

Abstract
Cancer immunotherapies that target adaptive immune checkpoints have significantly improved patient outcomes for multiple metastatic and treatment-refractory cancers. Recent studies, however, have demonstrated that innate immune checkpoints, which interfere with the detection and clearance of malignant cells and suppress innate sensing, also play a critical role in tumour-mediated immune escape and may be potential targets for cancer immunotherapy. In this review, we highlight the current understanding of how cancer cells evade the immune system by disrupting phagocytic clearance and the implications of phagocytosis checkpoint blockade on the activation of antitumour immune responses. A better understanding of the tumour-intrinsic processes that inhibit essential immune surveillance processes such as phagocytosis and innate sensing could pave the way for developing more effective combination immunotherapy strategies that incorporate both innate and adaptive immune responses to treat patients with cancer.
INTRODUCTION
T cell immune checkpoint inhibitors have drastically improved patient outcomes for multiple metastatic and treatment-refractory cancers.1–14 Although immune checkpoint inhibitors have demonstrated survival benefits for cancers previously considered terminal, patient response rates to these treatments remain suboptimal. As a result, there is a growing interest in finding a combination immunotherapy, where multiple immune checkpoint blockers or co-stimulatory agonists are given together in the hope of generating more potent antitumour responses.15–19 Although most of the effort thus far has focused on identifying the right mix of agents that can boost adaptive antitumour immune responses, agents that can stimulate innate immune cell activities are also increasingly being explored.20–22
As a major branch of the body’s immune defense, the innate immune system serves as the first line of defense against infection and malignant cell transformations.23 The cells of the innate immune system—such as monocytes, macrophages, and dendritic cells (DCs), all of which act as professional antigen presenting cells (APCs), and natural killer cells (NKs)—rely on germline-encoded pattern recognition receptors and other cell surface molecules to quickly detect microbial proteins or membrane molecules on tumour cells to orchestrate downstream inflammatory reactions.24 The innate immune response is also critical for activating the adaptive immune system through the processing and cross-presentation of antigens to T cells by APCs.23 Integral to this bridging of innate and adaptive immunity is APCs’ ability to engulf tumour cells through phagocytosis, a multi-step cellular process involving target cell recognition, cellular engulfment, and lysosomal digestion, regulated by receptor-ligand interactions between the target cell and the phagocyte (Figure 1).25 Although healthy normal tissues and cells have inherited the ability to avoid self-elimination by phagocytes through the expression of antiphagocytosis molecules,26–28 cancer cells depend even more on similar mechanisms to evade immune eradication.29 Therefore, identifying and targeting phagocytosis checkpoints in cancer will provide a new avenue for developing cancer immunotherapies to eliminate tumour immune escape.
Figure 1: Phagocytosis of cancer cells is regulated by pro- and anti-phagocytic signals.

Tumour cell phagocytosis by professional phagocytes is regulated by a host of prophagocytosis (“eat me”) and antiphagocytosis (“don’t eat me) signals through receptor-ligand interactions after the cell-cell interface. Identified eat me signals expressed by tumour cells include tumour antigens which when bound by antibodies can be recognized by the Fc-receptors (FcR) on phagocytes, as well as ER chaperon protein calreticulin and SLAMF7. On the other hand, tumour cells rely on the expression of PD-L1, CD47, β2 microglobulin and yet to be identified ligands that binds to LILRB2 to inhibit their phagocytotic clearance by phagocytes. Therapeutic antibodies targeting some of these receptor-ligand interactions have been investigated as potential immunotherapy for multiple types of cancers.
This review focuses on recent advances in the identification of phagocytosis checkpoints and stimulatory signals, as well as preclinical and early clinical evidence supporting the use of phagocytosis checkpoint blockade in cancer treatment (Box 1). We discuss the mechanistic processes governing innate and adaptive antitumour responses in the setting of phagocytosis checkpoint blockade and highlight how bridging the two branches of the immune system is critical for generating optimal antitumour immunity.
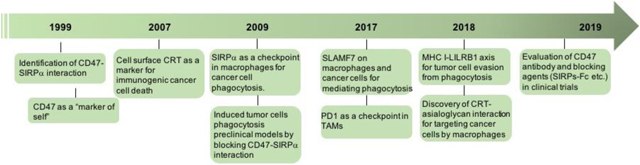
Box 1: History of phagocytosis checkpoint blockade in cancer.
The first phagocytosis checkpoint axis, CD47-SIRPa, was identified in the 1990s.41,42 CD47 was then identified as a “marker of self” on red blood cells (RBCs).26 Cell surface calreticulin (CRT) was shown to be a key protein marker defining immunogenic cancer cell death.92–94 In 2009, CD47 was found to be highly upregulated on malignant hematopoietic and non-hematopoietic cells. Blockade of CD47 induces tumour cell phagocytosis in vitro and in vivo, and inhibits tumour engraftment and growth in preclinical models.44,45 Since then, several CD47 blocking antibodies and agents have been developed and are currently being evaluated in clinical trials.138,140,141,197,203 In 2017 and 2018, PD-1 and LILRB1 were identified as additional regulators on macrophages for tumour cell phagocytosis through their interactions with PD-L1 and MHC I on tumour cells.56,57 In 2015, CRT generated by macrophages was found to be a critical effector for guiding macrophages for cancer cell recognition.47 Macrophage CRT was later found to interact with specific patterns of glycans on cancer cells to target them for phagocytosis.103 SLAMF7 was also shown to be a necessary component for phagocytosis of hematopoietic cancer cells during CD47 blockade, through not-yet-elucidated receptor interactions that include SLAMF7 homo-dimers on both cancer cells and phagocytes.111
PHAGOCYTOSIS CHECKPOINTS
CD47-Signal-regulatory protein α (SIRPα)
The first member identified in the signal-regulatory protein (SIRP) family, SIRPα, is expressed on myeloid cells, including all different types of macrophages.30–32 SIRPα has an immunoglobulin (Ig) domain in the extracellular region for ligand binding and a cytosolic domain that includes an immunoreceptor tyrosine-based inhibitory motif (ITIM) that allows association with Src homology-2 (SH2)-containing protein tyrosine phosphatases (SHP) for signal transduction.30,31 The other two members of the SIRP family, SIRPβ and SIRPγ, which are mainly expressed in macrophages and lymphocytes, respectively, also have Ig domains in the N-terminal regions with shorter cytosolic tails.30,33–35 SIRPβ binds to immunoreceptor tyrosine-based activation motif (ITAM)-containing protein DNAX activation protein 12 (DAP12) to transduce activating signals; SIRPγ is not directly involved in recruiting signaling proteins.35–38
CD47 was first cloned from ovarian cancer cells using a monoclonal antibody, OVTL3, that recognizes an epitope that is abundant in ovarian carcinomas but rarely expressed on normal tissues.39 At the same time, CD47 was also purified and cloned as a cell surface protein, named Integrin Associated Protein (IAP), that binds to αvβ3 integrin in the placenta and platelets.40 The interaction between CD47 and SIRPα, however, was not identified until later, when a monoclonal antibody, CC2C6, that inhibits the adhesion of hematopoietic cells to SIRPα was found to recognize CD47 as well, thus identifying CD47 as a potential ligand for SIRPα.41 Another study that probed a mouse brain cDNA library expressed in COS7 cells with murine SIRPα recombinant proteins also revealed CD47 as a SIRPα binding partner.42
The role of CD47 in phagocytosis and immune recognition was discovered when red blood cells (RBCs) from CD47−/− mice transfused into wild-type (WT) recipients were rapidly cleared, which was reversed when macrophages were depleted using clodronate liposome.26 Red pulp macrophages in the spleen were responsible for most of the RBC elimination through phagocytosis. Macrophage contact with WT but not CD47−/− RBCs triggered SIRPα phosphorylation, and blockade of SIRPα led to phagocytosis of WT RBCs, indicating that the CD47-SIRPα axis plays a key role in self-protecting RBCs from indiscriminate destruction in the circulation.26 CD47’s role in protecting against immune recognition and attack on self-tissues seems to be a conserved function across species. An M128L myxoma virus gene encoding a membrane protein that shares similar amino acid sequences with bovine CD47 is dispensable for replicating the myxoma virus but essential for inducing a lethal infection through the inhibition of phagocyte activities in the host.43
In addition to regulating normal tissue homeostasis, CD47 is also critical for cancer cells to evade immune clearance. Elevated expression of CD47 has been observed in a wide range of human cancers, including those from malignant hematopoietic and non-hematopoietic tumours.44,45 CD47 does not seem to be directly involved in regulating cell proliferation and viability for cancer cells, and CD47 knockout (KO) cancer cells are indistinguishable from their parental CD47-expressing cells in in vitro cell culture. Blockade of CD47 using monoclonal antibodies resulted in increased tumour cell phagocytosis by professional phagocytes and inhibited tumour engraftment and growth in mice that lack T, B and NK cells. Depletion of macrophages however, restore tumor growth, indicating that CD47 is critical for cancer cells to escape from macrophages attack and phagocytes play essential roles in immunosurveillance against cancer cells.44–48
At the transcriptional level, two upstream and downstream super enhancers, which consist of a set of constituent enhancers that are differentially expressed in different types of cancer cells, regulate the expression of CD47. Stimulating the tumour necrosis factor (TNF) inflammatory pathway activates NF-κB, which directly binds to a constituent enhancer of CD47 to regulate its expression in breast cancer cells.49 Hypoxia-inducible factor-1 (HIF-1) also binds to a CD47 promoter in breast cancer cells, and an analysis of a cohort of 1,040 primary human breast cancer specimens revealed a significant correlation between HIF-1 and CD47 expression.50 In melanoma cells, extracellular signal-regulated kinase (ERK) signaling upregulates transcriptional activation of CD47 through the transcriptional factor Nuclear respiratory factor 1 (NRF1).51 In human leukemia and lymphoma, the Myc oncogene directly binds to the promotors of CD47 genes in mouse and human tumour cells to regulate CD47 expression.52 Myc inactivation leads to inhibited CD47 expression, which is associated with an increased recruitment of CD4+ T cells and macrophages into tumours and improved survival.52 These effects were reversed when CD47 expression was restored. Taken together, these results suggest that oncogenic activation of CD47-SIRPα signaling enables cancer cells to evade immune detection and clearance by inhibiting phagocytosis by professional phagocytes.
Programmed death 1 (PD-1) – Programmed death-ligand 1 (PD-L1)
In addition to the CD47-SIRPα axis, other phagocytosis checkpoints that promote tumour cell evasion of phagocytic clearance have been discovered. Traditionally viewed as a T cell immune checkpoint,53–55 the PD-1/PD-L1 axis was recently found to play a role in regulating the phagocytic ability of tumour-associated macrophages (TAMs) as well.56 Similar to monocytes or macrophages from normal tissues such as spleen and peripheral blood, TAMs in early stage human and mouse tumours express minimal levels of PD-1. However, the expression of PD-1 in TAMs exponentially increases as the tumour grows.56 Phenotypically, most PD-1+ TAMs are M2-like macrophages, which constitute the predominant macrophage population in late-stage mouse and human colon cancers. However, the PD-1- macrophage populations displayed a greater phagocytic ability than PD-1+ TAMs against tumour cells. Disrupting the PD-1/PD-L1 axis using either an anti-mouse PD-1 antibody or a PD-L1 blocker (which lacked the Fc-domain) resulted in antitumour responses in mice that lacked T, B, and NK cells but still retained functional macrophages, indicating that the inhibition of the PD-1/PD-L1 axis in TAMs is responsible for the antitumour efficacy.56 When analyzing TAMs’ phagocytosis abilities against both PD-L1 over-expressing and KO tumour cells, no inhibition in the phagocytosis capacity of PD-1- TAM was noted. In contrast, PD-L1 KO significantly increased tumour cell phagocytosis by PD-1+ TAMs. Therefore, these observations strongly support the ideas that PD-L1 expression on cancer cells enables them to evade macrophage phagocytosis and that blockade of the PD-1/PD-L1 axis may induce both phagocyte- and T cell-mediated antitumour immunity.56 It is important to note that while the role of PD-1 in cytotoxic T lymphocyte (CTL) exhaustion is well-proven in mice, the demonstration that human responders have activated/amplified tumour-specific CTL clones is less clear, as is the evidence that such presence distinguishes responders from non-responders. It is therefore difficult to separate out as to whether the phagocytic system and/or the exhausted T cells are responsible for determining therapeutic outcomes. At the present time, the precise signaling events that promote the antiphagocytic function of PD-1 in TAMs are not well understood. It is worth noting that the macrophage receptors for tumour cell ‘don’t eat me’ signals identified to date - SIRPα, LILRB1, and PD144,56,57 - contain an ITIM domain which when activated, mediate signaling via their phosphorylation and activation of two tyrosine phosphatases, SHP1 and SHP2. It is however, unclear if both are stimulated in T cells and macrophages, as well as which factors promote the upregulation of PD-1 in TAM. Recent studies have suggested that cancer cell-derived exosomes from advanced tumours promote the differentiation of monocytes into PD-1+ TAMs with immune suppressive properties that impair effector T cell functions.58 However, it remains to be elucidated whether the induction of PD-1 expression follows a mechanistic pathway similar to exosome-mediated PD-L1 upregulation in monocytes.
MHC I-LILRB1
Major histocompability complex class I (MHC I) is expressed on many nucleated cells and presents antigens to T cells. Recently, it was discovered that the expression of MHC I on cancer cells correlates with their resistance to phagocytosis because of MHC I’s interaction with the leukocyte immunoglobulin-like receptor (LILRB1) on macrophages.57 LILRB1 is the most widely distributed member of the LILRB family, with expression on monocytes/macrophages, eosinophils, basophils, dendritic cells, certain NK cells, subsets of T cells, B cells,59–63 progenitor mast cells64 and osteoclasts.65 LILRB1 expression is also particularly abundant on TAMs. Further analysis revealed that the β2 microglobulin (B2M) subunit of the MHC I complex defines a species-specific interaction between MHC I and LILRB1. Disrupting the MHC I-LILRB1 interaction through genetic manipulation made cancer cells more vulnerable to phagocytic clearance. Co-implantation of MHC I+ and MHC I- cancer cells in immunocompromised mice showed a selective pressure against MHC I- cells in the engrafted tumour, despite the lack of adaptive immune cells. Deleting both MHC I and CD47 synergized to yield a stronger tumour inhibition in immunocompetent hosts that depend on the presence of macrophages.57 Given that antigen presentation by MHC I is required for T cell recognition of cancer cells and that MHC I downregulation has been commonly observed to render tumour cells resistant to T cell elimination,66–68 how cancer cells emerge by immunoselection given the pro- and anti-immunogenic role of MHC I remains an important yet unexplored question. Not all cancers or cancer types express high levels of B2M or PD-L1. Extensive testing of a full set of ‘don’t eat me’ signals on patient tumor cells as well as their cognate receptors on the TAM pools will enable the prediction of whether a patient will be more likely to respond to blockade of CD47 or other ‘don’t eat me’ signals. MHC I may be differentially regulated at different stages of tumour development or adapted to tumour microenvironments with a distinct composition of immune cells. Therapeutically, agents that block B2M without interfering with MHC I-T cell receptor binding or that inhibit LILRB1 may be explored to overcome phagocytosis resistance in tumour cells.
Another member of the LILBR family, LILRB2 was reported to be expressed on monocytes, macrophages, dendritic cells, basophils, hematopoietic stem cells,63,69–71 mast cell progenitors,64 activated endothelial cells,72 osteoclasts65 and activated CD4+ T cells.73 Therapeutic antibodies blocking LILRB2, which like LILRB1, also binds to HLA class I, promote the maturation of macrophages and enhanced their pro-inflammatory activation.74 Blockade of LILRB2 with monoclonal antibodies also increased the phagocytosis activities of TAMs and demonstrated enhanced antitumour effects when combined with anti-PD-L1 antibodies in transgenic mice that express human LILRB2 on CD11b+ cells. Mechanistically, it appears that LILRB2 blockade suppresses SHP1/2 phosphorylation, thus promoting M1-polarization of macrophages through the activation of ERK and p38 by suppressing SHP1/DUSP-mediated direct or indirect dephosphorylation (ERK at Y204 and p38 at Y182). This leads to the activation of NF-κB/STAT1, while suppressing the activities of the PI3K/AKT pathways downstream of M-CSF.74 At this point, it is unclear whether LILRB2 antagonism promotes phagocytosis activities directly or indirectly through phenotypic changes in macrophages. Although both LILRB1 and LILRB2 bind to HLA-I/MHC-I,69,75 whether the interaction for the latter also acts as a phagocytosis checkpoint remains to be seen. It is also unclear whether the reported enhanced Th2 response through Semaphorin-4A binding to LILRB4 on activated CD4+ T cells could have contribute to the in vivo effect of antagonist anti-LILRB2 antibodies.73
Finally, LILRB4 is another receptor that is well known to be expressed on monocytes, macrophages, dendritic cells,76–78 as well as plasmablasts,79 certain Tregs,80 activated endothelial cells72 and osteoclasts.65 Crosslinking of LILRB4 and other receptors, such as HLA-DR, CD11b, FcγRIII or FcγRI, inhibits monocyte activation.76,81 Naturally, LILRB4 is expressed on immunosuppressive myeloid cells such as myeloid-derived suppressor cells (MDSCs),82 tolerogenic DCs83 and TAMs,84 and inhibits T cell activity through LILRB4/SHP2/NFkB signaling axis in monocytic leukemia cells. Anti-LILRB4 antibodies reactivate T cells and blocked development of monocytic leukemia.77 Although it was demonstrated that ApoE binding can activate LILRB4,77 and LILRB4 interaction with CD166 mediates its inhibitory effect on tumor cells,85 further investigations are needed to clarify whether LILRB4 on monocytic cells can directly bind and act on a surface protein (ideally an immune inhibitory receptor) on T cells.
PHAGOCYTOSIS ACTIVATING PATHWAYS
Calreticulin
In many cancers, malignant cells express antiphagocytosis signals at higher levels than normal cells do, potentially to counter-balance the increased expression of prophagocytic “eat me” signals generated from oncogenic stresses.44,45
Calreticulin (CRT) and calnexin are members of the ER “lectin chaperone” family, and they bind to newly synthesized proteins to assist their folding and glycosylation.86–88 CRT plays important roles in inducing phagocytosis as well as downstream immune responses through different mechanisms where CRT can be resourced by target cells or macrophages. Apoptotic cells express calreticulin on their surface as a recognition ligand, which forms a bridging complex with C1Q and low density lipoprotein receptor-related protein 1 (LRP-1 or CD91) expressed on phagocytes to initiate phagocytic clearance.89–91 Cancer cell death may trigger an immune response during which dead or dying cancer cell proteins or antigens are taken up by DCs to process tumour antigens to activate adaptive immunity. While certain chemotherapies or radiotherapies, such as anthracyclines and gamma irradiation, induce immunogenic cancer cell death, other DNA damaging agents do not. In these cases, pro-apoptotic agents induce CRT translocation to the cell surface in dying cancer cells and enhanced cell surface CRT expression enables phagocytosis of these cells by DCs and trigger immunogenicity.92–95 Cell surface CRT, secreted ATP, and released high mobility group protein B1 define the damage-associated molecular patterns (DAMPs) for immunogenic cell death.96,97
Cell surface CRT is also found on living cells, and it mediates macrophage phagocytosis and elimination of these cells.98,99 For clearance of the living cells, activated macrophages are the major resources for CRT for recognition of target cells, including damaged, aged and malignant cells. During inflammation, neutrophils mature after infiltrating into the inflammation sites and are cleared by macrophages. While neutrophils undergo apoptosis before their clearance, the neutrophils from transgenic mice expressing anti-apoptotic protein Bcl2 become resistant to cell death but are as susceptible to phagocytosis as the apoptotic neutrophils, which suggests that their clearance by macrophages does not rely on the induction of cell death.100,101 Stimulating Toll-like receptor pathways in macrophages—especially TLR3, 4, and 7—activates and phosphorylates Bruton’s Tyrosine Kinase (BTK), which in turn phosphorylates CRT and activates its translocation to the cell surface on macrophages.47,102 How CRT, a soluble protein, is associated to macrophage cell surface remains to be explored. LPR-1 was identified to be a cell surface receptor for CRT but LRP-1 knockout mice need to be examined to fully understood its role in mediating this process. CRT interacts with its ligands on target cells to mediate their recognition and phagocytosis. This might be achieved by ligating cell surface CRT on macrophages with ligands on target cells, or by secreting CRT to directly decorate target cells for recognition.103 Therefore, phagocytosis of live cancer cells may not solely rely on CRT expression on cancer cells per se, but rather correlated with cell surface CRT exposure and secretion by macrophages. Importantly, altered patterns of carbohydrate expression were observed on apoptotic cells and a wide range of human cancer cells, including abnormal expression of glycans and branching of glycoproteins.104–107 The lectin-like domain in CRT enables its binding to an asialoglycan Tri-antennary and multivalent type II (Galβ1→4GlcNAc) chain epitopes (Tri-m/II) that are widely expressed on malignant cells as well as neutrophils with enforced expression of BCL2.103 This interaction enables recognition of these cells and stimulates phagocytosis by macrophages.103
Signaling lymphocytic activation molecule family (SLAMF) and Macrophage-1 (Mac-1)
The signaling lymphocytic activation molecule (SLAM) family receptors may be involved in mediating phagocytosis of hematopoietic malignant cells.108 SLAMF comprises nine receptors, seven of which are expressed in macrophages: CD48, Ly9, CD244, CD84, SLAMF7, SLAMF8, and SLAMF9.108–110 SLAMF receptors are single-pass transmembrane proteins with two to four extracellular Ig domains and tyrosine-rich intracellular tails.108 Knockout of SLAMF7 in the mice compromised the phagocytotic abilities, induced by CD47 blockade, of mouse bone marrow-derived macrophages against many B cell- and myeloid cell-derived cancer cell lines.111 SLAMF7-mediated cancer cell phagocytosis is independent of signaling lymphocyte activation molecule-associated protein (SAP) adaptors. Instead, SLAMF7 interacts with Mac-1 antigen, a heterodimeric complement receptor comprised by integrin CD11b and CD18 expressing on many innate immune cells including phagocytes. Mac-1 plays an essential role in phagocytosis of C3bi-opsonized pathogens and apoptotic cells112,113. SLAMF7 associates to Mac-1 to form a protein complex on the cell surface of macrophages. Through Mac-1, SLAMF7 interacts with two ITAM-motif containing receptors, FcRγ and DAP12, in macrophages to transmit an activation signaling cascade mediated by Src, Syk and Btk kinases to induce phagocytic machinery in macrophages.111 Importantly, while Mac-1 is a required component for SLAMF7-mediated phagocytosis of hematopoietic cancer cells, there is no evidence that SLAMF7 is reciprocally involved in the phagocytosis mediated by Mac-1 and C3bi.111
As a homotypic receptor, SLAMF7 on macrophages might interact with SLAMF7 on cancer cells for their recognition and phagocytosis. However, as showed in a recent study, the expression of SLAMF7 is missing from many diffuse large B-cell lymphoma (DLBCL) cell lines and primary cells, despite their susceptibility to anti-CD47 antibodies-induced phagocytosis by allogeneic human macrophages. In addition, the expression of SLAMF7 on different macrophage subpopulations in the tumor microenvironment is not correlated with their phagocytic ability against DLBCL cells induced by clinically relevant CD47 targeting antibodies.114 Since different macrophage models were used in these studies, further investigation needs to be performed to examine the molecular mechanisms of SLAMF7 in mediating tumor cell phagocytosis and it remains to be seen whether additional “eat me” signals are involved in binding to SLAMF7 to activate phagocytosis.
Fc Receptors
Fc receptors belong to a family of cell surface receptors that bind to the Fc domain of immunoglobulins and activate downstream signaling in different types of immune cells including macrophages, dendritic cells and granulocytes.115,116 Although classically known to exert antitumour effect via antibody-dependent cellular cytotoxicity (ADCC), IgG receptors (FcγR) also play a major role in mediating antibody-dependent cellular phagocytosis (ADCP) of tumour cells by professional phagocytes. Within the human FcγR family, FcγRIIB is the only inhibitory receptor and the others (FcγRI, FcγRIIA, FcγRC, FcγRIIIA, FcγRIIIB) are activating receptors.115–118 The FcγRs bind to immunoglobulin IgG1, IgG3 and IgG4, with the exception of FcγRIIIB, which does not bind to IgG4, and FcγRIIA, is the major receptor for IgG2.117 Binding of IgG induces phosphorylation of ITAM motifs which locate in the ligand-binding a-chain in FcγRIIA and FcγRIIC or in the signaling g-chain in FcγRI and FcγRIIIA.115 Phosphorylation of tyrosines in the ITAM motif by Src family kinases leads to the recruitment and activation of the Syk-family tyrosine kinases which in turn phosphorylate and activate downstream targets, including the Rac-GEFs (Guanine Nucleotide Exchange Factors).119,120 Rac-GEFs activate GTPases such as RhoA, Rac1 and Cdc42, leading to actin cytoskeleton rearrangement via actin-regulatory proteins to enable phagocytosis.120,121 FcγRIIB which is conserved in human and mice, contains an ITIM motif in its ligand-binding a-chain whose phosphorylation and activation result in the recruitment of phosphatases including SHP1 and SHIP1 (SH2-domain-containing inositol polyphosphate 5’ phosphatase 1) and thus transduce inhibitory signals.115,121 In addition, FcγRIIB promotes the cellular internalization of antibody-antigen complex on target cells to inhibit the interaction between antibodies and the activating FcγRs.122–124
Therapeutic antibodies targeting cancer cells engage both activating and inhibitory FcγRs on phagocytes. Antitumour effects of antibodies were largely diminished in FcγR−/− mice but were significantly enhanced in FcγRIIB−/− mice, indicating a direct role of FcγRs in mediating cytotoxicity of antibodies against cancer cells125. ADCP has been reported in many different types of cancers, which depends on activating FcγRs and was abolished when macrophages are depleted. Kupffer cells (liver macrophages) were shown to phagocytize cancer cells in circulation upon antibody treatment, in a FcγRI and FcγRIV dependent manner.126 Daratumumab, an anti-CD38 antibody, induced macrophage phagocytosis of multiple myeloma cells.127 In small cell lung cancer models, antibodies targeting several highly-expressing antigens enabled direct phagocytosis of the cancer cells by macrophages.48 On the other hand, antibodies engineered to have enhanced binding to activating FcγRs and diminished binding to FcγRIIB showed to improve the efficacy of cancer cell phagocytosis in mouse models of lymphoma and leukemia.128 In addition, antibodies that block FcγRIIB enhanced ADCP and therapeutic efficacy of rituximab129. Finally, glycoengineered CD20 antibody obinutuzumab induced phagocytosis of chronic lymphocytic leukemia (CLL) cells by neutrophils through binding to FcγRIIA and FcγRIIIB.130
TARGETING PHAGOCYTOSIS CHECKPOINTS
CD47-SIRPα axis
CD47 has a long N-terminal extracellular domain and five transmembrane domains, as well as a short cytosolic domain, which differs among CD47 isoforms. The extracellular V-type Immunoglobin-like (IgV-like) domain in CD47 is responsible for its interaction with SIRPα, as it binds to the N-terminal IgV-like domain in SIRPα.131,132 This interaction transduces a negative signal through the phosphorylation of two tyrosine residues in the intracellular ITIM motif of SIRPα.30,131,132 Tyrosine phosphorylated sites of SIRPα recruit and activate SHP-1 and SHP-2 phosphatases, which are hematopoietic-dominant and ubiquitously expressed, respectively.31,133,134 This negative signaling cascade leads to dephosphorylation of myosin-IIA and, thus, inhibits cytoskeleton rearrangement, which is a necessary step for macrophages to engulf target cells.135 A disulfide bond forms between Cys33 in the extracellular Ig domain and Cys263 in the loop between the fourth and fifth transmembrane domains of CD47.136 Disrupting this disulfide bond compromises SIRPα binding. A recent haploid genetic screen using a CC2C6 antibody that is known to bind to the SIRPα recognition site identified glutaminyl-peptide cyclotransferase-like protein (QPCTL) as a critical regulator for the CD47-SIRPα interaction.137 An N-terminal pyroglutamate in the CD47 protein, a posttranslational modification catalyzed by QPCTL, is dispensable for cell surface expression of CD47 but a necessary component for SIRPα binding. Deletion of CD47, inhibition of QPCTL, and blockade of the IgV-like domain in CD47 showed comparable effects in abolishing SIRPα binding and inducing target cell phagocytosis, indicating that the integrity of the IgV domain and the N-terminal pyroglutamate is necessary for maintaining SIRPα binding.
Blocking the CD47-SIRPα interaction leads to phagocytosis of live cancer cells and can be achieved in several ways: 1) CD47 or SIRPα antibodies that target their binding sites to block the CD47-SIRPα interaction44,45,138; 2) recombinant proteins of the extracellular regions of CD47 or SIRPα, which, when they reach the threshold concentrations, compete with the endogenous proteins for binding139–141; and 3) agents that target pathways that regulate the CD47 transcriptional/trafficking regulatory program to suppress cell surface expression of CD47 on cancer cells.49 In addition, while tumor specific monoclonal antibodies are able to target tumor cells and induce macrophage phagocytosis to some extent through IgG-FcγR interaction, therapies targeting CD47-SIRPα lower the threshold of macrophage activation142 and thus amplify the efficacy of ADCP induced by various therapeutic antibodies,143 which have shown to be effective in preclinical models of patient-derived cancer xenografts,44–46,139,143,144 fibrotic disorders,145 and atherosclerosis.146 It is worth noting that in mouse xenograft models of glioblastoma, anti-CD47 antibody treatment resulted in the polarization of TAMs towards a M1-like phenotype,147 suggesting that CD47-SIRPα pathway is likely involved in additional intracellular processes that regulate pro-tumour functions of TAMs. Interestingly, although M1 macrophages were reported to possess relatively stronger phagocytosis upon CD47 blockade,45,147 M2 macrophages which are considered as the major TAM population in established tumours, also maintain phagocytic ability against tumor cells. Therefore, more specific classification of subgroups of TAMs and functional assessments through genetic modulation including cell ablation or lineage tracing at the single cell level are required to better define the role CD47-SIRPα pathway play in TAM polarization.
In addition to macrophages and DCs, blockade of CD47-SIRPα interaction was found to promote neutrophil-mediated ADCC towards breast cancer cells induced by anti-HER2 antibody.148 similarly, SIRPα blockade induced macrophage- and neutrophil-mediated phagocytosis of several types of human cancer cells in vitro, and promoted neutrophil and macrophage infiltration in the lymphoma xenograft and augmented their antitumor effects.149 Recent evidence also suggest that CD47-SIRPα axis is involved in the regulation of neutrophil-mediated trogoptosis of cancer cells. Using high-power scanning electron microscopy, neutrophils were found to form intimate interactions with cancer cells coated by antibodies, allowing them to disrupt cancer cell plasma membrane to induce lytic cell death. Blockade of CD47-SIRPα axis was found to augment antibody-opsonized cancer cells trogoptosis by neutrophils both in vitro and in vivo.150
Clinically, CD47-SIRPα blocking agents are currently being evaluated in multiple ongoing trials as a monotherapy or in combination with other therapies for leukemia, lymphoma, and solid tumours (Table 1). Although majority of the studies are non-randomized phase I trials aimed at determining maximal tolerated dose and dose-limiting toxicities of the treatment regimen, early evidences suggest that CD47-SIRPα blockade is well tolerated in patients, despite the fact that CD47 is widely expressed on normal tissue. Severe toxicity from CD47-SIRPα blockade has not been reported from extensive preclinical testing and in clinical trials, unless the anti-CD47 reagent is of an immunoglobulin isotype that fixes complement proteins or activates ADCC (Box 2). The primary reason for this observation is that most CD47+ normal cells lack ‘eat me’ signals such as surface bound calreticulin.47,99,103 In addition, TTI-621, a protein that fuses the IgV domain of human SIRPα with the Fc region of human IgG1 and that is currently being tested in clinical trials, reported minimal binding to RBCs.141 This is probably due to different binding mechanisms between the blocking agents, as SIRPα preferentially binds to clustered CD47, while CD47 on RBCs is trapped by spectrin, resulting in limited mobility that prevents SIRPα binding.141,151 Another strategy is to take advantage of differential expression levels of phagocytosis checkpoints on “aged” and “young” RBCs. Administering a low, “priming dose” of anti-CD47 antibody leads to loss of less than 20% of RBC, likely those with calreticulin bound to their surface. This is followed by reticulocytosis process which generates new RBC that are more resistant to CD47-blockade mediated self-clearance by phagocytes.
Table 1:
Current clinical trials investigation phagocytosis checkpoint blockade for cancer treatment
| Clinical trial number |
Phase | Intervention | Trial design |
Number of patients |
Cancer type | Primary outcome |
|---|---|---|---|---|---|---|
| NCT02678338 | I | Anti-CD47 ab | Dose escalation | 20 | Hematological malignancies | Tolerability |
| NCT03717103 | I | Anti-CD47 ab | Dose escalation | 92 | Advanced malignancies | Safety, tolerability |
| NCT03763149 | I | Anti-CD47 ab | Dose escalation | 42 | Advanced malignancies, lymphoma | Safety, tolerability |
| NCT02216409 | I | Anti-CD47 ab | Dose escalation | 88 | Solid tumors | Safety, tolerability |
| NCT03248479 | Ib | Anti-CD47 ab, Azacitidine | Non-randomized | 96 | AML, MDS | Safety, tolerability |
| NCT02953782 | I/II | Anti-CD47 ab, cetuximab | Single arm, non-randomized | 112 | Solid tumour, advanced colorectal cancer | Safety, tolerability, efficacy |
| NCT02367196 | I | Anti-CD47 ab, rituximab | Dose escalation | 110 | Advanced solid and hematologic malignancies | Tolerability, safety |
| NCT02663518 | I | SIRPαFc, rituximab, nivolumab | Dose escalation | 260 | Relapsed/refractory hematologic and solid malignancies | Safety and tolerability |
| NCT03834948 | I | Anti-CD47 ab, | Dose escalation, expansion | 90 | Advanced solid tumours | Tolerability and safety |
| NCT02953509 | Ib/II | Anti-CD47 ab, rituximab | Single arm, non-randomized | 72 | Refractory/relapsed Non-Hodgkin’s lymphoma | Safety, tolerability, efficacy |
| NCT03512340 | I/Ib | Anti-CD47 ab | Dose escalation, expansion | 148 | Advanced solid cancer, hematologic cancer | Safety, tolerability |
| NCT02890368 | I | SIRPα-IgG1 Fc, anti-PD1/PD-L1, PEGylated IFN, T-Vec, radiation | Non-randomized, parallel assignment | 240 | Solid tumour and mycosis fungoides | Optimal delivery regimen |
| NCT03013218 | I | High affinity SIRPα | Dose escalation | 142 | Advanced solid tumours, lymphoma | DLT |
MDS, myelodysplastic syndrome; AML, acute myeloid leukemia; SIRPα, Signal-regulatory protein alpha; IFN, interferon; T-Vec, Talimogene laherparepvec; DLT, dose limiting toxicity.
Box 2: Potential toxicities of phagocytosis checkpoint blockade.
Because normal cells and tissues also employ phagocytosis checkpoints to evade immune clearance, there are concerns about potential toxicities associated with phagocytosis checkpoint blockade. This is especially the case for hematological tissues, where cells express CD47 to prevent programmed cell removal.26 As hematopoietic cells such as RBCs age, their expression of CD47 also declines, while prophagocytic signals increase, resulting in their eventual removal by tissue macrophages in the spleen, liver, and bone marrow. Administering a CD47-blocking antibody results in dose-dependent decreases in RBCs, hemoglobin, and hematocrit accompanied by reticulocytosis and recovery within 2–3 weeks.138 The ensuing blood pool exhibits a shift towards younger cells that are resistant to the effects of subsequent CD47 blockade. The primary reason that most CD47+ cells are not affected by blocking anti-CD47 reagents is that they lack “eat me” signals, providing tumour or pathogenic cell specificity of programmed cell removal. Beyond the hematologic system, the long-term effects of macrophage activation in the setting of phagocytosis checkpoint blockade within other tissues are unclear, although many patients have received sustained effective doses of 5F9 anti-CD47 antibodies without therapy associated toxicities other than red cell loss in the priming dose only. Anti-CD47 antibodies appear to be able to cross the blood–brain barrier and disrupting the CD47-SIRPα interaction promotes the phagocytosis activities of both macrophages and microglia within the central nervous system. In models where the antibody passes into the brain, drug related toxicities were not observed.204,205 As the survival of cancer patients continues to improve, toxicities that take a long time to manifest must be monitored and carefully evaluated to assess the true risks and benefits of phagocytosis checkpoint blockade.
BRIDGING INNATE AND ADAPTIVE ANTITUMOUR IMMUNITY
The ability to recognize pathogen associated molecular patterns (PAMPs) is characteristic of the innate immune system, which employs germline-encoded receptors to detect molecular patterns associated with extracellular or intracellular pathogens.152 Extracellular pathogens are mainly recognized by toll-like receptors (TLRs) or C-type lectin receptors (CLRs) anchored on the plasma membrane or within phagolysosomes (Figure 2).153 These transmembrane receptors contain a ligand-binding domain that detects conserved PAMPs and a signal transduction domain that projects into the cytosol, and they are responsible for initiating inflammatory responses by activating multiple pathways, including NF-κB, mitogen-activated protein kinases (MAPKs), JUN N-terminal kinases (JNKs), and interferon regulatory factor (IRF3, IRF5, and IRF7) (Figure 2).154 In the case of cancer, increased cellular turnover or stress might result in an increased release or exposure of endogenous molecules by tumour cells. These DAMPs are also picked up by pattern recognition receptors on phagocytes to initiate immune and inflammatory responses.155
Figure 2: Increased phagocytosis of tumour cells promotes activation of innate immune sensing pathways in APCs.

Following phagocytosis, degradation of tumour cells occur within phagolysosomes, which results in the recognition of tumour-derived DAMPs such as nuclear DNA (nDNA) and single stranded RNA (ssRNA) by TLRs, leading to subsequent activation of NFκB pathway. Alternatively, nucleic acids such as mitochondrial DNAs can escape from the phagosomes through a yet to be discovered mechanism into the cytosol, where they are detected by the cytosolic DNA sensor cGAS. cGAS then convert ATP and GTP into cGMP, which binds with STING to phosphorylate IRF3 and NFκB. This enables them to be translocated into the nucleus, where they act as transcription factors to promote the transcription of inflammatory cytokine genes such as type I IFNs and TNFα.
In contrast to extracellular signals, intracellular pathogens are detected by soluble proteins in the cytosol. Pattern recognition receptors, such as NOD-like receptors (NLRs) and retinoic acid-inducible gene-I-like receptors (RLRs), detect bacterial- and viral-derived signals, such as proteoglycans and double-stranded RNAs (dsRNA), to activate NF-κB and IRF, resulting in Type 1 interferon (IFN) induction.156,157 Recognition of intracellular DNA from microbes, by contrast, is mediated by the cytosolic DNA sensor cyclic GMP-AMP synthase (cGAS), which, upon DNA binding, catalyzes the synthesis of cyclic GMP-AMP (cGAMP) from ATP and GTP.158 The cGAMP then acts as a second messenger to bind and activate the ER-membrane adaptor, stimulator of interferon genes (STING), resulting in the activation of IRF3 and NF-kB to induce the expression of type 1 IFN, TNF, IL-1b, and IL-6, among others (Figure 2).159 In addition to foreign DNA, the cGAS-STING pathway is also critical for detecting self-nucleic acid within the cytosol. As DNA is bound within the confines of the nucleus and mitochondria in eukaryotic cells, self-DNA can accumulate within the cytosol during tumour cell phagocytosis by professional phagocytes, triggering the activation of cGAS-STING and antitumour immunity.159
Therefore, both extracellular and intracellular innate immune sensing pathways provide potential mechanisms for innate immune activation within APCs upon tumour cell uptake in the setting of phagocytosis checkpoint blockade. The subsequent production of type I IFNs through TLR or cGAS-STING activation may enhance the maturation of APCs and their ability to cross-present tumour-derived antigens by MHCs, to upregulate co-stimulatory molecules, and to prime effector T cell responses (Figure 3). In fact, cross-presentation of in vitro human tumors expressing transfected cytoplasmic ovalbumin can, in the presence of blocking anti-CD47, lead to massive phagocytosis and cross-presentation of the SIINFEKL peptide via MHC I to CTL precursors that express OT1 TCR that bind and are activated by this MHC I:SIINFEKL H2b complex.160 This bridging of innate and adaptive antitumour immune responses could be critical for generating long-term tumour control and provides a rationale for combining phagocytosis checkpoint blockade with other therapeutic approaches.
Figure 3: Increased phagocytosis of tumour cells promotes enhanced antigen cross-presentation and inflammatory cytokine release by APCs, both of which help prime effector cell responses against cancer cells.

In addition to induction of proinflammatory cytokines, phagocytosis of tumour cells also results in the release of neoantigens which are loaded onto MHCI molecules within the phagosomes. Alternative, tumour cell-derived proteins are degraded by proteasomes and shuttled back into the phagosomes or to the ER via transporter associated with antigen processing (TAP) for MHC loading. The antigen-loaded MHCs are then transported to the plasma membrane of the phagocytes, where they interact with T cell receptors (TCRs) expressed on T lymphocytes. With the help of inflammatory cytokines, the recognition of antigen-loaded MHC by TCR promotes the activation of T cells, thus enabling them to detect and eliminate tumour cells via cytotoxic responses.
Generation of antigen-specific T cell responses
Although the initial observation of phagocytosis checkpoint blockade using an anti-CD47 antibody largely attributed its effect to direct elimination of tumour cells by phagocytes, a growing body of evidence has emerged to support the idea that the adaptive immune system, particularly CD8 T cells, is also critical for the antitumour effect of CD47 blockade.160–163 Disrupting the SIRPα–CD47 interaction using monoclonal antibodies enhanced the phagocytosis activities of macrophages against human colon cancer cells. These macrophages also exhibited enhanced cross-presentation of tumour-derived antigens by MHC-I molecules on their surface, and they primed antigen-specific CD8+ T cell, but to date, not CD4+ T cell, responses in vitro and in vivo (Figure 3).160 Similarly, CD47 blockade was also shown to promote tumour cell phagocytosis by DCs, resulting in more efficient T cell cross-priming and antitumour effects.162 In this case, depleting CD8+ T cells, rather than CD4+ T cells, in vivo largely diminished the therapeutic response of CD47 blockade, suggesting an essential role of the adaptive immune system in mediating CD47 blockade’s antitumour effect.162 Finally, against syngeneic B16F10 melanomas, antibodies targeting both CD47 and TRP-1 synergized with an anti–PD-L1 antibody to produce a durable antitumour response, which was not achieved using CD47 blockade either alone or in combination with the anti–TRP-1 antibody.164 While these data further support the fact that CD47 blockade’s antitumour effect might also involve a functional adaptive immune system, the recent finding that PD-1 also serves as a phagocytosis checkpoint on tumour-associated macrophages complicates the conclusion, as PD-L1 and CD47 blockade provided better tumour control and longer survival in immunocompromised tumour-bearing mice than either treatment alone.56
Toll-like receptor (TLR) pathway
As the best characterized pattern recognition receptors, TLRs can detect tumour-associated DAMPs released within the tumour microenvironment or cancer cell engulfed within phagosomes, resulting in the activation of downstream inflammatory pathways.155 TLR activation in professional APCs is essential for key immunological processes, including processing and cross-presentation of antigens, upregulation of co-stimulatory molecules including CD80 and CD86, and T cell recruitment and activation.155 However, the role of TLR activation in determining the antitumour responses of phagocytosis checkpoint blockade is less defined. Anti-CD47 antibody treatments in MC38 tumours implanted in mice deficient in Myd88 and Trif, both of which are cytoplasmic Toll/Interleukin-1 receptor (TIR) domain-containing adaptor proteins,154 resulted in growth inhibition comparable to that in wild-type animals.162 Similarly, CD47−/− RBCs sufficed to activate splenic CD4+ DCs in Myd88−/− and Trif−/− mice, and they need to interact with CD18 (Integrin β2).165 Whether this holds true for non-hematopoietic cells or tumour cells is unclear. Although the TLR pathways may not be essential for generating antitumour adaptive responses, their activation nevertheless may augment the phagocytotic effect of CD47 blockade. Adding agonists to TLR-3, −4, and, −7 enhanced the antitumour activities of CD47 blockade in vivo. Activation of TLRs result in the phosphorylation of Btk in phagocytes, which then phosphorylates the ER chaperon CRT to promote its translocation to the plasma membrane.47 The CRT can be secreted by the macrophages or it may interact with other yet unidentified receptors expressed on tumour cells to promote their phagocytosis with CD47 blockade. However, it is unclear whether CRT upregulation by TLR occurs in other hematopoietic cell populations that express Btk. The potential for increased hematological toxicities with CD47 blockade is also unclear.
cGAS-STING pathway
Interferon production in host APCs can be triggered not only by TLRs, but also by the activation of the cytosolic DNA sensing cGAS-STING pathway. In tumour cells, increased genomic instability results in the formation of micronuclei, because DNA is mis-segregated during cell division.166,167 Breakdown of the micronuclear envelope associated with chromothripsis exposes the self-DNA to the cytosol, leading to rapid accumulation of cGAS,168 which binds to and is activated by the exposed nucleic acids,169 resulting in STING phosphorylation and interferon induction.169,170 Alternatively, tumour-derived DNA can activate cGAS-STING in host APCs after tumour cell phagocytosis.162 The T cell priming and antitumour effects of CD47 blockade appear to depend on the activation of cGAS-STING in APCs such as DCs. Treating MC38 tumours with an anti-CD47 antibody increased type I IFN production and antitumour responses in WT, but not STING-deficient, Tmem173GT mice.162 Similarly, DCs isolated from these animals showed a differential ability to prime CD8+ T cells ex vivo when co-cultured with tumour cells and anti-CD47 antibody. These results suggest that, at least in the context of CD47 blockade, host cell activation of the cGAS-STING pathway is critical for IFN production and antitumour adaptive immunity.162
Currently, it is unclear through which process tumour DNA escapes from phagosomes and enters the cytoplasm of APCs to trigger the cGAS-STING pathway. However, it appears that the origin of the DNA matters. After CD47 blockade-induced phagocytosis, an increase in the abundance of tumour-derived mitochondrial DNA (mtDNA), but not nuclear DNA (nDNA), in the cytosol of DCs was detected, despite the overall enrichment of both nucleic acids.171 The tumour mtDNA is recognized by cGAS in the cytosol, which leads to STING activation and type I IFN induction (Figure 2). Depleting mtDNA in tumour cells significantly reduced both type I IFN production and CD8 T cell priming capacities of DCs in the setting of CD47 blockade. This suggests that mtDNA, rather than nDNA, is the primary activator of innate immune sensing pathways in APCs after CD47 blockade-induced tumour cell phagocytosis.171 Mechanistically, it appears that CD47 blockade enabled the activation of NADPH oxidase NOX2 in DCs, which in turn inhibited phagosomal acidification and reduced the degradation of tumour mitochondrial DNA (mtDNA) in DCs. mtDNA was recognized by cyclic-GMP-AMP synthase (cGAS) in the DC cytosol, contributing to type I IFN production and antitumour adaptive immunity. The increase in cytosolic mtDNA content applied only to DCs, not to macrophages, despite the latter being more efficient in phagocytizing tumour cells after CD47 blockade. This discrepancy seems to be a result of the preferential inhibition of NOX2 within DCs and macrophages. The binding of CD47 with SIRPα recruits SHP-1 to dephosphorylate p47phox, which results in NOX2 inhibition. Blockade of CD47 restored the activities of NOX2 in DCs but not in macrophages, which de-acidify the phagosomes via reactive oxygen species (ROS)-mediated proton consumption to reduce DNA degradation. However, questions remain about which processes regulate NOX2 in macrophages, as phosphorylation of p47phox did not change in macrophages as it did in DCs after CD47 blockade.171 Similarly, it is unclear why CD47 blockade increased mtDNA, but not nDNA, content within the cytosol of DCs, though the linear structure of nDNA, which is more prone than circular mtRNA to enzymatic degradation, may have contributed to this effect.172 Finally, in addition to phagosomal degradation of DNA, cytosolic exonucleases including Trex1 can also dampen cGAS-STING activation and IFN induction.173 IFNγ stimulation upregulated Trex1 expression in macrophages but not in DCs, but IL4 had no effect. The upregulation of Trex1 not only increases DNA degradation in the cytosol, but also severely restricts antigen cross-presentation and T cell priming of macrophages.174 Therefore, multiple phagosomal and cytosolic regulators that perform unique functions within distinctive APC subpopulations probably determine the activation of innate immune sensing after tumour cell phagoytosis. A deeper understanding of these pathways should enable the development of more effective therapies that bridge innate and adaptive antitumour responses.
COMBINATION THERAPY WITH PHAGOCYTOSIS CHECKPOINT BLOCKADE
Therapeutic antibodies
Cytotoxicity towards cancer cells from blockade of the CD47-SIRPα axis occurs primarily through phagocytosis, instead of ADCC, as supported by the evidences that F(ab’)2 of CD47-blocking antibody induce effective phagocytosis and CD47 deficient cancer cells were dramatically more susceptible to phagocytosis as compared to the CD47-expressing counterparts.47,48,143 Blockade of the CD47-SIRPα axis can synergize with therapeutic antibodies to enhance the effect of ADCP or vice versa. An early example of this successful combination therapeutic strategy is when CD47 blocking antibody were given together with the anti-CD20 antibody Rituximab for the treatment of B cell non-Hodgkin’s lymphoma (NHL). In both localized and disseminated xenotransplantation models of NHL in NSG mice, combined Rituximab and CD47 blocking antibody inhibited tumour engraftment and growth, with 60% of mice achieving cancer elimination and long-term survival.143 Expanding on these results, a bispecific antibody that co-targets CD47 and CD20 also demonstrated improved efficacy and phagocytosis induction against lymphoma cells.175 Beyond hematological cancers, CD47-blocking reagents have also demonstrated enhanced phagocytosis induction and antitumour effects when combined with therapeutic antibodies in breast cancer (with trastuzumab),139 melanoma (with anti-CD271 antibody),176 small cell lung cancer (with anti-CD56 antibody),48 and colon cancer (with anti-EGFR or anti-EpCAM antibody).139
Cytokine therapy
Because phagocytosis checkpoint blockade relies on APCs to engulf cancer cells to generate antitumour responses, it may be complemented by strategies that either improve the phagocytosis process or enhance downstream immune activation. While the former can be accomplished by targeting multiple phagocytosis checkpoints or with co-stimulatory receptors, it may also be accomplished by modifying the properties of phagocytes. This hypothesis is supported by the observation that SIRPα−/− and CD47−/− mice, which exhibit minimal engulfment of normal self-cells, develop severe anemia in the setting of inflammation. Macrophages from these mice treated with IL-17, LPS, IL-6, IL-1β, and TNFα, but not IFNγ, initiated potent phagocytosis towards self-cells that is only restrained by CD47-SIRPα.177 In human glioma models, anti-CD47 treatment resulted in more prominent tumour cell phagocytosis by M1- than M2-polarized macrophages.147 Similarly, TTI-621, a soluble SIRPαFc fusion protein that blocks the CD47 signal, increased phagocytosis of lymphoma cells by multiple polarized macrophage subpopulations, with the greatest effect from macrophages stimulated by IFNγ, IFNγ + LPS, and IL-10 + TGFβ.140 Even the macrophages that have lower initial phagocytic capacities could be repolarized into more potent phagocytic phenotypes through the stimulation of cytokines or TLR agonists.140 Taken together, these results suggest that strategies that alter phagocyte phenotypes using cytokines, TLR agonists, chemokines or other growth factors may be combined with CD47 or other phagocytosis checkpoint blockades to induce tumour cell clearance.
Chemotherapy
Despite its potential toxicities, chemotherapy remains the backbone of the standard-of-care for many cancers, especially in the locally advanced and metastatic settings.178 With the emergence of immunotherapy, efforts have increasingly focused on identifying the optimal combination or ideal clinical scenarios in which chemotherapy can synergize with cancer immunotherapy to produce the maximal therapeutic benefit.179–182 However, this is complicated by the fact that many chemotherapeutic agents cause myelosuppression and are highly toxic to immune cells within the body.183 At the same time, chemotherapy can induce DNA damage in tumour cells, resulting in immunogenic cell death, which is associated with the translocation of the prophagocytosis signal CRT to the plasma membrane (Figure 4).92,184 As a result, chemotherapy administered after CD47 blockade was found to be detrimental to generating antitumour memory responses, whereas initiating chemotherapy before CD47 blockade improved the antitumour activities of anti-CD47 antibodies and preserved the host memory response against relapsing tumours.162 A separate study found that the synergistic antitumour effect of chemotherapy doxorubicin with SIRPα and PD-L1 dual blockade was abrogated when mice bearing MC38 tumours were first treated with Z-VAD-FMK, a caspase inhibitor that reduces the expression of membrane CRT.185 These results suggest that the induction of the prophagocytosis signal CRT is critical for synergizing chemotherapy with phagocytosis checkpoint blockade to produce antitumour effects. Regardless, the therapeutic responses of chemotherapy and phagocytosis checkpoint blockade will probably depend on multiple factors, including the type, timing, and dose of the agents used, the tumour types targeted, and perhaps the immunological status of the patients. A deeper understanding of the mechanisms that govern the potential synergism between cytotoxic agents and phagocytosis checkpoint blockers will provide a clearer insight into developing effective combination regimens for cancer treatment.
Figure 4: Combination therapy with phagocytosis checkpoint blockade.

Combined modality treatment can be utilized to promote more efficient tumour cell phagocytosis and innate immune sensing pathways to produce more potent antitumor responses. For example, ionizing radiation and certain classes of chemotherapeutic compounds, can enhance the translocation of prophagocytosis signal calreticulin from the ER to the tumour cell plasma membrane, where it may synergize with CD47 blockade. Radiation and chemotherapeutic agents can also induce nuclear DNA damage, thus induce cGAS-STING mediated type I IFN responses. Finally, in addition to acting as a T cell checkpoint, PD-L1 expression on tumour cells may also inhibit their phagocytosis by macrophages. Therefore, combination therapy of CD47 and PD-L1 blockade not only should improve the phagocytic clearance of tumour cells by phagocytes, but may also lead to more potent activation of antitumour T cell immunity through enhance priming effect.
Radiation therapy
CD47 blockade has also been investigated in combination with ionizing radiation to enhance antitumour effects. CD47 was found to interact with thrombospondin-1 (TSP-1) to limit recovery of normal tissues from cellular stress induced by radiation exposure. Accordingly, blockade of CD47 exerted a radioprotective effect on human endothelial cells and T cells in irradiated mice.186 In both endothelial and T cells, CD47 blockade promoted their survival in concert with the upregulation of autophagy pathways.187 In syngeneic B16 melanoma-bearing mice, CD47 blockade directly increased effector T cell infiltration into the tumour microenvironment and sensitized CD47-expressing tumours to ionizing radiation in a CD8+ T cell-dependent manner.161 Beyond promoting T cell recruitment and survival, radiation can also directly facilitate the phagocytosis of tumour cells by APCs.188 Exposure to ionizing radiation can promote the translocation of CRT within tumour cells from the ER to the plasma membrane where it acts as a prophagocytosis signal to phagocytes.189,190 Although radiation was found to upregulate the expression of MHC-I, which contains the B2m subunit via increased production of IFNβ, on tumour cells,191,192 it also makes tumour cells more recognizable to cytotoxic T cells.192 Furthermore, increased DNA damage from ionizing radiation can activate the cytosolic DNA sensing cGAS-STING pathway in both tumour cells169,170,173 and host APCs.193 Although the production of inflammatory cytokines after STING activation can induce tumour necrosis, it can also facilitate adaptive changes in tumours to promote immune resistance. TNFα, for example, through the activation of NF-κB, increases CD47 expression in at least in some breast cancers by promoting NF-κB binding to enhancers associated with the gene locus (Figure 4).49 Therefore, radiation-induced inflammatory responses may further sensitize tumours to phagocytosis checkpoint blockade, thus justifying further investigation of this combinatorial regimen for cancer treatment.
Adaptive checkpoint blockade
The observations that phagocytosis checkpoint blockade, including anti-CD47 therapy, stimulate both the innate and adaptive immune systems to generate antitumour responses provide a strong rationale for combining it with existing cancer immunotherapy strategies, such as T cell checkpoint inhibitors. The potential for such combinations was initially demonstrated when anti-CD47 antibody synergized with PD-L1 blockade to improve the therapeutic response rate against melanoma established in a syngeneic host.164 Similarly, using a fusion bispecific anti–PD-L1–SIRPa antibody that blocks both innate and adaptive checkpoints on tumour cells enhanced antitumour response.193 The generation of a bispecific antibody that simultaneously targets CD47 and a tumour-associated antigen increases the specificity and tolerability of CD47 blockade.194 While CD47 blockade can improve T cell priming and activation through IFN production, inhibiting PD-1 may also enhance the phagocytosis capabilities of intratumoural macrophages.56 It would be interesting to investigate the effect of CD47 blockade on the antitumour capabilities of T cells, as previous studies have suggested that CD47 negatively regulates the differentiation of Th1 CD4+ T cells195 and interacts with TSP-1 to inhibit T cell proliferation.196 Similarly, it is unclear what effect, if any, PD-1 blockade-induced tumour cell phagocytosis has on innate immune sensing pathway activation within APCs. Nonetheless, these results confirm the notion that the conventional divide between innate and adaptive checkpoints is becoming less obvious, as more of these checkpoints are found to function at both the innate and adaptive levels.
EARLY CLINICAL EVIDENCES
Malignant cells utilize multiple processes to evade host immune detection and clearance. Early clinical trial results have already generated exciting results for phagocytosis checkpoint blockade-based cancer immunotherapy. A phase 1b trial of treatment-refractory or relapsed non-Hodgkin’s lymphoma treated with 5F9, a humanized antibody that blocks CD47, and rituximab, an anti-CD20 monoclonal antibody, resulted in a 50% objective response and a 36% (~ 72% of objective response) complete response rate.197 Within the subset of patients with more aggressive, diffuse large B cell lymphoma, over 30% of patients had a complete response. Although promising, a longer follow up and larger numbers of patients are needed to confirm these findings. The concurrent administration of anti-CD47 antibody with rituximab also appeared to be well tolerated, with most of the toxicities reported as Grade 1–2 and anemia as the most common Grade 3 adverse event, which is expected given the expression of CD47 on red blood lifespan (Box 2).138 The severity of adverse reactions, however, can be reduced through a priming strategy, where a smaller dose of 5F9 is given one week before to remove aged RBC from circulation and to allow compensatory reticulocytosis to replenish the blood pool. This priming regimen, followed by a maintenance dose of 5F9, was also employed in a recent clinical trial in solid tumours.198 This trial also observed mild anemia in 50% of the patients, but they quickly recovered with no need for intervention. Though only two of the 62 patients (3%) had a partial response to 5F9 monotherapy, both patients had ovarian/fallopian tube primaries, which equates to a 10–15% overall response rate for these types of cancer. The generally favorable toxicity profile of anti-CD47 therapy, thus, offers promise for ongoing clinical trials of phagocytosis checkpoint blockade in multiple hematological and solid tumours, either as a single agent or in combination with other monoclonal antibodies or T cell immune checkpoint inhibitors (Table 1).
Blockade of CD47-SIRPα axis using the soluble SIRPαFc fusion protein TTI-621 has also demonstrated promising result in patients with Sézary syndrome, a leukemic variant of cutaneous T cell lymphoma.199 In this study, it was found that circulating Sézary cells have elevated expression of CD47, which is correlated with worse patient prognosis.199 Treatment with TTI-661 promoted phagocytosis of Sézary cells by macrophages, and significantly reduced tumour burden as measured by the level of lactate dehydrogenase in blood, decreased absolute lymphocyte counts and clinical involvement in skin erythema.199 Although the trial only included a very limited number of patients (n=5), these results nevertheless offer encouraging support for CD47-SIRPα blockade in the treatment of highly aggressive T cell malignancies.
CONCLUSION
It is becoming increasingly clear that tumour cell phagocytosis and subsequent immune recognition are governed by multiple inhibitory and stimulatory signals that must be considered to generate optimal antitumour responses. Innovative methods to identify key regulators of phagocytosis using genetic screening strategies under physiological conditions and non-neoplastic pathologies can potentially be expanded to the oncological setting. Recently studies using genome-wide CRISPR screens for example, have identified previously unknown regulators of phagocytosis with significant implications in amyloid-β clearance in Alzheimer’s disease.200 How these phagocytosis regulators work in concert to modulate tumour cell clearance by professional phagocytes, during various stages of tumourigenesis, among different cancer types, remains to be elucidated. From a clinical perspective, how phagocytosis checkpoint blockers/stimulators are to be incorporated into the current cancer immunotherapy paradigm needs to be further evaluated. At the onset, targeting phagocytosis checkpoints should complement existing T cell immune checkpoint inhibitors to maximize antitumour responses. For example, tumours with low levels of PD-L1, which are less sensitive to blockade of PD-1/PD-L1 axis, may in fact be more responsive to CD47-SIRPα disruption.56 Similarly, whereas adaptive immunotherapy relies on the generation of specific T cell clones that can recognize tumour-associated neoantigens, which is associated with the degree of genomic alterations in tumour cells, phagocytosis checkpoint blockade appears to be effective even against cancers with low mutational burdens such as AML.44,201 Therefore, phagocytosis checkpoints may provide an alternative strategy to treat tumours that are unresponsive or refractory to conventional cancer immunotherapies, or given concurrently with adaptive immune checkpoint inhibitors to improve the overall response rate in patients. Finally, it is also important to strike a balance between potency and toxicity.202 Unlike adaptive immune responses, which to a certain extent are bound by the limit of self-tolerance, innate responses are less specific and, thus, are more prone to normal tissue damage. This is especially important, as phagocytosis checkpoint inhibitors are likely to be used in conjunction with other immune modulators, such as TLR or STING agonists, cytokines, or systemic chemotherapies. Nevertheless, targeting phagocytosis checkpoints presents a new avenue to gain important insights into the mechanisms underlying tumour-mediated immune evasion and to develop more effective therapies that bridge the innate and adaptive immune systems to benefit patients with cancer.
Acknowledgements
This work was supported by grants from the Cancer Prevention and Research Institute of Texas RR180017 (W.J.), The National Cancer Institute (K08CA241070) (W.J.), the National Institute of Neurological Disorders and Stroke Grant R01 NS104315 (B.Y.S.K.), the National Cancer Institute Pathway to Independence Award R00CA201075 (to M.F.), the Damon Runyon–Dale F. Frey Award for Breakthrough Scientists DFS-22-16 (to M.F.) and the V Foundation for Cancer Research V Scholar Award V2018-012 (to M.F.). The authors thank Jonathan Feinberg of The University of Texas Southwestern Medical Center Department of Radiation Oncology for editorial assistance.
Competing interest statement
I.L.W. is a cofounder and director of and holds equity in Forty Seven, Inc. and has multiple patents licensed to Forty Seven Inc. M.F. declares patent applications pertaining to stimulating TLR/BTK signaling to promote CRT in macrophages assigned to the Stanford University and equity with Forty Seven, Inc.
Glossary
- Adaptive immune system
A major branch of the overall immune system that consists of highly specialized immune cell populations that recognize specific antigens to produce immune memory responses.
- Antigen presenting cells (APCs)
A collection of different immune cell populations that activate cellular immune responses by processing and presenting antigens that can be recognized by T cells. Classical professional APCs include dendritic cells and macrophages.
- Innate immune system
A major branch of the overall immune system that provides non-specific defense against pathogens immediately after an attack. The innate immune system is also responsible for educating the adaptive immune system through cross-priming.
- Cross-priming
The process by which naive T cells are activated by antigen presenting cells through antigen cross-presentation.
- Damage-associated molecular patterns (DAMPs)
Host cell derived biomolecules that can be recognized by pattern recognition receptors (PRRs) to initiate inflammatory responses.
- Fc-domain
The stem region of an antibody that interacts with the cell-surface–bound Fc receptors and proteins of the complement system.
- MHC class I (MHC I)
A complex of cell membrane proteins that is expressed by all nucleated cells to present antigens to be recognized by T cells.
- Pathogen associated molecular patterns (PAMPs)
Small molecular motifs derived from microbes that can be recognized by specialized pattern recognition receptors (PRRs).
- Phagocytosis
A cellular process by which cells or debris are engulfed by phagocytes.
- Phagolysosome
Cytoplasmic vesicular bodies formed by the fusion of a phagosome with a lysosome during the phagocytosis process.
- Tumour-associated macrophages (TAMs)
A class of macrophages found in high abundance in certain solid tumours that is often associated with immune suppressive properties within the tumour microenvironment.
References
- 1.Hodi FS et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363, 711–723, doi: 10.1056/NEJMoa1003466 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian SL et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366, 2443–2454, doi: 10.1056/NEJMoa1200690 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brahmer JR et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366, 2455–2465, doi: 10.1056/NEJMoa1200694 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reck M. et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med 375, 1823–1833, doi: 10.1056/NEJMoa1606774 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Carbone DP et al. First-Line Nivolumab in Stage IV or Recurrent Non-Small-Cell Lung Cancer. N Engl J Med 376, 2415–2426, doi: 10.1056/NEJMoa1613493 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balar AV et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet 389, 67–76, doi: 10.1016/S0140-6736(16)32455-2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garon EB et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 372, 2018–2028, doi: 10.1056/NEJMoa1501824 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Herbst RS et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387, 1540–1550, doi: 10.1016/S0140-6736(15)01281-7 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Antonia SJ et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N Engl J Med 377, 1919–1929, doi: 10.1056/NEJMoa1709937 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Morris VK et al. Nivolumab for previously treated unresectable metastatic anal cancer (NCI9673): a multicentre, single-arm, phase 2 study. Lancet Oncol 18, 446–453, doi: 10.1016/S1470-2045(17)30104-3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma P. et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol 18, 312–322, doi: 10.1016/S1470-2045(17)30065-7 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg JE et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 387, 1909–1920, doi: 10.1016/S0140-6736(16)00561-4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Motzer RJ et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 373, 1803–1813, doi: 10.1056/NEJMoa1510665 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nghiem PT et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N Engl J Med 374, 2542–2552, doi: 10.1056/NEJMoa1603702 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolchok JD et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med 377, 1345–1356, doi: 10.1056/NEJMoa1709684 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Motzer RJ et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med 378, 1277–1290, doi: 10.1056/NEJMoa1712126 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tawbi HA et al. Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain. N Engl J Med 379, 722–730, doi: 10.1056/NEJMoa1805453 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hellmann MD et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N Engl J Med 378, 2093–2104, doi: 10.1056/NEJMoa1801946 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wrangle JM et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: a non-randomised, open-label, phase 1b trial. Lancet Oncol 19, 694–704, doi: 10.1016/S1470-2045(18)30148-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gotwals P. et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer 17, 286–301, doi: 10.1038/nrc.2017.17 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Kranz LM et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534, 396–401, doi: 10.1038/nature18300 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Morvan MG & Lanier LL NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer 16, 7–19, doi: 10.1038/nrc.2015.5 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Iwasaki A & Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science 327, 291–295, doi: 10.1126/science.1183021 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer 4, 11–22, doi: 10.1038/nrc1252 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Jutras I & Desjardins M. Phagocytosis: at the crossroads of innate and adaptive immunity. Annu Rev Cell Dev Biol 21, 511–527, doi: 10.1146/annurev.cellbio.20.010403.102755 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Oldenborg PA et al. Role of CD47 as a marker of self on red blood cells. Science 288, 2051–2054 (2000). This important study for the first time showed the function of CD47 on red blood cells as a “marker of self” by interacting with SIRPα and inhibiting macrophages. [DOI] [PubMed] [Google Scholar]
- 27.Barclay AN & Van den Berg TK The interaction between signal regulatory protein alpha (SIRPalpha) and CD47: structure, function, and therapeutic target. Annu Rev Immunol 32, 25–50, doi: 10.1146/annurev-immunol-032713-120142 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Medzhitov R & Janeway CA Jr. Decoding the patterns of self and nonself by the innate immune system. Science 296, 298–300, doi: 10.1126/science.1068883 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Chao MP, Weissman IL & Majeti R. The CD47-SIRPalpha pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol 24, 225–232, doi: 10.1016/j.coi.2012.01.010 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kharitonenkov A. et al. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature 386, 181–186, doi: 10.1038/386181a0 (1997). This study identified the signal-regulatory proteins (SIRPs) family and their function in binding to SH2-domain-containing phsphotyrosine phosphotases for signal transduction. [DOI] [PubMed] [Google Scholar]
- 31.Fujioka Y. et al. A novel membrane glycoprotein, SHPS-1, that binds the SH2-domain-containing protein tyrosine phosphatase SHP-2 in response to mitogens and cell adhesion. Mol Cell Biol 16, 6887–6899 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Veillette A, Thibaudeau E & Latour S. High expression of inhibitory receptor SHPS-1 and its association with protein-tyrosine phosphatase SHP-1 in macrophages. J Biol Chem 273, 22719–22728 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Seiffert M. et al. Signal-regulatory protein alpha (SIRPalpha) but not SIRPbeta is involved in T-cell activation, binds to CD47 with high affinity, and is expressed on immature CD34(+)CD38(−) hematopoietic cells. Blood 97, 2741–2749 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Ichigotani Y. et al. Molecular cloning of a novel human gene (SIRP-B2) which encodes a new member of the SIRP/SHPS-1 protein family. J Hum Genet 45, 378–382, doi: 10.1007/s100380070013 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Barclay AN & Brown MH The SIRP family of receptors and immune regulation. Nat Rev Immunol 6, 457–464, doi: 10.1038/nri1859 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Dietrich J, Cella M, Seiffert M, Buhring HJ & Colonna M. Cutting edge: signal-regulatory protein beta 1 is a DAP12-associated activating receptor expressed in myeloid cells. J Immunol 164, 9–12 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Tomasello E. et al. Association of signal-regulatory proteins beta with KARAP/DAP-12. Eur J Immunol 30, 2147–2156, doi: (2000). [DOI] [PubMed] [Google Scholar]
- 38.Brooke G, Holbrook JD, Brown MH & Barclay AN Human lymphocytes interact directly with CD47 through a novel member of the signal regulatory protein (SIRP) family. J Immunol 173, 2562–2570 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Campbell IG, Freemont PS, Foulkes W & Trowsdale J. An ovarian tumor marker with homology to vaccinia virus contains an IgV-like region and multiple transmembrane domains. Cancer Res 52, 5416–5420 (1992). [PubMed] [Google Scholar]
- 40.Brown E, Hooper L, Ho T & Gresham H. Integrin-associated protein: a 50-kD plasma membrane antigen physically and functionally associated with integrins. J Cell Biol 111, 2785–2794 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seiffert M. et al. Human signal-regulatory protein is expressed on normal, but not on subsets of leukemic myeloid cells and mediates cellular adhesion involving its counterreceptor CD47. Blood 94, 3633–3643 (1999). [PubMed] [Google Scholar]
- 42.Jiang P, Lagenaur CF & Narayanan V. Integrin-associated protein is a ligand for the P84 neural adhesion molecule. J Biol Chem 274, 559–562 (1999). [DOI] [PubMed] [Google Scholar]
- 43.Cameron CM, Barrett JW, Mann M, Lucas A & McFadden G. Myxoma virus M128L is expressed as a cell surface CD47-like virulence factor that contributes to the downregulation of macrophage activation in vivo. Virology 337, 55–67, doi: 10.1016/j.virol.2005.03.037 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Majeti R. et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 138, 286–299, doi: 10.1016/j.cell.2009.05.045 (2009). This study identified CD47 as a “don’t eat me” signal on cancer cells for their self-protection, and revealed the therapeutic potential of CD47-blocking agents in inducing anti-tumor effects of macrophages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Willingham SB et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A 109, 6662–6667, doi: 10.1073/pnas.1121623109 (2012). This study examined the expression of CD47 in a wide range of solid tumor cells and evluated the anti-cancer effects of CD47 blocking antibodies in multiple solid tumor pre-clinical models. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan KS et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc Natl Acad Sci U S A 106, 14016–14021, doi: 10.1073/pnas.0906549106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feng M. et al. Macrophages eat cancer cells using their own calreticulin as a guide: roles of TLR and Btk. Proc Natl Acad Sci U S A 112, 2145–2150, doi: 10.1073/pnas.1424907112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weiskopf K. et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J Clin Invest 126, 2610–2620, doi: 10.1172/JCI81603 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Betancur PA et al. A CD47-associated super-enhancer links pro-inflammatory signalling to CD47 upregulation in breast cancer. Nat Commun 8, 14802, doi: 10.1038/ncomms14802 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang H. et al. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc Natl Acad Sci U S A 112, E6215–6223, doi: 10.1073/pnas.1520032112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu F. et al. BRAF/MEK inhibitors promote CD47 expression that is reversible by ERK inhibition in melanoma. Oncotarget 8, 69477–69492, doi: 10.18632/oncotarget.17704 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Casey SC et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231, doi: 10.1126/science.aac9935 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wei SC, Duffy CR & Allison JP Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov 8, 1069–1086, doi: 10.1158/2159-8290.CD-18-0367 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Pardoll DM The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12, 252–264, doi: 10.1038/nrc3239 (2012). This is an important review article summarizing the progress of identifying immue checkpoints in cancer and blocking them as cancer immunotherapies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baumeister SH, Freeman GJ, Dranoff G & Sharpe AH Coinhibitory Pathways in Immunotherapy for Cancer. Annu Rev Immunol 34, 539–573, doi: 10.1146/annurev-immunol-032414-112049 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Gordon SR et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499, doi: 10.1038/nature22396 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barkal AA et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol 19, 76–84, doi: 10.1038/s41590-017-0004-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li B. et al. Tumor-derived exosomal HMGB1 promotes esophageal squamous cell carcinoma progression through inducing PD1(+) TAM expansion. Oncogenesis 8, 17, doi: 10.1038/s41389-019-0126-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Borges L, Hsu ML, Fanger N, Kubin M & Cosman D. A family of human lymphoid and myeloid Ig-like receptors, some of which bind to MHC class I molecules. J Immunol 159, 5192–5196 (1997). [PubMed] [Google Scholar]
- 60.Colonna M. et al. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J Exp Med 186, 1809–1818, doi: 10.1084/jem.186.11.1809 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Samaridis J & Colonna M. Cloning of novel immunoglobulin superfamily receptors expressed on human myeloid and lymphoid cells: structural evidence for new stimulatory and inhibitory pathways. Eur J Immunol 27, 660–665, doi: 10.1002/eji.1830270313 (1997). [DOI] [PubMed] [Google Scholar]
- 62.Katz HR Inhibition of inflammatory responses by leukocyte Ig-like receptors. Adv Immunol 91, 251–272, doi: 10.1016/S0065-2776(06)91007-4 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Hunt JS et al. HLA-G in reproduction: studies on the maternal-fetal interface. Hum Immunol 61, 1113–1117 (2000). [DOI] [PubMed] [Google Scholar]
- 64.Tedla N, Lee CW, Borges L, Geczy CL & Arm JP Differential expression of leukocyte immunoglobulin-like receptors on cord-blood-derived human mast cell progenitors and mature mast cells. J Leukoc Biol 83, 334–343, doi: 10.1189/jlb.0507314 (2008). [DOI] [PubMed] [Google Scholar]
- 65.Mori Y. et al. Inhibitory immunoglobulin-like receptors LILRB and PIR-B negatively regulate osteoclast development. J Immunol 181, 4742–4751, doi: 10.4049/jimmunol.181.7.4742 (2008). [DOI] [PubMed] [Google Scholar]
- 66.Festenstein H & Garrido F. MHC antigens and malignancy. Nature 322, 502–503, doi: 10.1038/322502a0 (1986). [DOI] [PubMed] [Google Scholar]
- 67.Leone P. et al. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst 105, 1172–1187, doi: 10.1093/jnci/djt184 (2013). [DOI] [PubMed] [Google Scholar]
- 68.Garrido F, Ruiz-Cabello F & Aptsiauri N. Rejection versus escape: the tumor MHC dilemma. Cancer Immunol Immunother 66, 259–271, doi: 10.1007/s00262-016-1947-x (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Colonna M. et al. Human myelomonocytic cells express an inhibitory receptor for classical and nonclassical MHC class I molecules. J Immunol 160, 3096–3100 (1998). [PubMed] [Google Scholar]
- 70.Zheng J. et al. Inhibitory receptors bind ANGPTLs and support blood stem cells and leukaemia development. Nature 485, 656–660, doi: 10.1038/nature11095 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fanger NA et al. The MHC class I binding proteins LIR-1 and LIR-2 inhibit Fc receptor-mediated signaling in monocytes. European journal of immunology 28, 3423–3434, doi: (1998). [DOI] [PubMed] [Google Scholar]
- 72.Manavalan JS et al. Alloantigen specific CD8+CD28- FOXP3+ T suppressor cells induce ILT3+ ILT4+ tolerogenic endothelial cells, inhibiting alloreactivity. International immunology 16, 1055–1068, doi: 10.1093/intimm/dxh107 (2004). [DOI] [PubMed] [Google Scholar]
- 73.Lu N. et al. Human Semaphorin-4A drives Th2 responses by binding to receptor ILT-4. Nat Commun 9, 742, doi: 10.1038/s41467-018-03128-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen HM et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J Clin Invest 128, 5647–5662, doi: 10.1172/JCI97570 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jones DC et al. HLA class I allelic sequence and conformation regulate leukocyte Ig-like receptor binding. J Immunol 186, 2990–2997, doi: 10.4049/jimmunol.1003078 (2011). [DOI] [PubMed] [Google Scholar]
- 76.Cella M. et al. A novel inhibitory receptor (ILT3) expressed on monocytes, macrophages, and dendritic cells involved in antigen processing. J Exp Med 185, 1743–1751 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Deng M. et al. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature 562, 605–609 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.John S. et al. A Novel Anti-LILRB4 CAR-T Cell for the Treatment of Monocytic AML. Mol Ther 26, 2487–2495, doi: 10.1016/j.ymthe.2018.08.001 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Inui M. et al. Tolerogenic immunoreceptor ILT3/LILRB4 paradoxically marks pathogenic auto-antibody-producing plasmablasts and plasma cells in non-treated SLE. Int Immunol 28, 597–604, doi: 10.1093/intimm/dxw044 (2016). [DOI] [PubMed] [Google Scholar]
- 80.Ulges A. et al. Protein kinase CK2 enables regulatory T cells to suppress excessive TH2 responses in vivo. Nat Immunol 16, 267–275, doi: 10.1038/ni.3083 (2015). [DOI] [PubMed] [Google Scholar]
- 81.Kang X. et al. Inhibitory leukocyte immunoglobulin-like receptors: Immune checkpoint proteins and tumor sustaining factors. Cell Cycle 15, 25–40, doi: 10.1080/15384101.2015.1121324 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Goeje PL et al. Immunoglobulin-like transcript 3 is expressed by myeloid-derived suppressor cells and correlates with survival in patients with non-small cell lung cancer. Oncoimmunology 4, e1014242, doi: 10.1080/2162402X.2015.1014242 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chang CC et al. Tolerization of dendritic cells by T(S) cells: the crucial role of inhibitory receptors ILT3 and ILT4. Nat Immunol 3, 237–243, doi: 10.1038/ni760 (2002). [DOI] [PubMed] [Google Scholar]
- 84.Suciu-Foca N. et al. Soluble Ig-like transcript 3 inhibits tumor allograft rejection in humanized SCID mice and T cell responses in cancer patients. J Immunol 178, 7432–7441 (2007). [DOI] [PubMed] [Google Scholar]
- 85.Xu Z. et al. ILT3.Fc-CD166 Interaction Induces Inactivation of p70 S6 Kinase and Inhibits Tumor Cell Growth. J Immunol, doi: 10.4049/jimmunol.1700553 (2017). [DOI] [PubMed] [Google Scholar]
- 86.Zhang Q & Salter RD Distinct patterns of folding and interactions with calnexin and calreticulin in human class I MHC proteins with altered N-glycosylation. J Immunol 160, 831–837 (1998). [PubMed] [Google Scholar]
- 87.Harris MR, Yu YY, Kindle CS, Hansen TH & Solheim JC Calreticulin and calnexin interact with different protein and glycan determinants during the assembly of MHC class I. J Immunol 160, 5404–5409 (1998). [PubMed] [Google Scholar]
- 88.Krause KH & Michalak M. Calreticulin. Cell 88, 439–443 (1997). [DOI] [PubMed] [Google Scholar]
- 89.Basu S, Binder RJ, Ramalingam T & Srivastava PK CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity 14, 303–313 (2001). [DOI] [PubMed] [Google Scholar]
- 90.Ogden CA et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med 194, 781–795 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vandivier RW et al. Role of surfactant proteins A, D, and C1q in the clearance of apoptotic cells in vivo and in vitro: calreticulin and CD91 as a common collectin receptor complex. J Immunol 169, 3978–3986 (2002). [DOI] [PubMed] [Google Scholar]
- 92.Obeid M. et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 13, 54–61, doi: 10.1038/nm1523 (2007). This classic study identified the process of immunogenic cell death. [DOI] [PubMed] [Google Scholar]
- 93.Obeid M. et al. Ecto-calreticulin in immunogenic chemotherapy. Immunol Rev 220, 22–34, doi: 10.1111/j.1600-065X.2007.00567.x (2007). [DOI] [PubMed] [Google Scholar]
- 94.Obeid M. et al. Leveraging the immune system during chemotherapy: moving calreticulin to the cell surface converts apoptotic death from “silent” to immunogenic. Cancer Res 67, 7941–7944, doi: 10.1158/0008-5472.CAN-07-1622 (2007). [DOI] [PubMed] [Google Scholar]
- 95.Panaretakis T. et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ 15, 1499–1509, doi: 10.1038/cdd.2008.67 (2008). [DOI] [PubMed] [Google Scholar]
- 96.Krysko DV et al. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer 12, 860–875, doi: 10.1038/nrc3380 (2012). [DOI] [PubMed] [Google Scholar]
- 97.Garg AD et al. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim Biophys Acta 1805, 53–71, doi: 10.1016/j.bbcan.2009.08.003 (2010). [DOI] [PubMed] [Google Scholar]
- 98.Pang WW et al. Hematopoietic stem cell and progenitor cell mechanisms in myelodysplastic syndromes. Proc Natl Acad Sci U S A 110, 3011–3016, doi: 10.1073/pnas.1222861110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chao MP et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med 2, 63–94, doi: 10.1126/scitranslmed.3001375 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lagasse E & Weissman IL bcl-2 inhibits apoptosis of neutrophils but not their engulfment by macrophages. J Exp Med 179, 1047–1052 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chao MP, Majeti R & Weissman IL Programmed cell removal: a new obstacle in the road to developing cancer. Nat Rev Cancer 12, 58–67, doi: 10.1038/nrc3171 (2011). [DOI] [PubMed] [Google Scholar]
- 102.Byrne JC et al. Bruton’s tyrosine kinase is required for apoptotic cell uptake via regulating the phosphorylation and localization of calreticulin. J Immunol 190, 5207–5215, doi: 10.4049/jimmunol.1300057 (2013). [DOI] [PubMed] [Google Scholar]
- 103.Feng M. et al. Programmed cell removal by calreticulin in tissue homeostasis and cancer. Nat Commun 9, 3194, doi: 10.1038/s41467-018-05211-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Duvall E, Wyllie AH & Morris RG Macrophage recognition of cells undergoing programmed cell death (apoptosis). Immunology 56, 351–358 (1985). [PMC free article] [PubMed] [Google Scholar]
- 105.Witting A, Muller P, Herrmann A, Kettenmann H & Nolte C. Phagocytic clearance of apoptotic neurons by Microglia/Brain macrophages in vitro: involvement of lectin-, integrin-, and phosphatidylserine-mediated recognition. J Neurochem 75, 1060–1070 (2000). [DOI] [PubMed] [Google Scholar]
- 106.Hakomori S. Aberrant glycosylation in tumors and tumor-associated carbohydrate antigens. Adv Cancer Res 52, 257–331 (1989). [DOI] [PubMed] [Google Scholar]
- 107.Dube DH & Bertozzi CR Glycans in cancer and inflammation--potential for therapeutics and diagnostics. Nat Rev Drug Discov 4, 477–488, doi: 10.1038/nrd1751 (2005). [DOI] [PubMed] [Google Scholar]
- 108.Shachar I, Barak A, Lewinsky H, Sever L & Radomir L. SLAMF receptors on normal and malignant B cells. Clin Immunol, doi: 10.1016/j.clim.2018.10.020 (2018). [DOI] [PubMed] [Google Scholar]
- 109.Gautier EL et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol 13, 1118–1128, doi: 10.1038/ni.2419 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Miller JC et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat Immunol 13, 888–899, doi: 10.1038/ni.2370 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen J. et al. SLAMF7 is critical for phagocytosis of haematopoietic tumour cells via Mac-1 integrin. Nature 544, 493–497, doi: 10.1038/nature22076 (2017). This study demonstrated that SLAMF7 is critical for mediating CD47-blockaded induced phagocytosis of hematological cancer cells by phagocytes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Aderem A & Underhill DM Mechanisms of phagocytosis in macrophages. Annu Rev Immunol 17, 593–623, doi: 10.1146/annurev.immunol.17.1.593 (1999). [DOI] [PubMed] [Google Scholar]
- 113.Abram CL & Lowell CA The ins and outs of leukocyte integrin signaling. Annu Rev Immunol 27, 339–362, doi: 10.1146/annurev.immunol.021908.132554 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.He Y. et al. Cancer cell-expressed SLAMF7 is not required for CD47-mediated phagocytosis. Nat Commun 10, 533, doi: 10.1038/s41467-018-08013-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nimmerjahn F & Ravetch JV Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 8, 34–47, doi: 10.1038/nri2206 (2008). [DOI] [PubMed] [Google Scholar]
- 116.Bakema JE & van Egmond M. Fc receptor-dependent mechanisms of monoclonal antibody therapy of cancer. Curr Top Microbiol Immunol 382, 373–392, doi: 10.1007/978-3-319-07911-0_17 (2014). [DOI] [PubMed] [Google Scholar]
- 117.Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 119, 5640–5649, doi: 10.1182/blood-2012-01-380121 (2012). [DOI] [PubMed] [Google Scholar]
- 118.Daeron M. Fc receptor biology. Annu Rev Immunol 15, 203–234, doi: 10.1146/annurev.immunol.15.1.203 (1997). [DOI] [PubMed] [Google Scholar]
- 119.Crowley MT et al. A critical role for Syk in signal transduction and phagocytosis mediated by Fcgamma receptors on macrophages. J Exp Med 186, 1027–1039, doi: 10.1084/jem.186.7.1027 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mao Y & Finnemann SC Regulation of phagocytosis by Rho GTPases. Small GTPases 6, 89–99, doi: 10.4161/21541248.2014.989785 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Getahun A & Cambier JC Of ITIMs, ITAMs, and ITAMis: revisiting immunoglobulin Fc receptor signaling. Immunol Rev 268, 66–73, doi: 10.1111/imr.12336 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Beers SA et al. Antigenic modulation limits the efficacy of anti-CD20 antibodies: implications for antibody selection. Blood 115, 5191–5201, doi: 10.1182/blood-2010-01-263533 (2010). [DOI] [PubMed] [Google Scholar]
- 123.Bergtold A, Desai DD, Gavhane A & Clynes R. Cell surface recycling of internalized antigen permits dendritic cell priming of B cells. Immunity 23, 503–514, doi: 10.1016/j.immuni.2005.09.013 (2005). [DOI] [PubMed] [Google Scholar]
- 124.Budde P, Bewarder N, Weinrich V, Schulzeck O & Frey J. Tyrosine-containing sequence motifs of the human immunoglobulin G receptors FcRIIb1 and FcRIIb2 essential for endocytosis and regulation of calcium flux in B cells. J Biol Chem 269, 30636–30644 (1994). [PubMed] [Google Scholar]
- 125.Clynes RA, Towers TL, Presta LG & Ravetch JV Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med 6, 443–446, doi: 10.1038/74704 (2000). [DOI] [PubMed] [Google Scholar]
- 126.Gul N. et al. Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J Clin Invest 124, 812–823, doi: 10.1172/JCI66776 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Overdijk MB et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs 7, 311–321, doi: 10.1080/19420862.2015.1007813 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gul N & van Egmond M. Antibody-Dependent Phagocytosis of Tumor Cells by Macrophages: A Potent Effector Mechanism of Monoclonal Antibody Therapy of Cancer. Cancer Res 75, 5008–5013, doi: 10.1158/0008-5472.CAN-15-1330 (2015). [DOI] [PubMed] [Google Scholar]
- 129.Roghanian A. et al. Antagonistic human FcgammaRIIB (CD32B) antibodies have anti-tumor activity and overcome resistance to antibody therapy in vivo. Cancer Cell 27, 473–488, doi: 10.1016/j.ccell.2015.03.005 (2015). [DOI] [PubMed] [Google Scholar]
- 130.Golay J. et al. Glycoengineered CD20 antibody obinutuzumab activates neutrophils and mediates phagocytosis through CD16B more efficiently than rituximab. Blood 122, 3482–3491, doi: 10.1182/blood-2013-05-504043 (2013). [DOI] [PubMed] [Google Scholar]
- 131.Han X. et al. CD47, a ligand for the macrophage fusion receptor, participates in macrophage multinucleation. J Biol Chem 275, 37984–37992, doi: 10.1074/jbc.M002334200 (2000). [DOI] [PubMed] [Google Scholar]
- 132.Vernon-Wilson EF et al. CD47 is a ligand for rat macrophage membrane signal regulatory protein SIRP (OX41) and human SIRPalpha 1. Eur J Immunol 30, 2130–2137, doi: (2000). [DOI] [PubMed] [Google Scholar]
- 133.Noguchi T. et al. Characterization of a 115-kDa protein that binds to SH-PTP2, a protein-tyrosine phosphatase with Src homology 2 domains, in Chinese hamster ovary cells. J Biol Chem 271, 27652–27658 (1996). [DOI] [PubMed] [Google Scholar]
- 134.Neel BG, Gu H & Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci 28, 284–293, doi: 10.1016/S0968-0004(03)00091-4 (2003). [DOI] [PubMed] [Google Scholar]
- 135.Tsai RK & Discher DE Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J Cell Biol 180, 989–1003, doi: 10.1083/jcb.200708043 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rebres RA, Vaz LE, Green JM & Brown EJ Normal ligand binding and signaling by CD47 (integrin-associated protein) requires a long range disulfide bond between the extracellular and membrane-spanning domains. J Biol Chem 276, 34607–34616, doi: 10.1074/jbc.M106107200 (2001). [DOI] [PubMed] [Google Scholar]
- 137.Logtenberg MEW et al. Glutaminyl cyclase is an enzymatic modifier of the CD47- SIRPalpha axis and a target for cancer immunotherapy. Nat Med, doi: 10.1038/s41591-019-0356-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Liu J. et al. Pre-Clinical Development of a Humanized Anti-CD47 Antibody with Anti-Cancer Therapeutic Potential. PLoS One 10, e0137345, doi: 10.1371/journal.pone.0137345 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Weiskopf K. et al. Engineered SIRPalpha variants as immunotherapeutic adjuvants to anticancer antibodies. Science 341, 88–91, doi: 10.1126/science.1238856 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lin GHY et al. TTI-621 (SIRPalphaFc), a CD47-blocking cancer immunotherapeutic, triggers phagocytosis of lymphoma cells by multiple polarized macrophage subsets. PLoS One 12, e0187262, doi: 10.1371/journal.pone.0187262 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Petrova PS et al. TTI-621 (SIRPalphaFc): A CD47-Blocking Innate Immune Checkpoint Inhibitor with Broad Antitumor Activity and Minimal Erythrocyte Binding. Clin Cancer Res 23, 1068–1079, doi: 10.1158/1078-0432.CCR-16-1700 (2017). [DOI] [PubMed] [Google Scholar]
- 142.Mantovani A, Marchesi F, Malesci A, Laghi L & Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 14, 399–416, doi: 10.1038/nrclinonc.2016.217 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chao MP et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell 142, 699–713, doi: 10.1016/j.cell.2010.07.044 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pan Y. et al. Endoscopic molecular imaging of human bladder cancer using a CD47 antibody. Sci Transl Med 6, 260–148, doi: 10.1126/scitranslmed.3009457 (2014). [DOI] [PubMed] [Google Scholar]
- 145.Wernig G. et al. Unifying mechanism for different fibrotic diseases. Proc Natl Acad Sci U S A 114, 4757–4762, doi: 10.1073/pnas.1621375114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kojima Y. et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 536, 86–90, doi: 10.1038/nature18935 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhang M. et al. Anti-CD47 Treatment Stimulates Phagocytosis of Glioblastoma by M1 and M2 Polarized Macrophages and Promotes M1 Polarized Macrophages In Vivo. PLoS One 11, e0153550, doi: 10.1371/journal.pone.0153550 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhao XW et al. CD47-signal regulatory protein-alpha (SIRPalpha) interactions form a barrier for antibody-mediated tumor cell destruction. Proc Natl Acad Sci U S A 108, 18342–18347, doi: 10.1073/pnas.1106550108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ring NG et al. Anti-SIRPalpha antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc Natl Acad Sci U S A 114, E10578–E10585, doi: 10.1073/pnas.1710877114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Matlung HL et al. Neutrophils Kill Antibody-Opsonized Cancer Cells by Trogoptosis. Cell Rep 23, 3946–3959 e3946, doi: 10.1016/j.celrep.2018.05.082 (2018). [DOI] [PubMed] [Google Scholar]
- 151.Mouro-Chanteloup I. et al. Evidence that the red cell skeleton protein 4.2 interacts with the Rh membrane complex member CD47. Blood 101, 338–344, doi: 10.1182/blood-2002-04-1285 (2003). [DOI] [PubMed] [Google Scholar]
- 152.Pandey S, Kawai T & Akira S. Microbial sensing by Toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb Perspect Biol 7, a016246, doi: 10.1101/cshperspect.a016246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Brubaker SW, Bonham KS, Zanoni I & Kagan JC Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol 33, 257–290, doi: 10.1146/annurev-immunol-032414-112240 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Takeda K, Kaisho T & Akira S. Toll-like receptors. Annu Rev Immunol 21, 335–376, doi: 10.1146/annurev.immunol.21.120601.141126 (2003). [DOI] [PubMed] [Google Scholar]
- 155.Rakoff-Nahoum S & Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer 9, 57–63, doi: 10.1038/nrc2541 (2009). [DOI] [PubMed] [Google Scholar]
- 156.Meylan E, Tschopp J & Karin M. Intracellular pattern recognition receptors in the host response. Nature 442, 39–44, doi: 10.1038/nature04946 (2006). [DOI] [PubMed] [Google Scholar]
- 157.Kawai T & Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol 21, 317–337, doi: 10.1093/intimm/dxp017 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chen Q, Sun L & Chen ZJ Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol 17, 1142–1149, doi: 10.1038/ni.3558 (2016). [DOI] [PubMed] [Google Scholar]
- 159.Li T & Chen ZJ The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med 215, 1287–1299, doi: 10.1084/jem.20180139 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tseng D. et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci U S A 110, 11103–11108, doi: 10.1073/pnas.1305569110 (2013). The first study showing that CD47-blockade in tumour cells can promote more efficient CD8 T cell priming by APCs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Soto-Pantoja DR et al. CD47 in the tumor microenvironment limits cooperation between antitumor T-cell immunity and radiotherapy. Cancer Res 74, 6771–6783, doi: 10.1158/0008-5472.CAN-14-0037-T (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Liu X. et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med 21, 1209–1215, doi: 10.1038/nm.3931 (2015). An important study that demonstrated CD47-blockaded mediated antitumour response is dependent on CD8 T cells and innate immune sensing pathways in APCs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Veillette A & Chen J. SIRPalpha-CD47 Immune Checkpoint Blockade in Anticancer Therapy. Trends Immunol 39, 173–184, doi: 10.1016/j.it.2017.12.005 (2018). [DOI] [PubMed] [Google Scholar]
- 164.Sockolosky JT et al. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc Natl Acad Sci U S A 113, E2646–2654, doi: 10.1073/pnas.1604268113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yi T. et al. Splenic Dendritic Cells Survey Red Blood Cells for Missing Self-CD47 to Trigger Adaptive Immune Responses. Immunity 43, 764–775, doi: 10.1016/j.immuni.2015.08.021 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jdey W, Thierry S, Popova T, Stern MH & Dutreix M. Micronuclei Frequency in Tumors Is a Predictive Biomarker for Genetic Instability and Sensitivity to the DNA Repair Inhibitor AsiDNA. Cancer Res 77, 4207–4216, doi: 10.1158/0008-5472.CAN-16-2693 (2017). [DOI] [PubMed] [Google Scholar]
- 167.Bakhoum SF, Kabeche L, Murnane JP, Zaki BI & Compton DA DNA-damage response during mitosis induces whole-chromosome missegregation. Cancer Discov 4, 1281–1289, doi: 10.1158/2159-8290.CD-14-0403 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhang CZ et al. Chromothripsis from DNA damage in micronuclei. Nature 522, 179–184, doi: 10.1038/nature14493 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mackenzie KJ et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465, doi: 10.1038/nature23449 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Harding SM et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470, doi: 10.1038/nature23470 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Xu MM et al. Dendritic Cells but Not Macrophages Sense Tumor Mitochondrial DNA for Cross-priming through Signal Regulatory Protein alpha Signaling. Immunity 47, 363–373 e365, doi: 10.1016/j.immuni.2017.07.016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Li J. et al. Circular DNA: a stable probe for highly efficient mRNA imaging and gene therapy in living cells. Chem Commun (Camb) 54, 896–899, doi: 10.1039/c7cc08906f (2018). [DOI] [PubMed] [Google Scholar]
- 173.Vanpouille-Box C. et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun 8, 15618, doi: 10.1038/ncomms15618 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Pereira-Lopes S. et al. The exonuclease Trex1 restrains macrophage proinflammatory activation. J Immunol 191, 6128–6135, doi: 10.4049/jimmunol.1301603 (2013). [DOI] [PubMed] [Google Scholar]
- 175.Piccione EC et al. A bispecific antibody targeting CD47 and CD20 selectively binds and eliminates dual antigen expressing lymphoma cells. MAbs 7, 946–956, doi: 10.1080/19420862.2015.1062192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ngo M. et al. Antibody Therapy Targeting CD47 and CD271 Effectively Suppresses Melanoma Metastasis in Patient-Derived Xenografts. Cell Rep 16, 1701–1716, doi: 10.1016/j.celrep.2016.07.004 (2016). [DOI] [PubMed] [Google Scholar]
- 177.Bian Z. et al. Cd47-Sirpalpha interaction and IL-10 constrain inflammation-induced macrophage phagocytosis of healthy self-cells. Proc Natl Acad Sci U S A 113, E5434–5443, doi: 10.1073/pnas.1521069113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Verweij J & de Jonge MJ Achievements and future of chemotherapy. Eur J Cancer 36, 1479–1487 (2000). [DOI] [PubMed] [Google Scholar]
- 179.George S, Rini BI & Hammers HJ Emerging Role of Combination Immunotherapy in the First-line Treatment of Advanced Renal Cell Carcinoma: A Review. JAMA Oncol, doi: 10.1001/jamaoncol.2018.4604 (2018). [DOI] [PubMed] [Google Scholar]
- 180.Schmid P. et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med 379, 2108–2121, doi: 10.1056/NEJMoa1809615 (2018). [DOI] [PubMed] [Google Scholar]
- 181.Vanneman M & Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer 12, 237–251, doi: 10.1038/nrc3237 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Smyth MJ, Ngiow SF, Ribas A & Teng MW Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol 13, 143–158, doi: 10.1038/nrclinonc.2015.209 (2016). [DOI] [PubMed] [Google Scholar]
- 183.Wargo JA, Reuben A, Cooper ZA, Oh KS & Sullivan RJ Immune Effects of Chemotherapy, Radiation, and Targeted Therapy and Opportunities for Combination With Immunotherapy. Semin Oncol 42, 601–616, doi: 10.1053/j.seminoncol.2015.05.007 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Galluzzi L, Buque A, Kepp O, Zitvogel L & Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 17, 97–111, doi: 10.1038/nri.2016.107 (2017). [DOI] [PubMed] [Google Scholar]
- 185.Liu X. et al. Dual Targeting of Innate and Adaptive Checkpoints on Tumor Cells Limits Immune Evasion. Cell Rep 24, 2101–2111, doi: 10.1016/j.celrep.2018.07.062 (2018). [DOI] [PubMed] [Google Scholar]
- 186.Maxhimer JB et al. Radioprotection in normal tissue and delayed tumor growth by blockade of CD47 signaling. Sci Transl Med 1, 3–7, doi: 10.1126/scitranslmed.3000139 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Soto-Pantoja DR et al. CD47 deficiency confers cell and tissue radioprotection by activation of autophagy. Autophagy 8, 1628–1642, doi: 10.4161/auto.21562 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Vermeer DW et al. Radiation-induced loss of cell surface CD47 enhances immune-mediated clearance of human papillomavirus-positive cancer. Int J Cancer 133, 120–129, doi: 10.1002/ijc.28015 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Gameiro SR et al. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget 5, 403–416, doi: 10.18632/oncotarget.1719 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kono K, Mimura K & Kiessling R. Immunogenic tumor cell death induced by chemoradiotherapy: molecular mechanisms and a clinical translation. Cell Death Dis 4, e688, doi: 10.1038/cddis.2013.207 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Reits EA et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med 203, 1259–1271, doi: 10.1084/jem.20052494 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Wang X. et al. Suppression of Type I IFN Signaling in Tumors Mediates Resistance to Anti-PD-1 Treatment That Can Be Overcome by Radiotherapy. Cancer Res 77, 839–850, doi: 10.1158/0008-5472.CAN-15-3142 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Deng L. et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 41, 843–852, doi: 10.1016/j.immuni.2014.10.019 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Dheilly E. et al. Selective Blockade of the Ubiquitous Checkpoint Receptor CD47 Is Enabled by Dual-Targeting Bispecific Antibodies. Mol Ther 25, 523–533, doi: 10.1016/j.ymthe.2016.11.006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Bouguermouh S. et al. CD47 expression on T cell is a self-control negative regulator of type 1 immune response. J Immunol 180, 8073–8082 (2008). [DOI] [PubMed] [Google Scholar]
- 196.Li Z. et al. Interactions of thrombospondins with alpha4beta1 integrin and CD47 differentially modulate T cell behavior. J Cell Biol 157, 509–519, doi: 10.1083/jcb.200109098 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Advani R. et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N Engl J Med 379, 1711–1721, doi: 10.1056/NEJMoa1807315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sikic BI et al. First-in-Human, First-in-Class Phase I Trial of the Anti-CD47 Antibody Hu5F9-G4 in Patients With Advanced Cancers. J Clin Oncol, JCO1802018, doi: 10.1200/JCO.18.02018 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Johnson LDS et al. Targeting CD47 in Sezary syndrome with SIRPalphaFc. Blood Adv 3, 1145–1153, doi: 10.1182/bloodadvances.2018030577 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Haney MS et al. Identification of phagocytosis regulators using magnetic genome-wide CRISPR screens. Nat Genet 50, 1716–1727, doi: 10.1038/s41588-018-0254-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Martincorena I & Campbell PJ Somatic mutation in cancer and normal cells. Science 349, 1483–1489, doi: 10.1126/science.aab4082 (2015). [DOI] [PubMed] [Google Scholar]
- 202.Tang C, Jiang W & Yap TA Efficacy and Toxic Effects of Cancer Immunotherapy Combinations-A Double-edged Sword. JAMA Oncol 4, 1116–1117, doi: 10.1001/jamaoncol.2017.4606 (2018). [DOI] [PubMed] [Google Scholar]
- 203.Sikic BI et al. First-in-Human, First-in-Class Phase I Trial of the Anti-CD47 Antibody Hu5F9-G4 in Patients With Advanced Cancers. J Clin Oncol 37, 946–953, doi: 10.1200/JCO.18.02018 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lehrman EK et al. CD47 Protects Synapses from Excess Microglia-Mediated Pruning during Development. Neuron 100, 120–134 e126, doi: 10.1016/j.neuron.2018.09.017 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hutter G. et al. Microglia are effector cells of CD47-SIRPalpha antiphagocytic axis disruption against glioblastoma. Proc Natl Acad Sci U S A 116, 997–1006, doi: 10.1073/pnas.1721434116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]