

A state-of-the-art in vitro to in silico animal body simulation successfully predicts biodistribution with cellular resolution.
Abstract
Smart drug design for antibody and nanomaterial-based therapies allows optimization of drug efficacy and more efficient early-stage preclinical trials. The ideal drug must display maximum efficacy at target tissue sites, with transport from tissue vasculature to the cellular environment being critical. Biological simulations, when coupled with in vitro approaches, can predict this exposure in a rapid and efficient manner. As a result, it becomes possible to predict drug biodistribution within single cells of live animal tissue without the need for animal studies. Here, we successfully utilized an in vitro assay and a computational fluid dynamic model to translate in vitro cell kinetics (accounting for cell-induced degradation) to whole-body simulations for multiple species as well as nanomaterial types to predict drug distribution into individual tissue cells. We expect this work to assist in refining, reducing, and replacing animal testing, while providing scientists with a new perspective during the drug development process.
INTRODUCTION
Nanomedicines (NMs) such as antibodies and synthetic nanomaterials are an attractive development to complement conventional small-molecule medicines due to their active tissue targeting, variable circulation time, adjustable biodistribution, and added stability, all stemming from their inherent physical properties (1, 2). Current techniques to quantify delivered dose rely heavily on animal testing, with sacrifice and tissue homogenization being generally required for quantitative analysis (of NM per mass of tissue). This approach brings questions about ethics and surges in time and cost (3). From a purely scientific perspective, the destruction of tissue architecture eliminates critical knowledge of NM transport within the tissue cells and vasculature.
For a drug to reach the cells within an infected tissue, it must first exhibit optimal plasma pharmacokinetics and reach tissue vasculature. Fenestration and NM diameter dictate the critical transport of the drug through the microvessel wall into an infected or normal tissue cell environment. This complexity is compounded because of the fact that the body is a working system filled with tissues of variable vasculature porosity and diameter that are highly dependent on diseased state and tissue type. Simulation-based approaches provide a means of mechanistically tracking the transport of conventional small-molecule drugs in human and animal tissues (4) to guide a more efficient drug development process (5). However, NM transport within animal systems occurs differently than for small molecules, primarily through a flux within endothelial fenestrations and active uptake within tissue cells instead of conventional tissue-blood partitioning (6–10). Efforts have been made using simulations to account for paracellular transport processes through vascular reflection coefficients (σv) representative of fenestrations in endothelial vasculature but are constrained by their optimization to NMs of one particular size (11–13), assume only two tissue pore sizes (14), and often require fitting to animal datasets. This impedes predictive capabilities, as variations in NM diameter, changes in animal species, and differences in tissue or diseased states can alter the vessel diameters, which then cause the reflection coefficient to change, thereby requiring further animal testing (15). Moreover, the unique NM transport properties that dictate interactions within cells of a tissue (16–19) are currently addressed through time-dependent cell permeation (20) and in vitro macrophage rate kinetics (21), but current simulation approaches still treat tissues as a “black box” without the critical epithelial or endothelial cell content.
Here, we report a coupled in vitro/in silico approach to predict NM biodistribution in preclinical species at the single-cell level. Our work includes a variable reflection coefficient model that accounts for differences in nanoparticle size, animal species, and vasculature pore diameters, where the variable reflection coefficient is calculated through a fluid dynamic model. This represents a substantial improvement on the two-pore formalism. We also obtained increased resolution in our simulation compared to existing models by extracting kinetics of NM–tissue cell interaction in vitro and translating these data through mechanistic biology to a fully predictive animal simulation of drug biodistribution, which now affords cellular resolution. Our approach solves problems with current in vitro analytical approaches for quantitation of cellular NM content, where results are difficult to translate directly to animal studies and lack mechanistic perspective (22–25). We also addressed current issues with cell-induced degradation of NM fluorescence that is often overlooked, which can produce false negatives (26, 27). Validation of our in vitro work was performed through simulated lysosomal analysis coupled with atomic absorption spectroscopy (AAS). Our in vivo simulation was validated against published whole-body animal data for rats, mice, and nonhuman primates (NHPs). Quantum dots (QDs) were used as a model system because of their known potential in NM while also considering their limitations such as degradation under acidic conditions with cadmium leakage from the core. We show here that we are able to fully account for the complexities in such an NM platform, and we extended this capability to antibody- and metal/polymer-based NMs.
RESULTS
Concentration data determined from in vitro fluorescence assay yield kinetics of NM-cell interactions for preclinical predictions
Once an NM has circulated through animal blood supply and entered the tissue environment, it will interact with tissue cells via adsorption, desorption, and internalization, or active uptake processes (28). To quantify this transport and accumulation within tissue cells, we exposed a nontoxic QD to cells commonly encountered by NMs upon intravenous injection: macrophages, endothelial cells, and epithelial (liver and kidney) cells.
Fluorescence signal is highly compatible with high-throughput screening (29), and when it is used as an indicator of the number of particles present (30), it can be a powerful tool for kinetic analysis. In this study, the fluorescence of NMs is used to both quantify NM uptake by cells and give insight into cell- or medium-induced degradation. Tissue cell NM uptake was captured through a fluorescence assay that is composed of four compartments (Fig. 1A, schematic): the cell kinetic data (CKD; NM exposure to cells with 2× wash and trypsinization), cell system interactions (CSIs; NM exposure to cells but no wash), medium and protein effects (MPEs; NM present but no cells and no wash), and cell control (CC; cells in medium and trypsin and no NM added). From this design, quantitative concentrations [nanomolar (nM) quantities] of NM interacting with cells in vitro are obtained using Eq. 1. Figure 1A shows the raw fluorescence signal of the NM in medium only or NMs exposed to cells (IMPEt and ICSIt,, respectively). A significant difference between the fluorescence signal observed for IMPE,0ort and ICSI,t compartments suggests that cell- or medium-induced degradation is present, which we found to be present with QD for all cell lines. Quantitatively comparing ICKD,t to the fluorescence signal observed for these unwashed wells can yield a raw or calibrated uptake of NMs depending on IMPEt or ICSIt being used in the denominator, respectively, in Eq. 1 (see Materials and Methods for a more detailed explanation). Raw cell uptake signals (ICKD,t; Fig. 1A) were calibrated for QD degradative effects through Eq. 1 by inclusion of ICSIt in the denominator. The fraction of the total dose remaining, when multiplied by the total exposure dose ([Dose]), will yield a nanomolar (nM) concentration. This eliminates the need for a calibration curve, as Eq. 1 intrinsically accounts for any phenomena experienced by the NM upon exposure to the cell or medium environment. These results are entered into a cell kinetics simulation to obtain rate constants of NM-cell interaction that are translatable to our whole-body (in vivo) simulation (see below). All data were above the limits of detection (LOD) and quantitation (LOQ) with minimal trypsin interference (fig. S1).
| (1) |
where [Uptake]c,t is the total calibrated concentration of NM interacting with cells at time t and [Dose] is the applied in nM. All fluorescence data obtained in Fig. 1A were transformed to NM concentration profiles (calibrated or raw), shown in Fig. 1B.
Fig. 1. In vitro application of assay using multiple cell types with simulation and rate extraction.
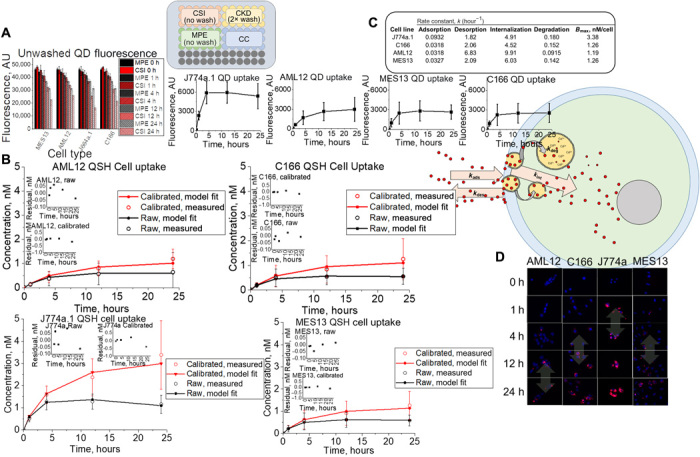
(A) Fluorescence data and assay setup representing unwashed QD (QSH) fluorescence for unwashed wells that contain kidney (MES13), liver (AML12), endothelial (C166), and macrophage (J774a.1) cell types. AU, arbitrary units. (B) Washed raw (black) and calibrated (red) fluorescence uptake concentrations (circles, measured) compared to simulation fits (lines) for liver, endothelial, macrophage, and kidney cell types. (C) Summary of rate constants optimized by the genetic algorithm for model fit to measured datasets. (D) Time-dependent confocal microscopy images of QD uptake, showing saturation (double-sided arrows) for each cell type in the study (blue and red represent cell nucleus Hoechst and QSH fluorescence, respectively). Cartoon is a visual representation of QD-cell interaction kinetics.
As Fig. 1B indicates, [Uptake]c,t values are considerably higher in macrophages (J774a.1) than in all other cell types (P < 0.05), with macrophages taking up approximately 34% of dose, followed by kidney and endothelial (12.59 and 12.56%, respectively) and liver (11.93%) cells. Overall, the assay displayed enough precision to detect differences in uptake for tissue types expected to interact with NMs in vivo.
Cell kinetics simulations were built to use the quantitative data obtained from the assay (Fig. 1B) reported here to extract rate kinetics of NM-cell interactions. The simulation consists of (i) medium, (ii) cell membrane, and (iii) cell space compartments interconnected through basic mass transfer equations and first-order rate constants. The cell kinetics simulation optimizes for adsorption, desorption, internalization, and degradation rate constants using the genetic algorithm (GA) (31) in MATLAB. All fluorescence-based concentration data shown in Fig. 1B were optimized in the cell kinetic model for parameters kads, kdes, and kint (calibrated datasets), then to raw datasets for kdeg optimization; see Supplementary Materials for details. As shown in Fig. 1C, the membrane adsorption rate constant was highest for macrophages and lowest for endothelial and liver cells. However, the internalization rate was highest for liver tissue cells and lowest for endothelial cells. Overall, each cell type had membrane adsorption as the rate-limiting step and exhibited different maximum capacities of uptake (Bmax; see Materials and Methods). Confocal imaging (Fig. 1D) visually supports these data, with rapid uptake and saturation by macrophages within 1 hour of exposure, and a more delayed uptake for the epithelial and endothelial tissue cells.
Animal tissue cell–induced degradation of NMs must be accounted for to accurately predict and translate in vitro kinetics to animal biodistribution
NMs can be prone to degradative processes within the cellular environment, which can cause fluctuations in fluorescent signal and result in inaccurate representation of tissue-level delivered dose. This issue has prevented fluorescence from being a useful tool for extracting rate kinetics. Here, we show that cellular degradative effects are present and accounted for in our in vitro assay and corresponding cell kinetics simulation. We ran QD (unstable under biological conditions) and PS (polystyrene; stable control) NMs against an aggressive murine liver Hepa1-6 cell line (32) and found that the NM prone to degradation (QD) exhibited substantial deviations in fluorescence, while PS fluorescence remained constant. Our findings in Fig. 2 show that degradative factors must be accounted for when aiming to extract rate kinetics for translation toward animal studies from in vitro fluorescence data. Degradation and stability of fluorescence signal were further observed for washed QD and PS cell samples in the well, respectively (fig. S2). Further evidence of cell-induced degradation is also supported by lysosomal colocalization studies (Fig. 2A, inset), observation of substantial Cd2+ core leakage under simulated lysosomal conditions (fig. S2) (33, 34), and fluorescence spectral data under cell exposure (fig. S2). In spite of minimal export of free Cd2+ from cells to the supernatant (fig. S2), we noticed minimal Cd2+ induced toxicity in cell culture (fig. S2) at doses performed in this study. We account for this effect through the design of our assay in conjunction with Eq. 1.
Fig. 2. In vitro assay development and validation to AAS.

(A and B) In vitro assay setup with its coupled unwashed fluorescence signal for (A) QD and (B) PS. Images show Hepa1-6 colocalization of QD in lysosomal compartments as well as snapshots of fluorescence under ultraviolet illumination under simulated lysosomal exposure conditions with pH 2.5, 3.0, 3.5, 4.0, 4.5, and 7.4 (1 to 6, respectively). (C to E) QD uptake studies using the in vitro technique with (C) fluorescence, (D) AAS, (E) combined, and the (F) statistical results using two-tailed t test when comparing raw and calibrated fluorescence uptake to AAS data. The asterisks in figure represent significance at the * (P < 0.05), ** (P < 0.01), and *** (P < 0.001) levels.
Raw QD concentrations exhibited a saturable cell uptake profile, with a maximum concentration at approximately 12 hours after cell exposure (0.228 ± 0.0852 nM) (Fig. 2C). When calibrated for degradation, QD concentrations (Fig. 2C) showed a completely different profile, with a nonsaturable uptake trend as a function of time and significant deviation between calibrated and raw concentrations at approximately 4 hours when cell-induced degradation begins (Fig. 2A). In comparison, the calibrated and raw PS uptake profiles are not statistically different (P > 0.05) and reach saturability within 1 hour of exposure (Fig. 2C, blue, solid and dashed). Overall, we find that 4.78 ± 1.22% QD and 1.07 ± 0.085% PS were adsorbed to/internalized by cells after 24 hours with respect to the initial applied dose.
Validation by AAS shows that the calibrated fluorescence data delivered by the in vitro assay are critical for accurate quantitation of cell uptake
AAS analysis validated the quantification of QD uptake through our fluorescence assay. AAS data obtained from the CSI and MPE compartments show that the cadmium concentration in both scenarios remained relatively constant at concentrations of approximately 3.60 ± 0.0602 mg/liter and 3.54 ± 0.0841 mg/liter (fig. S3D), respectively, since no cadmium is removed from the system for these samples (unwashed). Parallel studies using a sample vial of QD stock diluted equally showed no significant difference (P > 0.05; fig. S3G), indicating quantitative collection of Cd2+ from the 96-well plates. Extraction and harvest efficiencies for each time point were also determined to understand if the full dose of cadmium was extracted from the cells and harvested from the wells, with all results showing full extraction and harvest efficiency (fig. S3G). AAS data obtained from the CKD compartments (Fig. 2E) showed a gradual increase in total Cd2+ content, up to an average of 0.164 ± 0.0332 mg/liter, which corresponds to 4.56 ± 0.925% of the applied dose. Cadmium concentrations from AAS were converted to nanomolar concentrations of QD through a linear correlation of the slopes of the QD and Cd(NO3)2 AAS calibration curves (fig. S3C). We also performed a standard addition method and six-point calibration method in parallel for the 24-hour time point (fig. S3, E and F) for assay quality assurance. Results did not differ significantly (P > 0.05; fig. S3F), indicative of minimal cell matrix interference on AAS data.
Data in Fig. 2E indicate similar QD uptake for calibrated, raw, and AAS methods for up to 4 hours (P > 0.05; Fig. 2F), suggesting that no significant degradation occurs. After 4 hours, as cell-induced degradation takes effect, raw QD concentrations obtained from the in vitro assay begin to saturate and deviate from AAS (Fig. 2E). When calibrated for cell- and medium-induced degradation, AAS and fluorescence concentration uptake profiles are equivalent for all time points and do not differ with statistical significance, indicating the critical importance of calibrating for cell- and medium-induced degradation when using fluorescence as a tool for quantitative analysis.
In vitro kinetics coupled with mechanistic vasculature and tissue modeling can accurately predict tissue and individual cell uptake within an animal body for NMs of different sizes
An NM located within tissues can accumulate inside the interstitia, vasculature, or within variable cell types. To account for accumulation within variable cell types in this complex architecture, in vitro cellular kinetics were translated to our in silico animal simulation. To determine the quality of the rate constants and simulation resolution, we conducted two pilot studies in which we (i) assessed trends in whole-tissue uptake as a function of size and (ii) quantified NM uptake at the single-cell level. The architecture of the animal simulation can be seen in Fig. 3, where each tissue compartment contains four subcompartments (epithelial, endothelial, interstitial, and macrophage). For an NM to transport to the interstitia of a certain tissue, we assumed that it travels through the fenestrations unique to the capillaries of that particular tissue (Fig. 3, inset), each with variable sizes found in literature. These fenestrations were represented by reflection coefficients (σv) computed here to include the effects of particle drag and frictional hindrance (35) (Fig. 4A) akin to that of a sphere through an artificial porous membrane (36). The calculated vascular reflection coefficient was then held as a constant in a series of ordinary differential equations representing mass transfer kinetics from the blood supply to interstitial space where the NM will react with tissue cells (Fig. 4B) through first-order rate constants optimized to our in vitro data.
Fig. 3. An overall schematic of the animal NM simulation.

Scheme of the overall tissue and blood compartments that make up the full animal. Each tissue compartment is interconnected through blood flow rates and contains subcompartments (zoomed-in area), where an NM will distribute upon internalization through endothelial fenestrae.
Fig. 4. NM tracking inside tissues for NM of various sizes.

(A) Paracellular transport of NM through the endothelial fenestrae. Frictional hindrance and drag force are considered when calculating for the reflection coefficient, which is performed for each tissue type. (B) Schematic of the NM transport process through the endothelial lining into the interstitial space from where it will interact with tissue cells through rate constants. (C) Tissue-level (liver) simulation predictions for variable-sized NMs compared to observed. (D and E) Examples (D) of cellular-level simulation outputs for variable-sized NMs (E) compared to observed splenic macrophage data for validation of rate constants and simulation output. Raw simulated data using rate constants with degradative effects (dashed) show increased error >2-fold for 20 and 100 nm.
Because of the liver and spleen being common targets for NM sequestration, we used these tissues as case studies to understand and capture the sensitivity and accuracy of the simulation’s tissue and cellular outputs to changes in NM size (37, 38). Figure 4C shows that a 33 and 83% increase in particle size will produce an increase of 15 and 37% uptake in total liver tissue, respectively. When compared to observed data (37), this lies within twofold error and follows the same observed positive correlative trends between size and uptake. This phenomenon can be explained through the fact that as NM size increases, such that it exceeds other tissue pore sizes, the NMs from those tissues should then funnel toward tissues with larger pores—in this case, the liver. Variable (20 to 500 nm) diameter biodistribution analysis using Chen 2015 datasets (38) (fig. S4) supports this principle. Upon reaching the target tissue, the NM will interact with cells through rate constants determined in vitro. Here, we can see that liver endothelial cells (Fig. 4D) have direct exposure to the blood supply and thus have immediate interaction with the NMs at the tissue site, with rapid membrane binding and eventual saturation at approximately 24 hours (Alalaiwe study). As the NM enters the interstitia of the tissue, it quickly interacts with epithelial (liver, Alalaiwe study) and macrophage (spleen, Chen study) tissue cells through our in vitro rate constants. To validate overall model quantification at the single-cell level, we used splenic macrophage uptake data from the Chen study (38). When using the in vitro rate constants calibrated for degradative effects, all simulations show accurate predictions (<2-fold error) for NMs ranging from 2 to 500 nm in individual macrophages when compared to observed harvested splenic macrophages (Fig. 4E). If simulations used rate constants optimized to raw datasets, then all simulation predictions would severely underpredict NM uptake in macrophages, leading to >2-fold error for most outputs (Fig. 4E). These results build confidence in the predictive power of the simulation and capability to predict variable-sized drug content at the single-cell level for animals for the first time, strictly from in vitro data.
Tissue-level uptake of NMs can be predicted for multiple species
To further assess the simulation’s predictive power and translational capabilities, predictions were compared against measured tissue-level content for multiple NM types and species. Complete sets of physiological values (tissue volumes and blood flow rates) were obtained (39) for three species including rats, mice, and NHP to scale cellular content (epithelial, endothelial, and macrophage). The NM animal simulation was validated to 15 preclinical datasets, which included different dosing scenarios (0.029 to 64.3 mg/kg body weight), NM types (polymer, QD, metal, and antibody), and NM sizes (4 to 197 nm diameter), all shown in Fig. 5 (A to E). As most laboratory biodistribution data only capture total uptake at the tissue level, validation of our simulation included total tissue (sum of macrophage, epithelial, endothelial, vascular, and interstitial) content. Evaluations of predictive performance were performed according to World Health Organization guidelines (40) as well as standards accepted by pharmaceutical and academic consortiums (41) involved in drug development and safety. Specifically, 49.78, 33.31, and 16.90% of datasets lay within <2-fold, <3-fold, and >3-fold error, respectively, demonstrating reasonable model performance.
Fig. 5. Simulation outputs for validation to various animal studies.

(A) Tissue-level predicted (line) versus observed (points) examples for all species types (rats, mice, and NHP) for visual evidence of model capabilities. (B) Heat map of fold error analysis calculated for all tissue, NM, and species types in simulation. Green, yellow, and red represent <2-fold, <3-fold, and >3-fold errors. Fold error was calculated according to equations given in Materials and Methods, where simulated dataset averages were compared to observed. (C) All data point averages specific to tissue types for simulations and observed were plotted against each other to yield a linear fit (R2 = 0.861). (D) Tissue-specific fold errors are shown to visualize model accuracy for each tissue in this study. (E) NM-simulated outputs were ranked (1 to 4) according to fold error analysis.
Antibodies (13, 42, 43) exhibited the highest percentage of distributions lying within twofold error, followed by metal (37, 44–46), polymer (47–50), and QD-based (51–54) NMs (Fig. 5E). Simulated plasma outputs generally underpredicted uptake, most likely due to lack of immunoglobulin G (IgG)–endothelial FcRn receptor binding (55). Simulation outputs were slightly overestimated for tissues with high FcRn expression, where IgG catabolism is prominent (Fig. 5A, skin, muscle, and liver) (56). Major reticuloendothelial system tissues (liver and spleen) show differing uptake profiles and relative quantities, highly dependent on tissue macrophage content. Overall, simulations mostly fell within twofold error of observed values for many of these studies, but generally underpredict total tissue content primarily due to transcellular transport not being accounted for in our simulation (figs. S5 and S6). Peripheral tissues like the brain and lungs experienced quick saturation within the first hour, with a slight clearance profile most likely due to NM accumulation within tissue vasculature and limited permeation through the blood-brain barrier and capillary fenestrations (fig. S7) (45–47). Observed transmission electron microscope evidence supports localization within tissue vasculature, and simulation data predict and substantiate this finding (46).
For studies where metabolism was observed, our simulation either exhibited overestimations or did not fit the observed tissue profiles (fig. S8). Although reasonably matched to observed data (<2-fold error), simulations underpredicted liver and kidney uptake of 21-nm CdSeS-SiOH QD (fig. S9). Contrary to the 21-nm QD, 3.8-nm mercaptosuccinic acid (MSA)–QD exhibited minimal renal and biliary clearance (fig. S9, C and D), primarily due to considerable QD accumulation within interstitial space (30 min) and gradual accumulation within mesangial cells (53) after prolonged periods (fig. S9E). Moreover, electrostatic repulsion from the proteoglycans in the basement membrane of the glomerulus fenestrae on the small QD is believed to influence minimal renal elimination, which can be addressed through additional computational approaches using surface potential of endothelial cell membranes (57) and subsequent (58) repulsion. Overall, deviations between simulation and empirical datasets are due to some factors not being accounted for yet, such as additional physiological transport processes (transcellular and electrostatic interactions). Future work may address NM size distribution effects by including a fractional NM population approach, which would more accurately capture particle transport through vasculature fenestrae when NM physicochemical data are available. This would be mostly applicable to deliberate exposure scenarios where NMs are fully characterized, e.g., therapeutics.
Our simulation reasonably predicts biodistribution for polyethylene glycol, molecular weight 2000 (PEG-2000)– and PEG-5000–coated Au, with plasma concentrations lying within 1.45 and 1.52 average fold error (AFE), respectively (fig. S10). However, our simulation does not account for blood half-life as a function of trends in PEG chain length (resistance to aggregation by providing minimal protein adsorption), so this effect is not expressed in simulation outputs thus far (59). We expect that, as transcellular transport is included as an additional tissue uptake mechanism, NM should escape plasma and enter the tissues, thereby correcting the 66.67% simulated overestimations.
Overall, NMs between 10 and 100 nm diameter (n = 65) exhibited the best predictive capabilities for our simulation (>50% below twofold error for all tissues), most likely due to the fact that (i) cellular rate constants obtained in vitro translated well to the live animal simulation, (ii) paracellular transport through endothelial fenestrations was the primary means of tissue transport, and (iii) biliary and/or renal clearance of NM minimally affected biodistribution. Approximately 50% of tissues for datasets containing NMs with diameters <10 nm (n = 10) lie above threefold error, and 40% lie below twofold error. For NMs >100 nm in diameter (n = 16), the simulation demonstrated similar errors within all error ranges (36.43, 25.71, and 37.86%; <2-fold, <3-fold, and >3-fold error, respectively). Here, we suspect NM size to exceed pore diameter, limiting tissue uptake, so transcellular transport would be necessary to fully capture tissue uptake. Simulated and observed datasets were compared through log analysis of the averages of the datasets and plotted against each other, as shown in Fig. 5C. Model-predicted averages and observed averages were fairly linearly correlated, indicative of reasonable model predictivity across all species and NM types (R2 = 0.861).
DISCUSSION
We have demonstrated a viable platform for reduction or refinement of animal testing during NM development. The in vitro concentration data from our fluorescence assay coupled with our cell kinetics simulation deliver rate constants for cell-NM interaction (60) that show excellent translatability to our mechanistic predictive in silico whole-body animal simulation. Our approach does not require fitting to animal biodistribution datasets, as all parameters are optimized to in vitro data or calculated (in the case of the variable reflection coefficient) through a fluid dynamic model. We have also shown that the process works under conditions where the NM experiences degradation by the biological environment. Our work expands on extensive simulation efforts completed for biologics (39) and conventional small molecules, for which a more mechanistic simulation can be beneficial to inform proper dosing scenarios for effective treatment. More recent simulations have started to allow modeling of biodistribution in animals for NMs but often fit tissue and blood partition coefficients to animal data to describe equilibrium partitioning between the two phases. Our work makes substantial advances on the current state of the field through the implementation of a mechanistic fluid dynamic simulation coupled with in vitro cell interaction kinetics to provide a much higher resolution of NM distribution and quantitation.
The combination of in vitro and in silico methods discussed here is expected to assist future smart drug design and can be a platform to help scientists make better informed decisions while reducing live animal testing. For example, NMs sequestered in the interstitia of a tissue alone may not have a therapeutic or toxic effect on the cells, so it is critical to track cell uptake phenomena. As we can see from our simulation results, epithelial tissue cells require substantial time to internalize drug, in some cases up to 48 hours. Furthermore, only 0.94% of the total tissue dose may be located within the epithelial cells, even though the total liver tissue would have taken up 14.0% of the injected intravenous dose. In this case, perhaps the scientist may want to increase the injected dose, change the size of the NM, or completely alter the surface chemistry and run the in vitro assay/simulations again to obtain higher rate constants.
This work is also important in understanding unintended sequestration of NM in peripheral organs, especially when pore sizes are influential factors. Fenestrae diameter can change across species types as well as differ for normal and diseased states of the same tissue (61). Care was taken to ensure that reasonable fenestra diameters were chosen for all tissues in the normal state, especially those where NMs are commonly known to sequester (e.g., liver and spleen). In cases where NM diameter was substantially less than tissue fenestrae (e.g., splenic sinusoids), more flexibility was allowed when choosing the pore size for the fluid dynamic model, as there is limited drag and friction effects present when there are two or more order of magnitude difference between the NM and pore diameters. Overall, the majority of fenestrae diameters chosen were rodent specific, especially for the liver, as liver capillary diameters can differ substantially (e.g., 280- and 110-nm upper limits for rodents and humans, respectively) (62). For the majority of tissue types, upper limits were chosen, as NMs are expected to filter through the largest pore, when present (62).
The simulations generally underpredicted the total tissue content. This can be addressed through future inclusion of transcellular transport of materials through the endothelial cell lining. When this effect is accounted for, we expect the simulations to increase predictions and match observed total tissue content. Future work is also necessary to consider additional mechanistic processes within whole-body simulations. Processes like antibody catabolism, metabolic (phase I and II) breakdown and clearance of NMs, and transcellular transport of NMs through the vascular lining will need to be included to provide additional model flexibility and predictive power. NM or antibody changes in physicochemistry (e.g., through conjugation of drugs to the antibody surface) can be further captured through the fluorescence assay via rate constant optimization. Inclusion of charge and lipophilicity effects may assist in development of a more accurate transport model. Overall, we expect this work to serve as a new approach for predictive simulation of NM transport and distribution in animal species.
MATERIALS AND METHODS
In vitro statistical analysis
Statistical analysis was performed on Microsoft Excel 2010. All calculated statistical evaluations were performed using the Student’s two-tailed t test at the P < 0.05, P < 0.01, or P < 0.001 level.
In vitro quantitation of cellular uptake
To ensure repeatability, the fluorescence assay was completed approximately 2 months apart with a new batch of thawed cells for both NM types (n = 2), with reproducible results. Comprehensive analysis on toxicity and stability (figs. S1 and S2) revealed minimal toxicity and sedimentation for a 10 nM dose of the NMs chosen for this study, which indicated minimal cell stress and constant exposure dose. All data were within the linear dynamic range, as well as within the LOD and quantitation (fig. S1, C and D). The fluorescence assay was built starting from the application of cells on a 96-well plate in three “compartments”: the CSI compartment [cells + NM (unwashed), accounts for cell-induced NM degradation], the CKD compartment [cells + NM (washed), measurement of NM uptake], and the CC compartment [cells in medium + no NM (unwashed), control with untreated cells to subtract background signal]. The MPE compartment [no cells + NM in medium (unwashed)] accounted for medium- and protein-induced degradative effects on the NM in the absence of exposure to cells. Note that the CSI and MPE compartments are never washed and therefore maintain the initial applied dose of NM (10 nM). The CKD compartment was washed at each time, t, to remove NMs that are not cell membrane bound or internalized by cells. Control experiments on blank wells showed minimal NM adhesion to the sides and surface of wells, indicating that all fluorescence should strictly come from NM interacting with cells. For assay development and validation to AAS for QD, we used 18 wells per compartment, which resulted in one 96-well plate per time point. For the application of this assay to cell types for rate extraction that we used in animal simulations, we applied QD at a dose of 10 nM to each compartment in triplicate. At the time of application, we allowed cells to reach 90% confluence and establish membrane integrity (48 hours) (63). At time 0, the CSI, CKD, and MPE compartments were dosed with 10 nM QSH or PS [10% fetal bovine serum (FBS)/Dulbecco’s Modified Eagle’s medium (DMEM) suspension], with one NM type per plate. Comparing (by t test) the fluorescence signal for wells in the CSI compartment at time t with the fluorescence signal from wells in the MPE compartment at time t gave insight into cell-induced degradation. If they were statistically different, we concluded that cell-induced NM degradation was present and the quantity of fluorescence signal loss due to this effect was determined from the difference of CSI and MPE at time t. Similarly, comparing the fluorescence signal (by t test) from wells in the MPE compartment at time t with respect to MPE at time 0 gave a description of medium-induced degradation. These critical steps guided NM uptake calculations, especially if degradation was present.
In vitro quantitation of degradation
Signals obtained from in vitro assay included
1) ICSI,0
2) ICSI,t
3) IMPE,0
4) IMPE,t
5) ICKDt
6) ICC
Overall, raw fluorescence descriptive of cell uptake (ICKD,t) was taken relative to raw fluorescence of unwashed cells at time t (ICSI,t) to obtain a calibrated fraction of uptake (fcell,c)
| (2) |
Raw fluorescence descriptive of cell uptake (ICKD,t) was also taken relative to raw fluorescence of unwashed cells at time 0 (ICSI,0) to obtain a raw fraction of uptake (fcell,r)
| (3) |
These two fractions were then used to obtain concentration of NM uptake using the general equation
| (4) |
where fcell,x was the fraction of uptake for x = raw or corrected, [Uptake]t was the concentration of NM taken up by cells (nM), and [Dose] was the applied dose in nM. To determine if cell-induced degradation was present, a two-tailed t test was performed between unwashed CSI and MPE compartments at time t (ICSIt and IMPEt, respectively). To determine if medium-induced degradation was present, a two-tailed t test was performed between unwashed MPE at time 0 and time t. Cell-induced degradation (Icdegt) was taken as the difference between unwashed without (IMPEt) and with (ICSIt) cell exposure
| (5) |
If medium degradation was present, the intensity of this degradation type was taken as the difference between unwashed wells without cell exposure from time 0 to time t.
| (6) |
Together, the sum of these values equals the total degradation that an NM can undergo for the in vitro assay (Idegt)
| (7) |
Assessment of NM toxicity
MTS (CellTiter 96 Aqueous Non-Radioactive Cell Proliferation, VWR) assay was run to determine toxicity of a variety of NMs at different doses (QSH and PS) for optimal NM exposure conditions. NMs were applied to murine Hepa1-6 cells for a period of 24 hours. Briefly, cells were seeded in triplicate onto wells of a clear flat-bottom 96-well plate at a density of 34,700 cells per well and left 24 hours for attachment. After 24 hours, medium was aspirated, and 100 μl of all NM solutions was applied to wells, except controls, for a period of 24 hours in 37°C and CO2 incubator. Negative controls were kept in medium to retain complete viability, and positive controls were kept in water for cell death. All NMs were diluted in DMEM supplemented with 10% FBS at various doses, ranging from 5 nM to approximately 250 nM. After 24 hours of exposure, cells were washed twice with complete growth medium and reapplied with 100 μl of DMEM with 10% FBS. Aliquots (20 μl) of MTS were added, and background absorbance was captured at 490 nm. Plates were then incubated for 2 hours, and absorbance was checked again. Sample absorption values were normalized to that of cells exposed to complete growth medium.
AAS quantification of Cd content within cells
Samples were collected from all compartments in the fluorescence assay at each time t, degraded equally in 33% (v/v) aqua regia (AR), and measured for absolute cadmium content. AAS measurements were referenced to a six-point cadmium calibration curve (fig. S3, A to C) constructed with equal % (v/v) AR, as well as lay above LOD and LOQ.
Lysosomal colocalization analysis
Cells were seeded onto 35-mm-diameter tissue-coated petri dishes (35mm TC-Treated Culture Dish, Corning) with 2 ml of solution (347,000 cells/ml) and left in an incubator at 37°C and 5% CO2 for 24 hours. Cells were washed once with complete growth medium, and 2 ml of 10 nM QSH solutions was added. After 24 hours, petri dishes were removed from the incubator and washed twice with complete growth medium. LysoTracker Green (DND-26, Thermo Fisher Scientific) was added at 1 μM concentration, and confocal images were obtained. Lysosomal colocalization studies were performed using a spinning-disk confocal imaging system. Z-stacks were taken at 2-μm step sizes, with a total distance of 40 μm.
Cell kinetic confocal microscopy
A paralleled series of cell kinetic samples containing QSH were analyzed for uptake using confocal microscopy. At each time point in the study, cells were washed twice, trypsinized, and transferred to 35-mm petri dishes containing 2 ml of complete growth medium. After 24 hours, cells were washed twice with complete growth medium, and Hoechst 33257 was applied. Cells were then imaged for NM uptake with twenty 2-μm step sizes.
Simulated lysosomal buffer analysis
Cellular lysosomal environment was mimicked to determine stress induced on fluorescence through lysosomal material exposure. The citric acid (>99.5%, ACS Reagent, Sigma-Aldrich)–simulated lysosome chelator buffer at pH 2.5 to 5.0 was created and used as the solvent for QSH and PS. Controls contained pH 7.4 Dulbecco’s Phosphate-Buffered Saline (DPBS) buffer solutions. More specifically, stock solutions of 0.25 and 0.19 mM solutions of sodium citrate monobasic (Anhydrous, Sigma-Aldrich) and dibasic (sesquihydrate, Sigma-Aldrich), respectively, were made. Stock solutions of 50 and 20 mM citric acid stock solution were also made in separate vials. Then, six solutions of equal concentrations of 10 nM QSH were made in either sodium citrate monobasic/dibasic with citric acid. To achieve the desired pH of 2.5, 3.0, 3.5, 4.5, or 5.0, pH was adjusted by combination of dibasic or monobasic sodium citrate stock solution with small aliquots of citric acid solutions. For size analysis, Zetasizer (Malvern) DLS measurements were obtained. Here, samples were diluted in situ in solvents of desired pH and measurements were obtained immediately after. Fluorescent plate readings were run in triplicate of 100 μl of solutions applied to wells of a 96-well plate system. Fluorescence was taken with 580- or 525-nm excitation and 595- or 620-nm emission, respectively, for QSH or PS, using a Tecan M200 plate reader. To check for Cd2+ core leakage, 10 nM QSH and PS were analyzed for fluorescence in phosphate-buffered solution (PBS), water, and simulated lysosomal buffer at pH 2.5, 4.5, and 5.0 at 0- and 24-hour exposure. For each time point, samples were collected and centrifuged at 15,000g for 20 min through an Amicon Ultra 10-kDa filter to separate possible cations from QSH. Filtrate was then analyzed for free cadmium content using a PerkinElmer atomic absorption spectrometer with a cadmium hollow cathode lamp with a wavelength of 288.65 nm. Flow rate was adjusted to 4 ml/min, and samples were run in triplicate.
Prolonged cell exposure analysis
Prolonged exposure to intracellular environment analysis was performed after washing at time t. Here, QSH or PS washed samples at time t were left to incubate to an additional 12 − x and 24 − x hours, where x is the time of wash for each particular sample. At total experimental time of 12 and 24 hours, previously washed plates were mixed and measured for fluorescence changes from their original time t. An example is shown below:
2-hour wash fluorescence ➔ 10-hour post-wash (12 − 2 hours) fluorescence ➔ 22-hour post-wash (24 − 2 hours) fluorescence.
Twelve-hour washed sample only contained 24 − x prolonged cell exposure data, and 24-hour washed sample contains no prolonged exposure, given that cell exposure was only allowed for the duration of a total time of 24 hours.
Calculation of fluorescent plate reader LOD and quantitation
LOD and LOQ were calculated from the construction of an eight-point calibration curve with concentration ranging from 0.10 to 10 or 15 nM for QSH or PS. LOD and LOQ were calculated on the basis of the SD of the response signal of the blank and slope of the linear curve through zero (equations below)
| (8) |
| (9) |
where is the SD of the blank (NM suspension in trypsin) and S is the slope of the calibration curve. All readings were performed on a Tecan M200 Pro microplate reader.
In vitro cell kinetics simulation
The overall simulation was a three-compartment model in which the laws of mass transfer kinetics were used.
Medium compartment
This compartment included the medium environment from which cells receive their respective NM dose. The initial dose condition was taken as the 10 nM applied in the fluorescence assay. The medium compartment NM dose evolution with time was then described as
| (10) |
where [Med] was the concentration (nM) of NM in medium, [Mem] was the concentration (nM) of NM adhered to the cell membrane, and kads and kdes were the first-order rate constants for adsorption and desorption to and from the cell membrane, respectively.
Cell membrane compartment
The cell membrane compartment was defined as the outer boundary of the cell with which the NM reversibly binds. This compartment separated the medium from the internal space of the cell. NMs that were internalized by the cell first adsorbed to this compartment through the adsorption rate constant, kads. Once adsorbed, NMs (i) left this compartment through desorption, kdes, or (ii) entered the cell via kint as expressed by
| (11) |
with parameters described above.
Cell space compartment
The cell space compartment received NMs that have transported inside the cell via the first-order rate constant for internalization (kint). Here, NMs can become degraded if the process occurs (determined through the fluorescence assay). The cell space compartment NM dose evolution as a function of time was then described as
| (12) |
where [Cell] was the concentration (nM) of NM in cell interior at time t and kdeg was the first-order rate constant for degradation of QD obtained from optimization to raw datasets (see below). The degradation rate constant was used to track quantities of NM degraded over the course of the experiment.
In vitro rate constant structure
First-order rate constants are variables that describe a proportion (fraction) of drug entering or leaving a specific compartment at any point in time (units of time−1). For purposes of this study, in vitro rate constants were scaled according to methods used by Li et al. (8, 64), where a total maximum first-order uptake rate (hour−1) is optimized to animal datasets according to a general equation
| (13) |
where k is a net rate of uptake in tissue (hour−1), kmax is a maximum optimized rate in tissue (hour−1), At is the amount of nanoparticles captured by tissue at time t (mg), Bmax is the maximum cell uptake capacity for nanoparticles per organ weight (mg/g tissue), and Wt is the weight of the tissue (g). Thus, the net uptake rate will decrease as the amount of nanoparticles in cells approaches the total Bmax * Wt. We used a version of this method by optimizing maximum cell membrane binding and internalization rates (hour−1) to in vitro cell uptake data according to
| (14) |
and
| (15) |
where kads and kint are the net adsorption and internalization (respectively) first-order rates of uptake similar to k from Eq. 12 (hour−1), ka and ki are the maximum GA optimized adsorption and internalization rates (hour−1), [Celltot] is the total simulated NM uptake in cells (nM), and Bmax is the total cell capacity [maximum concentration of NM taken up by cells measured in vitro (nM)]. The value of the net first-order uptake is heavily dependent on the maximum total capacity of uptake. As more NM is taken up by cells, [Celltot] approaches Bmax and drives kads toward 0, thus pushing the total uptake toward an asymptotic maximum. These maximum rates were optimized by the GA and held as constant for extrapolation to in vivo simulations, where they determined total rates of uptake (kep,ads, kep,int) based on total tissue cell composition. Specifically
| (16) |
and
| (17) |
where ka and ki are the maximum adsorption and internalization rate constants determined in vitro (hour−1); Aep is the total amount in, e.g., epithelial tissue cells (mg); and Bmax is the maximum cell uptake capacity per tissue (mg/tissue) as calculated by
| (18) |
where Bmax, vitro is the maximum cell uptake capacity in vitro (nM), Ncells, w is the number of cells per well (cells/well), Vwell is the applied volume of NM solution per well (liter), MW is the NM molecular weight (g/mol), and Ncells, t is the number of cell types per tissue type. By usage of this equation, we accounted for maximal saturable captured in in vitro when using animal simulations. Furthermore, the maximal rates of uptake were determined in vitro and are in units of hour−1. When accounting for total cell compositions in tissues as well as exposure doses, the net rates of uptake will change depending on tissue and cell types in animals. The rate constants representing desorption (kdes) and degradation (kdeg) were first order (hour−1) and are not directly involved in the internalization process, so these maximum constants were directly implemented into the animal simulation as optimized in vitro. However, we expect the total rate of desorption and degradation to change based on exposure dose as well as blood flow rates in animals. However, constant proportions (fractions) are expected to desorb and degrade as determined in vitro.
In vitro rate constant optimization
In vitro rate constants (ka, kdes, ki, and kdeg) were optimized using the GA, originally developed by Holland in the early 1970s. This was typically used as an artificial intelligence algorithm [e.g., it is used for training artificial neural networks (65)] for a more robust optimization of parameters based on evolutionary ideas of natural selection where it does not rely heavily on initial input and often leads to a global minimum (66). For optimization, rate constants were supplied by the GA to produce simulation outputs at particular time points. When fed to the model equations (described above), membrane ([Mem](t)) and cell space ([Cell](t)) simulated outputs were summed at each time point and compared to in vitro measured values using the residual sum of squares described below. To supply model equations (Eqs. 11 and 12) with rate constants, initial vectors (“chromosomes”) composed of rate constants (“genes”) were randomly populated by the GA, fed to the cell kinetic model, calculated for fitness, and underwent selection, crossover, and mutations to maximize diversity and produce better fitness at each iteration (“generation”). Specifically, Eqs. 11 and 12 were first optimized to the calibrated datasets where no degradation was present, holding kdeg at 0. Once optimized, we reconsidered QD raw concentration values containing degradation effects to determine the rate of degradation (kdeg), holding the previously optimized adsorption, desorption, and internalization rates (kads, kdes, and kint) as constant. All simulations were performed in MATLAB v2015b. Parameter optimization was implemented with the GA optimization function from the Optimization Toolbox. Parameters for estimation included the following: initial population, 300; population size, 50; generations, 100; mutation rate, mutation Gaussian; crossover rate, 0.80; selection function, stochastic uniform.
The GA was evaluated using the residual sum squares as the fitness function (Eq. 19)
| (19) |
where RSS represented the residual sum of squares from model output (mi) at time (i) to measured data (yi) for n time points. Standard error was computed as
| (20) |
where S is the standard error, RSS is the residual sum of squares, and n is the total time points.
Model output upper and lower bounds were evaluated at the 95% confidence interval through
| (21) |
where CL(95%) represented 95% confidence limit. The GA was run for 100 generations, enough to allow convergence at a fitness value representative of measured data.
Animal simulation
Nanomaterial transport to cells of a particular tissue was captured through the whole-body NM physiologically based pharmacokinetic simulation. For a nanomaterial to reach target tissue cells, it must first bypass the endothelial cell lining primarily through paracellular transport. Here, intercellular gaps are represented by a reflection coefficient, σv. The reflection coefficient built on previous studies by Bungay and Brenner (67), Lightfoot et al. (68), and Lewellen (69), where hydrodynamic transport was captured, which included steric exclusion (ϕ); hindrances to diffusion, drag, and pressure drop across the sphere [G′(α)]; and frictional interactions with the wall [F′(α)]. The NM must travel from the blood vasculature (Cv) to the interstitial space (CIS) through equation
| (22) |
where the tissue blood flow (Qt) and lung NM concentration CLUV serve as inputs to this compartment. The NM will interact with the endothelial cell membrane (Amem) of that tissue’s compartment via the adsorption (kadendo) and desorption (kdesendo) rate constants determined from in vitro data. The vasculature reflection coefficient (σV) serves as guidance for the NM to enter the interstitial space given by
| (23) |
where the individual tissue cells (epithelial and macrophages) will interact with the NM through the predetermined in vitro rate constants kad and kdes for their cell membranes (Cmem). The flow rate into the interstitial space was set to the lymphatic flow rate, and vasculature interstitial volumes (VIS) guided the concentration for this compartment. Once the NM enters the tissue cell membrane compartment
| (24) |
it will desorb via the desorption rate constant (kdes) or will be internalized into the cell space via the internalization rate constant (kint). Once inside the cell space, the NM can be thus degraded or sequestered within the cellular environment (Ccell).
| (25) |
All tissue compartments here were described as a series of differential equations designed to solve for concentrations using the MATLAB ODE solver.
NMs analyzed in animal studies for simulation
The metal-based particles were composed of PEG-2000–coated (32 nm) (37) or PEG-5000–coated Au (28 nm) (45) and PEG-2000–coated (33 nm) (46) or Poly 2 Hydroxyethyl Aspartamide (PHEA)–coated (66 nm) (44) Super paramagnetic iron oxide (SPIO) NMs. Polymer-based NMs consisted of Polyarylamide–PEG-2000 (35 nm) (47), Poly Lactic-co-Glycolic Acid (PLGA) (197 nm) (48), poly(glycolic acid) (PGA) (112 nm) (49), and PS-Polyethylene oxide (PEO) (107 nm) (50). QD studies varied substantially, as we analyzed hydroxide-coated [CdSeS-SiOH, 21 nm (52)], mercaptoundecanoic acid–coated [CdSe/ZnS-LM, 25 nm (51)], mercaptosuccinic acid–coated [CdTe/CdS-MSA, 3.8 nm (53)], and mercaptoproprionic acid–coated [CdTe-MPA, 4 nm (54)] NMs.
NM transport to tissue interstitia from blood
The transport of a nanoparticle from the blood supply to the tissue cells was simulated through fluid dynamic theory, primarily obtained from a hydrodynamic simulation of solutes through pores captured by modeling a reflection coefficient, σ, originally derived by Curry in 1974 (35). The published and validated fluid dynamic approach was folded into the body simulation to provide predictive, mechanistic method of transport of any nanoparticle from the blood supply through pores located within an impenetrable membrane to the tissues. Any nanoparticle with a particular size can be predicted to enter all tissues of the body based on transport through capillary walls of these tissues. In theory, a nanoparticle must travel through a pore to enter the interstitial space of a particular tissue. Upon encountering a pore, a nanoparticle cannot occupy positions smaller than one nanoparticle radius from the pore’s edge in blood vessel fenestrae. Nanoparticle entry through pores becomes sterically restricted as the nanoparticle approaches the size of the pore (70). This effect was described by the solute partition coefficient below
| (26) |
| (27) |
where α is the ratio of solute radius to pore radius. The partition coefficient above (∅) is the ratio of the area available to the solute to the total pore surface area, accounting for the steric hindrance the particle has upon entering the pores of the fenestrae in blood vessels. However, the particle experiences a frictional hindrance from the pore walls upon entering. The frictional hindrance factor F(α) is a factor that defines this phenomenon, capturing reduction in diffusion due to hindrance that the wall exerts on the particle through the viscosity of the fluid and was captured through the following equation
| (28) |
Additionally, the difference between the solute velocity and water velocity (drag force) was captured through the hydrodynamic function G(α) according to the following equation
| (29) |
The reflection coefficient accounts for the hydrodynamics of convection and diffusion of hard spheres within a right cylindrical pore. The reflection coefficient is independent of the number of channels. When the reflection coefficient approaches 0, the nanoparticle enters the pathways in the membrane. When it is approaching 1, the pore excludes the nanoparticle and it remains outside the pores. The equation for the variable reflection coefficient accounted for both the frictional force and drag
| (30) |
The above equation for the reflection coefficient builds on previous studies by Bungay and Brenner (67), Lightfoot et al. (68), and Lewellen (69), where hydrodynamic transport was captured, where they accounted for full hydrodynamics including steric exclusion; hindrances to diffusion, drag, and pressure drop across the sphere; and torque and rotation produced by viscous interactions with the wall. These studies all validated the assumptions to measured data for permeability across animal tissue. This formula assumes no interactions between solute particles.
Supplementary Material
Acknowledgments
We thank M. Miller for support and interest in the animal simulation project as well as T. Maxwell for sample preparation and assistance on confocal imaging and J. Cruz for her assistance with cell kinetic studies. Funding: None. Author contributions: E.P. performed the laboratory work and wrote the simulation code, and A.J.G. supervised and directed the whole project, giving needed input and direction. E.P. and A.J.G. both wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/4/eaax2642/DC1
Fig. S1. Quality assurance on fluorescence signal obtained from NMs in biological solutions.
Fig. S2. Cellular and lysosomal degradation studies supporting evidence of cellular-induced degradation on fluorescence in kinetic assay.
Fig. S3. Validation of QSH data by AAS.
Fig. S4. Cellular analysis of NM uptake in varying tissues for NM with different diameters.
Fig. S5. Liver and spleen total and cellular uptake of NM for multiple species and NM types.
Fig. S6. Total tissue content for mouse intravenous dosing studies.
Fig. S7. Brain and lung NM subtissue content.
Fig. S8. Analysis of tissues involved in NM metabolism.
Fig. S9. Liver and kidney total NM content for 21- and 3.8-nm-sized QDs.
Fig. S10. Plasma NM content and correlation with PEG chain length.
REFERENCES AND NOTES
- 1.Ventola C. L., Progress in nanomedicine: Approved and investigational nanodrugs. P T 42, 742–755 (2017). [PMC free article] [PubMed] [Google Scholar]
- 2.Rudramurthy G. R., Swamy M. K., Potential applications of engineered nanoparticles in medicine and biology: An update. J. Biol. Inorg. Chem. 23, 1185–1204 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Burden N., Aschberger K., Chaudhry Q., Clift M. J. D., Fowler P., Johnston H., Landsiedel R., Rowland J., Stone V., Doak S. H., Aligning nanotoxicology with the 3Rs: What is needed to realise the short, medium and long-term opportunities? Regul. Toxicol. Pharmacol. 91, 257–266 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Brown R. P., Delp M. D., Lindstedt S. L., Rhomberg L. R., Beliles R. P., Physiological parameter values for physiologically based pharmacokinetic models. Toxicol. Ind. Health 13, 407–484 (1997). [DOI] [PubMed] [Google Scholar]
- 5.Wagner C., Zhao P., Pan Y., Hsu V., Grillo J., Huang S. M., Sinha V., Application of physiologically based pharmacokinetic (PBPK) modeling to support dose selection: Report of an FDA public workshop on PBPK. CPT Pharmacometrics Syst. Pharmacol. 4, 226–230 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McSweeney M. D., Wessler T., Price L. S. L., Ciociola E. C., Herity L. B., Piscitelli J. A., Zamboni W. C., Forest M. G., Cao Y., Lai S. K., A minimal physiologically based pharmacokinetic model that predicts anti-PEG IgG-mediated clearance of PEGylated drugs in human and mouse. J. Control. Release 284, 171–178 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carlander U., Li D., Jolliet O., Emond C., Johanson G., Toward a general physiologically-based pharmacokinetic model for intravenously injected nanoparticles. Int. J. Nanomedicine 11, 625–640 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li D., Johanson G., Emond C., Carlander U., Philbert M., Jolliet O., Physiologically based pharmacokinetic modeling of polyethylene glycol-coated polyacrylamide nanoparticles in rats. Nanotoxicology 8 ( suppl. 1), 128–137 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Lin Z., Monteiro-Riviere N. A., Riviere J. E., A physiologically based pharmacokinetic model for polyethylene glycol-coated gold nanoparticles of different sizes in adult mice. Nanotoxicology 10, 162–172 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Lin Z., Monteiro-Riviere N. A., Kannan R., Riviere J. E., A computational framework for interspecies pharmacokinetics, exposure and toxicity assessment of gold nanoparticles. Nanomedicine 11, 107–119 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Baxter L. T., Zhu H., Mackensen D. G., Jain R. K., Physiologically based pharmacokinetic model for specific and nonspecific monoclonal antibodies and fragments in normal tissues and human tumor xenografts in nude mice. Cancer Res. 54, 1517–1528 (1994). [PubMed] [Google Scholar]
- 12.Covell D. G., Barbet J., Holton O. D., Black C. D. V., Parker R. J., Weinstein J. N., Pharmacokinetics of monoclonal immunoglobulin G1, F(ab′)2, and Fab′ in mice. Cancer Res. 46, 3969–3978 (1986). [PubMed] [Google Scholar]
- 13.Garg A., Balthasar J. P., Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J. Pharmacokinet. Pharmacodyn. 34, 687–709 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Niederalt C., Kuepfer L., Solodenko J., Eissing T., Siegmund H.-U., Block M., Willmann S., Lippert J., A generic whole body physiologically based pharmacokinetic model for therapeutic proteins in PK-Sim. J. Pharmacokinet. Pharmacodyn. 45, 235–257 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen T., Guan S., Gan Z., Zhang G., Yu Q., Polymeric micelles with uniform surface properties and tunable size and charge: Positive charges improve tumor accumulation. Biomacromolecules 17, 1801–1810 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Treuel L., Jiang X., Nienhaus G. U., New views on cellular uptake and trafficking of manufactured nanoparticles. J. R. Soc. Interface 10, 20120939 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Praetorius A., Tufenkji N., Goss K.-U., Scheringer M., von der Kammer F., Elimelech M., The road to nowhere: Equilibrium partition coefficients for nanoparticles. Environ. Sci. Nano 1, 317–323 (2014). [Google Scholar]
- 18.Beddoes C. M., Case C. P., Briscoe W. H., Understanding nanoparticle cellular entry: A physicochemical perspective. Adv. Colloid Interface Sci. 218, 48–68 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Ahn S., Seo E., Kim K., Lee S. J., Controlled cellular uptake and drug efficacy of nanotherapeutics. Sci. Rep. 3, 1997 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bachler G., Losert S., Umehara Y., von Goetz N., Rodriguez-Lorenzo L., Petri-Fink A., Rothen-Rutishauser B., Hungerbuehler K., Translocation of gold nanoparticles across the lung epithelial tissue barrier: Combining in vitro and in silico methods to substitute in vivo experiments. Part. Fibre Toxicol. 12, 18 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henrique Silva A., Lima E. Jr., Vasquez Mansilla M., Zysler R. D., Mojica Pisciotti M. L., Locatelli C., Reddy Rajoli R. K., Owen A., Creczynski-Pasa T. B., Siccardi M., A physiologically based pharmacokinetic model to predict the superparamagnetic iron oxide nanoparticles (SPIONs) accumulation in vivo. Eur. J. Nanomed. 9, 79–90 (2017). [Google Scholar]
- 22.Vanhecke D., Rodriguez-Lorenzo L., Clift M. J. D., Blank F., Petri-Fink A., Rothen-Rutishauser B., Quantification of nanoparticles at the single-cell level: An overview about state-of-the-art techniques and their limitations. Nanomedicine 9, 1885–1900 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Monteiro-Riviere N. A., Inman A. O., Zhang L. W., Limitations and relative utility of screening assays to assess engineered nanoparticle toxicity in a human cell line. Toxicol. Appl. Pharmacol. 234, 222–235 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Collins A. R., Annangi B., Rubio L., Marcos R., Dorn M., Merker C., Estrela-Lopis I., Cimpan M. R., Ibrahim M., Cimpan E., Ostermann M., Sauter A., El Yamani N., Shaposhnikov S., Chevillard S., Paget V., Grall R., Delic J., Goñi-de-Cerio F., Suarez-Merino B., Fessard V., Hogeveen K. N., Fjellsbø L. M., Pran E. R., Brzicova T., Topinka J., Silva M. J., Leite P. E., Ribeiro A. R., Granjeiro J. M., Grafström R., Prina-Mello A., Dusinska M., High throughput toxicity screening and intracellular detection of nanomaterials. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 9, e1413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gutiérrez L., Romero S., da Silva G. B., Costo R., Vargas M. D., Ronconi C. M., Serna C. J., Veintemillas-Verdaguer S., del Puerto Morales M., Degradation of magnetic nanoparticles mimicking lysosomal conditions followed by AC susceptibility. Biomed. Tech. 60, 417–425 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Soenen S., Abe S., Manshian B., Aubert T., Hens Z., De Smedt S., Braeckmans K., The effect of intracellular degradation on cytotoxicity and cell labeling efficacy of inorganic ligand-stabilized colloidal CdSe/CdS quantum dots. J. Biomed. Nanotechnol. 11, 631–643 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Kelf T. A., Sreenivasan V. K. A., Sun J., Kim E. J., Goldys E. M., Zvyagin A. V., Non-specific cellular uptake of surface-functionalized quantum dots. Nanotechnology 21, 285105 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Feng Q., Liu Y., Huang J., Chen K., Huang J., Xiao K., Uptake, distribution, clearance, and toxicity of iron oxide nanoparticles with different sizes and coatings. Sci. Rep. 8, 2082 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rizk M. A., El-Sayed S. A. E., AbouLaila M., Eltaysh R., Yokoyama N., Igarashi I., Performance and consistency of a fluorescence-based high-throughput screening assay for use in Babesia drug screening in mice. Sci. Rep. 7, 12774 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Faria M., Björnmalm M., Thurecht K. J., Kent S. J., Parton R. G., Kavallaris M., Johnston A. P. R., Gooding J. J., Corrie S. R., Boyd B. J., Thordarson P., Whittaker A. K., Stevens M. M., Prestidge C. A., Porter C. J. H., Parak W. J., Davis T. P., Crampin E. J., Caruso F., Minimum information reporting in bio–nano experimental literature. Nat. Nanotechnol. 13, 777–785 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.J. H. Holland, Adaptation in Natural and Artificial Systems (University of Michigan Press, 1975). [Google Scholar]
- 32.Tang Y., Han S., Liu H., Chen X., Huang L., Li X., Zhang J., The role of surface chemistry in determining in vivo biodistribution and toxicity of CdSe/ZnS core-shell quantum dots. Biomaterials 34, 8741–8755 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Zhu Z.-J., Yeh Y.-C., Tang R., Yan B., Tamayo J., Vachet R. W., Rotello V. M., Stability of quantum dots in live cells. Nat. Chem. 3, 963–968 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Derfus A. M., Chan W. C. W., Bhatia S. N., Probing the cytotoxicity of semiconductor quantum dots. Nano Lett. 4, 11–18 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Curry F. E., A hydrodynamic description of the osmotic reflection coefficient with application to the pore theory of transcapillary exchange. Microvasc. Res. 8, 236–252 (1974). [DOI] [PubMed] [Google Scholar]
- 36.Pappenheimer J. R., Renkin E. M., Borrero L. M., Filtration, diffusion and molecular sieving through peripheral capillary membranes. Am. J. Physiol. 167, 13–46 (1951). [DOI] [PubMed] [Google Scholar]
- 37.Alalaiwe A., Roberts G., Carpinone P., Munson J., Roberts S., Influence of PEG coating on the oral bioavailability of gold nanoparticles in rats. Drug Deliv. 24, 591–598 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen K.-H., Lundy D. J., Toh E. K.-W., Chen C.-H., Shih C., Chen P., Chang H.-C., Lai J. J., Stayton P. S., Hoffman A. S., Hsieh P. C.-H., Nanoparticle distribution during systemic inflammation is size-dependent and organ-specific. Nanoscale 7, 15863–15872 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Shah D. K., Betts A. M., Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J. Pharmacokinet. Pharmacodyn. 39, 67–86 (2012). [DOI] [PubMed] [Google Scholar]
- 40.World Health Organization , Characterization and application of physiologically based pharmacokinetic models in risk assessment. Int. Prog. Chem. Safety 9, 1–91 (2010). [Google Scholar]
- 41.Poulin P., Jones H. M., Do Jones R., Yates J. W., Gibson C. R., Chien J. Y., Ring B. J., Adkison K. K., He H., Vuppugalla R., Marathe P., Fischer V., Dutta S., Sinha V. K., Björnsson T., Lavé T., Ku M. S., PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 1: Goals, properties of the PhRMA dataset, and comparison with literature datasets. J. Pharm. Sci. 100, 4050–4073 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Bazin-Redureau M. I., Renard C. B., Scherrmann J.-M. G., Pharmacokinetics of heterologous and homologous immunoglobulin G, F(ab’)2 and Fab after intravenous administration in the rat. J. Pharm. Pharmacol. 49, 277–281 (1997). [DOI] [PubMed] [Google Scholar]
- 43.Vasicek L. A., Spellman D. S., Hsieh S., Seghezzi W., Zhang S., Santostefano M., Bateman K. P., Quantitation of a therapeutic antibody in serum using intact sequential affinity capture, trypsin digestion, and LC-MS/MS. Anal. Chem. 90, 866–871 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Park J., Cho W., Park H. J., Cha K. H., Ha D. C., Choi Y. W., Lee H. Y., Cho S. H., Hwang S. J., Biodistribution of newly synthesized PHEA-based polymer-coated SPION in Sprague Dawley rats as magnetic resonance contrast agent. Int. J. Nanomed. 8, 4077–4089 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cho W.-S., Cho M., Jeong J., Choi M., Cho H.-Y., Han B. S., Kim S. H., Kim H. O., Lim Y. T., Chung B. H., Jeong J., Acute toxicity and pharmacokinetics of 13 nm-sized PEG-coated gold nanoparticles. Toxicol. Appl. Pharmacol. 236, 16–24 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Chiarelli P. A., Revia R. A., Stephen Z. R., Wang K., Jeon M., Nelson V., Kievit F. M., Sham J., Ellenbogen R. G., Kiem H.-P., Zhang M., Nanoparticle biokinetics in mice and nonhuman primates. ACS Nano 11, 9514–9524 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wenger Y., Schneider R. J. II, Reddy G. R., Kopelman R., Jolliet O., Philbert M. A., Tissue distribution and pharmacokinetics of stable polyacrylamide nanoparticles following intravenous injection in the rat. Toxicol. Appl. Pharmacol. 251, 181–190 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tosi G., Vergoni A. V., Ruozi B., Bondioli L., Badiali L., Rivasi F., Costantino L., Forni F., Vandelli M. A., Sialic acid and glycopeptides conjugated PLGA nanoparticles for central nervous system targeting: In vivo pharmacological evidence and biodistribution. J. Control. Release 145, 49–57 (2010). [DOI] [PubMed] [Google Scholar]
- 49.Abellan-Pose R., Rodríguez-Évora M., Vicente S., Csaba N., Évora C., Alonso M. J., Delgado A., Biodistribution of radiolabeled polyglutamic acid and PEG-polyglutamic acid nanocapsules. Eur. J. Pharm. Biopharm. 112, 155–163 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Jennings L., Ivashchenko O., Marsman I. J., Laan A. C., Denkova A. G., Waton G., Beekman F. J., Schosseler F., Mendes E., In vivo biodistribution of stable spherical and filamentous micelles probed by high-sensitivity SPECT. Biomater. Sci. 4, 1202–1211 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Fischer H. C., Liu L., Pang K. S., Chan W. C. W., Pharmacokinetics of nanoscale quantum dots: In vivo distribution, sequestration, and clearance in the rat. Adv. Funct. Mater. 16, 1299–1305 (2006). [Google Scholar]
- 52.Chen Z., Chen H., Meng H., Xing G., Gao X., Sun B., Shi X., Yuan H., Zhang C., Liu R., Zhao F., Zhao Y., Fang X., Bio-distribution and metabolic paths of silica coated CdSeS quantum dots. Toxicol. Appl. Pharmacol. 230, 364–371 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Liang X., Wang H., Zhu Y., Zhang R., Cogger V. C., Liu X., Xu Z. P., Grice J. E., Roberts M. S., Short- and long-term tracking of anionic ultrasmall nanoparticles in kidney. ACS Nano 10, 387–395 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Su Y., Peng F., Jiang Z., Zhong Y., Lu Y., Jiang X., Huang Q., Fan C., Lee S.-T., He Y., In vivo distribution, pharmacokinetics, and toxicity of aqueous synthesized cadmium-containing quantum dots. Biomaterials 32, 5855–5862 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Brambell F. W. R., Hemmings W. A., Morris I. G., A theoretical model of γ-globulin catabolism. Nature 203, 1352–1355 (1964). [DOI] [PubMed] [Google Scholar]
- 56.Henderson L. A., Baynes J. W., Thorpe S. R., Identification of the sites of IgG catabolism in the rat. Arch. Biochem. Biophys. 215, 1–11 (1982). [DOI] [PubMed] [Google Scholar]
- 57.Tan Q., Ferrier G. A., Chen B. K., Wang C., Sun Y., Quantification of the specific membrane capacitance of single cells using a microfluidic device and impedance spectroscopy measurement. Biomicrofluidics 6, 34112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Filippov A. N., Iksanov R. K., Volkov A. V., Interaction of a charged spherical particle with a pore of a charged hydrophobic membrane in an electrolyte solution. Petrol. Chem. 51, 536–541 (2011). [Google Scholar]
- 59.Lipka J., Semmler-Behnke M., Sperling R. A., Wenk A., Takenaka S., Schleh C., Kissel T., Parak W. J., Kreyling W. G., Biodistribution of PEG-modified gold nanoparticles following intratracheal instillation and intravenous injection. Biomaterials 31, 6574–6581 (2010). [DOI] [PubMed] [Google Scholar]
- 60.Price E., Gesquiere A. J., An in vitro assay and artificial intelligence approach to determine rate constants of nanomaterial-cell interactions. Sci. Rep. 9, 13943 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Braet F., Wisse E., Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: A review. Comp. Hepatol. 1, 1 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sarin H., Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J. Angiogenesis Res. 2, 14 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Serdiuk T., Alekseev S., Lysenko V., Skryshevsky V., Géloën A., Trypsinization-dependent cell labeling with fluorescent nanoparticles. Nanoscale Res. Lett. 9, 568 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li D., Morishita M., Wagner J. G., Fatouraie M., Wooldridge M., Eagle W. E., Barres J., Carlander U., Emond C., Jolliet O., In vivo biodistribution and physiologically based pharmacokinetic modeling of inhaled fresh and aged cerium oxide nanoparticles in rats. Part. Fibre Toxicol. 13, 45 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fernandez M., Caballero J., Fernandez L., Sarai A., Genetic algorithm optimization in drug design QSAR: Bayesian-regularized genetic neural networks (BRGNN) and genetic algorithm-optimized support vectors machines (GA-SVM). Mol. Divers. 15, 269–289 (2011). [DOI] [PubMed] [Google Scholar]
- 66.Kuo H.-C., Lin C.-H., A directed genetic algorithm for global optimization. Appl. Math Comput. 219, 7348–7364 (2013). [Google Scholar]
- 67.Bungay P. M., Brenner H., Pressure drop due to the motion of a sphere near the wall bounding a Poiseuille flow. J. Fluid Mech. 60, 81–96 (2006). [Google Scholar]
- 68.Lightfoot E. N., Bassingthwaighte J. B., Grabowski E. F., Hydrodynamic Models for Diffusion in Microporous Membranes. Ann. Biomed. Eng. 4, 78–90 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.P. Lewellen, Hydrodynamic Analysis of Microporous Mass Transport, thesis, University of Wisconsin–Madison (1982). [Google Scholar]
- 70.Bassingthwaighte J. B., A practical extension of hydrodynamic theory of porous transport for hydrophilic solutes. Microcirculation 13, 111–118 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fujita T., Tokunaga J., Edanaga M., Scanning electron microscopy of the glomerular filtration membrane in the rat kidney. Cell Tissue Res. 166, 299–314 (1976). [DOI] [PubMed] [Google Scholar]
- 72.Korneliussen H., Fenestrated blood capillaries and lymphatic capillaries in rat skeletal muscle. Cell Tissue Res. 163, 169–174 (1975). [DOI] [PubMed] [Google Scholar]
- 73.Weihe E., Kalmbach P., Ultrastructure of capillaries in the conduction system of the heart in various mammals. Cell Tissue Res. 192, 77–87 (1978). [DOI] [PubMed] [Google Scholar]
- 74.Simionescu M., Simionescu N., Palade G. E., Morphometric data on the endothelium of blood capillaries. J. Cell Biol. 60, 128–152 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Okada H., Takemura G., Suzuki K., Oda K., Takada C., Hotta Y., Miyazaki N., Tsujimoto A., Muraki I., Ando Y., Zaikokuji R., Matsumoto A., Kitagaki H., Tamaoki Y., Usui T., Doi T., Yoshida T., Yoshida S., Ushikoshi H., Toyoda I., Ogura S., Three-dimensional ultrastructure of capillary endothelial glycocalyx under normal and experimental endotoxemic conditions. Crit. Care 21, 261 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Imayama S., Scanning and transmission electron microscope study on the terminal blood vessels of the rat skin. J. Invest. Dermatol. 76, 151–157 (1981). [DOI] [PubMed] [Google Scholar]
- 77.Townsley M. I., Structure and composition of pulmonary arteries, capillaries, and veins. Comp. Physiol. 2, 675–709 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/4/eaax2642/DC1
Fig. S1. Quality assurance on fluorescence signal obtained from NMs in biological solutions.
Fig. S2. Cellular and lysosomal degradation studies supporting evidence of cellular-induced degradation on fluorescence in kinetic assay.
Fig. S3. Validation of QSH data by AAS.
Fig. S4. Cellular analysis of NM uptake in varying tissues for NM with different diameters.
Fig. S5. Liver and spleen total and cellular uptake of NM for multiple species and NM types.
Fig. S6. Total tissue content for mouse intravenous dosing studies.
Fig. S7. Brain and lung NM subtissue content.
Fig. S8. Analysis of tissues involved in NM metabolism.
Fig. S9. Liver and kidney total NM content for 21- and 3.8-nm-sized QDs.
Fig. S10. Plasma NM content and correlation with PEG chain length.