

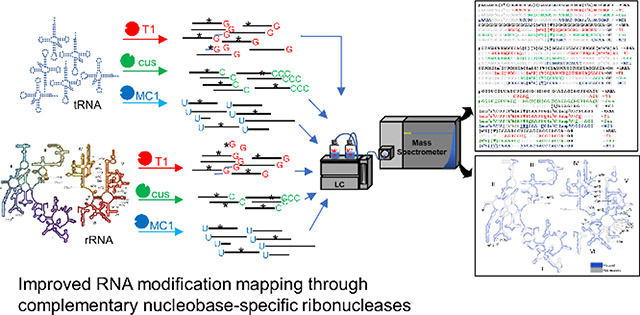
Abstract
Locating ribonucleoside modifications within an RNA sequence requires digestion of the RNA into oligoribonucleotides of amenable size for subsequent analysis by LC-MS (liquid chromatography-mass spectrometry). This approach, widely referred to as RNA modification mapping, is facilitated through ribonucleases (RNases) such as T1 (guanosine-specific), U2 (purine-selective) and A (pyrimidine-specific) among others. Sequence coverage by these enzymes depends on positioning of the recognized nucleobase (such as guanine or purine or pyrimidine) in the sequence and its ribonucleotide composition. Using E. coli transfer RNA (tRNA) and ribosomal RNA (rRNA) as model samples, we demonstrate the ability of complementary nucleobase-specific ribonucleases cusativin (C-specific) and MC1 (U-specific) to generate digestion products that facilitate confident mapping of modifications in regions such as G-rich and pyrimidine-rich segments of RNA, and to distinguish C to U sequence differences. These enzymes also increase the number of oligonucleotide digestion products that are unique to a specific RNA sequence. Further, with these additional RNases, multiple modifications can be localized with high confidence in a single set of experiments with minimal dependence on the individual tRNA abundance in a mixture. The sequence overlaps observed with these complementary digestion products and that of RNase T1 improved sequence coverage to 75% or above. A similar level of sequence coverage was also observed for the 2904-nt long 23S rRNA indicating their utility has no dependence on RNA size. Wide-scale adoption of these additional modification mapping tools could help expedite the characterization of modified RNA sequences to understand their structural and functional role in various living systems.
Keywords: RNA modification mapping, nucleoside substitutions, overlapping digestion products, RNase T1, MC1, cusativin, LC-MS
Graphical Abstract
Introduction
RNA is decorated with post-transcriptional nucleoside modifications in almost all organisms.1 Such additional chemical groups on canonical ribonucleosides can affect RNA stability,2 folding,3, 4 and decoding during ribosome mediated mRNA translation.5, 6 Not surprisingly, these modifications have been implicated in regulation of gene expression,7, 8 immunomodulation,9, 10 stem cell identity,11 development,12, 13 cancer,14, 15 metabolic health of offspring16 and other human diseases.17–19 Identification of the potential locations of modification in the RNA of interest is critical to understanding its structure-based function in a given biochemical event that lead to disease. The vast majority of location-specific information of RNA modifications is obtained through deep sequencing20 or mass spectrometric technologies.
The high-throughput techniques based on next-generation sequencing (NGS) were initially developed for rapid characterization of m6A (N6-methyladenosine) sites by MeRIP21 or m6A-seq22 in mRNAs. Subsequently, the NGS-based deep sequencing methodology has been expanded to the detection of 1-methyladenosine (m1A)23, pseudouridine (Ψ or Y)24–26, 5-methylcytidine (m5C)27, 5-hydroxymethylcytidine (hm5C)28, ribose methylations (Nm)29, N4-acetylcytidine (ac4C)30, 7-methylguanosine (m7G)31, and 3-methylcytidine (m3C)32. These methods generally depend on antibody-based enrichment and immunocapturing, reverse transcriptase-signatures (either arrest or misincorporation at m3C, m1A, 1-methylinosine (m1I), N2,N2-dimethylguanosine (m22G), and 1-methylguanosine (m1G) sites, engineered enzymes and substrates (KlenTaq DNA polymerase for 2’-O-methylations) for error-prone cDNA synthesis33, specific chemical reactivity to the modified nucleoside (such as m7G, Ψ) or protection from cleavage (Nm) (reviewed in ref34). Thus, these methodologies are designed to locate modifications one (or two) at a time in an indirect fashion using small amounts of RNA (~10 ng). While these methodologies are being widely adopted for mRNA analysis, limitations such as frequent occurrence of false positives and negatives due to systematic errors and base-calling qualities were noticed.35 Moreover, detection is limited to those nucleosides where antibodies or selective chemical reactivity is available.36 Apart from mapping, NGS-based deep sequencing has also been used for identification and measurement of changes in abundance of RNA species following dealkylation with enzymes, such as AlkB, a method referred to as ARM-seq37 or DM-tRNA-seq38.
Mass spectrometry (MS) provides direct readout of the resident modifications in an unambiguous manner as the modification yields an increase in the mass of the canonical ribonucleoside, which is readily detected by liquid chromatography coupled with mass spectrometry (LC-MS) analysis. The McCloskey group performed initial identification and location of modified ribonucleosides in transfer RNA (tRNA)39, 40 and ribosomal RNA(rRNA)41–44 through a process referred to as RNA modification mapping.45 In this bottom-up approach, the modified RNA sequence is reconstructed from the constituent modified nucleosides (determined from separate analysis) and oligonucleotide digestion products generated by treatment with nucleobase-specific ribonucleases.46, 47, 48 Selective cleavage of RNA has an advantage of restricting the compositional value of one of the four ribonucleotides to a single residue in an oligonucleotide digestion product. Such restriction simplifies the MS data analysis due to a decrease in number of allowable base compositions for a given mass measurement.45 Such an approach is highly suited for RNAs that have high density of modifications as they generally interfere with deep-sequencing procedures.
In practice, the RNA is treated with RNases such as T1, A or U2 and the resulting digestion products are separated on a reverse phase column in combination with ion pairing reagent to reduce the complexity of analytes entering the mass spectrometer. The oligonucleotide anions in the chromatographic eluent enter into the gas phase through electrospray ionization and are further resolved based on their mass-to-charge (m/z) values. Mass-based selection and collision-induced dissociation (CID) of oligonucleotide anions (referred to as tandem mass spectrometry or MS/MS) yield sequence-informative product (fragment) ions49 with the attached chemical group. Matching oligonucleotide digestion products to the specific RNA sequence depend on their length and uniqueness. This is in turn determined by the nucleotide composition, distribution of the recognized nucleobase in the sequence. If the nucleotide distribution contained multiple pyrimidines or purines in the sequence, ambiguities and uncertainties arise during modified ribonucleotide sequence construction. To increase sequence coverage and enhance the placement of modified nucleosides, the use of more than one ribonuclease is common. For example, RNase T1 recognizes guanosine (rG), and if the RNA is rich in G-residues, T1 yield monomers (GMP) to smaller oligomers (< 4-mers) that may not be uniquely matched to the RNA sequence. Therefore, the RNA is generally digested with multiple enzymes such as pyrimidine-specific RNase A and/or purine-selective U2, so that the information from these digestion products could fill the gaps left by RNase T1 coverage.
The complexity of analysis further increases when dealing with mixtures such as total tRNA. For example, E. coli and yeast have 40–50 different tRNA sequences (referred to as isodecoders), and humans could have as many as 274 isodecoders. To decrease the complexity (and the associated uncertainty), Suzuki’s lab has developed chromatography techniques to purify one tRNA sequence at a time using sequence-specific probes as demonstrated for mitochondrial tRNA (with 22 isodecoders).50, 51 Our laboratory has been developing global approaches for identification of tRNA sequences by employing tandem mass spectrometry52, comparative analysis with well-characterized organism53, and multiple ribonucleases54. Similarly, characterization of modifications in rRNA (with >2900 nt) is challenging. Therefore, these RNAs are analyzed in a segmented fashion where the resident modifications are characterized on domain basis as shown in case of E. coli55, Clostridium sporogens56, Haloarcula marismortui57, Thermus thermophilus58, Schizosaccharomyces pombe59 and Homo sapiens60.
In an effort to increase the repertoire of biochemical tools for 4-lane sequencing (one lane for each nucleoside-specific enzymatic digestion products) of modified RNAs, we have previously characterized cytidine (C)-specific cusativin61, and uridine(U)-specific62 MC1 ribonucleases to complement the guanosine(G)-specific ribonuclease T1. RNase MC1 exhibited high specificity while cleaving the phosphodiester bond at 5’-end of uracil residues, where a mere replacement of oxygen with sulfur atom at position 4 on uracil nitrogenous base (in other words 4-thiouracil or s4U) inhibits cleavage of RNA. RNase cusativin recognizes cytidine to cleave the phosphodiester bond at its 3’-side. However, the phosphodiester bond between consecutive cytidines is not a substrate for this enzyme leading to generation of longer digestion products. In the current study, we demonstrate the combined utility of RNases T1, cusativin and MC1 in securing improved sequence coverage, when RNA modification mapping is performed on global scale without prior purification of individual tRNAs. These enzymes generated data leading to improved sequence coverage and modification information in G-rich and pyrimidine-rich sequences of cellular noncoding RNA. Further, the combined sequence coverage of RNases T1, cusativin and MC1 reached 75–100% for each of the resident tRNAs (70–90 nt) when total tRNA (with >40 different sequences) was analyzed. A similar strategy adopted for 2904 nt long 23S rRNA yielded >60% coverage through the LC-MS/MS based RNA modification mapping procedures.
Materials and Methods
Transfer RNA isolation and digestion
Transfer RNA was purified from K12 strain of E. coli using Tri-reagent by differential precipitation with sodium salts as described before.63 Isolated tRNA was reprecipitated with 3M ammonium acetate to replace the sodium ions at the final purification step before dissolving it in water. RNase T1 and bacterial alkaline phosphatase were procured from Worthington Biochemical Corporation. The RNases MC162 and cusativin61 were overexpressed in BL21 and Rosetta™ (DE3) (Novagen) cells, respectively, and purified on a nickel column using His-tag protein purification kit (EMD Millipore) as per the manufacturer’s guidelines. Each batch of ribonuclease protein preparation was evaluated for optimal RNA:protein ratio to ensure appropriate digestion of substrate RNA. In general, optimal digestion of the tRNA mixture or rRNA was carried out by mixing 0.5–1.0 μg of ribonuclease protein for each μg of RNA in the presence of 120 mM ammonium acetate (pH not adjusted) for 90 min at 37 °C for MC1 and 62 °C for cusativin.
Ribosomal RNA isolation and digestion
Total RNA was initially isolated from SQ171 strain of E. coli using Tri-reagent as per the manufacturer’s instructions. The total RNA was subsequently electrophoresed on 1.2 % low melting point agarose gel. The 23S rRNA band was excised and purified using Zymoclean Gel RNA recovery kit (Zymo Research). The rRNA was digested separately with each of the three ribonucleases, and dried in SpeedVac (Thermo Fisher Scientific, Inc.) at low temperature and stored at −20 oC.
LC-MS/MS analysis of tRNA
The LC-MS/MS analysis of tRNA digests was performed using a Poroshell 120 EC-C18 column (1.0X750 mm, Agilent) with chromatographic conditions similar to those described earlier.62 Briefly, the speed-vac dried RNA digest was resuspended in 2 mM EDTA and mobile phase A (200 mM hexafluoroisopropanol [HFIP] [Sigma], 8.15 mM triethylamine [TEA, Fisher Scientific] in water [Burdick and Jackson, Bridgeport], pH 7.0). The gradient liquid chromatography was performed with a combination of mobile phase A and mobile phase B (100 mM HFIP, 4.08 mM TEA in 70% methanol [Burdick and Jackson], pH 7.0) at a flow rate of 40 μL min−1 at 50 °C by using Finnigan Surveyor pump and autosampler system. The gradient consists of 1% B for initial two minutes to enable sample loading, ramped to 20% B in 10 min, 70% B in 62 min, 98% B in 2 min with a hold for 2 min and equilibration step for initial conditions. The chromatographic eluent containing the resolved digestion products were analyzed by LTQ-XL (Thermo Fisher Scientific) mass spectrometer with the acquisition settings identical to those described before.64 In general, the sheath gas, auxiliary gas, and sweep gas at the ionization source were set to 25, 14, and 10 arbitrary units (au), respectively. The spray voltage was 4 kV, capillary temperature-275°C, capillary voltage at −23 V and the tube lens was set to −80 V. The theoretical m/z (mass/charge) values of putative digestion products (both U-specific and nonspecific) and the corresponding collision-induced dissociated (CID) fragment ions were computed using Mongo Oligo (http://mods.rna.albany.edu/masspec/Mongo-Oligo).
LC-MS/MS analysis of rRNA
The LC-MS/MS analysis of rRNA was performed on an Ultimate 3000 (Thermo scientific) UHPLC system using a Waters XBridge C18 column (1.0 × 150 mm, 3.5 μm particle size) at 60 °C. Mobile phase A consisted of 8 mM TEA and 200 mM HFIP (pH 7.8) in H2O, and mobile phase B consisted of 8 mM TEA and 200 mM HFIP in 1:1 H2O:methanol. A chromatographic gradient consisting of a 5 min hold at 5% B, ramping to 55% B at 70 min, with a 5 min hold at 100% B and 30 min re-equilibration at 5% B was used at a flow rate of 65 μL min−1 for all separations. Mass spectrometric detection of the chromatographic eluent was performed in negative ion mode through electrospray ionization on a Waters Synapt G2-S (Quadrupole time-of-flight, Q-TOF) mass spectrometer operating in sensitivity mode (V-mode). The ESI source parameters consisted of 2.5 kV source voltage, 30 V sample cone, source and desolvation temperatures at 120 °C and 400 °C, cone and desolvation gas flow rates at 5 and 800 L h−1, respectively. A scan range of 400 to 2000 m/z (0.5 sec scan time) for MS acquisition and 200 to 2000 m/z (1.0 sec scan time) for MS/MS acquisition was used. MS/MS spectra were collected under data-dependent acquisition mode using an m/z dependent collision energy profile.
Data processing
The template set of modified E. coli tRNA and 23S rRNA sequences were obtained from Modomics database.1 The m/z values of the theoretically expected oligonucleotide digestion products (and their fragment ions in MS/MS spectra) of each tRNA and rRNA and for each ribonuclease were computed using the Mongo Oligo mass calculator and custom-designed software. Identification of the modified oligonucleotides and their assignment to the RNA sequences was initially carried out with RNAModMapper65 following conversion of the RAW data files to MGF files using the MSConvert feature of ProteoWizard. The data processing settings varied depending on the instrument used to acquire data. For LTQ-XL data, the settings include mass tolerance of 1 Da for both precursor and fragment ions, the 3’ end settings include -OH for RNase T1 (used in combination with bacterial alkaline phosphatase66, and 3’-PO4 or cyclic phosphate for MC1 and cusativin. The number of missed cleavages were set to 0 for T1 and 4 for MC1 and cusativin. The thresholds of P score and dot product score were set at 70 and 0.8, respectively as described before.67 A fixed sequence approach was used, with known modifications reported for E. coli tRNA1 included in the sequence file.
For processing Synapt G2 data, the mass tolerance was set at 0.4 and 0.6 Da for precursor and product ions, respectively. A higher product ion mass tolerance is required because the lock-spray correction can only be performed on MS data.68 The 3’ end settings include -OH for RNase T1 (used in combination with bacterial alkaline phosphatase66), and 3’-PO4 or cyclic phosphate for MC1 and cusativin. The number of missed cleavages were set to 0 for T1 and 4 for MC1 and cusativin. The P score and dot product score thresholds were set at 55 and 0.7 and the obtained oligonucleotide hits were manually verified. As described above for the tRNA analysis, a fixed sequence approach with known modifications of 23S rRNA 69, 70 were included in the sequence file. The data processing methodology requires detection of ≥80% of the expected fragmented ions whose relative abundance (not absolute intensity) is 5% or above for positive scoring of an oligonucleotide. Oligonucleotides smaller than 5-mers identified by RNAModMapper were not considered for sequence coverage unless they contained unique modifications (such as anticodon or variable region-specific). Manual processing of RAW data files was performed on respective platforms, Xcalibur (for LTQ-XL data, Thermo scientific) and MassLynx v4.1 (for Synapt G2 data, Waters Corporation).
Result and Discussion
Improved coverage of G-rich and pyrimidine-rich sequences during modification mapping
The success of RNA modification mapping procedures by LC-MS/MS, in general, depends on the length, nucleobase composition, and complexity of oligonucleotides resulting from digestion of the RNA sample with a specific RNase. Previously we demonstrated that RNase digestion and MS analysis of tRNAs can take advantage of these factors through an approach denoted as the Signature Digestion Product (SDP) method.71 SDPs are oligonucleotide sequences that are unique to a particular tRNA and can used to identify specific tRNAs in a mixture. Beyond simply identifying tRNAs, increasing the number of SDPs through the use of multiple enzymes enhances the accuracy of RNA modification mapping through improved sequence coverage.72
To demonstrate how RNases MC1 and cusativin could play a complementary role to other RNases, especially in generating sequence-specific information for G-rich regions of RNA, we first analyzed a mixture of E. coli total tRNAs. Digestion of E. coli total tRNA with MC1 yielded the digestion product, [D]GGGAGAGCGCC>p from position 17–28 (Figure 1A, Supplemental Figure S1, recognized nucleotide is underlined) that can be uniquely matched to tRNAAla[cmo5U]GC but not to the tRNAAla(GGC). The latter contained a similar sequence but yielded a shorter product upon MC1 treatment, [D]GGGAGAGCGC>p from position 17–27 due to the substitution of C with U at position 28 (Figure 1B, Supplemental Figure S2). Thus, MC1 is able to distinguish the two different isoacceptors of tRNAAla (cmo5UGC vs GGC anticodons) based on this single base difference at position 28 in each sequence. This information complements that obtained from RNase T1, which can distinguish the two sequences based on the digestion products from the variable loop ([m7G]UCUGp vs [m7G]UCAGp) and amino acid acceptor loop (CAUAGp vs CUUAGp). Similarly, those digestion products complement RNase A, which could distinguish the sequences through digestion products of variable region (AGGAG[m7G]Up vs AAGAG[m7G]Up).
Figure 1:

Characterization of G-rich and pyrimidine-rich sequences from tRNA digests by tandem mass spectrometry. Sequence informative fragment ion series (cn, with common 5’-end and yn with common 3’-end) are illustrated in the spectra. (A) CID spectrum of MC1 digestion product with m/z 1316.82 corresponding to [D]GGGAGAGCGCC>p from tRNAAla-[cmo5U]GC. (B) CID spectrum of MC1 digestion product with m/z 1215.32 corresponding to [D]GGGAGAGCGC>p from tRNAAla-GGC. (C) CID spectrum of cusativin digestion product with m/z 1249.4 corresponding to AAGUCCCCCCCC>p from tRNALeu-CAG.
It was also found that cusativin generates longer digestion products that could easily be assigned to specific RNA sequences. One such product observed from the total tRNA digest revealed a sequence of AAGUCCCCCCCC>p (Figure 1C, Supplemental Figure S3), which can be uniquely assigned to tRNALeu(CAG). The inability of cusativin to cleave the bond between cytidines61 provides an added advantage to generate longer digestion product for unique assignment.
RNA-specific unique digestion products for RNases MC1 and cusativin
As RNases T1, MC1 and cusativin exhibit complementary nucleobase specificity, we examined the utility of these enzymes to generate digestion products that are both unique and overlapping for various RNA molecules of a mixture. Figure 2 illustrates the observed digestion products against the full sequence of tRNALys as a representative example (spectra shown in Supplemental Figures S4–S22) following LC-MS/MS analysis of a total tRNA digest. Examination of the observed oligonucleotide digestion products indicate that RNase T1 generated only one unique digestion product harboring the anticodon region. Digestion of same tRNA mixture by cusativin generated as many as 6 unique products (Table 1) that are specific to tRNALys. Of these, 4 are unmodified products with 5 to 9 nucleotides in length arising from 5’-end, D-stem loop, anticodon, variable loop and amino acid acceptor regions of RNA sequence. Similarly, MC1 generated 3 unique products (from D-loop, anticodon, amino acid acceptor), of which one is unmodified (Table 1). Consistent with previous observations,62 hypermodified or methylated uridine (mnm5s2U or acp3U or m5U) residues were not recognized as substrate by MC1 presumably, because of the absence of cleavage of RNA at these residues (Supplemental Figures S18 & S20). However, simple modifications such as dihydrouridine and pseudouridines were indistinguishable from uridine substrate and those digestion products were clearly detected (DAGAGCAG>p, Supplemental Figure S17, 19 & S21). Similarly, the phosphodiester bonds between two tandem cytidines is not cleaved (for example, GAAUCCp, Supplemental Figure S15) by cusativin,61 presumably due to the inefficient binding of these residues in the bipartite active site of cusativin enzyme (our own unpublished observations). Further, even the bonds between cytidine and adenosine resist the cleavage, if those residues exist at the sequence termini (GACCCACCA, Supplemental Figure S16). Nevertheless, inclusion of cusativin and MC1 into the RNA modification mapping toolbox can provide rich and unique information for different regions of tRNA, thus facilitating confident sequence assignment.
Figure 2.

RNA modification and sequence mapping of E. coli tRNALys from the digestion products of RNases T1, cusativin, and MC1. (A) The observed digestion products of each ribonuclease are matched against the known sequence of tRNALys. The cleavage sites for each enzyme is underlined in an oligomer. Note the overlapping regions between the sequences of digestion products. (B) A schematic view of looking at the overlaps between the observed digestion products of the three ribonucleases. (C) The clover-leaf model of 2D structure of E. coli tRNALys is depicted.
Table 1:
Detected digestion products considered for sequence coverage of tRNALys
| RNase T1 | cusativin | MC1 |
|---|---|---|
| CUCAG ACU[mnm5s2U]UU[t6A]A[Ψ]CAAUUG* [m7G][acp3U]CG AAUCCUG ACCCACCA |
pGGGUC* GUUAGC* AG[D][D]GG[D]AGAGC AGUUGAC* U[mnm5s2U]UU[t6A]A[Ψ]C* AAUUG[m7G][acp3U]C* AGG[m5U][Ψ]C GAAUCC GACCCACCA* |
[D]AGAGCAG* U[mnm5s2U]U [Ψ]CAA UG[m7G][acp3U]CGCAGG[m5U]* [Ψ]CGAA UGCACGACCCACCA* |
denotes unique digestion products
Overlapping digestion products and sequence coverage
Next, we examined the possibility of reconstructing the modified tRNALys sequence from unique and overlapping digestion products. A total of 61 nucleotides (nt) out of 77 nt (~79%) was covered by the detected enzyme specific (T1, cusativin and MC1) unique digestion products of tRNALys (Supplemental Figure S23A). Of these, the sequence from positions 20–54 and 63–76 had overlapping regions encompassing 49 out of 76 nt (64%). The D-loop, T-loop and acceptor stem are the regions that were left out of sequence coverage from this exercise. Inclusion of other digestion products with or without modifications improved the coverage to 93 and 100% sequence coverage, respectively, (compared to 51% for T1 alone), with overlaps for >60 nt (Supplemental Figure S23B, Figure 2). About 64% of the sequence was consistently observed in any two of the three LC-MS/MS datasets generated by the ribonucleases. In other words, 64% of the sequence is corroborated by digestion products from any two ribonucleases. Thus, an expanded number of RNA-specific unique products and their overlapping nature improves modification mapping and sequence interpretation.
Encouraged by the observations with tRNALys, we examined the sequence coverage of all the tRNAs in these enzymatic digests. There are 43 isodecoder tRNA sequences (belonging to 39 isoacceptor families) deposited in Modomics database1 for E. coli. Just like the data analysis for tRNALys, all the tRNA-specific unique digestion products for each enzyme (Supplemental excel file1), other modified and unmodified oligonucleotides that exhibit overlaps and match with the target RNA sequence were considered for computing sequence coverage. Approximately, 126 out of 168 unique digestion products (from 43 isodecoders) were detected (~75%) for cusativin in the current LC-MS method. In the MC1 digest, the detection was up to 47% (90/191). However, another 20–24% of the expected digestion products either did not trigger MS/MS or the sequence-informative fragment ion coverage (and abundance) is below the set threshold criteria (<70%) to be counted. Such products were not considered for mapping and sequence coverage considerations in the present study. The LC-MS/MS data ambiguity due to contamination of rRNA fragments in tRNA fraction is minimized based on the differences in density and types of modifications in these two types of RNA. The characteristic hypermodifications and unique sequence features of tRNA are not found in any rRNA. Therefore, the oligonucleotides bearing such modifications can only be assigned to tRNA. Further, assignment of the digestion products starts with the detection of sequences that are unique to specific tRNA, which in turn minimizes the assignment of rRNA oligonucleotides to tRNA.
Multiple factors could be responsible for incomplete characterization. A significant number of these uncharacterized digestion products contained m7G or were longer than 15-mers. During collision-induced dissociation (CID), loss of m7G predominates the other phosphodiester cleavages64, 73 leading to very low abundance of sequence informative fragment ions in the tandem MS/MS spectrum. Potential strategies to circumvent this problem could include targeted ion monitoring, improved chromatographic resolution, or an exclusion list strategy.74
Longer oligomers (>15 mers) are poorly resolved in the current chromatographic gradient conditions, thereby resulting in coelution, competitive ionization and ion suppression. Complete MS/MS coverage of such longer oligonucleotides is also challenging due to the inability to find conditions that generate all necessary product ions of sufficient abundance with traditional CID-based methods and hardware. It remains to be seen whether novel chromatographic methods75 or other mobile phase additives/modifiers76, 77 could aid in resolution of longer oligomers. Similarly, RNA backbone cleavage by electron detachment78, higher-energy collisional dissociation (HCD)79 or photodissociation80 could help alleviate the problems of poor fragmentation of longer oligomers. In spite of these challenges, the overall sequence coverage varied from 75–100% for specific tRNAs in the complex mixture, where oligonucleotide sequences are detected by at least one enzyme (Table 2). The observed coverage was 39–88% when the sequence is required to be covered by digestion products of any two enzymes (Table 2).
Table 2:
Observed sequence coverage of E. coli tRNAs by the detected digestion products of RNases T1, cusativin and MC1.
| tRNA | length (nt) | % Sequence Coverage | ||||
|---|---|---|---|---|---|---|
| RNase T1 | cusativin | MC1 | Any two enzymes# | Total* | ||
| Ala([cmo5U]GC) | 76 | 58 | 58 | 61 | 78 | 91 |
| Ala(GGC) | 76 | 51 | 61 | 72 | 87 | 93 |
| Arg([I]CG) | 77 | 69 | 73 | 58 | 66 | 95 |
| Arg|CCG | 77 | 39 | 74 | 39 | 56 | 97 |
| Arg([mnm5U]CU) | 77 | 49 | 53 | 48 | 52 | 91 |
| Asn([Q]UU) | 76 | 61 | 63 | 38 | 42 | 82 |
| Asp([gluQ]UC) | 77 | 44 | 66 | 45 | 48 | 84 |
| Cys(GCA) | 74 | 59 | 69 | 72 | 81 | 100 |
| Gln([cmnm5s2U]UG) | 75 | 77 | 80 | 48 | 81 | 100 |
| Gln(CUG) | 75 | 76 | 71 | 52 | 75 | 100 |
| Glu([mnm5s2U]UC) | 76 | 72 | 75 | 36 | 63 | 96 |
| Glu([mnm5U]UC) | 76 | 72 | 75 | 49 | 67 | 97 |
| Gly(CCC) | 74 | 64 | 58 | 30 | 43 | 85 |
| Gly([mnm5U]CC) | 75 | 65 | 67 | 59 | 76 | 100 |
| Gly(GCC) | 76 | 63 | 79 | 55 | 67 | 97 |
| His([Q]UG) | 77 | 75 | 78 | 43 | 74 | 100 |
| Ile|GAU | 77 | 57 | 58 | 62 | 70 | 90 |
| Ile([k2C]AU) | 76 | 74 | 63 | 25 | 66 | 91 |
| Leu(CAG) | 87 | 57 | 64 | 53 | 68 | 95 |
| Leu(GAG) | 87 | 67 | 53 | 43 | 48 | 89 |
| Leu([cmnm5Um]AA) | 87 | 56 | 49 | 47 | 51 | 91 |
| Leu([Cm]AA) | 85 | 54 | 79 | 65 | 80 | 93 |
| Leu([cmo5U]AG) | 85 | 55 | 60 | 58 | 59 | 98 |
| Lys([mnm5s2U]UU) | 76 | 51 | 88 | 61 | 64 | 100 |
| Met([ac4C]AU) | 77 | 69 | 62 | 65 | 73 | 91 |
| Phe(GAA) | 76 | 61 | 68 | 42 | 72 | 88 |
| Pro(CGG) | 77 | 53 | 62 | 34 | 45 | 90 |
| Sec(UCA) | 89 | 46 | 56 | 39 | 45 | 82 |
| Ser([cmo5U]GA) | 88 | 58 | 60 | 39 | 70 | 89 |
| Ser(CGA) | 88 | 53 | 50 | 25 | 45 | 75 |
| Ser(GCU) | 93 | 52 | 58 | 20 | 46 | 87 |
| Ser(GGA) | 88 | 47 | 50 | 72 | 52 | 99 |
| Thr1(GGU) | 76 | 63 | 55 | 43 | 47 | 91 |
| Thr3(GGU) | 76 | 66 | 50 | 39 | 54 | 88 |
| Thr([cmo5U]GU) | 76 | 62 | 62 | 47 | 58 | 96 |
| Trp(CCA) | 76 | 70 | 55 | 28 | 40 | 93 |
| Tyr(QUA) | 85 | 74 | 52 | 33 | 49 | 89 |
| Tyr(QUA) | 85 | 73 | 56 | 27 | 47 | 92 |
| Val([cmo5U]AC) | 76 | 68 | 57 | 54 | 70 | 89 |
| Val(GAC) | 77 | 66 | 52 | 52 | 69 | 92 |
| Val(GAC) | 77 | 57 | 42 | 51 | 48 | 92 |
| Ini(CAU) | 77 | 56 | 64 | 74 | 61 | 96 |
| Ini(CAU) | 77 | 56 | 56 | 78 | 62 | 100 |
refers to the detection and sequence coverage reported by at least two of the three enzymes
refers to the total coverage by all three enzymes where detection was made by at least one enzyme.
Escherichia coli is known to exhibit co-variation of tRNA abundance and codon usage at different growth rates.81, 82 However, tRNAs such as tRNAThr(GGU) and tRNASec(UCA) were reported at lower abundance (0.16–1.7% of total tRNA) irrespective of the growth rates.82 Examination of sequence coverage for these tRNAs indicate >80% coverage (Table 2) (Supplemental Figures S24–S26), suggesting that this approach is capable of characterizing the entire modified RNA sequence in a mixture independent of tRNA abundance. Similar types of overlapping digestion products can be generated with a variant of RNase U2 with non-specific ribonuclease activity, although those priori results were limited to analyses of only a single tRNA.83
Analysis of 23S rRNA and its digestion product pattern complexity
Encouraged by the performance with tRNA, we tested the suitability of enzymes for modification mapping of longer RNAs such as 23S rRNA of E. coli. Digestion of 23S rRNA with RNase T1 generates oligomers ranging from dinucleotides to 18-mers apart from monomers (Gp). Of these, there are 203 oligonucleotides with 5 nt or higher in size covering ~30% of sequence. Of these, 57 (ranging from 5–16 nt) exhibited unique mass and sequence, therefore, the inferred oligonucleotide can be assigned to specific sequence locations (Supplemental excel file 2). There are 36 isomers of various lengths (5–16 nt), nucleobase composition, and even identical sequences. While the majority of sequence isomeric forms could be differentiated at the MS/MS level based on the pattern of sequence informative fragment ions52, 84, the identical sequences cannot be assigned in a similar manner. They can be considered, however, for sequence coverage, if digestion products generated by other enzymes have overlaps.
Assuming complete digestion, cusativin and MC1, independently, are expected to yield 276 and 231 oligonucleotides with 5 nucleotides or more in the sequence. Cusativin is expected to generate highest number of unique digestion products (132) followed by MC1 (97). They are also expected to generate 46 (5–11 nt) and 40 (5–12 nt) different types of isomer sequences, respectively. (Table 3) The unique digestion products exhibited length of 16 nt to as much as 35 nt (Supplemental excel file 2).
Table 3:
Expected pattern of digestion products for RNases T1, cusativin, and MC1 from 23S rRNA of E. coli.
| Digestion Products | RNase T1 | cusativin | MC1 |
|---|---|---|---|
| ≥ 5 nt | 203 | 276 | 231 |
| Unique | 57 (5–16 nt) | 132 (5–25 nt) | 97 (5–35 nt) |
| Isomers | 36 (5–18 nt) | 46 (5–11 nt) | 40 (5–12 nt) |
Modification mapping of 23S rRNA
Initially, we looked for digestion products that are rich in G content or pyrimidines in RNase MC1 and cusativin digests. Figure 3 illustrates the detection of a few such representative digestion products from the LC-MS data. For example, UGCGGCAGCGACGC>p could easily be discerned from the RNase MC1 digest of purified 23S rRNA (Figure 3, Supplemental Figures S27) and uniquely assigned to a specific segment (Domain II, position 1159–1172) of 23S rRNA. Similarly, UG[Gm]GGC>p could be identified from cusativin digest and assigned to domain V (position 2249–2254) (Figure 3, Supplemental Figure S28).
Figure 3:

Characterization of G-rich and pyrimidine-rich sequences from specific regions of 23S rRNA. Sequence informative fragment ion series (cn and yn) are illustrated in the spectra. (A) Tandem mass spectrum (MS/MS) of MC1 digestion product m/z 1518.86 corresponding to UGCGGCAGCGACGC>p (position 1159–1172). (B) Tandem mass spectrum of cusativin digestion product with m/z 1001.62 corresponding to UG[Gm]GGC>p (position 2249–2254).
.23S rRNA exhibits 25 known post-transcriptional modifications with pseudouridylations and methylations of the base and sugar.85 These modifications are important to stabilize the conformation of the rRNA, enhance ribosome interaction with ligands, prevent rRNA degradation and antibiotic resistance.55, 70 Digestion of 23S rRNA with RNase T1 generated oligonucleotides that could map mass-shift generating modifications such as m1G(745) and m5U(747) in ACUAAU[m1G]Ψ[m5U]G, m6A(1618) in ACAC[m6A]G, m2G(1835) in CCU[m2G], m3Ψ (1915) in ΨAAC[m3Ψ]AΨAACG, m5U(1939) in AAA[m5U]UCCUUG, m5C(1962) in AC[m5C]UG, m7G(2069) in U[m7G]AACCUUUACUAUAG, and D(2449) in A[D]AACAG (data not shown). Treatment of RNA with cusativin enabled us to identify other modifications, such as m6A(2030) in UGUG[m6A]AGAUGC, Gm(2251) in UG[Gm]GGC, m2G(2445) and D(2449) in G[m2G]GGA[D]AAC (Supplemental Figures S29–S30). The oligomers with Um (UGGC[Um]G)and Cm (CAC[Cm]UCG) were observed only at the MS1 level (data not shown).
Sequence coverage for 23S rRNA
From the 2904 nt long 23S rRNA, the detected oligonucleotide digestion products by RNase T1, cusativin and MC1 covered various regions of 23S rRNA that correspond to a total of 819, 748 and 721 nt, respectively. A representative example of the observed digestion products and their sequence overlaps is illustrated for the region between 1900–2027 nt of 23Sr RNA (Figure 4). Overall, about 28% of the entire sequence was covered by RNase T1 digestion products. However, the domain-specific sequence coverage by this enzyme varied from 19–36% (Table 4). Digestion products of cusativin yielded a coverage of 26% with domain specific coverage varying from 22–29%. RNase MC1 provided coverage of about 24% with the domain-specific coverage varied from 15–35%. Compilation of all digestion products by three enzymes and matching with the expected modified sequence revealed a total coverage of 61%. About 48% sequence coverage was corroborated by digestion with any two of the three enzymes employed. Representation of all the observed digestion products with MS/MS information were overlaid on the secondary structure of 23S rRNA using RiboVision suite86 (Figure 5).
Figure 4:

A portion of 23S rRNA sequence matched against the complementary digestion products of RNases T1, cusativin and MC1. Note the sequence overlaps observed between the digestion products of RNase T1 (red colored font), cusativin (green), and MC1 (blue). The digestion products depicted in brown did not generate the MS/MS spectrum. However, their m/z values exhibited mass error of less than 5 ppm.
Table 4:
Observed sequence coverage of 23S rRNA domains following LC-MS analysis
| 23S rRNA domain | Total Nucleotides | Sequence coverage (%) | |||||
|---|---|---|---|---|---|---|---|
| T1 | Cus | MC1 | Any two RNases# | Total* | MS1 only (with or without MS/MS) | ||
| I | 541 | 33 | 22 | 28 | 57 | 67 | 88 |
| II | 739 | 19 | 27 | 15 | 43 | 54 | 83 |
| III | 376 | 34 | 26 | 27 | 54 | 64 | 89 |
| IV | 371 | 36 | 23 | 35 | 58 | 71 | 89 |
| V | 608 | 28 | 29 | 22 | 50 | 60 | 76 |
| VI | 269 | 27 | 29 | 35 | 52 | 61 | 89 |
| Total | 2904 | 28 | 26 | 25 | 48 | 61 | 85 |
refers to the sequence coverage reported by at least two of three enzymes
refers to the total coverage by all three enzymes
Figure 5:

Overall coverage of 23S rRNA sequence from the sequence confirmed oligonucleotide digestion products of RNases T1, cusativin and MC1 and their sequence overlaps. (A). Column plot was generated using excel, where the mapped nucleotide (given number 1) was plotted against the nucleotide position (x-axis) of rRNA sequence. The digestion products are color coded, T1 products by red, cusativin by green, and MC1 by blue. (B) Sequence coverage is laid out on the secondary structure of 23S rRNA. The figure was generated by using the secondary structure (phylogeny) format of large ribosomal subunit of E. coli by employing RiboVision suite. The dashed line represents a continuity of the sequence between Domains III and IV. The mapped nucleotides are colored in blue, nucleotides not mapped are colored in gray. The nucleotide numbering and 5S of rRNA were hidden for clarity (5S not mapped in the current work). The Italian numbers I, II, III, IV, V and VI represent each domain of 23S rRNA.
A deeper examination of the LC-MS data revealed that a number of oligonucleotides did not trigger MS/MS or did not meet the minimum threshold of coverage by sequence informative fragment ions (>70%) presumably because of low precursor ion abundance. A number of such oligonucleotide anions exhibited m/z values highly similar to the theoretically expected values with mass errors ranging from 5–20 PPM. This might suggest further potential to improve sequence coverage, provided the MS/MS spectra could be used to confirm the sequence. If the MS1 data with unconfirmed sequence information is included, the sequence coverage could theoretically reach >75% (Table 4, supplemental Figures S31–S32) for various domains.
In general, longer oligonucleotides (>15 nt) are not easily characterized by bottom-up LC-MS/MS methods as their ionization and mass spectrometric analysis requirements differ significantly compared to smaller ones. The combination of longer digestion products and missed cleavages would lead to longer segments of RNA in the digests that could be poorly ionizable under the tested conditions leading to incomplete detection and characterization. Moreover, multiple oligonucleotides of similar length exhibit similar retention time on the chromatographic column leading to coelution and ion suppression to the point where the abundance falls below the MS/MS triggering threshold. As described above, improvement of the chromatographic setup and/or inclusion of two-dimensional liquid chromatography87 and other multiplexing techniques88 could improve the MS signal to trigger MS/MS. Resolving the longer oligonucleotide digestion products generated by MC1 or cusativin through offline fractionation and subsequent treatment with RNase T1 could also improve the MS/MS sequence coverage. A mass spectrometer with a faster duty cycle, using a mass exclusion list74 and alternate fragmentation methods such as photodissociation80, 89, 90 could potentially improve the characterization further.
Utility of these novel tools in characterization of modified RNA and their implications
The ability to generate unmodified but unique digestion products for specific tRNAs allows a determination of the baseline levels of transcripts in a given sample. This in turn would enable accurate evaluation of the modification status at a given location, due to xenobiotic or intracellular conditions, with reference to the transcript level. Thus, alterations in modification levels vs the changes in transcript levels could be differentiated through multiple unique oligonucleotides of a target tRNA. Similarly, the status of interdependent modifications or complex modification codependency can be investigated by these base specific enzymes in situations such as the impact of mutations in one modifying enzyme on the status of a second modification. For example, the hypermodified G nucleobase, yW37, formation requires 2’-O-methylation of C32 and N34 by Trm7/Trm732 and Trm7/Trm734, respectively for generation of modified sequences of yeast and human tRNAPhe. 91 Other examples include interdependency of modifications at positions 37 (i6A) and 32 (m3C) of eukaryotic tRNASer 92, and 7-methyl-G at position 46 affecting the status of Gm and m1G at positions 18 and 37, respectively, in Thermus thermophilus. 93 In cases, where location information of modifications in RNA are unknown, the modified oligonucleotide sequence information obtained by one type of digestion products (for example, RNase T1) can be corroborated by the oligonucleotides generated by at least one of the two enzymes (i.e. cusativin or MC1). Such a feasibility has been recently documented during modification mapping of the anticodon regions of tRNA from Methanocaldococcus jannaschii.67 Availability of DNA sequence information for this archaeal species enabled the modification mapping through the utilization of the nucleobase-specific enzymes. More focused bioinformatic effort is required for de novo mapping analysis, where the nucleotide sequence information of transcripts is unknown.
In summary, we have demonstrated the utility of C-specific cusativin and U-specific MC1 ribonucleases in providing location-specific information of nucleoside modifications in G-rich or pyrimidine-rich regions of RNA. Additionally, these RNases enable one to detect C to U sequence changes, facilitate unique digestion product patterns for different regions of tRNA, and provide improved sequence coverage when used with other RNases. Taken together, as part of the modification mapping toolbox, these enzymes can play an important role in the characterization of complex mixtures of cellular tRNAs or ribosomal RNA for their resident nucleoside modifications.
Supplementary Material
Acknowledgments
This work is financially supported by funding from the National Institutes of Health (NIGMS R01 058843 to P.A.L.). The generous support of the Rieveschl Eminent Scholar Endowment and the University of Cincinnati for these studies is also appreciated. The authors thank Lalit Kumar for developing the customized software used to predict large oligonucleotide digestion products.
Abbreviations
- LC
liquid chromatography
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- TEAA
triethylammonium acetate
- HFIP
hexafluoroisopropanol
- CID
collision-induced dissociation
- E. coli
Escherichia coli
- PTM
posttranscriptional modification
- rRNA
ribosomal RNA
- ESI
electrospray ionization
- m6A
N6-methyladenosine
- m1A
1-methyladenosine
- t6A
N6-threonylcarbamoyladenosine
- m5C
5-methylcytidine
- hm5C
5-hydroxymethylcytidine
- Nm
ribose methylations
- ac4C
N4-acetylcytidine
- m3C
3-methylcytidine
- m7G
7-methylguanosine
- m1G
1-methylguanosine
- m1I
1-methylinosine
- m22G
N2,N2-dimethylguanosine
- D
dihydrouridine
- Ψ/Y
pseudouridine
- m5U
5-methyluridine
- acp3U
3-(3-amino-3-carboxypropyl)uridine
- mnm5s2U
5-methylaminomethyl-2-thiouridine
Footnotes
Conflict of interest: The authors declare no conflict of interest.
Literature cited
- 1.Boccaletto P, Machnicka MA, Purta E, Piatkowski P, Baginski B, Wirecki TK, de Crecy-Lagard V, Ross R, Limbach PA, Kotter A, Helm M and Bujnicki JM, Nucleic Acids Res, 2018, 46, D303–D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lorenz C, Lunse CE and Morl M, Biomolecules, 2017, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Motorin Y and Helm M, Biochemistry, 2010, 49, 4934–4944. [DOI] [PubMed] [Google Scholar]
- 4.Wang X, Matuszek Z, Huang Y, Parisien M, Dai Q, Clark W, Schwartz MH and Pan T, Rna, 2018, 24, 1305–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agris PF, Narendran A, Sarachan K, Vare VYP and Eruysal E, The Enzymes, 2017, 41, 1–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nilsson EM and Alexander RW, IUBMB life, 2019, DOI: 10.1002/iub.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song J and Yi C, ACS chemical biology, 2017, 12, 316–325. [DOI] [PubMed] [Google Scholar]
- 8.Duechler M, Leszczynska G, Sochacka E and Nawrot B, Cellular and molecular life sciences : CMLS, 2016, 73, 3075–3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freund I, Eigenbrod T, Helm M and Dalpke AH, Genes, 2019, 10, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karikó K, Buckstein M, Ni H and Weissman D, Immunity, 2005, 23, 165–175. [DOI] [PubMed] [Google Scholar]
- 11.Morena F, Argentati C, Bazzucchi M, Emiliani C and Martino S, Genes, 2018, 9, 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanchez-Vasquez E, Alata Jimenez N, Vazquez NA and Strobl-Mazzulla PH, Mechanisms of development, 2018, DOI: 10.1016/j.mod.2018.04.002. [DOI] [PubMed] [Google Scholar]
- 13.Frye M, Harada BT, Behm M and He C, Science (New York, N.Y.), 2018, 361, 1346–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lian H, Wang QH, Zhu CB, Ma J and Jin WL, Trends in cancer, 2018, 4, 207–221. [DOI] [PubMed] [Google Scholar]
- 15.Goldman SL, Hassan C, Khunte M, Soldatenko A, Jong Y, Afshinnekoo E and Mason CE, Frontiers in genetics, 2019, 10, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Shi J, Rassoulzadegan M, Tuorto F and Chen Q, Nature reviews. Endocrinology, 2019, 15, 489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torres AG, Batlle E and Ribas de Pouplana L, Trends Mol Med, 2014, 20, 306–314. [DOI] [PubMed] [Google Scholar]
- 18.Jonkhout N, Tran J, Smith MA, Schonrock N, Mattick JS and Novoa EM, Rna, 2017, 23, 1754–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dorn L, Tual-Chalot S, Stellos K and Accornero F, Journal of molecular and cellular cardiology, 2019, 129, 272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Xiong X and Yi C, Nature methods, 2016, 14, 23–31. [DOI] [PubMed] [Google Scholar]
- 21.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE and Jaffrey SR, Cell, 2012, 149, 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, Sorek R and Rechavi G, Nature, 2012, 485, 201–206. [DOI] [PubMed] [Google Scholar]
- 23.Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, Peer E, Kol N, Ben-Haim MS, Dai Q, Di Segni A, Salmon-Divon M, Clark WC, Zheng G, Pan T, Solomon O, Eyal E, Hershkovitz V, Han D, Dore LC, Amariglio N, Rechavi G and He C, Nature, 2016, 530, 441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, Bartoli KM and Gilbert WV, Nature, 2014, 515, 143–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, Leon-Ricardo BX, Engreitz JM, Guttman M, Satija R, Lander ES, Fink G and Regev A, Cell, 2014, 159, 148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang W, Eckwahl MJ, Zhou KI and Pan T, Rna, 2019, 25, 1218–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Squires JE, Patel HR, Nousch M, Sibbritt T, Humphreys DT, Parker BJ, Suter CM and Preiss T, Nucleic Acids Res, 2012, 40, 5023–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delatte B, Wang F, Ngoc LV, Collignon E, Bonvin E, Deplus R, Calonne E, Hassabi B, Putmans P, Awe S, Wetzel C, Kreher J, Soin R, Creppe C, Limbach PA, Gueydan C, Kruys V, Brehm A, Minakhina S, Defrance M, Steward R and Fuks F, Science (New York, N.Y.), 2016, 351, 282–285. [DOI] [PubMed] [Google Scholar]
- 29.Birkedal U, Christensen-Dalsgaard M, Krogh N, Sabarinathan R, Gorodkin J and Nielsen H, Angewandte Chemie (International ed. in English), 2015, 54, 451–455. [DOI] [PubMed] [Google Scholar]
- 30.Thomas JM, Bryson KM and Meier JL, in Methods in enzymology, ed. Shukla AK, Academic Press, 2019, vol. 621, pp. 31–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Enroth C, Poulsen LD, Iversen S, Kirpekar F, Albrechtsen A and Vinther J, Nucleic Acids Research, 2019, 47, e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marchand V, Ayadi L, Ernst FGM, Hertler J, Bourguignon-Igel V, Galvanin A, Kotter A, Helm M, Lafontaine DLJ and Motorin Y, Angewandte Chemie International Edition, 2018, 57, 16785–16790. [DOI] [PubMed] [Google Scholar]
- 33.Aschenbrenner J, Werner S, Marchand V, Adam M, Motorin Y, Helm M and Marx A, Angewandte Chemie (International ed. in English), 2018, 57, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Motorin Y and Helm M, Genes, 2019, 10, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Helm M and Motorin Y, Nat Rev Genet, 2017, 18, 275–291. [DOI] [PubMed] [Google Scholar]
- 36.Liu H, Begik O, Lucas MC, Ramirez JM, Mason CE, Wiener D, Schwartz S, Mattick JS, Smith MA and Novoa EM, Nature communications, 2019, 10, 4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cozen AE, Quartley E, Holmes AD, Hrabeta-Robinson E, Phizicky EM and Lowe TM, Nature methods, 2015, 12, 879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng G, Qin Y, Clark WC, Dai Q, Yi C, He C, Lambowitz AM and Pan T, Nature methods, 2015, 12, 835–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edmonds CG, Crain PF, Gupta R, Hashizume T, Hocart CH, Kowalak JA, Pomerantz SC, Stetter KO and McCloskey JA, Journal of bacteriology, 1991, 173, 3138–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kowalak JA, Dalluge JJ, McCloskey JA and Stetter KO, Biochemistry, 1994, 33, 7869–7876. [DOI] [PubMed] [Google Scholar]
- 41.Bakin A, Kowalak JA, McCloskey JA and Ofengand J, Nucleic Acids Res, 1994, 22, 3681–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bruenger E, Kowalak JA, Kuchino Y, McCloskey JA, Mizushima H, Stetter KO and Crain PF, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 1993, 7, 196–200. [DOI] [PubMed] [Google Scholar]
- 43.Kowalak JA, Bruenger E, Hashizume T, Peltier JM, Ofengand J and McCloskey JA, Nucleic Acids Res, 1996, 24, 688–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kowalak JA, Bruenger E and McCloskey JA, The Journal of biological chemistry, 1995, 270, 17758–17764. [DOI] [PubMed] [Google Scholar]
- 45.Kowalak JA, Pomerantz SC, Crain PF and McCloskey JA, Nucleic Acids Research, 1993, 21, 4577–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gaston KW and Limbach PA, RNA biology, 2014, 11, 1568–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ross R, Cao X, Yu N and Limbach PA, Methods (San Diego, Calif.), 2016, 107, 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pomerantz SC, Kowalak JA and McCloskey JA, Journal of the American Society for Mass Spectrometry, 1993, 4, 204–209. [DOI] [PubMed] [Google Scholar]
- 49.McLuckey SA, Van Berker GJ and Glish GL, Journal of the American Society for Mass Spectrometry, 1992, 3, 60–70. [DOI] [PubMed] [Google Scholar]
- 50.Miyauchi K, Ohara T and Suzuki T, Nucleic Acids Res, 2007, 35, e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suzuki T and Suzuki T, Methods in enzymology, 2007, 425, 231–239. [DOI] [PubMed] [Google Scholar]
- 52.Wetzel C and Limbach PA, The Analyst, 2013, 138, 6063–6072. [DOI] [PubMed] [Google Scholar]
- 53.Li S and Limbach PA, The Analyst, 2013, 138, 1386–1394. [DOI] [PubMed] [Google Scholar]
- 54.Puri P, Wetzel C, Saffert P, Gaston KW, Russell SP, Cordero Varela JA, van der Vlies P, Zhang G, Limbach PA, Ignatova Z and Poolman B, Molecular microbiology, 2014, 93, 944–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Havelund JF, Giessing AM, Hansen T, Rasmussen A, Scott LG and Kirpekar F, Journal of molecular biology, 2011, 411, 529–536. [DOI] [PubMed] [Google Scholar]
- 56.Kirpekar F, Hansen LH, Mundus J, Tryggedsson S, Teixeira Dos Santos P, Ntokou E and Vester B, RNA biology, 2018, DOI: 10.1080/15476286.2018.1486662, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kirpekar F, Hansen LH, Rasmussen A, Poehlsgaard J and Vester B, Journal of molecular biology, 2005, 348, 563–573. [DOI] [PubMed] [Google Scholar]
- 58.Mengel-Jorgensen J, Jensen SS, Rasmussen A, Poehlsgaard J, Iversen JJ and Kirpekar F, The Journal of biological chemistry, 2006, 281, 22108–22117. [DOI] [PubMed] [Google Scholar]
- 59.Taoka M, Nobe Y, Hori M, Takeuchi A, Masaki S, Yamauchi Y, Nakayama H, Takahashi N and Isobe T, Nucleic Acids Res, 2015, 43, e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taoka M, Nobe Y, Yamaki Y, Sato K, Ishikawa H, Izumikawa K, Yamauchi Y, Hirota K, Nakayama H, Takahashi N and Isobe T, Nucleic Acids Res, 2018, DOI: 10.1093/nar/gky811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Addepalli B, Venus S, Thakur P and Limbach PA, Anal Bioanal Chem, 2017, 409, 5645–5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Addepalli B, Lesner NP and Limbach PA, Rna, 2015, 21, 1746–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Addepalli B and Limbach PA, Journal of the American Society for Mass Spectrometry, 2011, 22, 1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wong SY, Javid B, Addepalli B, Piszczek G, Strader MB, Limbach PA and Barry CE, Antimicrobial Agents and Chemotherapy, 2013, 57, 6311–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu N, Lobue PA, Cao X and Limbach PA, Anal Chem, 2017, 89, 10744–10752. [DOI] [PubMed] [Google Scholar]
- 66.Krivos KL, Addepalli B and Limbach PA, Rapid communications in mass spectrometry : RCM, 2011, 25, 3609–3616. [DOI] [PubMed] [Google Scholar]
- 67.Yu N, Jora M, Solivio B, Thakur P, Acevedo-Rocha CG, Randau L, de Crecy-Lagard V, Addepalli B and Limbach PA, Journal of bacteriology, 2019, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lobue PA, Yu N, Jora M, Abernathy S and Limbach PA, Methods (San Diego, Calif.), 2019, 156, 128–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gao H, Sengupta J, Valle M, Korostelev A, Eswar N, Stagg SM, Van Roey P, Agrawal RK, Harvey SC, Sali A, Chapman MS and Frank J, Cell, 2003, 113, 789–801. [DOI] [PubMed] [Google Scholar]
- 70.Sergiev PV, Golovina AY, Prokhorova IV, Sergeeva OV, Osterman IA, Nesterchuk MV, Burakovsky DE, Bogdanov AA and Dontsova OA, in Ribosomes: Structure, Function, and Dynamics, eds. Rodnina MV, Wintermeyer W and R. Green, Springer; Vienna, Vienna, 2011, DOI: 10.1007/978-3-7091-0215-2_9, pp. 97–110. [DOI] [Google Scholar]
- 71.Hossain M and Limbach PA, Rna, 2007, 13, 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hossain M and Limbach PA, Analytical and Bioanalytical Chemistry, 2009, 394, 1125–1135. [DOI] [PubMed] [Google Scholar]
- 73.Ross RL, Cao X and Limbach PA, Biomolecules, 2017, 7, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cao X and Limbach PA, Anal Chem, 2015, 87, 8433–8440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lobue PA, Jora M, Addepalli B and Limbach PA, Journal of chromatography. A, 2019, 1595, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li N, El Zahar NM, Saad JG, van der Hage ERE and Bartlett MG, Journal of chromatography. A, 2018, 1580, 110–119. [DOI] [PubMed] [Google Scholar]
- 77.Liu R, Ruan Y, Liu Z and Gong L, Rapid communications in mass spectrometry : RCM, 2019, 33, 697–709. [DOI] [PubMed] [Google Scholar]
- 78.Taucher M and Breuker K, Angewandte Chemie (International ed. in English), 2012, 51, 11289–11292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jora M, Burns AP, Ross RL, Lobue PA, Zhao R, Palumbo CM, Beal PA, Addepalli B and Limbach PA, J Am Soc Mass Spectrom, 2018, 29, 1745–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cleland TP, DeHart CJ, Fellers RT, VanNispen AJ, Greer JB, LeDuc RD, Parker WR, Thomas PM, Kelleher NL and Brodbelt JS, Journal of proteome research, 2017, 16, 2072–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Berg OG and Kurland CG, Journal of molecular biology, 1997, 270, 544–550. [DOI] [PubMed] [Google Scholar]
- 82.Dong H, Nilsson L and Kurland CG, Journal of molecular biology, 1996, 260, 649–663. [DOI] [PubMed] [Google Scholar]
- 83.Solivio B, Yu N, Addepalli B and Limbach PA, Anal Chim Acta, 2018, 1036, 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wetzel C and Limbach PA, J Proteomics, 2012, 75, 3450–3464. [DOI] [PubMed] [Google Scholar]
- 85.Sergeeva OV, Bogdanov AA and Sergiev PV, Biochimie, 2015, 117, 110–118. [DOI] [PubMed] [Google Scholar]
- 86.Bernier CR, Petrov AS, Waterbury CC, Jett J, Li F, Freil LE, Xiong X, Wang L, Migliozzi BLR, Hershkovits E, Xue Y, Hsiao C, Bowman JC, Harvey SC, Grover MA, Wartell ZJ and Williams LD, Faraday Discussions, 2014, 169, 195–207. [DOI] [PubMed] [Google Scholar]
- 87.Roussis SG, Cedillo I and Rentel C, Analytical biochemistry, 2018, 556, 45–52. [DOI] [PubMed] [Google Scholar]
- 88.Porebski PA and Lynen F, Journal of chromatography. A, 2014, 1336, 87–93. [DOI] [PubMed] [Google Scholar]
- 89.Cammarata MB, Macias LA, Rosenberg J, Bolufer A and Brodbelt JS, Anal Chem, 2018, 90, 6385–6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Greer SM, Bern M, Becker C and Brodbelt JS, Journal of proteome research, 2018, 17, 1340–1347. [DOI] [PubMed] [Google Scholar]
- 91.Guy MP and Phizicky EM, Rna, 2015, 21, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Arimbasseri AG, Iben J, Wei FY, Rijal K, Tomizawa K, Hafner M and Maraia RJ, Rna, 2016, 22, 1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tomikawa C, Yokogawa T, Kanai T and Hori H, Nucleic Acids Res, 2010, 38, 942–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.