

Abstract
DNA polymerase (pol) κ, one of the Y-family polymerases, is thought to function in the error-free translesion DNA synthesis (TLS) opposite the bulky N2-guanyl DNA lesions induced by many carcinogens such as polycyclic aromatic hydrocarbons. We analyzed the biochemical properties of eight pol κ variants positioned in the polymerase core domain, using the recombinant pol κ (residues 1–526) protein and the DNA template containing an N2-CH2(9-anthracenyl)G (N2-AnthG). The truncation R219X was devoid of polymerase activity, and the E419G and Y432S variants showed much lower polymerase activity than wild-type pol κ. In steady-state kinetic analyses, E419G and Y432S displayed 20- to 34-fold decreases in kcat/Km for dCTP insertion opposite G and N2-AnthG compared to wild-type. The L21F, I39T, and D189G variants, as well as E419G and Y432S, displayed 6- to 22-fold decreases in kcat/Km for next-base extension from C paired with N2-AnthG, compared to wild-type pol κ. The defective Y432S variant had a 4- to 5-fold lower DNA-binding affinity than wild-type, while a slightly more efficient S423R variant possessed a 2- to 3-fold higher DNA-binding affinity. These results suggest that R219X abolishes—and the E419G, Y432S, L21F, I39T, and D189G variations substantially impair—TLS ability of pol κ opposite bulky N2-G lesions in the insertion step opposite the lesion and/or the subsequent extension step, raising the possibility that certain non-synonymous pol κ genetic variations translate into individual differences in susceptibility to genotoxic carcinogens.
Keywords: DNA polymerase κ, translesion DNA synthesis, carcinogen, DNA adduct, genetic variation
Graphical Abstract
(INTRODUCTION)
The cellular genome is continuously damaged by endogenous and exogenous agents. The various cellular transactions dealing with DNA damage determine the level of residual DNA lesions and their final biological outcome. The cellular capacity for efficient and faithful repair and replication against DNA damage can be important determinants for genome maintenance and cell survival.1 Cells utilize robust DNA repair systems to remove the potentially harmful DNA lesions, but these are often imperfect and can leave unfixed lesions in DNA. Unrepaired DNA lesions can induce replication errors and/or blockage of DNA polymerases during DNA replication, which can lead to genetic mutation and cell death.2 Cells are equipped with DNA damage tolerance system involving specialized DNA polymerases, mainly belonging to the Y-Family, to carry out translesion DNA synthesis (TLS) at DNA lesions that block replicative polymerases and thus avoid the permanent cell-cycle arrest and cell death in the face of potential mutagenic risks.3 Each of the Y-Family polymerases can perform its unique TLS, differing in both the efficiency (e.g. high to low) and fidelity (e.g. error-free to error-prone) depending on the lesion type, and thus they can play potentially dual roles in both the lesion bypass and mutagenesis in cells.4
The exocyclic N2-amino group of guanine in the DNA minor groove is often attacked by various potential carcinogens, to form guanyl N2-adducts varying in size and shape.5 Bulky N2-G adducts are commonly formed from covalent interaction of activated bulky carcinogens, including the oxidation metabolites of estrogens, heterocyclic amines, polycyclic aromatic hydrocarbons, and tamoxifen catalyzed by cytochrome P450 enzymes.6 These bulky N2-G adducts can frequently induce DNA replication errors and mutations in bacterial and mammalian cells.7 We have previously addressed the biochemical details of replicative bypass across various N2-G adducts by various DNA polymerases. Y-Family DNA polymerases could bypass bulky N2-G adducts that block replicative DNA polymerases, and the lesion bulk could be an important factor for determining the extent of replication blockage and errors (e.g. misincorporation) by each DNA polymerase.8–16
DNA polymerase (pol) κ is one of the four human Y-Family DNA polymerases and is specialized in bypassing bulky minor-groove N2-G adducts very efficiently in an error-free manner.8 Prokaryotic and archaeal orthologs of pol κ, Escherichia coli dinB (pol IV) and Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4), are also able to copy DNA relatively efficiently and faithfully past some N2-G adducts.15,17 However, some subtle differences are apparent in that replicative bypass of benzo[a]pyrene-derived N2-G adducts is error-free by pol κ but is mutagenic by Dpo4,18–20 which might be related to their distinct structural features, for example, an additional N-terminal extension and an enlarged structural gap between the core and little finger domains of pol κ, compared to Dpo4.20,21 Multiple lines of in vitro and in vivo evidence have suggested that a group of bulky N2-G adducts such as N2-benzo[a]pyrene diol epoxide (BPDE)-G, N2-(1-carboxyethyl)-G, N2-(estradiol-6-yl)-G, and α-(N2-deoxyguanosyl) tamoxifen, as well as an acrolein-mediated N2-G interstrand cross-link, might be among the cognate or most-favored lesion substrates of pol κ.3, 22–28 Pol κ-deficient mouse embryonic fibroblast cells show a high sensitivity to both killing and mutagenesis to benzo[a]pyrene23 and an impaired TLS (i.e. decrease in both bypass efficiency and fidelity) across an N2-benzo[a]pyrene diol epoxide-G on a shuttle vector plasmid.27 Pol κ is specifically required for recovery from S-phase checkpoint arrest in mouse cells following exposure to BPDE.28 One haplotype involving a noncoding variation in the POLK gene was significantly associated with a reduced risk of lung cancer,29 although its specific mechanism has not been elucidated. In these respects, it can be postulated that pol κ might play a role in preventing mutagenesis from bulky carcinogen-bound N2-G adducts in cells, and thus the altered functional status of pol κ by genetic variations might modify an individual’s risk of mutation and cancer from exposure to certain chemical carcinogens.
The human POLK gene encodes the pol κ protein consisting of 870 amino acids. Its N-terminal polymerase core region (amino acids 19–526) is known to be indispensable for catalytic activity of pol κ and its ternary complex crystal structure has been determined.30,31 To the present time, a total of ~136 germline variations in POLK gene have been described for human individuals in dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP), but the functional effects of these genetic variations have not been biochemically evaluated yet. Biochemical investigations to assess the direct effects of germline genetic variations on the pol κ function are important for understanding and predicting their biochemical mechanisms and biological consequences prior to or after exploring their clinical association. In the present study, we paid particular attention to the non-synonymous coding POLK gene variations located in the polymerase core domains and have been predicted to have deleterious effects by in silico prediction tools such as SIFT and PolyPhen,32–34 in that they would be likely to affect the catalytic property of pol κ and thus quantitatively and/or qualitatively modify its TLS function. In order to identify and characterize the functional genetic variations of human pol κ, we investigated the biochemical impact of eight selected missense or nonsense genetic variations on the enzymatic properties of human pol κ regarding normal incorporation and bulky N2-G adduct bypass. We performed the experiments with full-length primer extensions, steady-state kinetics, and pol-DNA binding, using the recombinant catalytic core (1–526 amino acids) proteins of wild-type and eight variant human pol κ enzymes with oligonucleotide DNA templates containing an undamaged G or a bulky N2-CH2(9-anthracenyl)G (N2-AnthG) adduct. Here we describe that seven germline genetic variations could alter enzyme function for DNA synthesis upon normal and/or N2-AnthG-adducted DNA substrates, some of which also alter DNA-binding affinity. These findings are discussed in the context of understanding the likely mechanistic and functional consequences of altered TLS with pol κ variants.
EXPERIMENTAL PROCEDURES
Materials.
T4 polynucleotide kinase and dNTPs were purchased from New England Biolabs (Ipswich, MA). [γ−32P]ATP (specific activity 3 × 103 Ci/mmol) was purchased from PerkinElmer Life Sciences (Boston, MA). Bio-spin columns were purchased from Bio-Rad (Hercules, CA). A protease inhibitor cocktail was obtained from Roche Applied Science (Indianapolis, IN). The pBAD-TOPO TA expression kit was from Invitrogen (Carlsbad, CA) and the QuickChange mutagenesis kit was from Stratagene (La Jolla, CA). FPLC columns were purchased from GE Healthcare (Uppsala, Sweden).
DNA Substrates.
24-mer (5′−GCC TCG AGC CAG CCG CAG ACG CAG−3′) and 25-C-mer (5′−GCC TCG AGC CAG CCG CAG ACG CAG C−3′) oligonucleotides were obtained from Midland Certified Reagent Co. (Midland, TX). Two 36-mer oligonucleotides (3′−CGG AGC TCG GTC GGC GTC TGC GTC XCT CCT GCG GCT−5′; X = G or N2-CH2(9-anthracenyl)G (N2-AnthG)) containing a G or N2-AnthG (Figure 1) were prepared as previously described.9, 10 An 18-FAM-mer (5′−(FAM)-AGC CAG CCG CAG ACG CAG−3′; FAM = 6-carboxyfluorescein) was prepared from Bioneer (Daejeon, Korea). 24-mer and 25-mer were used as primers, and 36-mers were used as templates. Primers were 5′ end-labeled using T4 polynucleotide kinase with [γ−32P]ATP and annealed with template to make duplex primer-template DNA substrates.
Figure 1. N2-CH2(9-anthracenyl)G (N2-AnthG).

Structures of N2-AnthG and normal G are shown.
Selection of Human POLK Gene Variations Having Potentially Functional Impact.
We made an effort to search for human POLK gene variations that might be highly probable to alter enzyme function. We first looked for the naturally occurring genetic variations in the coding region of the POLK gene from the public database dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP). We then narrowed these to the likely dysfunctional variations based on three criteria: i) non-synonymous coding variations that result in amino acid change (missense) or premature termination of translation (nonsense), ii) variations located in the polymerase core domain (amino acid residues 19 to 526), and iii) missense variations expected to exert deleterious or damaging effects on protein function by SIFT and Polyphen prediction programs.32–34 We thus selected seven missense and one nonsense genetic variants and performed detailed biochemical analyses using recombinant protein pol κ1−526 proteins purified from Escherichia coli. Current information for the eight POLK gene variations included in this study is summarized in Table 1, based on public databases such as dbSNP, HapMap (http://www.hapmap.org), and 1000 genomes (http://browser.1000genomes.org).
Table 1.
POLK gene variations studied.
| rs IDa | nucleotide change | amino acid change | protein domain | minor allele frequency | predictionb |
|
|---|---|---|---|---|---|---|
| SIFT | PolyPhen-2 | |||||
| rs3104729 | c.61C>T | L21F | N-clasp | 0.006c | damaging | probably damaging |
| rs3094258 | c.116T>C | I39T | N-clasp | N/Ad | damaging | probably damaging |
| rs111689950 | c.566A>G | D189G | Palm | N/A | damaging | probably damaging |
| rs3094265 | c.655A>T | R219Xe | Palm | N/A | N/A | N/A |
| rs3104717 | c.656G>T | R219I | Palm | 0.008c | damaging | probably damaging |
| rs111584802 | c.1256A>G | E419G | PAD | N/A | damaging | probably damaging |
| rs35257416 | c.1267A>C | S423R | PAD | 0.004f | damaging | possibly damaging |
| rs77612491 | c.1295A>C | Y432S | PAD | N/A | damaging | probably damaging |
Construction of Expression Vectors for Catalytic Cores of Eight Pol κ Variants.
The gene fragment covering the catalytic core of wild-type pol κ was obtained by PCR amplification from the vector pAcHLT/HPOLK8 as template using AccuTaq LA DNA polymerase (Sigma Aldrich, St. Louis, MO) with a pair of primers: 5′-GCC ATG GGA CAT CAC CAC CAT CAT CAC ATG GAT AGC ACA AAG GAG-3′ and 5′-CTA TTG TTG GTG TTT CCT GTC-3′. The resulting PCR products of 1.6 kb POLK core were cloned into the vector pBAD-TOPO. The N-terminal leader sequence encoding 12 extra amino acids in the vector was removed by NcoI digestion and the resulting vector was relegated to form the pBAD-wtPOLK1−526 vector, which can generate the pol κ core protein (amino acids 1–526) with an N-terminal MGHHHHHH tag. Following sequence confirmation of the wild type pol κ core gene insert, each of the eight different substitutions in the POLK gene was created with a QuickChange mutagenesis kit with the pBAD-wtPOLK1−526 vector as template. The oligonucleotide primers for introducing the point mutations in POLK were 5′-CTT CTG CTT AGG ATG GGA TTT AAT GAT AAT AAA GCA GGA ATG G-3′ for L21F, 5′-GAA AAT TAA CAA AAC TAT AAT GGA AGC CAC GAA GGG GTC C-3′ for I39T, 5′-GCT GAT TAT GGT CCC AAT TTT ATG GCC ATG AGT CTT-3′ for D189G, 5′-GGC CTG AGG ATA AAA TAA GGT ATT TCA TCA AAA TGG GAA GC-3′ for R219I, 5′-GCC TGA GGA TAA ATG AAG GTA TTT CAT CAA AAT GGG AAG C-3′ for R219X, 5′-GGA AAA GTA TGA GCG TTG GGA GGA CAT TCA GTG AG-3′ for E419G, 5′-CGT TGA GAG GAC ATT CCG TGA GAT AAA TAA AGC GG-3′ for S423R, 5′-CGG AAG AGC AAT CCA GCC TAT GTC AAG AAC TTT GC-3′ for Y432S (and its corresponding antiparallel primer for each mutation). All eight substitutions were confirmed by nucleotide sequence analyses of the constructed vectors.
Expression and Purification of Recombinant Proteins.
The wild type and variant forms of recombinant pol κ core proteins were expressed in E. coli strain TOP10 cells. E. coli TOP10 harboring each vector for the recombinant protein was grown in Luria-Bertani broth supplemented with ampicillin (100 μg ml−1) at 37 °C, with aeration, to an OD600 of 0.6. L-Arabinose was added to 0.02% (w/v), and incubation was continued for 14 h at 16 °C. The cells were harvested by centrifugation and resuspended in lysis buffer (50 mM Tris-HCl, pH 7.4, containing 300 mM NaCl, 10% glycerol (v/v), 5 mM β-mercaptoethanol, 1 mg lysozyme mL−1, and protease inhibitor cocktail (Roche Applied Sciences, Indianapolis, IN)), cooled on ice for 30 min, and then lysed by sonication (12 × 10 s duration with a Branson digital sonifier microtip, (VWR, West Chester, PA), 45% amplitude, with intervening cooling time). The cell lysate was clarified by centrifugation at 4 × 104 × g for 60 min at 4 °C. The resulting supernatant was loaded onto a 1-mL HisTrap HP column and the column was washed with 20 mL of Buffer A (50 mM Tris-HCl (pH 7.4), 300 mM NaCl, 10% glycerol (v/v), and 5 mM β-mercaptoethanol) containing 40 mM imidazole. His-tagged pol κ core fractions (eluted with 400 mM imidazole in buffer A) were collected and diluted 6-fold with buffer B (50 mM Tris-HCl (pH 7.4), 1 mM EDTA, 10% glycerol (v/v), and 5 mM β-mercaptoethanol). Pol κ core was further purified to near homogeneity with the use of a Mono S column and a 50 mM to 2 M NaCl gradient in buffer B. Pol κ core was eluted at ~400 mM NaCl. Protein concentrations were estimated using a Bradford protein assay, and the quality of purified proteins was assessed by SDS-polyacrylamide gel electrophoresis and Coomassie Brilliant Blue staining (Figure S1, Supporting Information).
DNA Polymerization Assays and Steady-State Kinetic Analysis.
Standard DNA polymerase reactions of 8 μL were performed in 50 mM Tris-HCl (pH 7.5) buffer containing 50 mM NaCl, 5 mM dithiothreitol, 100 μg mlL−1 bovine serum albumin (BSA) (w/v), and 10% glycerol (v/v) with 100 nM primer-template substrate at 37 °C. Reactions were initiated by the addition of dNTPs and MgCl2 (5 mM final concentration) to preincubated enzyme/DNA mixtures and terminated with six volumes of a solution of 20 mM EDTA in 95% formamide (v/v). For steady-state kinetic analysis, the primer-template was extended in the presence of 0.1–10 nM pol κ with increasing concentrations of individual dNTPs for 5 or 10 min, when the maximum amount of extension products was ≤20% of total DNA substrates. Products were resolved using a 16% polyacrylamide (w/v) gel electrophoresis system containing 8 M urea and visualized using a Bio-Rad Personal Molecular Imager and Quantity One™ software (Bio-Rad). The product formation rates (as a function of dNTP concentration) were plotted to estimate the kinetic parameters Km and kcat by non-linear regression fitting to the Michaelis-Menten equation using Graph Pad Prism 5.0 software (GraphPad, San Diego, CA). Misinsertion frequency (f) opposite G or N2-AnthG was calculated as f = (kcat/Km)dNTP/(kcat/Km)dCTP .35
Fluorescence Anisotropy Experiments.
The 5 nM 5’−6-carboxyfluorescein-labeled 18-FAM-mer primer annealed with 36-mer template DNA containing an unmodified G or N2-AnthG was incubated with varying concentrations (0 – 500 nM) of pol κ for 20 min at 25 °C. The polymerase-DNA binding reaction was performed in the presence of 50 mM HEPES buffer (pH 7.5) containing 10 mM potassium acetate, 10 mM KCl, 0.1 mM ZnCl2, 5 mM MgCl2, 0.1 mM EDTA, 2 mM β-mercaptoethanol and 0.1 mg mL−1 BSA as modified from a previous study.36 Fluorescence anisotropy was measured with a Cary Eclipse Fluorescence Spectrophotometer (Agilent Technologies, Santa Clara, CA) equipped with a manual polarizer, using excitation and emission wavelengths of 471 and 525 nm, respectively. The anisotropy data were plotted vs. enzyme concentration and fit to a quadratic equation to estimate Kd,DNA using the equation: A = A0 + (Amax –A0)((Dt + Et + Kd) – ((Dt + Et + Kd)2 – (4DtEt))½)/(2Dt), where A is measured anisotropy, A0 is initial anisotropy (DNA alone), Amax is maximum anisotropy, Dt is the concentration of fluorescein-labeled DNA, Et is enzyme concentration, and Kd is the equilibrium dissociation constant for enzyme binding to DNA.
RESULTS
Overall Strategy.
The objective of this study was to characterize the functional germ-line genetic variations in human pol κ. For this, we first chose human POLK genetic variations likely to impair the enzyme function of pol κ, from the dbSNP database. We selected eight functional candidate variations by searching for non-synonymous coding variations that reside in the polymerase core domains and also are predicted to be dysfunctional by prediction programs or a premature translation termination site. Thereafter, we investigated the biochemical effects of eight variations on the catalytic properties of pol κ in both normal and translesion DNA replication across G and N2-AnthG. A set of experiments, including full-length primer extension, steady-state kinetics of nucleotide insertion opposite the lesion and the subsequent extension, and pol-DNA binding assay, was performed sequentially using the recombinant catalytic core (1–526 amino acids) of these pol κ1−526 enzymes and oligonucleotides containing a normal G or a cognate N2-AnthG adduct at a defined site.
Primer Extension Across G and N2-AnthG with All Four dNTPs by Wild-type and Variant Pol κ Enzymes.
In order to assess the possible alterations in activities for normal and translesional DNA synthesis by wild-type and eight pol κ variants, we extended the 24-mer primers annealed to 36-mer templates containing unmodified G or N2-AnthG (Figure 1) at the 25th position in the presence of all four dNTPs using the wild-type and variant pol κ proteins (Figure 2). N2-AnthG was chosen as a model bulky minor-groove N2-G DNA adduct, which was found to be bypassed efficiently and accurately by human pol κ.8 Wild-type pol κ readily extended the primers past G and yielded products up to about 33-mers in proportion to enzyme concentration, and this pattern was also observed with the L21F, I39T, D189G, R219I, and S423R variants. However, the E419G and Y432S variants showed much less activity than wild type pol κ, indicating the severe impairment of polymerase activities due to those substitutions. For TLS polymerization across N2-AnthG, wild-type pol κ readily extended primers past N2-AnthG and yielded substantial products up to about 33-mers, which was similarly observed with the R219I and S423R variants (although the extent of bypass synthesis past N2-AnthG was less than that past G). However, the L21F, I39T, and D189G variants generated substantially reduced amounts of extended products compared to wild-type pol κ, indicating considerable attenuation of their lesion-bypass activities. The E419G and Y432S variants generated only a trace of one-base or almost no extension at N2-AnthG with 1 nM enzyme concentrations, indicating the severe diminution of their lesion-bypass activities. As expected, the R219X variant did not show any activity with either DNA substrate, indicating the total loss of polymerase activity due to its premature translation termination at the codon 219.
Figure 2. Extension of a 32P-labeled 24-mer primer across G or N2-AnthG at position 25 by human wild-type and variant pol κ1−526 enzymes.

Primer (24-mer) was annealed with each of the two different 36-mer templates, containing an unmodified G or N2-AnthG placed at the 25th position from the 3′-end. Reactions were done for 15 min with increasing concentrations of pol κ1−526 (0 − 1 nM) and DNA substrate (100 nM primer/template) as indicated. 32P-labeled 24-mer primer was extended in the presence of all four dNTPs (50 μM each). The reaction products were analyzed by denaturing gel electrophoresis and phosphorimaging. (A) Extension across G. (B) Extension across N2-AnthG.
Primer Extension Beyond an N2-AnthG:C Pair with All Four dNTPs by Wild-type and Variant Pol κ Enzymes.
Primer extension (“next-base”) experiments were done with a 25-C-mer primer containing a 3′-end C positioned opposite an N2-AnthG of a 36-mer template in the presence of all four dNTPs by the wild-type and eight variant pol κ proteins to qualitatively evaluate the subsequent extension activities of pol κ variants from the N2-AnthG:C pair in comparison with wild-type pol κ (Figure 3). The R219I and S423R variants extended the primers past N2-AnthG to a similar extent as wild-type pol κ; however the L21F, I39T, and D189G variants generated extension products less efficiently than wild type pol κ, indicating considerable attenuation of their extension ability following C insertion opposite an N2-AnthG due to amino acid substitution. The E419G and Y432S variants yielded only trace extension products past N2-AnthG, while the truncated R219X showed no extension, as expected.
Figure 3. Extension of a 32P-labeled 25-mer primer beyond an N2-AnthG:C pair by human wild-type and variants pol κ1−526 enzymes.

Primer (25-C-mer) containing a C at the 3’end was annealed with the 36-mer template (36-N2-AnthG-mer) containing an N2-AnthG placed at the 25th position from the 3′-end. Reactions were done for 15 min with increasing concentrations of pol κ1−526 (0 − 1 nM) and DNA substrate (100 nM primer/template) containing an N2-AnthG:C pair as indicated. 32P-labeled 25-mer primer was extended in the presence of all four dNTPs (50 μM each). The reaction products were analyzed by denaturing gel electrophoresis and phosphorimaging.
Steady-State Kinetics of Nucleotide Incorporation Opposite N2-AnthG by the Wild-type and Variant Pol κ Enzymes.
Steady-state kinetic parameters were determined for incorporation of single nucleotides into 24-mer/36-mer duplexes opposite a G or N2-AnthG by pol κ variants in comparison to wild-type (Tables 2 and 3). The specificity constant (kcat/Km) and misinsertion frequency, f = (kcat/Km)incorrect dNTP/(kcat/Km)correct dNTP, were utilized as semi-quantitative indicators for the catalytic efficiency and fidelity of a distributive enzyme, pol κ, as in previous work.37 For correct dCTP insertion opposite undamaged G, the L21F, I39T, D189G, and R219I variants showed kcat/Km values similar to wild-type pol κ, with relative efficiencies of 0.5 to 1.0 (Table 2). Those variants also had relatively low misinsertion frequencies (for incorrect nucleotides), comparable to wild-type pol κ. However, the E419G and Y432S variants showed 22- and 34-fold reductions in kcat/Km values for correct dCTP insertion opposite G compared to wild-type pol κ, which was accompanied by about 4- to 20-fold increase in misinsertion frequencies for dGTP and dATP. Similar trends of results were observed with the N2-AnthG template (Table 3). The E419G and Y432S variations caused about 20-fold reductions in the kcat/Km values for correct dCTP insertion opposite N2-AnthG compared to the wild-type enzyme, as well as 4- to 17-fold increases in misinsertion frequencies for dGTP, dATP, and dTTP opposite N2-AnthG. Noticeably, the L21F, I39T, and D189G variants showed 2- to 3-fold decreases in kcat/Km for correct dCTP insertion opposite N2-AnthG compared to wild-type pol κ, which might in part explain the substantially diminished bypass at N2-AnthG with those variants. On the other hand, the S423R variant displayed 2- to 3-fold increases in kcat/Km for correct dCTP insertion opposite G and N2-AnthG compared to wild-type pol κ, while showing interesting alterations in nucleotide insertion fidelity, 3- to 4-fold decreases in dATP and dGTP misinsertion frequencies opposite N2-AnthG but 3- to 4-fold increases in dTTP and dATP misinsertion frequencies opposite G.
Table 2.
Steady-state kinetic parameters for dNTP incorporation opposite G by human wild-type and variant pol κ1−526 enzymes
| pol κ1−526 | dNTP | Km (μM) | kcat (s−1) | kcat/Km (s−1M−1) | finsa | relative efficiencyb |
|---|---|---|---|---|---|---|
| wild-type | A | 470 ± 70 | 0.0074 ± 0.0003 | 0.016 | 0.00055 | |
| T | 1600 ± 100 | 0.19 ± 0.01 | 0.12 | 0.0041 | ||
| G | 150 ± 9 | 0.029 ± 0.0004 | 0.19 | 0.0066 | ||
| C | 21 ± 3 | 0.61 ± 0.026 | 29 | 1 | 1 | |
| L21F | A | 520 ± 90 | 0.0025 ± 0.0002 | 0.0048 | 0.00024 | |
| T | 630 ± 160 | 0.023 ± 0.002 | 0.037 | 0.0019 | ||
| G | 200 ± 40 | 0.013 ± 0.001 | 0.065 | 0.0033 | ||
| C | 55 ± 9.6 | 1.1 ± 0.06 | 20 | 1 | 0.69 | |
| I39T | A | 820 ± 60 | 0.0078 ± 0.0002 | 0.0095 | 0.00043 | |
| T | 980 ± 400 | 0.059 ± 0.01 | 0.060 | 0.0027 | ||
| G | 250 ± 40 | 0.014 ± 0.001 | 0.056 | 0.0025 | ||
| C | 27 ± 4 | 1.1 ± 0.06 | 22 | 1 | 0.76 | |
| D189G | A | 1000 ± 300 | 0.0067 ± 0.001 | 0.0067 | 0.00048 | |
| T | 1900 ± 310 | 0.033 ± 0.003 | 0.017 | 0.0012 | ||
| G | 740 ± 110 | 0.010 ± 0.001 | 0.014 | 0.0010 | ||
| C | 81 ± 11 | 1.1 ± 0.05 | 14 | 1 | 0.48 | |
| R219I | A | 530 ± 80 | 0.018 ± 0.001 | 0.034 | 0.0012 | |
| T | 840 ± 60 | 0.18 ± 0.01 | 0.21 | 0.0072 | ||
| G | 170 ± 30 | 0.013 ± 0.001 | 0.076 | 0.0026 | ||
| C | 38 ± 3 | 1.1 ± 0.03 | 29 | 1 | 1.0 | |
| E419G | A | 900 ± 140 | 0.0084 ± 0.001 | 0.0093 | 0.0072 | |
| T | 2200 ± 380 | 0.017 ± 0.002 | 0.0077 | 0.0059 | ||
| G | 600 ± 110 | 0.020 ± 0.002 | 0.033 | 0.025 | ||
| C | 150 ± 34 | 0.19 ± 0.018 | 1.3 | 1 | 0.045 | |
| S423R | A | 140 ± 30 | 0.016 ± 0.001 | 0.11 | 0.0023 | |
| T | 530 ± 60 | 0.28 ± 0.01 | 0.53 | 0.011 | ||
| G | 93 ± 20 | 0.016 ± 0.001 | 0.17 | 0.0036 | ||
| C | 17 ± 1.7 | 0.80 ± 0.02 | 47 | 1 | 1.6 | |
| Y432S | A | 1500 ± 200 | 0.014 ± 0.001 | 0.0093 | 0.011 | |
| T | 1200 ± 300 | 0.057 ± 0.003 | 0.0048 | 0.0056 | ||
| G | 500 ± 60 | 0.020 ± 0.001 | 0.040 | 0.047 | ||
| C | 130 ± 19 | 0.11 ± 0.0066 | 0.85 | 1 | 0.029 | |
Misinsertion frequency, calculated by dividing kcat/Km for each dNTP incorporation by the kcat/Km for dCTP incorporation opposite G.
Relative efficiency, calculated by dividing kcat/Km of each pol κ1−526 for dCTP incorporation opposite G by kcat/Km of wild type pol κ1–526 for dCTP incorporation opposite undamaged G.
Table 3.
Steady-state kinetic parameters for dNTP incorporation opposite N2-AnthG by human wild-type and variant pol κ1−526 enzymes
| pol κ1−526 | dNTP | Km (μM) | kcat (s−1) | kcat/Km (s−1M−1) | finsa | relative efficiencyb |
|---|---|---|---|---|---|---|
| wild-type | A | 160 ± 30 | 0.00074 ± 0.00003 | 0.0046 | 0.0014 | |
| T | 360 ± 70 | 0.0015 ± 0.0001 | 0.0042 | 0.0013 | ||
| G | 32 ± 3 | 0.00029 ± 0.000003 | 0.0091 | 0.0028 | ||
| C | 380 ± 30 | 1.2 ± 0.03 | 3.2 | 1 | 0.11 | |
| L21F | A | 400 ± 90 | 0.00020 ± 0.00001 | 0.00050 | 0.00055 | |
| T | 510 ± 90 | 0.0013 ± 0.0001 | 0.0025 | 0.0027 | ||
| G | 73 ± 10 | 0.00023 ± 0.00001 | 0.0032 | 0.0035 | ||
| C | 1100 ± 68 | 1.0 ± 0.03 | 0.91 | 1 | 0.031 | |
| I39T | A | 390 ± 90 | 0.00039 ± 0.00002 | 0.0010 | 0.00059 | |
| T | 330 ± 10 | 0.0012 ± 0.00002 | 0.0036 | 0.0021 | ||
| G | 69 ± 10 | 0.00020 ± 0.00001 | 0.0029 | 0.0017 | ||
| C | 820 ± 230 | 1.4 ± 0.17 | 1.7 | 1 | 0.059 | |
| D189G | A | 450 ± 80 | 0.000067 ± 0.000003 | 0.00015 | 0.00015 | |
| T | 520 ± 120 | 0.0010 ± 0.0001 | 0.0019 | 0.0019 | ||
| G | 510 ± 40 | 0.00067 ± 0.00002 | 0.0013 | 0.0013 | ||
| C | 1000 ± 200 | 1.0 ± 0.06 | 1.0 | 1 | 0.034 | |
| R219I | A | 230 ± 40 | 0.00053 ± 0.00003 | 0.0023 | 0.00068 | |
| T | 330 ± 30 | 0.0024 ± 0.0001 | 0.0073 | 0.0021 | ||
| G | 85 ± 10 | 0.00053 ± 0.00001 | 0.0062 | 0.0018 | ||
| C | 590 ± 180 | 2.0 ± 0.27 | 3.4 | 1 | 0.12 | |
| E419G | A | 760 ± 36 | 0.0025 ± 0.0001 | 0.0033 | 0.024 | |
| T | 1100 ± 220 | 0.0015 ± 0.0002 | 0.0014 | 0.010 | ||
| G | 77 ± 14 | 0.00011 ± 0.00001 | 0.0014 | 0.010 | ||
| C | 1600 ± 290 | 0.23 ± 0.01 | 0.14 | 1 | 0.0048 | |
| S423R | A | 120 ± 10 | 0.00053 ± 0.00002 | 0.0044 | 0.00050 | |
| T | 230 ± 40 | 0.0035 ± 0.0001 | 0.015 | 0.0017 | ||
| G | 27 ± 3 | 0.00018 ± 0.00001 | 0.0067 | 0.00076 | ||
| C | 170 ± 11 | 1.5± 0.03 | 8.8 | 1 | 0.30 | |
| Y432S | A | 600 ± 100 | 0.00057 ± 0.00003 | 0.00095 | 0.0059 | |
| T | 520 ± 50 | 0.0011 ± 0.00003 | 0.0021 | 0.013 | ||
| G | 81 ± 9 | 0.00029 ± 0.00001 | 0.0036 | 0.023 | ||
| C | 320 ± 110 | 0.050 ± 0.004 | 0.16 | 1 | 0.0055 | |
Misinsertion frequency, calculated by dividing kcat/Km for each dNTP incorporation by the kcat/Km for dCTP incorporation opposite N2-AnthG.
Relative efficiency, calculated by dividing kcat/Km of each pol κ1−526 for dCTP incorporation opposite N2-AnthG by kcat/Km of wild type pol κ1−526 for dCTP incorporation opposite undamaged G in Table 2.
Steady-State Kinetics of Next-Base Extension from an N2-AnthG:C Pair by Wild-type and Variant Pol κ Enzymes.
Our finding that the primer extensions past N2-AnthG were considerably retarded with many pol κ variants (Figures 2 and 3) led to further steady-state kinetic analysis for the subsequent extension step following dCTP insertion opposite N2-AnthG by pol κ enzymes. Steady-state kinetic parameters were determined for incorporation of the next complementary nucleotide, dGTP, opposite template C following the adducted N2-AnthG:C base pair into 36-N2-AnthG-mer/25-C-mer template-primer DNA by wild-type pol κ and the variants, in comparison with next-base extension from the normal G:C pair of a 36-G-mer/25-C-mer substrate (Table 4). Concerning the kcat/Km values for next-base extension from the G (or N2-AnthG):C pair, the pol κ variants showed similar trends as with correct nucleotide insertion opposite G (or N2-AnthG) in Tables 2 and 3. For instance, the E419G and Y432S variations caused severe (~20-fold) reductions in kcat/Km values in next-base extension from the N2-AnthG:C pair compared to wild-type pol κ , which was similarly observed in the corresponding cases for dCTP incorporation opposite N2-AnthG with those variants. However, the L21F, I39T, and D189G variants showed large (6- to13-fold) decreases in kcat/Km for next-base extension only from N2-AnthG:C pair (but not from G:C pair) compared to wild-type pol κ, indicating that those variations might significantly impair the subsequent extension step beyond N2-AnthG (but not G), which might in large part explain the exceptionally reduced bypass only at N2-AnthG but not at undamaged G with those variants in Figure 2B.
Table 4.
Steady-state kinetic parameters for next base extension from G (or N2-AnthG): C pair template:primer termini by human wild-type and variant pol κ1−526 enzymes
| base-pair at 3′ primer termini (template:primer) | pol κ1−526 | extension with dGTP (the correct nucleotide opposite next template C) |
||||
|---|---|---|---|---|---|---|
| Km (μM) | kcat (s−1) | kcat/Km (mM−1 s−1) | relative extension efficiencya | |||
| G:C | wild-type | 5.0 ± 0.9 | 0.28 ± 0.02 | 56 | 1 | |
| L21F | 9.0 ± 1.3 | 0.30 ± 0.02 | 33 | 0.59 | ||
| I39T | 6.0 ± 1.3 | 0.16 ± 0.01 | 27 | 0.48 | ||
| D189G | 8.2 ± 0.7 | 0.40 ± 0.01 | 49 | 0.88 | ||
| R219I | 6.0 ± 1.0 | 0.50 ± 0.03 | 83 | 1.5 | ||
| E419G | 23 ± 7 | 0.30 ± 0.03 | 13 | 0.23 | ||
| S423R | 1.4 ± 0.2 | 0.27 ± 0.01 | 190 | 3.4 | ||
| Y432S | 25 ± 4 | 0.080 ± 0.004 | 3.2 | 0.057 | ||
| N2-AnthG:C | wild-type | 14 ± 2 | 0.54 ± 0.02 | 39 | 0.70 | |
| L21F | 65 ± 18 | 0.41 ± 0.06 | 6.3 | 0.11 | ||
| I39T | 97 ± 12 | 0.60 ± 0.03 | 6.2 | 0.11 | ||
| D189G | 200 ± 10 | 0.60 ± 0.02 | 3.0 | 0.054 | ||
| R219I | 7.3 ± 3.0 | 0.20 ± 0.02 | 27 | 0.48 | ||
| E419G | 27 ± 6 | 0.048 ± 0.004 | 1.8 | 0.032 | ||
| S423R | 3.7 ± 0.4 | 0.12 ± 0.03 | 32 | 0.57 | ||
| Y432S | 24 ± 3 | 0.051 ± 0.003 | 2.1 | 0.038 | ||
Relative extension efficiency, calculated by dividing kcat/Km of each pol κ1−526 for each dGTP incorporation opposite the next template C from the G (or N2-AnthG):C pair by kcat/Km of wild type pol κ1−526 for dGTP incorporation opposite the next template C from the undamaged G:C pair.
Binding of the Wild-type and Variant Pol κ Enzymes to DNA Containing G or N2-AnthG.
The effects of eight genetic variations on DNA binding affinity of pol κ were evaluated using fluorescence anisotropy experiments. The equilibrium dissociation constants (Kd,DNA) of the wild type and variant pols κ were estimated by fitting the anisotropy of fluorescein-labeled primer-template DNA (18-FAM-mer/36-mer) as a function of increasing concentration of pol κ to a quadratic equation (Table 5). The Kd,DNA of the wild-type pol κ for unmodified DNA was similar to that for N2-AnthG-adducted DNA, indicating that DNA-binding affinity of pol κ is not affected by an N2-CH2(9-anthracenyl) group at G, which was also similarly observed in most of variant pol κ enzymes. The Kd,DNA values of Y432S were 4- to 5-fold higher for both N2-AnthG-adducted and unmodified DNA substrates than for wild-type pol κ, indicating a relatively large decrease in DNA-binding affinity of pol κ by the Y432S substitution. In contrast, the S423R variant showed 2- to 3-fold increases in binding affinity for DNA substrates, which might partly be related to slight increases in relative efficiencies for correct nucleotide insertion opposite G and N2-AnthG with this variant compared to wild-type pol κ in Tables 2 and 3. It was also observed that the L21F variation caused a slight (2-fold) reduction in the Kd,DNA of pol κ only for unmodified DNA but not for N2-AnthG-adducted DNA (compared to wild-type). However, this was not a large change, which might in part explain a slightly less diminution of relative efficiency in dCTP insertion opposite G compared to N2-AnthG with this variant in Tables 2 and 3.
Table 5.
Binding affinities of wild-type and variant pol κ enzymes for DNA substrates
| pol κ1−526 |
Kd (nM) |
|
|---|---|---|
| 18-FAM-mer/36-G-mer | 18-FAM-mer/36-N2-AnthG-mer | |
| wild-type | 50 ± 12 | 52 ± 16 |
| L21F | 27 ± 5 | 60 ± 9 |
| I39T | 42 ± 8 | 45 ± 15 |
| D189G | 58 ± 15 | 54 ± 11 |
| R219I | 50 ± 12 | 50 ± 8 |
| E419G | 40 ± 9 | 44 ± 9 |
| S423R | 21 ± 5 | 19 ± 3 |
| Y432S | 270 ± 60 | 220 ± 30 |
DISCUSSION
In this work, we have investigated the biochemical properties of eight non-synonymous coding variants of human pol κ, a key player in the error-free TLS of minor-groove DNA adducts. Seven missense and a nonsense variations were selected for study because they are presumed to possibly cause dysfunctional impacts on pol κ on the basis of their locations, types, and predicted effects. Our biochemical data reveal that the E419G and Y432S variations severely impair both the efficiency and fidelity of pol κ for both normal and translesion DNA synthesis across G and N2-AnthG, while the L21F, I39T, and D189G variations considerably impair pol κ efficiency chiefly for translesion DNA synthesis (particularly in the subsequent extension step) past N2-AnthG. As anticipated, the truncated R219X variant, which loses large parts of the palm, thumb, and PAD domains, lacked polymerase activity. Two variations positioned in the PAD domain were also found to substantially influence DNA binding of pol κ but in opposite ways, in that the Y432S variation reduces DNA-binding affinity by 4- to 5-fold, while S423R increases it by 2- to 3-fold.
This is the first study, to our knowledge, to analyze functional alterations in germline genetic variants of human pol κ using biochemical approaches. Seven among the eight studied POLK genetic variations were found to at least partially affect the enzymatic function of pol κ in translesion DNA synthesis and/or DNA binding. These functional genetic variations seem to be rare, in that their minor allele frequencies are < 1% or not available yet in human populations (beyond their reported occurrence in a human) (Table 1). Recent articles in the literature propose the possible relevance of rare genetic variants to human disease risks. The rare genetic variations that can be really abundant in human populations and more likely to be functional than common variations, have been proposed to be potential sources of unexplained heritability in complex human diseases, while only a small fraction of the known disease heritability have been explained by common genetic variants in genome-wide association studies.38–41 Even spontaneous new (de novo) germline mutations, i.e. the most extreme form of rare genetic variations, have been implicated in rare and common forms of neurodevelopmental diseases.42 Three somatic POLK mutations, which might result in a missense change at codon 205 or abnormal splicing, were reported in prostate tumors, although their functional impacts are not revealed yet.43 Rare variants are being increasingly identified with technological progress and application of the high-throughput next-generation sequencing. The number of non-synonymous POLK gene variations reported in dbSNP increased from 21 in 2008 into 58 in 2011 and 91 in 2013, all of which are likely to be rare because their minor allele frequencies are < 1% or unavailable. Our biochemical approach is a good initial step to identify the functional pol κ variants and reveal their mechanistic insights, although we have focused on only eight selected “probably functional” coding variants in this study. Nine additional non-synonymous coding POLK variations, which are putatively deleterious, have been added in dbSNP since our study began and are worth exploring further.
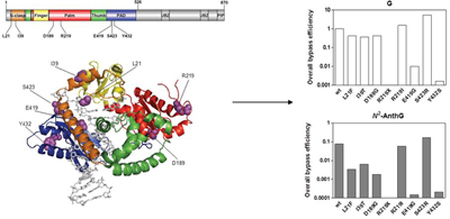
Seven functional pol κ variants identified in this study can be categorized into four types according to the changes of polymerase efficiencies represented by overall bypass efficiencies44 across G and N2-AnthG (Fig. 4), which are the variant devoid of activity (R219X), variants severely impaired of both normal and TLS efficiency (E419G and Y432S), variants impaired mainly in TLS efficiency (L21F, I39T, and D189G), and the variant showing slight enhancement of polymerase efficiency (S423R). We also note that the values for overall bypass efficiencies with each pol κ variants are in good agreement with the qualitative results of full-length primer extension experiments in Fig 2. The fidelity of nucleotide insertion opposite G and N2-AnthG did not seems to be affected in most variants but was severely impaired with the E419G and Y432S variants (Tables 2 and 3), due to a greater reduction of insertion efficiency for correct dCTP than for the other nucleotides with those defective variants.
Figure 4. Overall bypass efficiencies of human variant pol κ1−526 enzymes for G and N2-AnthG compared to the wild-type.

The overall bypass efficiency was calculated by multiplying the relative efficiency for dCTP insertion opposite G (or N2-AnthG) and the relative efficiency for the subsequent next-base extension by each pol κ1−526, based on the insertion and extension kinetic parameters in Tables 2–4 as described previously.44
Structural views of pol κ may be helpful in understanding the mechanistic aspects of genetic variations (Fig. 5). Pol κ possesses the classic polymerase domains (finger, palm, and thumb), the Y-family specific “polymerase associated domain (PAD)” (also known as little finger) which makes the majority of DNA contacts, and the unique N-clasp domain which fully encircles DNA to allow the efficient extension of mismatched or distorted primer termini.30 In the crystal structures with DNA containing bulky N2-G adducts at the post-insertion position, Dpo4, which is an archaeal ortholog of pol κ but lacks the N-clasp domain, accommodates an N2-CH2(2-naphthyl) group of G in the syn (or anti in a disordered nonproductive conformation) orientation between the template DNA and the little finger domain in a productive conformation,15 while flipping/looping-out bulkier N2-BPDE-G extrahelically into the gap between the little finger and core domains in productive conformation (or intercalating the benzo[a]pyrene ring into the DNA helix in a nonproductive conformation).20 Computational modeling of human pol κ postulates a good accommodation of an N2-BPDE-G in the minor-groove side and a structural role of the N-clasp domain in near error-free TLS of N2-BPDE-G.45 In these circumstances, it can be postulated that pol κ might accommodate an N2-AnthG, which is smaller than N2-BPDE-G but bulkier than N2-CH2(2-naphthyl)G, well in the active site by means of N-clasp and PAD domains. We can also assume that non-synonymous POLK genetic variations in the N-clasp and PAD domains as well as in classic polymerase domains seem to be likely to affect the lesion bypass capability of pol κ from the available structural information.30,31,46
Figure 5. Locations of genetic pol κ variations.

Structure of human pol κ21−517 (PDB code, 2OH2) bound to primer/template DNA and incoming nucleotide is shown using Pymol. Pol κ21−517 is shown in cartoon ribbons, and the primer/template DNA and dNTP are shown in gray sticks. The N-clasp, finger, palm, thumb, and PAD domains are colored orange, yellow, red, green, and blue, respectively. The amino acid residues (in purple spheres) of genetic pol κ variants are indicated. The structural domains of pol κ are shown in the upper schematic diagram using DOG (version 2.0),54 where locations of amino acids related to eight studied variations are indicated.
It is noteworthy that L21F and I39T variations, located in the N-clasp domain, caused considerable catalytic impairment of N2-AnthG bypass, particularly in the subsequent extension step from C inserted opposite N2-AnthG, which might be due to local perturbation at the N-clasp domain (proposed to tolerate distortions at the primer terminus).30,45 Interestingly the D189G variation at a loop between αE helix and β5 strand of the catalytic palm domain also seemed to impair the subsequent extension step of lesion bypass, which might come from the local structural alteration of that loop positioned near the 3′ terminal nucleotide of the primer at the post-insertion position. It is also remarkable that two E419G and Y432S variations (though not S423R) positioned at the PAD domain caused severe reductions in general polymerase activity of pol κ, indicating the necessity of an intact PAD domain to hold DNA near the active site for full catalytic efficiency of pol κ. A strong catalytic defect of the E419G variant, in spite of its normal DNA binding affinity, seems to be related to the loss of a long side chain with negative charge in a Glu residue lying in the template DNA side across the active site residues, which might perhaps induce a conformational deformation in the active site cleft. Differing DNA binding affinities between the Y432S and S423R variants might in part explain the opposite functional changes in these two variants. A severe catalytic defect of the Y432S variant, which loses an aromatic benzene ring in a Tyr residue at the solvent-facing αQ helix supporting the four-stranded β sheet of the PAD domain, is likely to be attributable to a moderate reduction in DNA binding affinity, as well as possible structural destabilization of the PAD domain. In contrast, a slight increase in polymerase efficiency with the S423R variant might be related to its higher DNA binding affinity, possibly due to gain of a positively charged Arg residue near the phosphosugar backbone at 5′ unpaired region of template DNA.
How would these functional POLK genetic variations influence TLS process across bulky N2-G DNA adducts relating to mutagenesis in cells? Eukaryotic TLS processing of DNA lesions is supposed to involve polymerase switching between replicative and TLS polymerases, mediated by the protein-protein interactions including PCNA and the additional post-translational modifications such as ubiquitination at a site of DNA damage, where a set of Y-family polymerases are recruited and utilized.47, 48 Accordingly, the mutation rate and mutational spectrum of a specific DNA lesion in cells might be influenced by overall kinetic behavior of various DNA polymerases, mainly TLS polymerases, utilized for the lesion. When encountering a bulky N2-G adduct such as N2-BPDE-G during DNA replication, normal cells might best utilize pol κ, which is most kinetically favored (i.e. most efficient) and, moreover, most accurate at bulky N2-G lesion bypass among Y-family pols,8 and thus enable largely error-free TLS. If a cell possesses only kinetically defective pol κ variants, e.g., E419G and Y432S, we can speculate that other less accurate TLS pols (e.g., pols η and ι) can be more frequently employed at bulky N2-G lesions, which would increase error-prone TLS events and thus result in more mutations in cells. Several observations support this view, especially a possible alternative role of pol η in pol κ-deficient cells: pol κ-deficient mouse embryonic fibroblasts show markedly elevated G to T transversions after benzo[a]pyrene exposure23 and preferential dATP misinsertion opposite a site-specific N2-BPDE-G in a shuttle vector,27 which is in good agreement with the proclivity of pol η to preferentially misincorporate dATP opposite N2-BPDE-G in vitro.19,49 Conversely, if the cell has inherently more efficient pol κ variants with similar or higher accuracy, e.g. S423R, and utilize it correctly at bulky N2-G damage sites, more error-free TLS events at bulky N2-G lesions would result, with and fewer mutations in cells. It is also worth examining other non-synonymous coding variations located in protein interaction domains of pol κ to bind other proteins such as PCNA, ubiquitin, and REV1, in that they might alter cellular TLS events such as polymerase switching and coordination. It is also plausible that noncoding functional variations in regulatory regions for pol κ might be able to alter pol κ expression levels in cells and thus influence TLS events, although this aspect was not examined here. Pol κ has been also suggested to be involved in nucleotide excision repair,50,51 homologous recombination repair,52 and replication checkpoint response,53 all of which have a role in preventing DNA damage-induced mutagenesis, and assuring the maintenance of genome stability. Collectively, it is likely that functional genetic variations (either coding or noncoding) of pol κ might alter the multiple cellular pathways of TLS, DNA repair, and checkpoint control of genotoxic stress in cells, which might affect in turn overall genomic stability and thus modify an individual’s risk to cancer. In these aspects, it is desirable to perform further studies to verify the in vivo impact of those functional pol κ variants in both cellular and organismal contexts.
In conclusion, our results suggest that seven germline genetic variations in human POLK gene may either hamper or facilitate the error-free TLS capability of pol κ across bulky N2-G DNA lesions, generating different mutation phenotypes in the affected individuals, i.e. predisposing or protecting from mutagenesis following exposure to certain genotoxic carcinogens, e.g. polycyclic aromatic hydrocarbons. The identification of dysfunctional genetic variations for human pol κ in this work may provide insight into our understanding of individual differences in mutation phenotypes to specific mutagens and related disease risks in human populations and in future investigations for deciphering genetic markers on various TLS DNA polymerases for individual mutation risks related to cancer.
Supplementary Material
Acknowledgments
Funding Sources. This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (Grant 2012R1A1A2042391) (to J.-Y. C.), Korea Healthcare technology R & D project Grant, Ministry for Health, Welfare, and Family Affairs, Republic of Korea (A084187) (to J.-Y. C.), Samsung Biomedical Research Institute grant, #SBRI GL1B33111 (to J.-Y. C.), and United States Public Health Service (USPHS) Grant R01 ES010375 (to F. P. G.).
Abbreviations:
- A
adenine
- BPDE
benzo[a]pyrene diol epoxide
- BSA
bovine serum albumin
- C
cytosine
- dNTP
deoxynucleoside triphosphate
- EDTA
ethylenediaminetetraacetic acid
- G
guanine
- N2-Anth
N2-CH2(9-anthracenyl)
- PCR
polymerase chain reaction
- pol
DNA polymerase
- SDS
sodium dodecyl sulfate
- T
thymine
- TLS
translesion DNA synthesis
Footnotes
Supporting Information. Analysis of human pol κ1−526 wild-type and variant proteins by SDS-polyacrylamide gel electrophoresis (Figure S1). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, and Ellenberger T (2006) DNA Repair And Mutagenesis. 2nd ed., American Society for Microbiology Press, Washington, D. C. [Google Scholar]
- (2).Guengerich FP (2006) Interactions of carcinogen-bound DNA with individual DNA polymerases. Chem. Rev. 106, 420–452. [DOI] [PubMed] [Google Scholar]
- (3).Waters LS, Minesinger BK, Wiltrout ME, D’Souza S, Woodruff RV, and Walker GC (2009) Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 73, 134–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Choi J-Y, Eoff RE, and Guengerich FP (2011) Bypass DNA polymerases, In Chemical Carcinogenesis (Penning TM, Ed.) pp 345–373, Humana Press, New York, NY. [Google Scholar]
- (5).Geacintov NE, and Broyde S (2010) The Chemical Biology of DNA Damage. Wiley-VCH Verlag GmbH, Weinheim, Germany. [Google Scholar]
- (6).Rendic S, and Guengerich FP (2012) Contributions of human enzymes in carcinogen metabolism. Chem. Res. Toxicol. 25, 1316–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Basu AK (2011) Mutagenesis: The outcome of faulty replication of DNA, In Chemical Carcinogenesis (Penning TM, Ed.) pp 375–399, Humana Press, New York, NY. [Google Scholar]
- (8).Choi J-Y, Angel KC, and Guengerich FP (2006) Translesion synthesis across bulky N2-alkyl guanine DNA adducts by human DNA polymerase κ. J. Biol. Chem. 281, 21062–21072. [DOI] [PubMed] [Google Scholar]
- (9).Choi J-Y, and Guengerich FP (2004) Analysis of the effect of bulk at N2-alkylguanine DNA adducts on catalytic efficiency and fidelity of the processive DNA polymerases bacteriophage T7 exonuclease- and HIV-1 reverse transcriptase. J. Biol. Chem. 279, 19217–19229. [DOI] [PubMed] [Google Scholar]
- (10).Choi J-Y, and Guengerich FP (2005) Adduct size limits efficient and error-free bypass across bulky N2-guanine DNA lesions by human DNA polymerase η. J. Mol. Biol. 352, 72–90. [DOI] [PubMed] [Google Scholar]
- (11).Choi J-Y, and Guengerich FP (2006) Kinetic evidence for inefficient and error-prone bypass across bulky N2-guanine DNA adducts by human DNA polymerase ι. J. Biol. Chem, 281, 12315–12324. [DOI] [PubMed] [Google Scholar]
- (12).Choi J-Y, and Guengerich FP (2008) Kinetic analysis of translesion synthesis opposite bulky N2- and O6-alkylguanine DNA adducts by human DNA polymerase REV1. J. Biol. Chem. 283, 23645–23655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Choi J-Y, Stover JS, Angel KC, Chowdhury G, Rizzo CJ, and Guengerich FP (2006) Biochemical basis of genotoxicity of heterocyclic arylamine food mutagens: Human DNA polymerase η selectively produces a two-base deletion in copying the N2-guanyl adduct of 2-amino-3-methylimidazo[4,5-f]quinoline but not the C8 adduct at the NarI G3 site. J. Biol. Chem. 281, 25297–25306. [DOI] [PubMed] [Google Scholar]
- (14).Lim S, Song I, Guengerich FP, and Choi J-Y (2012) Effects of N2-alkylguanine, O6-alkylguanine, and abasic lesions on DNA binding and bypass synthesis by the euryarchaeal B-family DNA polymerase vent (exo-). Chem. Res. Toxicol. 25, 1699–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhang H, Eoff RL, Kozekov ID, Rizzo CJ, Egli M, and Guengerich FP (2009) Versatility of Y-family Sulfolobus solfataricus DNA polymerase Dpo4 in translesion synthesis past bulky N2-alkylguanine adducts. J. Biol. Chem. 284, 3563–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Choi J-Y, Zang H, Angel KC, Kozekov ID, Goodenough AK, Rizzo CJ, and Guengerich FP (2006) Translesion synthesis across 1,N2-ethenoguanine by human DNA polymerases. Chem. Res. Toxicol. 19, 879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Jarosz DF, Godoy VG, Delaney JC, Essigmann JM, and Walker GC (2006) A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature 439, 225–228. [DOI] [PubMed] [Google Scholar]
- (18).Perlow-Poehnelt RA, Likhterov I, Scicchitano DA, Geacintov NE, and Broyde S (2004) The spacious active site of a Y-family DNA polymerase facilitates promiscuous nucleotide incorporation opposite a bulky carcinogen-DNA adduct: elucidating the structure-function relationship through experimental and computational approaches. J. Biol. Chem, 279, 36951–36961. [DOI] [PubMed] [Google Scholar]
- (19).Rechkoblit O, Zhang Y, Guo D, Wang Z, Amin S, Krzeminsky J, Louneva N, and Geacintov NE (2002) trans-Lesion synthesis past bulky benzo[a]pyrene diol epoxide N2-dG and N6-dA lesions catalyzed by DNA bypass polymerases. J. Biol. Chem. 277, 30488–30494. [DOI] [PubMed] [Google Scholar]
- (20).Bauer J, Xing G, Yagi H, Sayer JM, Jerina DM, and Ling H (2007) A structural gap in Dpo4 supports mutagenic bypass of a major benzo[a]pyrene dG adduct in DNA through template misalignment. Proc. Natl. Acad. Sci. U.S.A. 104, 14905–14910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Liu Y, Yang Y, Tang TS, Zhang H, Wang Z, Friedberg E, Yang W, and Guo C (2014) Variants of mouse DNA polymerase κ reveal a mechanism of efficient and accurate translesion synthesis past a benzo[a]pyrene dG adduct. Proc. Natl. Acad. Sci. U.S.A, 111, 1789–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Minko IG, Harbut MB, Kozekov ID, Kozekova A, Jakobs PM, Olson SB, Moses RE, Harris TM, Rizzo CJ, and Lloyd RS (2008) Role for DNA polymerase κ in the processing of N2-N2-guanine interstrand cross-links. J. Biol. Chem. 283, 17075–17082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ogi T, Shinkai Y, Tanaka K, and Ohmori H (2002) Pol κ protects mammalian cells against the lethal and mutagenic effects of benzo[a]pyrene. Proc. Natl. Acad. Sci. U.S.A. 99, 15548–15553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Poon K, Itoh S, Suzuki N, Laxmi YR, Yoshizawa I, and Shibutani S (2008) Miscoding properties of 6α- and 6β-diastereoisomers of the N2-(estradiol-6-yl)-2-deoxyguanosine DNA adduct by Y-family human DNA polymerases. Biochemistry 47, 6695–6701. [DOI] [PubMed] [Google Scholar]
- (25).Yasui M, Suzuki N, Laxmi YR, and Shibutani S (2006) Translesion synthesis past tamoxifen-derived DNA adducts by human DNA polymerases η and κ. Biochemistry 45, 12167–12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Yuan B, Cao H, Jiang Y, Hong H, and Wang Y (2008) Efficient and accurate bypass of N2-(1-carboxyethyl)-2´-deoxyguanosine by DinB DNA polymerase in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 105, 8679–8684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Avkin S, Goldsmith M, Velasco-Miguel S, Geacintov N, Friedberg EC, and Livneh Z (2004) Quantitative analysis of translesion DNA synthesis across a benzo[a]pyrene-guanine adduct in mammalian cells: the role of DNA polymerase κ. J. Biol. Chem. 279, 53298–53305. [DOI] [PubMed] [Google Scholar]
- (28).Bi X, Slater DM, Ohmori H, and Vaziri C (2005) DNA polymerase κ is specifically required for recovery from the benzo[a]pyrene-dihydrodiol epoxide (BPDE)-induced S-phase checkpoint. J. Biol. Chem, 280, 22343–22355. [DOI] [PubMed] [Google Scholar]
- (29).Michiels S, Danoy P, Dessen P, Bera A, Boulet T, Bouchardy C, Lathrop M, Sarasin A, and Benhamou S (2007) Polymorphism discovery in 62 DNA repair genes and haplotype associations with risks for lung and head and neck cancers. Carcinogenesis 28, 1731–1739. [DOI] [PubMed] [Google Scholar]
- (30).Lone S, Townson SA, Uljon SN, Johnson RE, Brahma A, Nair DT, Prakash S, Prakash L, and Aggarwal AK (2007) Human DNA polymerase κ encircles DNA: implications for mismatch extension and lesion bypass. Mol. Cell 25, 601–614. [DOI] [PubMed] [Google Scholar]
- (31).Uljon SN, Johnson RE, Edwards TA, Prakash S, Prakash L, and Aggarwal AK (2004) Crystal structure of the catalytic core of human DNA polymerase κ. Structure 12, 1395–1404. [DOI] [PubMed] [Google Scholar]
- (32).Ng PC, and Henikoff S (2001) Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ramensky V, Bork P, and Sunyaev S (2002) Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 30, 3894–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, and Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Boosalis MS, Petruska J, and Goodman MF (1987) DNA polymerase insertion fidelity: gel assay for site-specific kinetics. J. Biol. Chem. 262, 14689–14696. [PubMed] [Google Scholar]
- (36).Ketkar A, Zafar MK, Maddukuri L, Yamanaka K, Banerjee S, Egli M, Choi JY, Lloyd RS, and Eoff RL (2013) Leukotriene biosynthesis inhibitor MK886 impedes DNA polymerase activity. Chem. Res. Toxicol. 26, 221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Choi J-Y, Lim S, Kim EJ, Jo A, and Guengerich FP (2010) Translesion synthesis across abasic lesions by human B-family and Y-family DNA polymerases α, δ, η, ι, κ, and REV1. J. Mol. Biol. 404, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, and Visscher PM (2009) Finding the missing heritability of complex diseases. Nature 461, 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Nelson MR, Wegmann D, Ehm MG, Kessner D, St Jean P, Verzilli C, Shen J, Tang Z, Bacanu SA, Fraser D, Warren L, Aponte J, Zawistowski M, Liu X, Zhang H, Zhang Y, Li J, Li Y, Li L, Woollard P, Topp S, Hall MD, Nangle K, Wang J, Abecasis G, Cardon LR, Zollner S, Whittaker JC, Chissoe SL, Novembre J, and Mooser V (2012) An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science 337, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Tennessen JA, Bigham AW, O’Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, Kang HM, Jordan D, Leal SM, Gabriel S, Rieder MJ, Abecasis G, Altshuler D, Nickerson DA, Boerwinkle E, Sunyaev S, Bustamante CD, Bamshad MJ, Akey JM, Broad GO, Seattle GO, and Project NES (2012) Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zhu Q, Ge D, Maia JM, Zhu M, Petrovski S, Dickson SP, Heinzen EL, Shianna KV, and Goldstein DB (2011) A genome-wide comparison of the functional properties of rare and common genetic variants in humans. Am. J. Hum. Genet. 88, 458–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Veltman JA, and Brunner HG (2012) De novo mutations in human genetic disease. Nat. Rev. Genet. 13, 565–575. [DOI] [PubMed] [Google Scholar]
- (43).Makridakis NM, Caldas Ferraz LF, and Reichardt JK (2009) Genomic analysis of cancer tissue reveals that somatic mutations commonly occur in a specific motif. Hum. Mutat. 30, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Levine RL, Miller H, Grollman A, Ohashi E, Ohmori H, Masutani C, Hanaoka F, and Moriya M (2001) Translesion DNA synthesis catalyzed by human pol η and pol κ across 1,N6-ethenodeoxyadenosine. J. Biol. Chem. 276, 18717–18721. [DOI] [PubMed] [Google Scholar]
- (45).Jia L, Geacintov NE, and Broyde S (2008) The N-clasp of human DNA polymerase κ promotes blockage or error-free bypass of adenine- or guanine-benzo[a]pyrenyl lesions. Nucleic Acids Res. 36, 6571–6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Irimia A, Eoff RL, Guengerich FP, and Egli M (2009) Structural and functional elucidation of the mechanism promoting error-prone synthesis by human DNA polymerase κ opposite the 7,8-dihydro-8-oxo-2’-deoxyguanosine adduct. J. Biol. Chem. 284, 22467–22480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Goodman MF, and Woodgate R (2013) Translesion DNA polymerases. Cold Spring Harb. Perspect. Biol, 5, a010363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Sale JE, Lehmann AR, and Woodgate R (2012) Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 13, 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Zhang Y, Yuan F, Wu X, Rechkoblit O, Taylor JS, Geacintov NE, and Wang Z (2000) Error-prone lesion bypass by human DNA polymerase η. Nucleic Acids Res. 28, 4717–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Ogi T, and Lehmann AR (2006) The Y-family DNA polymerase κ (pol κ) functions in mammalian nucleotide-excision repair. Nat. Cell Biol. 8, 640–642. [DOI] [PubMed] [Google Scholar]
- (51).Ogi T, Limsirichaikul S, Overmeer RM, Volker M, Takenaka K, Cloney R, Nakazawa Y, Niimi A, Miki Y, Jaspers NG, Mullenders LH, Yamashita S, Fousteri MI, and Lehmann AR (2010) Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol. Cell 37, 714–727. [DOI] [PubMed] [Google Scholar]
- (52).Sebesta M, Burkovics P, Juhasz S, Zhang S, Szabo JE, Lee MY, Haracska L, and Krejci L (2013) Role of PCNA and TLS polymerases in D-loop extension during homologous recombination in humans. DNA Repair (Amst.) 12, 691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Betous R, Pillaire MJ, Pierini L, van der Laan S, Recolin B, Ohl-Seguy E, Guo C, Niimi N, Gruz P, Nohmi T, Friedberg E, Cazaux C, Maiorano D, and Hoffmann JS (2013) DNA polymerase κ-dependent DNA synthesis at stalled replication forks is important for CHK1 activation. EMBO J. 32, 2172–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Ren J, Wen L, Gao X, Jin C, Xue Y, and Yao X (2009) DOG 1.0: illustrator of protein domain structures. Cell Res. 19, 271–273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.