

Abstract
Fermentation, also known as aging, is vital for enhancing the quality of flue‐cured tobacco leaves (FTLs). Aged FTLs demonstrate high‐quality sensory characteristics, while unaged FTLs do not. Microbes play important roles in the FTL fermentation process. However, the eukaryotic microbial community diversity is poorly understood, as are microbial associations within FTLs. We aimed to characterize and compare the microbiota associated with two important categories, fresh and strong flavor style FTLs, and to reveal correlations between the microbial taxa within them. Based on 16S and 18S rRNA Illumina MiSeq sequencing, the community richness and diversity of prokaryotes were almost as high as that of eukaryotes. The dominant microbes of FTLs belonged to seven genera, including Pseudomonas, Bacillus, Methylobacterium, Acinetobacter, Sphingomonas, Neophaeosphaeria, and Cladosporium, of the Proteobacteria, Firmicutes, and Ascomycota phyla. According to partial least square discriminant analysis (PLS‐DA), Xanthomonas, Franconibacter, Massilia, Quadrisphaera, Staphylococcus, Cladosporium, Lodderomyces, Symmetrospora, Golovinomyces, and Dioszegia were significantly positively correlated with fresh flavor style FTLs, while Xenophilus, Fusarium, unclassified Ustilaginaceae, Tilletiopsis, Cryphonectria, Colletotrichum, and Cyanodermella were significantly positively correlated with strong flavor style FTLs. Network analysis identified seven hubs, Aureimonas, Kocuria, Massilia, Brachybacterium, Clostridium, Dietzia, and Vishniacozyma, that may play important roles in FTL ecosystem stability, which may be destroyed by Myrmecridium. FTL microbiota was found to be correlated with flavor style. Species present in lower numbers than the dominant microbes might be used as microbial markers to discriminate different flavor style samples and to stabilize FTL microbial communities. This research advances our understanding of FTL microbiota and describes a means of discriminating between fresh and strong flavor FTLs based on their respective stable microbiota.
Keywords: co‐occurrence patterns, flue‐cured tobacco leaf, Illumina MiSeq sequencing, microbial community, network analysis, partial least squares discriminant analysis
In this study, we use 16S and 18S rRNA Illumina MiSeq sequencing data to characterize and compare the microbiota associated with fresh and strong flavor style flue‐cured tobacco leaves and reveal correlations between the microbial taxa within them. Flue‐cured tobacco leaf microbiota are correlated with flavor style, and species present at low numbers within the microbial communities can be used as microbial markers to discriminate between samples of different flavor styles and to stabilize flue‐cured tobacco leaf microbial communities.

1. INTRODUCTION
Tobacco (Nicotiana tabacum L.) is one of the largest economic nonfood crops in the world. In China, the most important type of tobacco is the flue‐cured tobacco (Su et al., 2011; Zhao et al., 2007). The flavor of flue‐cured tobacco leaf (FTL) changes throughout the process of fermentation, gradually aging over long periods (typically at least 12 months). The aging process results in FTL of a high commercial quality and causes a change in color to a darker yellow, elimination of harmful odors, degradation of harmful substances, reduction of incentive odor, and development of tobacco‐specific flavors (Yu & Gong, 2009). According to flavor styles, Chinese aged FTL could be traditionally divided into three categories: fresh flavor style, middle flavor style, and strong flavor style.
Microbes have been found to play important roles during the FTL fermentation process (Reid, McKinstry, & Haley, 1937), which include the production of tobacco‐specific flavors (English, Bell, & Berger, 1967) and degradation of nicotine and tobacco‐specific nitrosamines (Gong et al., 2009; Liu, He, et al., 2015; Liu, Ma, et al., 2015). Studies based on traditional culture‐dependent methods have isolated Bacillus, Streptomyces, Aspergillus, and Penicillium from FTLs, identifying these species as dominant microbes in FTL fermentations (Qiu, Zhao, Yue, Qi, & Zhang, 2000; Zhao, Qiu, Zhang, Qi, & Yue, 2000). To better characterize the microbial community associated with FTLs, prokaryotic diversity has been investigated using culture‐independent molecular biology techniques such as polymerase chain reaction denaturing gradient gel electrophoresis (PCR‐DGGE) (Huang et al., 2010; Zhao et al., 2007), 16S rRNA gene libraries (Su et al., 2011), Roche 454 bar‐coded pyrosequencing (Liu, He, et al., 2015; Liu, Ma, et al., 2015), and Illumina MiSeq sequencing (Wang et al., 2018). However, little is known about the eukaryotic community structure of FTL based on culture‐independent molecular biology techniques.
Partial least squares discriminant analysis (PLS‐DA) can be used to construct discrimination and classification models by reduction of data dimensionality (Berrueta, Alonso‐Salces, & Héberger, 2007; Vaclavik, Lacina, Hajslova, & Zweigenbaum, 2011), which is a powerful means of discriminating samples with different characteristics (Ramadan, Jacobs, Grigorov, & Kochhar, 2006; Wiklund et al., 2008). PLS‐DA has been used in distinguishing different kinds of Chinese liquors (Zhang, Yuan, Zeng, et al., 2017) and pit muds (Zhang, Yuan, Liao, & Zhang, 2017). It may therefore be of use in distinguishing different FTL flavor styles and in identifying microbes, which contribute significantly to desirable tobacco characteristics.
The network interface, in the form of a set of nodes and edges, carries meaningful statistical and structural features that shed light on the underlying rules guiding the community components and functions of the system being described (Newman, 2006). Recently, network analysis has been widely applied to reveal ecological linkages among microorganisms in complex ecosystems, such as marine water (Steele et al., 2011), soil (Barberán, Bates, Casamayor, & Fierer, 2012), and pit mud (Hu, Du, Ren, & Xu, 2016). To our knowledge, the existence of direct or indirect interactions among microbial taxa coexisting in FTLs has not been reported. Identifying and describing such interactions could clarify the ecological rules guiding community assembly within the FTL ecosystem.
2. MATERIALS AND METHODS
2.1. FTLs sampling
Samples of FTL were collected from a tobacco warehouse located in Shifang city, Sichuan province of China. The FTLs labeled as fresh flavor style and strong flavor style by sensory assessors were marked accordingly (F1, F2, F3, S1, S2, and S3). FTLs from three well‐known planting origins located in China were randomly selected for each style, and triplicate subsamples were collected and placed into each tobacco leaf storage box. Tobacco leaves approximately 20 cm from the top of the tobacco leaf storage box was removed and discarded. In total, 2 kg of leaf samples were taken from the four corners and the center of the storage box using the five‐point method. All samples were well‐mixed, transferred into sterile bags, and stored at −20°C.
2.2. DNA extraction and illumina MiSeq sequencing
Tobacco (25 g) was suspended in 500 ml of sterile saline and shaken for 2 hr at 200 rpm, after which the supernatant was centrifuged at 10,000 g for 20 min. Genomic DNA was extracted from the resulting pellet using an EZNA® Soil DNA Kit (Omega). The genomic DNA was sent to GENEWIZ Inc. for PCR amplification and sequencing of the V3‐V4 hypervariable region of 16S rRNA genes (primers: 5′‐CCT ACG GRR BGC ASC AGK VRV GAA/T3′ and 5′‐GGA CTA CNV GGG TWT CTA ATC C‐3′) and the V7‐V8 hypervariable region of 18S rRNA genes (forward primers containing the sequence: 5′‐CGW TAA CGA ACG AG‐3′ and reverse primers containing 5′‐AIC CAT TCA ATC GG‐3′).
DNA libraries, validated by Agilent 2100 Bioanalyzer (Agilent) and quantified by Qubit 2.0 Fluorometer (Invitrogen), were multiplexed and loaded on an Illumina MiSeq sequencing system according to manufacturer's instructions (Illumina). Sequencing was performed using a 2 × 250 paired‐end (PE) configuration. Image analysis and base calling were performed using the MiSeq Control Software (MCS) of the MiSeq instrument.
2.3. Sequence processing and data analysis
The sequencing data were processed using the QIIME platform version 1.9.1 (http://qiime.org/). The forward and reverse reads were merged according to the unique sample barcode sequence, followed by quality control processing (for 16S rRNA gene lengths between 430 and 470 bp, average length 455 bp, and for 18S rRNA gene lengths between 340 and 380 bp, average length 355 bp), and then truncated by removing the barcode and primer sequences. Qualified sequences were classified into operational taxonomic units (OTUs) at a 97% sequence identity using the clustering program VSEARCH version 1.9.6 and the Silva 132 database (https://www.arb-silva.de/) (Rognes, Flouri, Nichols, Quince, & Mahé, 2016). The phylogenetic affiliation of each sequence was analyzed by Ribosomal Database Program (RDP) Classifier at the 80% confidence level. Significant differences between the fresh flavor style and strong flavor style FTL groups were determined using SPSS software, version 19 (IBM), by one‐way analysis of variance and Duncan's multiple comparison test (p < .05). Alpha diversity, including Chao1 and Shannon values, was analyzed using QIIME version 1.9.1 (Caporaso et al., 2010). To reduce potential confounding effects due to uneven sampling, we randomly rarefied the OTU table to an even depth for alpha diversity analysis.
PLS‐DA and hierarchical cluster analysis (HCA) were conducted using SIMCA‐P version 13.0 (UMETRICS, Sweden) to discriminate different flavor styles of FTLs and reveal specific markers according to microbial compositions. The cross‐validated coefficient of determination, Q2, which indicates the variance captured in cross‐validation, was used as an indicator of overfitting. R2 was used to indicate the variance captured with the model (Pantsar‐Kallio, Reinikainen, & Oksanen, 2001). CoeffCS are coefficients used for interpreting how strongly Y is correlated with the systematic part of each of X‐variable. PLS‐DA of various FTLs is represented as a two‐dimensional representation of the scores (t[1] and t[2]) on the first and second PLS‐DA components.
Pairwise Spearman's rank correlations among genera with relative abundances higher than 0.1% were performed using SPSS Statistics version 19 (IBM, America). Spearman's correlation coefficients with statistical significance (p < .01) were considered valid co‐occurrence (or negative) events for a robust correlation (Barberán et al., 2012; Hu et al., 2016; Zhao et al., 2014). Networks were explored and visualized using the interactive platform Gephi (Bastian, Heymann, & Jacomy, 2009) based on the correlation matrix constructed by Spearman's correlations, with each node and edge representing one genus and a strong and significant correlation, respectively.
2.4. Nucleotide sequence accession number
The MiSeq sequences determined in this study have been deposited in the GenBank under the following accession number: PRJNA498896 and release date: 2019–11–19.
3. RESULTS
3.1. Prokaryotic Community Diversity and Structure
In total, 344,929 qualified reads were obtained from all FTLs. Each sample contained 102 to 122 OTUs, based on 97% similarity of 16S rRNA sequences (Table 1). The rarefaction curves all reached the saturation plateau (Figure A1 a) with coverage of more than 99%, indicating that Illumina MiSeq sequencing was deep enough to represent all bacterial communities detected. The Chao1 and Shannon values in the two different groups suggested similar bacterial species richness and diversity in all samples.
Table 1.
Diversity indices of prokaryotic and eukaryotic communities in FTLs
| Sample | Prokaryotic diversity | Eukaryotic diversity | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Qualified Sequences | OTUs | Chao1 | Shannon | Coverage | Qualified Sequences | OTUs | Chao1 | Shannon | Coverage | |
| F1 | 69017 | 118 | 119.00 | 5.23 | 0.99 | 60332 | 83 | 83.00 | 3.94 | 0.99 |
| F2 | 58868 | 115 | 120.00 | 4.87 | 0.99 | 61357 | 83 | 83.00 | 3.82 | 0.99 |
| F3 | 75999 | 122 | 124.00 | 4.30 | 0.99 | 61380 | 92 | 92.50 | 4.22 | 0.99 |
| S1 | 49342 | 118 | 121.33 | 5.16 | 0.99 | 55261 | 89 | 90.00 | 4.43 | 0.99 |
| S2 | 42724 | 118 | 123.00 | 4.47 | 0.99 | 59396 | 83 | 88.00 | 4.46 | 0.99 |
| S3 | 48979 | 102 | 104.14 | 2.50 | 0.99 | 69611 | 87 | 87.17 | 4.14 | 0.99 |
F1 to F3 denote fresh flavor style FTLs; S1 to S3 denote strong flavor style FTLs.
The phylogenetic structure analysis indicated that the identified sequences were affiliated with five bacterial phyla: Proteobacteria, Firmicutes, Cyanobacteria, Actinobacteria, and Bacteroidetes (Figure 1a). Based on the average relative abundance, the dominant bacterial phyla were Proteobacteria (68.93 ± 25.16%) and Firmicutes (24.49 ± 25.81%). A total of 54 bacterial genera were detected, of which 27 had a relative abundance higher than 1.0% in at least one sample (Figure 1b). The dominant bacterial genera, with relative abundance higher than 5.0% in at least one sample, were Pseudomonas, Acinetobacter, Rhizobium, Pantoea, Weissella, Bacillus, Methylobacterium, Sphingomonas, Aureimonas, and Ralstonia, which represented between 45.51% and 82.66% of the total abundance of each FTL sample. Compared with the proportions of the dominant genera in the strong flavor style FTLs (S1, S2, and S3), the proportions of most groups decreased slightly (p > .05) in the fresh flavor style FTLs (F1, F2, and F3).
Figure 1.
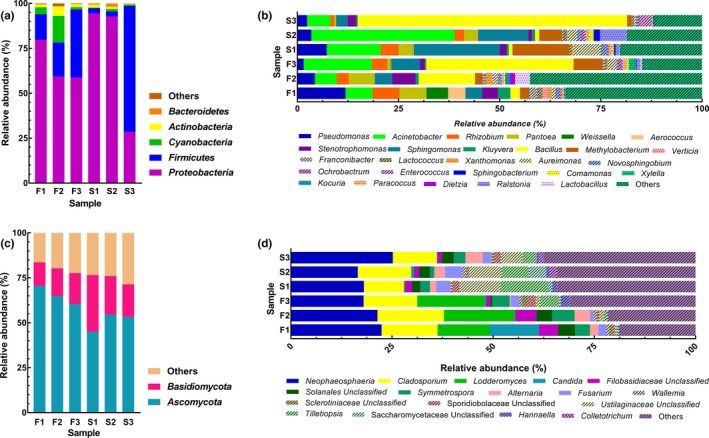
Plot of phylum and genus level relative abundances of prokaryotic and eukaryotic communities in FTLs. (a) and (b) represent prokaryotic communities at the level of phylum and genus; (c) and (d) represent eukaryotic communities at the level of phylum and genus. F1 to F3 denote fresh flavor style FTLs; S1 to S3 denote strong flavor style FTLs
3.2. Eukaryotic community diversity and structure
In total, 367,337 qualified reads were obtained from all FTLs (Table 1). Each sample contained 83 to 92 OTUs, based on 97% similarity of 18S rRNA sequences. Based on the rarefaction curves (Figure A1 b) and coverage values, Illumina MiSeq sequencing was found to represent all fungal communities. The diversity and richness of the fungal communities were generally lower than those of the bacterial communities.
Figure 1c shows the two main eukaryotic phyla, Ascomycota and Basidiomycota, which had average relative abundances of 58.16 ± 9.06% and 19.43 ± 6.53%, respectively. Ascomycota had higher relative abundances (p < .05) in fresh flavor style FTLs (F1, F2, and F3) than in strong flavor style FTLs (S1, S2, and S3). There were 53 eukaryotic genera across all samples, with 17 genera having a relative abundance of higher than 1.0% in at least one sample (Figure 1d). Lodderomyces and Symmetrospora had higher relative abundances (p < .05) in the fresh flavor style FTLs than in the strong flavor style FTLs, while unclassified Ustilaginaceae displayed the opposite trend. The dominant genera were Neophaeosphaeria, Cladosporium, Lodderomyces, Tilletiopsis, unclassified Ustilaginaceae, and Symmetrospora represented between 67.61% and 84.66% of the total abundance in each FTL sample.
3.3. PLS‐DA and HCA
PLS‐DA was used to construct a statistical model for FTL discrimination and classification, and two significant principal components of the total variance in data matrix were extracted. The R2 and Q2 were 0.997 and 0.775, respectively, which meant that a total of 99.7% dummy Y variable per class, and 77.5% overall cross‐validated R 2 for these two components. The data indicated that the PLS‐DA model was suitable for this research. Fresh flavor style and strong flavor style FTL groups were clearly separated on the score scatter plot, with the fresh flavor style group located on the left side of the plot and the strong flavor style group located on the right side (Figure 2a). The coefficients refer to the PLS‐DA model being rewritten as a regression model. CoeffCS (coefficient values between variables and samples of significant first and second principal components) are shown in Table 2. Xanthomonas, Franconibacter, Massilia, Quadrisphaera, Staphylococcus, Cladosporium, Lodderomyces, Symmetrospora, Golovinomyces, and Dioszegia were significantly positively correlated with DA(1) (fresh flavor style FTL group). Xenophilus, Fusarium, unclassified Ustilaginaceae, Tilletiopsis, Cryphonectria, Colletotrichum, and Cyanodermella were significantly positively correlated with DA(2) (strong flavor style FTL group). HCA also identified two groups of samples (Figure 2b), the first consisting of samples F1, F2, and F3, and the second of samples S1, S2, and S3, which was consistent with the PLS‐DA.
Figure 2.
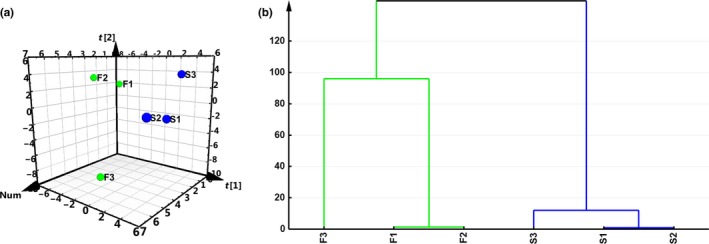
Score scatter plot of PLS‐DA (a) and dendrogram of HCA (b). PLS‐DA of various FTLs is represented as a two‐dimensional representation of the scores (t[1] and t[2]) on the first and second PLS‐DA components. F1 to F3 denote the fresh flavor style FTLs; S1 to S3 denote strong flavor style FTLs
Table 2.
Coefficient values between variables and group†
| Variable | CoeffCS[1] | CoeffCS[2] | ||
|---|---|---|---|---|
| DA(1) | DA(2) | DA(1) | DA(2) | |
| Stenotrophomonas | 0.020164* | −0.020164‡ | 0.014278 | −0.014280 |
| Xanthomonas | 0.022872‡ | −0.022872‡ | 0.018785‡ | −0.018785‡ |
| Franconibacter | 0.022677‡ | −0.022677‡ | 0.023343‡ | −0.023343‡ |
| Brachybacterium | 0.020946‡ | −0.020946‡ | 0.017670 | −0.017670‡ |
| Massilia | 0.028344‡ | −0.028344‡ | 0.032326‡ | −0.032326‡ |
| Aeromicrobium | 0.020618 | −0.020620 | 0.024559‡ | −0.024559‡ |
| Roseomonas | −0.019860‡ | 0.019860‡ | −0.018810 | 0.018809 |
| Quadrisphaera | 0.022342‡ | −0.022342‡ | 0.020474‡ | −0.020474‡ |
| Xenophilus | −0.028989‡ | 0.028989‡ | −0.039998‡ | 0.039998‡ |
| Staphylococcus | 0.027194‡ | −0.027194‡ | 0.034612‡ | −0.034612‡ |
| Sporolactobacillus | 0.023550 | −0.023550 | 0.033789‡ | −0.033789‡ |
| Cladosporium | 0.025311‡ | −0.025311‡ | 0.026734‡ | −0.026734‡ |
| Lodderomyces | 0.032772‡ | −0.032772‡ | 0.039731‡ | −0.039731‡ |
| unclassified Filobasidiaceae | 0.021517‡ | −0.021517‡ | 0.016375 | −0.016380 |
| Symmetrospora | 0.027974‡ | −0.027974‡ | 0.030922‡ | −0.030922‡ |
| Fusarium | −0.024946‡ | 0.024946‡ | −0.024955‡ | 0.024955‡ |
| Vishniacozyma | 0.021165‡ | −0.021165‡ | 0.019052 | −0.019050 |
| Aureobasidium | 0.015633‡ | −0.015633‡ | 0.007527 | −0.007530 |
| Golovinomyces | 0.027221‡ | −0.027221‡ | 0.026030‡ | −0.026030‡ |
| unclassified Ustilaginaceae | −0.030978‡ | 0.030978‡ | −0.036197‡ | 0.036197‡ |
| Dioszegia | 0.025596‡ | −0.025596‡ | 0.028663‡ | −0.028663‡ |
| Tilletiopsis | −0.022887‡ | 0.022887‡ | −0.022104‡ | 0.022104‡ |
| Taphrina | 0.025651 | −0.025650‡ | 0.036222‡ | −0.036220‡ |
| Cryphonectria | −0.024840‡ | 0.024837‡ | −0.02856‡ | 0.028563‡ |
| Rhizoctonia | −0.025910‡ | 0.025914‡ | −0.027720 | 0.027717 |
| Colletotrichum | −0.023750‡ | 0.023760‡ | −0.028350‡ | 0.028349‡ |
| Cyanodermella | −0.029790‡ | 0.029789‡ | −0.034680‡ | 0.034678‡ |
CoeffCS are coefficients used for interpreting the influence of the variables X on Y. CoeffCS[1] and CoeffCS[2] represent significant principal component 1 and 2, respectively. DA(1) denotes the fresh flavor style FTL group (F1 to F3); DA(2) denotes strong flavor style FTL group (S1 to S3).
Coefficient is significant (above the noise).
3.4. Correlation network description
The co‐occurrence patterns of FTL microorganisms were explored based on strong and significant correlations (p < .01), and a total of 64 nodes (genera) and 72 edges (pairs of significant and robust correlations) were found (Figure 3a). The modularity index was 0.797 (>0.4), suggesting that the network had a modular structure. At the phylum level, Proteobacteria, Ascomycota, Basidiomycota, Firmicutes, and Actinobacteria accounted for 35.94%, 15.62%, 14.06%, 12.5%, and 12.5% of all nodes, respectively. The average degree (edges per node) was 2.25. There were seven hubs (highly connected genera, degree ≥ 5), including Aureimonas, Kocuria, Massilia, Brachybacterium, Clostridium, Dietzia, and Vishniacozyma. Genera from different phyla (interphylum) had a high co‐occurrence incidence (69.4%, ratio of targeted edges to total edges). Between all pairs of any two phyla, the incidence of co‐occurrence between Proteobacteria and Firmicutes, and between Proteobacteria and Actinobacteria, was the highest, at up to 11.1%. Massilia was significantly and positively correlated with Kocuria, and Clostridium with Dietzia. The incidence of co‐occurrence within a phylum (intraphylum) was 30.6% and was observed among genera from the phyla Proteobacteria (22.2%) and Actinobacteria (5.6%). Methylobacterium was significantly and positively correlated with Sphingomonas.
Figure 3.
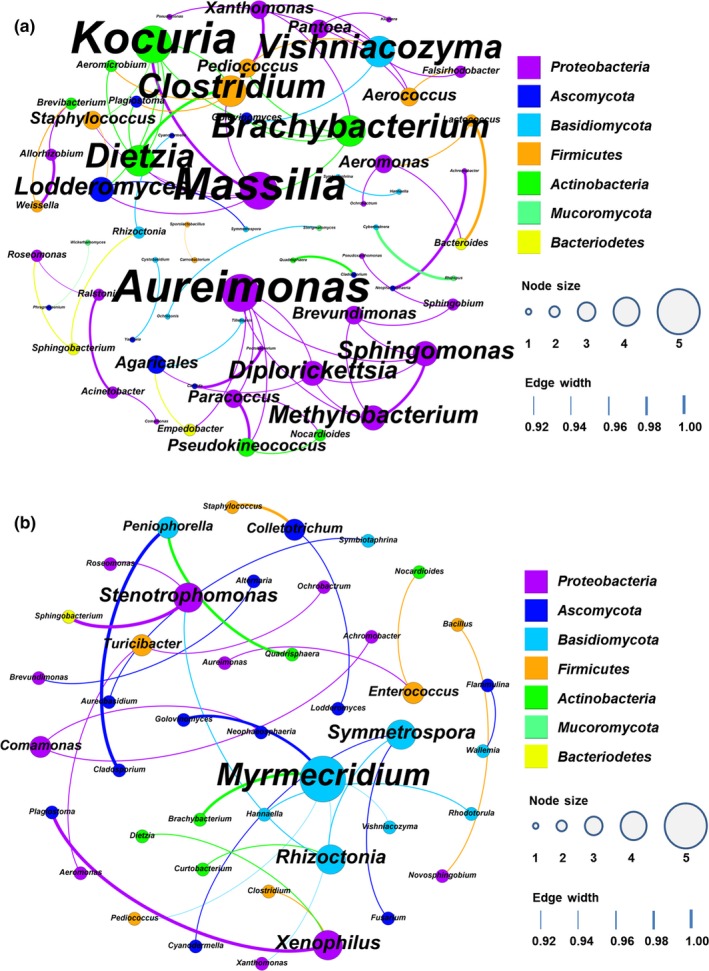
Network of co‐occurring microbial genera in FTLs based on correlation analysis, positive and negative correlation. (a) positive correlation network; (b) negative correlation network. For each panel, the size of each node is proportional to the number of connections, nodes of the same color are affiliated with the same phylum, and the thickness of each connection between two nodes is proportional to the value of Spearman's correlation coefficients with statistical significance (p < .01)
Moreover, a total of 30 pairs of significant and robust negative correlations were identified from 43 genera (Figure 3b). The modularity index was 0.88 (>0.4), suggesting that the network had a modular structure. At the phylum level, Proteobacteria, Ascomycota, and Basidiomycota accounted for 25.58%, 25.58%, and 20.93% of all nodes, respectively. The average degree (edges per node) was 1.395, and the highest degree was 5 (hubs, Myrmecridium). Genera from different phyla (interphylum) had a high incidence of negative occurrence (80.0%, ratio of targeted edges to total edges).
4. DISCUSSION
In this study, Illumina MiSeq sequencing based on 16S rRNA and 18S rRNA genes was used to investigate the diversity and structure of the prokaryotic and eukaryotic communities associated with FTLs. Based on their microbial community compositions, the differences between fresh and strong flavor style FTLs were explored, as along with the co‐occurrence patterns of FTL microorganisms in the individual communities. Firmicutes, Proteobacteria, Actinobacteria, and Bacteroidetes were abundant in FTLs. However, the most dominant phylum was either Proteobacteria or Firmicutes, corroborating the results of previous studies (Huang et al., 2010; Su et al., 2011; Wang et al., 2018). Specifically, Huang et al. (2010) and Su et al. (2011) indicated that Proteobacteria was the dominant phylum of Zimbabwe FTLs and K326 FTLs, while Wang et al. (2018) found that Firmicutes was the dominant phylum of FTLs.
Compared with PCR‐DGGE, 16S rRNA gene clone libraries sequencing, and pyrosequencing, Illumina MiSeq sequencing with a higher sequence output could unveil more information about microbial community (Hirai, Nagai, & Hidaka, 2017). In our study, the Illumina MiSeq sequencing results showed that 61.72% of the prokaryotes belonged to the genera Pseudomonas, Bacillus, Methylobacterium, Acinetobacter, and Sphingomonas. These genera are reported to be important contributors to nicotine degradation, or to the formation of representative flavor compounds. They are also used as biocontrol agents. Although bacterial genera varied from different resources and sequencing methods (Liu, He, et al., 2015; Liu, Ma, et al., 2015), Pseudomonas and Bacillus are the dominant genera in most of the FTL samples (Huang et al., 2010; Su et al., 2011; Wang et al., 2018; Zhao et al., 2007), as shown in our study, and functionally important contributors in the process of FTL aging (Huang et al., 2010; Wang et al., 2018). Pseudomonas strains isolated from tobacco leaves and tobacco soils have been reported to be nicotine‐degrading. Isolates include Pseudomonas sp. HF‐1 (Ruan, Min, Peng, & Huang, 2005), P. putida S16 (Wang, Liu, Tang, Meng, & Xu, 2007), Pseudomonas sp. Nic22 (Chen et al., 2008), Pseudomonas sp. ZUTSKD (Zhong et al., 2010), and P. stutzeri ZCJ (Zhao et al., 2012). Nicotine degradation pathways in Pseudomonas species can be classified into two categories, depending on whether metabolites are directed into the tricarboxylic acid cycle (TAC). Nicotine can be converted into N‐methylmyosmine, cotinine, or nornicotine, which is then converted into maleamic acid, and finally fumaric acid in the TAC. Alternatively, nicotine may be converted into nicotyrine, which is not directed into the TAC (Li, Duan, Zhang, & Yang, 2010; Ruan et al., 2005; Tang et al., 2008, 2009, 2011; Wang, Yang, Min, & Lv, 2009).
Bacillus species fulfill different functions in FTLs. Some species are considered endophytic and/or beneficial to plants, including tobacco, and have been reported to be the functional microorganism in the promotion of tobacco fermentation and formation of aged flavor compounds (English et al., 1967; Huang et al., 2010). English et al. (1967) revealed that B. subtilis and B. circulans could hasten the development of desirable flavors and improve the smoking qualities of cigar tobacco. B. thuringiensis, which produces bipyramidal crystals, is present on the tobacco leaf surface and can control insect pests that affect stored tobacco (Kaelin & Gadani, 2000). B. subtilis has a strong ability to control the effects of tobacco black shank (Han et al., 2016) and displayed an antagonistic effect against Verticillium dahliae, which causes verticillium wilt (Berg & Ballin, 1994).
Similar to B. subtilis, Sphingomonas has an antagonistic effect on Verticillium dahliae (Berg & Ballin, 1994), but has the additional ability to degrade a wide variety of dimeric lignin compounds into a series of flavor compounds (Masai, Katayama, Nishikawa, & Fukuda, 1999). Sphingomonas abundance was 4% in the 16S rRNA clone library of Zimbabwe tobacco (Su et al., 2011). Acinetobacter and Sphingomonas isolated from soil tobacco waste are able to degrade nicotine (Wang et al., 2011). Methylobacterium strains, which are frequently encountered as endophytes, degrade one‐carbon compounds such as methanol and methylamine, and are capable of forming biofilms, producing quorum‐sensing signals, and resisting heavy metal and other stresses (Ardanov, Sessitsch, Häggman, Kozyrovska, & Pirttila, 2012; Rossetto et al., 2011).
Neophaeosphaeria and Cladosporium, from the Ascomycota, were dominant fungal genera in FTLs, although Lodderomyces, Candida, unclassified Ustilaginaceae, and Tilletiopsis were also present at high relative abundances (>10%) in some samples. However, the specific functions of these genera during tobacco leaf fermentation and flavor formation are not well‐understood. Cladosporium can produce γ‐decalactone (Berger, 2015). It can also contribute to lignin and cellulose‐degradation, as it produces laccase and cellulase (Jin et al., 2012), and may consequently play a flavor‐enhancing role during FTL fermentation. Tilletiopsis produce hydrolytic enzymes and antifungal compounds, which are effective against powdery mildew fungi (Urquhart & Punja, 2002); this genus may therefore serve as a biocontrol agent.
The relative abundance of microorganisms varied between different FTL samples, and even within the same flavor style FTLs, relative abundance changed. The planting area (including edaphic factors, climatic factors, and biologic factors) may directly cause differences in the microbial communities among the same flavor style FTLs. By one‐way analysis of variance and Duncan's multiple comparison test, there was only one phylum (Ascomycota) and three (Lodderomyces, Symmetrospora, and unclassified Ustilaginaceae) out of 107 genera that were significantly different (p < .05) between fresh flavor style FTLs and strong flavor style FTLs. However, the boundary separating fresh and strong flavor style FTLs was not obvious and did not allow for straightforward discrimination or classification. Therefore, in order to distinguish between fresh and strong flavor style FTLs, the microbial data had to be analyzed using metrology tools. Analysis of the microbial relative abundances by PLS‐DA and HCA indicated that samples could be separated into fresh and strong flavor groups and that some genera could be used as markers for the discrimination of samples. The results of our study agree with those of the sensory evaluation methods traditionally used to classify FTLs.
Further investigation was conducted by correlation‐based network analysis. The less‐related interphylum genera had much higher co‐occurrence ratios than intraphylum genera in FTLs, a finding that has also been observed in activated sludge (Ju, Xia, Guo, Wang, & Zhang, 2014) and pit muds (Hu et al., 2016). This phenomenon can be attributed to two factors: phylogenetic overdispersion of all biological communities, and the effect of negative interactions on the community assembly (Horner‐Devine & Bohannan, 2006; Ju et al., 2014; Slingsby & Verboom, 2006). The co‐occurrence patterns revealed that community members may share niche spaces and have synergetic relationships in FTLs (Barberán et al., 2012). Based on co‐occurrence analysis there existed eight main hubs that may play important roles in ecosystem stability: Aureimonas, Kocuria, Massilia, Brachybacterium, Clostridium, Dietzia, Vishniacozyma, and Myrmecridium (Peura, Bertilsson, Jones, & Eiler, 2015). Genera from the main hubs were endophytes (Aureimonas, Ikeda et al., 2010; Myrmecridium, Tan et al., 2012) and plant‐growth‐promoting rhizobacteria (Brachybacterium, Gontia, Kavita, Schmid, Hartmann, & Jha, 2011, Jiang et al., 2018; Dietzia, Bharti, Pandey, Barnawal, Patel, & Kalr, 2016). It is possible that Massilia (Myeong, Seong, Kim, & Sul, 2016; Ofek, Hadar, & Minz, 2012; Xu et al., 2016), Vishniacozyma (Gramiscia, Lutzb, Lopesa, & Sangorrína, 2018), and Clostridium (Doi et al., 1998; Murashima, Kosugi, & Doi, 2003) produce antibiotic compounds or enzymes to maintain their own niches in FTLs, but this requires further confirmation.
Although the number of samples (n = 3) was statistically significant and results of previous studies (Huang et al., 2010; Su et al., 2011; Wang et al., 2018) were corroborated, the sample size is still small in this study. More samples of fresh and strong flavor style FTLs from different batches and sources should be considered to verify the reliability and reproducibility. Illumina MiSeq sequencing based on 16S rRNA and 18S rRNA genes provided the diversity and structure of the prokaryotic and eukaryotic communities associated with FTLs; however, amplicon‐based studies could not identify viable organisms. Thus, omics technologies, including genomics, transcriptomics, proteomics, metabolomics, and fluxomics, should be utilized to intensively study genetic, protein, and product information related to the microbial metabolism, and the mechanisms producing patterns of community coexistence, as the complex structure and community of microbes in FTLs is complex, and several correlations of the microbial taxa. Moreover, some specific species, including endophytes, plant‐growth‐promoting rhizobacteria, and other abundance microbes isolated from FTLs, should be studied for their function in FTLs, as they might play essential roles in stabilizing inhabiting microbial community and producing beneficial substances for FTLs.
5. CONCLUSION
We used Illumina MiSeq sequencing to analyze the microbiota associated with fresh and strong flavor style FTLs. Bacterial and fungal community compositions and diversities were analyzed, distinctions among FTLs of different types were revealed, and correlations between microbial taxa within FTL ecosystems were described. The dominant microbes came from seven genera, including Pseudomonas, Bacillus, Methylobacterium, Acinetobacter, Sphingomonas, Neophaeosphaeria, and Cladosporium, belonging to bacterial phyla Firmicutes and Proteobacteria, and the fungal phyla Ascomycota. Xanthomonas, Franconibacter, Massilia, Quadrisphaera, Staphylococcus, Cladosporium, Lodderomyces, Symmetrospora, Golovinomyces, and Dioszegia were significantly positively correlated with the fresh flavor style FTL group, while Xenophilus, Fusarium, unclassified Ustilaginaceae, Tilletiopsis, Cryphonectria, Colletotrichum, and Cyanodermella were significantly positively correlated with the strong flavor style FTL group. Additionally, endophytes and rhizobacteria, including Aureimonas, Kocuria, Massilia, Brachybacterium, Clostridium, Dietzia, and Vishniacozyma, combined multiple niches in FTLs, which might be destroyed by Myrmecridium. These findings represent a step forward in revealing microbial diversities, understanding differences in microbial community structure, and uncovering the stable microbial communities associated with fresh and strong flavor FTLs.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
Dongliang Li and Zhongyang Ding designed the concept. Qianying Zhang performed formal analysis. Dongliang Li and Zhongyang Ding contributed to funding acquisition. Qianying Zhang and Zongze Geng performed investigation. Qianying Zhang and Zongze Geng prepared the original draft. Qianying Zhang reviewed and edited the manuscript.
ETHICS STATEMENT
None required.
ACKNOWLEDGEMENTS
This work was funded by Sichuan Science and Technology Program (No. 2019YJ0264), and China Tobacco Sichuan Industrial Co., Ltd. (No. rtx201820).
APPENDIX 1.
Figure A1.
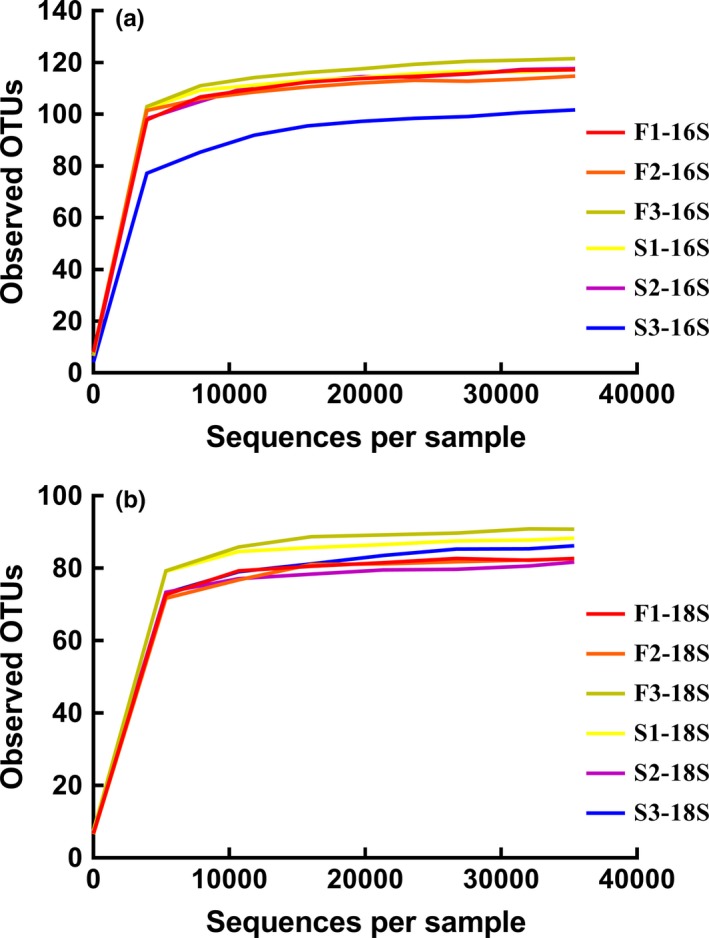
Rarefaction curve of the observed number of OTUs from samples of 16S rRNA gene (a) and 18S rRNA gene (b) at a genetic distance of 3%
Zhang Q, Geng Z, Li D, Ding Z. Characterization and discrimination of microbial community and co‐occurrence patterns in fresh and strong flavor style flue‐cured tobacco leaves. MicrobiologyOpen. 2020;9:e965 10.1002/mbo3.965
Contributor Information
Dongliang Li, Email: 360188228@qq.com.
Zhongyang Ding, Email: bioding@163.com.
DATA AVAILABILITY STATEMENT
The datasets generated for this study can be found in GenBank under the accession number PRJNA498896: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA498896.
REFERENCES
- Ardanov, P. , Sessitsch, A. , Häggman, H. , Kozyrovska, N. , & Pirttila, A. M. (2012). Methylobacterium‐induced endophyte community changes correspond with protection of plants against pathogen attack. PLoS ONE, 7, e46802 10.1371/journal.pone.0046802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberán, A. , Bates, S. T. , Casamayor, E. O. , & Fierer, N. (2012). Using network analysis to explore co‐occurrence patterns in soil microbial communities. The ISME Journal, 6, 343–351. 10.1038/ismej.2011.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastian, M. , Heymann, S. , & Jacomy, M. (2009). Gephi: An open source software for exploring and manipulating networks. In: Third International AAAI Conference on Weblogs and Social Media, 361–362. http://www.aaai.org/ocs/index.php/ICWSM/09/paper/view/154
- Berg, G. , & Ballin, G. (1994). Bacterial antagonist to Verticillium dahlia Kleb. Journal of Phytopathology, 141, 99–110. 10.1111/j.1439-0434.1994.tb01449.x [DOI] [Google Scholar]
- Berger, R. G. (2015). Biotechnology as a source of natural volatile flavours. Current Opinion in Food Science, 1, 38–43. 10.1016/j.cofs.2014.09.003 [DOI] [Google Scholar]
- Berrueta, L. A. , Alonso‐Salces, R. M. , & Héberger, K. (2007). Supervised pattern recognition in food analysis. Journal of Chromatography A, 1158, 196–214. 10.1016/j.chroma.2007.05.024 [DOI] [PubMed] [Google Scholar]
- Bharti, N. , Pandey, S. S. , Barnawal, D. , Patel, V. K. , & Kalr, A. (2016). Plant growth promoting rhizobacteria Dietzia natronolimnaea modulates the expression of stress responsive genes providing protection of wheat from salinity stress. Scientific Reports, 6, 34768 10.1038/srep34768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso, J. G. , Kuczynski, J. , Stombaugh, J. , Bittinger, K. , Bushman, F. D. , Costello, E. K. , … Knight, R. (2010). QIIME allows analysis of high‐throughput community sequencing data. Nature Methods, 7, 335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. M. , Li, X. M. , Yang, J. K. , Gong, X. W. , Li, B. , & Zhang, K. Q. (2008). Isolation of nicotine‐degrading bacterium Pseudomonas sp. Nic22, and its potential application in tobacco processing. International Biodeterioration & Biodegradation, 62, 226–231. 10.1016/j.ibiod.2008.01.012 [DOI] [Google Scholar]
- Doi, R. H. , Park, J. S. , Liu, C. C. , Malburg, L. M. , Tamaru, Y. , Ichiishi, A. , & Ibrahim, A. (1998). Cellulosome and noncellulosomal cellulases of Clostridium cellulovorans . Extremophiles, 2, 53–60. 10.1007/s007920050 [DOI] [PubMed] [Google Scholar]
- English, C. F. , Bell, E. J. , & Berger, A. J. (1967). Isolation of thermophiles from broadleaf tobacco and effect of pure culture inoculation on cigar aroma and mildness. Applied and Environmental Microbiology, 15, 117–119. 10.1002/jobm.3630080514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, X. W. , Yang, J. K. , Duan, Y. Q. , Dong, J. Y. , Zhe, W. , Wang, L. , … Zhang, K. Q. (2009). Isolation and characterization of Rhodococcus sp. Y22 and its potential application to tobacco processing. Research in Microbiology, 160, 200–204. 10.1016/j.resmic.2009.02.004 [DOI] [PubMed] [Google Scholar]
- Gontia, I. , Kavita, K. , Schmid, M. , Hartmann, A. , & Jha, B. (2011). Brachybacterium saurashtrense sp. nov., a halotolerant root‐associated bacterium with plant growth‐promoting potential. International Journal of Systematic and Evolutionary Microbiology, 61, 2799–2804. 10.1099/ijs.0.023176-0 [DOI] [PubMed] [Google Scholar]
- Gramiscia, B. R. , Lutzb, M. C. , Lopesa, C. A. , & Sangorrína, M. P. (2018). Enhancing the efficacy of yeast biocontrol agents against postharvest pathogens through nutrient profiling and the use of other additives. Biological Control, 121, 151–158. 10.1016/j.biocontrol.2018.03.001 [DOI] [Google Scholar]
- Han, T. , You, C. , Zhang, C. , Feng, C. , Zhang, J. , Wang, F. , & Kong, F. (2016). Biocontrol potential of antagonist Bacillus subtilis Tpb55 against tobacco black shank. BioControl, 61, 195–205. 10.1007/s10526-015-9705-0 [DOI] [Google Scholar]
- Hirai, J. , Nagai, S. , & Hidaka, K. (2017). Evaluation of metagenetic community analysis of planktonic copepods using Illumina MiSeq: Comparisons with morphological classification and metagenetic analysis using Roche 454. PLoS ONE, 12(7), e0181452 10.1371/journal.pone.0181452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner‐Devine, M. C. , & Bohannan, B. J. M. (2006). Phylogenetic clustering and overdispersion in bacterial communities. Ecology, 87, 100–108. 10.1890/0012-9658(2006)87[100:PCAOIB]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Hu, X. L. , Du, H. , Ren, C. , & Xu, Y. (2016). Illuminating anaerobic microbial community and cooccurrence patterns across a quality gradient in Chinese liquor fermentation pit muds. Applied and Environmental Microbiology, 82, 2506–2515. 10.1128/AEM.03409-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, J. W. , Yang, J. K. , Duan, Y. Q. , Gu, W. , Gong, X. W. , Zhe, W. , … Zhang, K. Q. (2010). Bacterial diversities on unaged and aging flue‐cured tobacco leaves estimated by 16S rRNA sequence analysis. Applied Microbiology and Biotechnology, 88, 553–562. 10.1007/s00253-010-2763-4 [DOI] [PubMed] [Google Scholar]
- Ikeda, S. , Okubo, T. , Anda, M. , Nakashita, H. , Yasuda, M. , Sato, S. , … Minamisawa, K. (2010). Community‐ and genome‐based views of plant‐associated bacteria: Plant‐bacterial interactions in soybean and rice. Plant & Cell Physiology, 51, 1398–1410. 10.1093/pcp/pcq119 [DOI] [PubMed] [Google Scholar]
- Jiang, Z. K. , Tu, L. , Huang, D. L. , Osterman, I. A. , Tyurin, A. P. , Liu, S. W. , … Sun, C. H. (2018). Diversity, novelty, and antimicrobial activity of endophytic actinobacteria from mangrove plants in Beilun Estuary National Nature Reserve of Guangxi. China. Frontiers in Microbiology, 9, 868 10.3389/fmicb.2018.00868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, R. , Liao, H. D. , Liu, X. M. , Zheng, M. , Xiong, X. Q. , Liu, X. W. , … Zhu, Y. H. (2012). Identification and characterization of a fungal strain with lignin and cellulose hydrolysis activities. African Journal of Microbiology Research, 6, 6545–6550. 10.5897/AJMR12.476 [DOI] [Google Scholar]
- Ju, F. , Xia, Y. , Guo, F. , Wang, Z. P. , & Zhang, T. (2014). Taxonomic relatedness shapes bacterial assembly in active sludge of globally distributed wastewater treatment plants. Environmental Microbiology, 16, 2421–2432. 10.1111/1462-2920.12355 [DOI] [PubMed] [Google Scholar]
- Kaelin, P. , & Gadani, F. (2000). Occurrence of Bacillus thuringiensis on cured tobacco leaves. Current Microbiology, 40, 205–209. 10.1007/s002849910041 [DOI] [PubMed] [Google Scholar]
- Li, H. J. , Li, X. M. , Duan, Y. Q. , Zhang, K. Q. , & Yang, J. K. (2010). Biotransformation of nicotine by microorganism: The case of Pseudomonas spp. Applied Microbiology and Biotechnology, 86, 11–17. 10.1007/s00253-009-2427-4 [DOI] [PubMed] [Google Scholar]
- Liu, H. , He, H. , Cheng, C. , Liu, J. , Shu, M. , Jiao, Y. , … Zhong, W. (2015). Diversity analysis of the bacterial community in tobacco waste extract during reconstituted tobacco process. Applied Microbiology and Biotechnology, 99, 469–476. 10.1007/s00253-014-5960-8 [DOI] [PubMed] [Google Scholar]
- Liu, J. , Ma, G. , Chen, T. , Hou, Y. , Yang, S. , Zhang, K. , & Yang, J. (2015). Nicotine‐degrading microorganisms and their potential applications. Applied Microbiology and Biotechnology, 99, 3775–3785. 10.1007/s00253-015-6525-1 [DOI] [PubMed] [Google Scholar]
- Masai, E. , Katayama, Y. , Nishikawa, S. , & Fukuda, M. (1999). Characterization of Sphingomonas paucimobilis SYK‐6 genes involved in degradation of lignin‐related compounds. Journal of Industrial Microbiology and Biotechnology, 23, 364–373. 10.1038/sj.jim.2900747 [DOI] [PubMed] [Google Scholar]
- Murashima, K. , Kosugi, A. , & Doi, R. H. (2003). Synergistic effects of cellulosomal xylanase and cellulases from Clostridium cellulovorans on plant cell wall degradation. Journal of Bacteriology, 185, 1518–1524. 10.1128/JB.185.5.1518-1524.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myeong, N. Y. , Seong, H. J. , Kim, H. J. , & Sul, W. J. (2016). Complete genome sequence of antibiotic and anticancer agent violacein producing Massilia sp. strain NR 4–1. Journal of Biotechnology, 223, 36–37. 10.1016/j.jbiotec.2016.02.027 [DOI] [PubMed] [Google Scholar]
- Newman, M. E. J. (2006). Modularity and community structure in networks. Proceedings of the National Academy of Sciences of the United States of America, 103, 8577 10.1073/pnas.0601602103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofek, M. , Hadar, Y. , & Minz, D. (2012). Ecology of root colonizing Massilia (Oxalobacteraceae). PLoS ONE, 7, e40117 10.1371/journal.pone.0040117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantsar‐Kallio, M. , Reinikainen, S. P. , & Oksanen, M. (2001). Interactions of soil components and their effects on speciation of chromium in soils. Analytica Chimica Acta, 439, 9–17. 10.1016/S0003-2670(01)00840-6 [DOI] [Google Scholar]
- Peura, S. , Bertilsson, S. , Jones, R. I. , & Eiler, A. (2015). Resistant microbial co‐occurrence patterns inferred by network topology. Applied and Environmental Microbiology, 81, 2090–2097. 10.1128/AEM.03660-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, L. , Zhao, M. , Yue, X. , Qi, W. , & Zhang, W. (2000). Isolation and identification of the microflora on tobacco leaves during the natural fermentation of flue‐cured tobacco. Tobacco Science & Technology, 3, 14–17. 10.3969/j.issn.1002-0861.2000.03.006 [DOI] [Google Scholar]
- Ramadan, Z. , Jacobs, D. , Grigorov, M. , & Kochhar, S. (2006). Metabolic profiling using principal component analysis, discriminant partial least squares, and genetic algorithms. Talanta, 68, 1683–1691. 10.1016/j.talanta.2005.08.042 [DOI] [PubMed] [Google Scholar]
- Reid, J. J. , McKinstry, D. W. , & Haley, D. E. (1937). The fermentation of cigar‐leaf tobacco. Science, 2235, 404 10.1126/science.86.2235.404 [DOI] [PubMed] [Google Scholar]
- Rognes, T. , Flouri, T. , Nichols, B. , Quince, C. , & Mahé, F. (2016). VSEARCH: A versatile open source tool for metagenomics. PeerJ, 4, e2584 10.7717/peerj.2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetto, P. B. , Dourado, M. N. , Quecine, M. C. , Andreote, F. D. , Araújo, W. L. , Azevedo, J. L. , & Pizzirani‐Kleiner, A. A. (2011). Specific plant induced biofilm formation in Methylobacterium species. Brazilian Journal of Microbiology, 42, 878–883. 10.1590/S1517-83822011000300006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan, A. D. , Min, H. , Peng, X. H. , & Huang, Z. (2005). Isolation and characterization of Pseudomonas sp strain HF‐1, capable of degrading nicotine. Research in Microbiology, 156, 700–706. 10.1016/j.resmic.2005.02.010 [DOI] [PubMed] [Google Scholar]
- Slingsby, J. A. , & Verboom, G. A. (2006). Phylogenetic relatedness limits co‐occurrence at fine spatial scales: Evidence from the Schoenoid sedges (Cyperaceae: Schoeneae) of the Cape Floristic Region, South Africa. The American Naturalist, 168, 14–27. 10.1086/505158 [DOI] [PubMed] [Google Scholar]
- Steele, J. A. , Countway, P. D. , Xia, L. , Vigil, P. D. , Beman, J. M. , Kim, D. Y. , … Fuhrman, J. A. (2011). Marine bacterial, archaeal and protistan association networks reveal ecological linkages. The ISME Journal, 5, 1414–1425. 10.1038/ismej.2011.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, C. , Gu, W. , Zhe, W. , Zhang, K. Q. , Duan, Y. Q. , & Yang, J. K. (2011). Diversity and phylogeny of bacteria on Zimbabwe tobacco leaves estimated by 16S rRNA sequence analysis. Applied Microbiology and Biotechnology, 92, 1033–1044. 10.1007/s00253-011-3367-3 [DOI] [PubMed] [Google Scholar]
- Tan, X. M. , Chen, X. M. , Wang, C. L. , Jin, X. H. , Cui, J. L. , Chen, J. , … Zhao, L. F. (2012). Isolation and Identification of endophytic fungi in roots of nine Holcoglossum plants (Orchidaceae) collected from Yunnan, Guangxi, and Hainan Provinces of China. Current Microbiology, 64, 140–147. 10.1007/s00284-011-0045-8 [DOI] [PubMed] [Google Scholar]
- Tang, H. Z. , Wang, L. J. , Meng, X. Z. , Ma, L. Y. , Wang, S. N. , He, X. F. , … Xu, P. (2009). Novel nicotine oxidoreductase‐encoding gene involved in nicotine degradation by Pseudomonas putida strain S16. Applied and Environmental Microbiology, 75, 772–778. 10.1128/AEM.02300-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, H. , Wang, S. , Ma, L. , Meng, X. , Deng, Z. , Zhang, D. , … Xu, P. (2008). A novel gene, encoding 6‐hydroxy‐3‐succinoylpyridine hydroxylase, involved in nicotine degradation by Pseudomonas putida strain S16. Applied and Environmental Microbiology, 74, 1567–1574. 10.1128/AEM.02529-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, H. , Yao, Y. , Zhang, D. , Meng, X. , Wang, L. , Yu, H. , … Xu, P. (2011). A Novel NADH‐dependent and FAD‐containing hydroxylase is crucial for nicotine degradation by Pseudomonas putida . Journal of Biological Chemistry, 286, 39179–39187. 10.1074/jbc.M111.283929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urquhart, E. J. , & Punja, Z. K. (2002). Hydrolytic enzymes and antifungal compounds produced by Tilletiopsis species, phyllosphere yeasts that are antagonists of powdery mildew fungi. Canadian Journal of Microbiology, 48, 219–229. 10.1139/w02-008 [DOI] [PubMed] [Google Scholar]
- Vaclavik, L. , Lacina, O. , Hajslova, J. , & Zweigenbaum, J. (2011). The use of high performance liquid chromatography‐quadrupole time‐of‐flight mass spectrometry coupled to advanced data mining and chemometric tools for discrimination and classification of red wines according to their variety. Analytica Chimica Acta, 685, 45–51. 10.1016/j.aca.2010.11.018 [DOI] [PubMed] [Google Scholar]
- Wang, F. , Zhao, H. , Xiang, H. , Wu, L. , Men, X. , Qi, C. , … Xian, M. (2018). Species diversity and functional prediction of surface bacterial communities on aging flue‐cured tobaccos. Current Microbiology, 75, 1306–1315. 10.1007/s00284-018-1525-x [DOI] [PubMed] [Google Scholar]
- Wang, M. , Yang, G. , Min, H. , & Lv, Z. (2009). A novel nicotine catabolic plasmid pMH1 in Pseudomonas sp. strain HF‐1. Canadian Journal of Microbiology, 55, 228–233. 10.1139/W08-135 [DOI] [PubMed] [Google Scholar]
- Wang, M. , Yang, G. , Wang, X. , Yao, Y. , Min, H. , & Lu, Z. (2011). Nicotine degradation by two novel bacterial isolates of Acinetobacter sp. TW and Sphingomonas sp. TY and their responses in the presence of neonicotinoid insecticides. World Journal of Microbiology and Biotechnology, 27, 1633–1640. 10.1007/s11274-010-0617-y [DOI] [Google Scholar]
- Wang, S. N. , Liu, Z. , Tang, H. Z. , Meng, J. , & Xu, P. (2007). Characterization of environmentally friendly nicotine degradation by Pseudomonas putida biotype A strain S16. Microbiology, 153, 1556–1565. 10.1099/mic.0.2006/005223-0 [DOI] [PubMed] [Google Scholar]
- Wiklund, S. , Johansson, E. , Sjöström, L. , Mellerowicz, E. J. , Edlund, U. , Shockcor, J. P. , … Trygg, J. (2008). Visualization of GC/TOF‐MS‐based metabolomics data for identification of biochemically interesting compounds using OPLS class models. Analytical Chemistry, 80, 115–122. 10.1021/ac0713510 [DOI] [PubMed] [Google Scholar]
- Xu, B. , Dai, L. , Li, J. , Deng, M. , Miao, H. , Zhou, J. , … Huang, Z. (2016). Molecular and biochemical characterization of a novel xylanase from Massilia sp. RBM26 isolated from the feces of Rhinopithecus bieti . Journal of Microbiology and Biotechnology, 26, 9–19. 10.4014/jmb.1504.04021 [DOI] [PubMed] [Google Scholar]
- Yu, J. J. , & Gong, C. R. (2009). Tobacco raw materials preliminary processing (pp. 203–239). Beijing: China Agriculture Press. [Google Scholar]
- Zhang, Q. Y. , Yuan, Y. J. , Liao, Z. M. , & Zhang, W. X. (2017). Use of microbial indicators combined with environmental factors coupled with metrology tools for discrimination and classification of Luzhou‐flavoured pit muds. Journal of Applied Microbiology, 123, 933–943. 10.1111/jam.13544 [DOI] [PubMed] [Google Scholar]
- Zhang, Q. , Yuan, Y. , Zeng, L. , Wang, S. , Tang, Q. , Wu, Z. , & Zhang, W. (2017). Discrimination of Luzhou‐flavoured fresh raw liquor distilled from Zaopei fermented in new, trend to‐be aged and aged pit mud based on their aroma and flavour compounds. Journal of the Institute of Brewing, 123, 242–251. 10.1002/jib.411 [DOI] [Google Scholar]
- Zhao, D. , Huang, R. , Zeng, J. , Yu, Z. , Liu, P. , Cheng, S. , & Wu, Q. L. (2014). Pyrosequencing analysis of bacterial community and assembly in activated sludge samples from different geographic regions in China. Applied Microbiology and Biotechnology, 98, 9119–9128. 10.1007/s00253-014-5920-3 [DOI] [PubMed] [Google Scholar]
- Zhao, L. , Zhu, C. , Gao, Y. , Wang, C. , Li, X. , Shu, M. , … Zhong, W. (2012). Nicotine degradation enhancement by Pseudomonas stutzeri ZCJ during aging process of tobacco leaves. World Journal of Microbiology and Biotechnology, 28, 2077–2086. 10.1007/s11274-012-1010-9 [DOI] [PubMed] [Google Scholar]
- Zhao, M. , Qiu, L. , Zhang, W. , Qi, W. , & Yue, X. (2000). Study on the changes of biological activity of flue‐cured tobacco leaves during aging. Journal of Huazhong Agricultural University, 19, 537–542. 10.3321/j.issn:1000-2421.2000.06.006 [DOI] [Google Scholar]
- Zhao, M. , Wang, B. , Li, F. , Qiu, L. , Li, F. , Wang, S. , & Cui, J. (2007). Analysis of bacterial communities on aging flue‐cured tobacco leaves by 16S rDNA PCR‐DGGE technology. Applied Microbiology and Biotechnology, 73, 1435–1440. 10.1007/s00253-006-0625-x [DOI] [PubMed] [Google Scholar]
- Zhong, W. , Zhu, C. , Shu, M. , Sun, K. , Zhao, L. , Wang, C. , … Chen, J. (2010). Degradation of nicotine in tobacco waste extract by newly isolated Pseudomonas sp. ZUTSKD. Bioresource Technology, 101, 6935–6941. 10.1016/j.biortech.2010.03.142 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated for this study can be found in GenBank under the accession number PRJNA498896: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA498896.