377 (4) |
| C6—H6 | 0.9500 | C414—C418 | 1.514 (4) |
| C7—C8 | 1.365 (4) | C415—C416 | 1.390 (4) |
| C7—H7 | 0.9500 | C415—H415 | 0.9500 |
| C8—C8A | 1.417 (4) | C416—H416 | 0.9500 |
| C8—H8 | 0.9500 | C417—H41D | 0.9800 |
| C321—C322 | 1.391 (4) | C417—H41E | 0.9800 |
| C321—C326 | 1.391 (4) | C417—H41F | 0.9800 |
| C322—C323 | 1.387 (4) | C418—H41A | 0.9800 |
| C322—H322 | 0.9500 | C418—H41B | 0.9800 |
| C323—C324 | 1.410 (4) | C418—H41C | 0.9800 |
| C2—N1—C8A | 117.2 (2) | C326—C325—C324 | 122.7 (3) |
| C31—N32—C321 | 126.6 (2) | C326—C325—H325 | 118.7 |
| C31—N32—H32 | 119 (2) | C324—C325—H325 | 118.7 |
| C321—N32—H32 | 114 (2) | C325—C326—C321 | 119.2 (3) |
| C4—N41—C411 | 125.7 (2) | C325—C326—H326 | 120.4 |
| C4—N41—H41 | 112 (2) | C321—C326—H326 | 120.4 |
| C411—N41—H41 | 115 (2) | C323—C327—H32A | 109.5 |
| N1—C2—C3 | 125.9 (3) | C323—C327—H32B | 109.5 |
| N1—C2—H2 | 111.9 (17) | H32A—C327—H32B | 109.5 |
| C3—C2—H2 | 122.2 (17) | C323—C327—H32C | 109.5 |
| C4—C3—C2 | 117.6 (3) | H32A—C327—H32C | 109.5 |
| C4—C3—C31 | 121.3 (2) | H32B—C327—H32C | 109.5 |
| C2—C3—C31 | 120.9 (2) | C324—C328—H32D | 109.5 |
| N41—C4—C3 | 121.9 (3) | C324—C328—H32E | 109.5 |
| N41—C4—C4A | 120.6 (2) | H32D—C328—H32E | 109.5 |
| C3—C4—C4A | 117.4 (2) | C324—C328—H32F | 109.5 |
| C6—C5—C4A | 120.9 (3) | H32D—C328—H32F | 109.5 |
| C6—C5—H5 | 119.5 | H32E—C328—H32F | 109.5 |
| C4A—C5—H5 | 119.5 | C412—C411—C416 | 119.5 (3) |
| C8A—C4A—C5 | 118.3 (3) | C412—C411—N41 | 119.0 (3) |
| C8A—C4A—C4 | 118.3 (2) | C416—C411—N41 | 121.5 (2) |
| C5—C4A—C4 | 123.3 (3) | C411—C412—C413 | 121.2 (3) |
| C5—C6—C7 | 120.4 (3) | C411—C412—H412 | 119.4 |
| C5—C6—H6 | 119.8 | C413—C412—H412 | 119.4 |
| C7—C6—H6 | 119.8 | C414—C413—C412 | 118.8 (3) |
| C8—C7—C6 | 120.3 (3) | C414—C413—C417 | 121.4 (3) |
| C8—C7—H7 | 119.9 | C412—C413—C417 | 119.8 (3) |
| C6—C7—H7 | 119.9 | C415—C414—C413 | 119.6 (3) |
| C7—C8—C8A | 120.3 (3) | C415—C414—C418 | 120.7 (3) |
| C7—C8—H8 | 119.9 | C413—C414—C418 | 119.6 (3) |
| C8A—C8—H8 | 119.9 | C414—C415—C416 | 121.5 (3) |
| N1—C8A—C4A | 122.8 (3) | C414—C415—H415 | 119.3 |
| N1—C8A—C8 | 117.7 (3) | C416—C415—H415 | 119.3 |
| C4A—C8A—C8 | 119.5 (3) | C411—C416—C415 | 119.4 (3) |
| O31—C31—N32 | 121.4 (3) | C411—C416—H416 | 120.3 |
| O31—C31—C3 | 121.1 (2) | C415—C416—H416 | 120.3 |
| N32—C31—C3 | 117.4 (2) | C413—C417—H41D | 109.5 |
| C322—C321—C326 | 118.8 (3) | C413—C417—H41E | 109.5 |
| C322—C321—N32 | 116.8 (2) | H41D—C417—H41E | 109.5 |
| C326—C321—N32 | 124.3 (2) | C413—C417—H41F | 109.5 |
| C323—C322—C321 | 122.1 (3) | H41D—C417—H41F | 109.5 |
| C323—C322—H322 | 119.0 | H41E—C417—H41F | 109.5 |
| C321—C322—H322 | 119.0 | C414—C418—H41A | 109.5 |
| C322—C323—C324 | 119.1 (3) | C414—C418—H41B | 109.5 |
| C322—C323—C327 | 120.1 (3) | H41A—C418—H41B | 109.5 |
| C324—C323—C327 | 120.8 (3) | C414—C418—H41C | 109.5 |
| C325—C324—C323 | 118.1 (3) | H41A—C418—H41C | 109.5 |
| C325—C324—C328 | 120.7 (3) | H41B—C418—H41C | 109.5 |
| C323—C324—C328 | 121.2 (3) | ||
| C8A—N1—C2—C3 | −5.5 (4) | C2—C3—C31—N32 | 5.0 (4) |
| N1—C2—C3—C4 | 1.1 (4) | C31—N32—C321—C322 | −150.3 (3) |
| N1—C2—C3—C31 | −174.0 (3) | C31—N32—C321—C326 | 31.7 (4) |
| C411—N41—C4—C3 | 141.0 (3) | C326—C321—C322—C323 | 0.2 (4) |
| C411—N41—C4—C4A | −41.6 (4) | N32—C321—C322—C323 | −177.9 (2) |
| C2—C3—C4—N41 | −175.7 (3) | C321—C322—C323—C324 | 0.8 (4) |
| C31—C3—C4—N41 | −0.6 (4) | C321—C322—C323—C327 | −178.3 (3) |
| C2—C3—C4—C4A | 6.8 (4) | C322—C323—C324—C325 | −0.9 (4) |
| C31—C3—C4—C4A | −178.1 (2) | C327—C323—C324—C325 | 178.1 (3) |
| C6—C5—C4A—C8A | −4.1 (4) | C322—C323—C324—C328 | 179.8 (3) |
| C6—C5—C4A—C4 | 179.2 (3) | C327—C323—C324—C328 | −1.2 (4) |
| N41—C4—C4A—C8A | 172.4 (3) | C323—C324—C325—C326 | 0.1 (4) |
| C3—C4—C4A—C8A | −10.1 (4) | C328—C324—C325—C326 | 179.4 (3) |
| N41—C4—C4A—C5 | −10.8 (4) | C324—C325—C326—C321 | 0.8 (4) |
| C3—C4—C4A—C5 | 166.7 (3) | C322—C321—C326—C325 | −1.0 (4) |
| C4A—C5—C6—C7 | −1.5 (4) | N32—C321—C326—C325 | 176.9 (2) |
| C5—C6—C7—C8 | 4.7 (5) | C4—N41—C411—C412 | 155.0 (3) |
| C6—C7—C8—C8A | −2.2 (4) | C4—N41—C411—C416 | −27.8 (4) |
| C2—N1—C8A—C4A | 1.7 (4) | C416—C411—C412—C413 | 2.3 (4) |
| C2—N1—C8A—C8 | −175.7 (2) | N41—C411—C412—C413 | 179.6 (3) |
| C5—C4A—C8A—N1 | −170.9 (3) | C411—C412—C413—C414 | −1.1 (4) |
| C4—C4A—C8A—N1 | 6.0 (4) | C411—C412—C413—C417 | 178.3 (3) |
| C5—C4A—C8A—C8 | 6.4 (4) | C412—C413—C414—C415 | −0.5 (4) |
| C4—C4A—C8A—C8 | −176.7 (2) | C417—C413—C414—C415 | −179.9 (3) |
| C7—C8—C8A—N1 | 174.1 (3) | C412—C413—C414—C418 | 178.7 (3) |
| C7—C8—C8A—C4A | −3.4 (4) | C417—C413—C414—C418 | −0.6 (4) |
| C321—N32—C31—O31 | −3.1 (4) | C413—C414—C415—C416 | 1.0 (4) |
| C321—N32—C31—C3 | 173.8 (2) | C418—C414—C415—C416 | −178.3 (3) |
| C4—C3—C31—O31 | 6.9 (4) | C412—C411—C416—C415 | −1.8 (4) |
| C2—C3—C31—O31 | −178.1 (3) | N41—C411—C416—C415 | −179.1 (3) |
| C4—C3—C31—N32 | −170.0 (3) | C414—C415—C416—C411 | 0.2 (4) |
Hydrogen-bond geometry (Å, º)
Cg is the centroid of the N1/C2–C4/C4A/C8A ring.
| D—H···A | D—H | H···A | D···A | D—H···A |
| N41—H41···O31 | 0.84 (4) | 1.93 (3) | 2.635 (3) | 142 (3) |
| C326—H326···O31 | 0.95 | 2.40 | 2.887 (3) | 112 |
| N32—H32···N1i | 0.90 (4) | 2.07 (4) | 2.891 (3) | 150 (3) |
| C2—H2···N1i | 0.96 (3) | 2.55 (3) | 3.477 (4) | 163 (2) |
| C416—H416···O31ii | 0.95 | 2.39 | 3.252 (4) | 150 |
| C326—H326···Cgiii | 0.95 | 2.82 | 3.398 (3) | 120 |
Symmetry codes: (i) x+1/2, −y+3/2, −z+1; (ii) x−1, y, z; (iii) x+1, y, z.
Funding Statement
This work was funded by FEDER grant UID/QUI/00081. Fundação para a Ciência e a Tecnologia grant PTDC/ASP-PES/28397/2017.
References
- Allen, F. R., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (2006). International Tables for Crystallography, Vol. C, ch. 9.5, pp. 790–811. Chester: IUCr
- Antony, H. A. & Parija, S. C. (2016). Trop. Parasitol. 6, 30–41. [DOI] [PMC free article] [PubMed]
- Benzerka, S., Bouraiou, A., Bouacida, S., Roisnel, T. & Belfaitah, A. (2010). Acta Cryst. E66, o2304–o2305. [DOI] [PMC free article] [PubMed]
- Boyd, M., Boyd, P. D. W., Atwell, G. J., Wilson, W. R. & Denny, W. A. (1992). J. Chem. Soc. Perkin Trans. 2, pp. 579–585.
- Cagide, F., Silva, T., Reis, J., Gaspar, A., Borges, F., Gomes, L. R. & Low, J. N. (2015). Chem. Commun. 51, 2832–2835. [DOI] [PubMed]
- Coles, S. J. & Gale, P. A. (2012). Chem. Sci. 3, 683–689.
- Florke, U. & Egold, H. (2016). Private Communication (refcode DAMIOT). CCDC, Cambridge, England.
- Gavezzotti, A. (2003). J. Phys. Chem. B, 107, 2344–2353.
- Gavezzotti, A. (2008). Mol. Phys. 106, 1473–1485.
- Gomes, L. R., Low, J. N., Cagide, F., Chavarria, D. & Borges, F. (2015a). Acta Cryst. E71, 547–554. [DOI] [PMC free article] [PubMed]
- Gomes, L. R., Low, J. N., Cagide, F., Gaspar, A. & Borges, F. (2015b). Acta Cryst. E71, 1270–1277. [DOI] [PMC free article] [PubMed]
- Gomes, L. R., Low, J. N., Fonseca, A., Matos, M. J. & Borges, F. (2016). Acta Cryst. E72, 926–932. [DOI] [PMC free article] [PubMed]
- Govender, H., Mocktar, C. & Koorbanally, N. A. (2018). J. Heterocycl. Chem. 55, 1002–1009.
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed]
- Haile, P. A., Votta, B. J., Marquis, R. W., Bury, M. J., Mehlmann, J. F., Singhaus, R. Jr, Charnley, A. K., Lakdawala, A. S., Convery, M. A., Lipshutz, D. B., Desai, B. M., Swift, B., Capriotti, C. A., Berger, S. B., Majahan, M. K., Reilly, M. A., Rivera, E. J., Sun, H. H., Nagilla, R., Beal, A. M., Finger, J. N., Cook, M. N., King, B. W., Ouellette, M. T., Totoritis, R. D., Pierdomenico, M., Negroni, A., Stronati, L., Cucchiara, S., Ziolkowski, B., Vossenkamper, A., MacDonald, T. T., Gough, P. J., Bertin, J. & Casillas, L. N. (2016). J. Med. Chem. 59, 4876–4880.
- Hübschle, C. B., Sheldrick, G. M. & Dittrich, B. (2011). J. Appl. Cryst. 44, 1281–1284. [DOI] [PMC free article] [PubMed]
- Katritzky, A. R., Huang, T.-B., Voronkov, M. V. & Steel, P. J. (2000). J. Org. Chem. 65, 8069–8073. [DOI] [PubMed]
- Lokaj, J., Kettmann, V., Černuchová, P., Milata, V. & Fronc, M. (2007). Acta Cryst. E63, o1164–o1166.
- Macrae, C. F., Edgington, P. R., McCabe, P., Pidcock, E., Shields, G. P., Taylor, R., Towler, M. & van de Streek, J. (2006). J. Appl. Cryst. 39, 453–457.
- McArdle, P., Gilligan, K., Cunningham, D., Dark, R. & Mahon, M. (2004). CrystEngComm, 6, 303–309.
- McKinnon, J. J., Spackman, M. A. & Mitchell, A. S. (2004). Acta Cryst. B60, 627–668. [DOI] [PubMed]
- Mphahlele, M. J. & Mphahlele, M. M. (2011). Tetrahedron, 67, 4689–4695.
- Mugnaini, C., Pasquini, S. & Corelli, F. (2009). Curr. Med. Chem. 16, 1746–1767. [DOI] [PubMed]
- Musiol, R. (2017). Exp. Opin. Drug. Discov. 12, 583–597. [DOI] [PubMed]
- Nainwal, L. M., Tasneem, S., Akhtar, W., Verma, G., Khan, M. F., Parvez, S., Shaquiquzzaman, M., Akhter, M. & Alam, M. M. (2019). Eur. J. Med. Chem. 164, 121–170. [DOI] [PubMed]
- Parsons, S., Flack, H. D. & Wagner, T. (2013). Acta Cryst. B69, 249–259. [DOI] [PMC free article] [PubMed]
- Rigaku OD (2018). CrysAlis PRO. Rigaku Corporation, Tokyo, Japan.
- Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8.
- Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8.
- Spek, A. L. (2020). Acta Cryst. E76, 1–11. [DOI] [PMC free article] [PubMed]
- Wolff, S. K., Grimwood, D. I., McKinnon, J. J., Turner, M. J., Jayatilaka, D. & Spackman, M. A. (2012). Crystal Explorer. The University of Western Australia.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S2056989020000298/zl2767sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989020000298/zl2767Isup2.hkl
Spectra. DOI: 10.1107/S2056989020000298/zl2767sup3.tif
CCDC reference: 1879928
Additional supporting information: crystallographic information; 3D view; checkCIF report