

Abstract
Layer-by-layer nanoparticles are modular drug delivery vehicles that incorporate multiple functional materials through sequential deposition of polyelectrolytes onto charged nanoparticle cores. Here, we combined the multicomponent features and tumor targeting capabilities of layer-by-layer assembly with functional biosensing peptides to create a new class of nanotheranostics. These nanoparticles encapsulate a high weight percentage of siRNA while also carrying a synthetic biosensing peptide on the surface that is cleaved into a urinary reporter upon exposure to specific proteases overexpressed in the tumor microenvironment. Importantly, this biosensor reports back on molecular signatures characteristic to metastatic tumors and associated with poor prognosis – MMP9 protease overexpression. We show this nanotheranostic mediates noninvasive urinary-based diagnostics in mouse models of three different cancers with simultaneous gene silencing in flank and metastatic mouse models of ovarian cancer. This work provides a modular nanotheranostic platform to both perturb and characterize tumors, allowing for simultaneous treatment and monitoring of disease.
Keywords: theranostic, nanoparticle, layer-by-layer, gene silencing, tumor targeting, self-assembly, gene delivery
Graphical Abstract
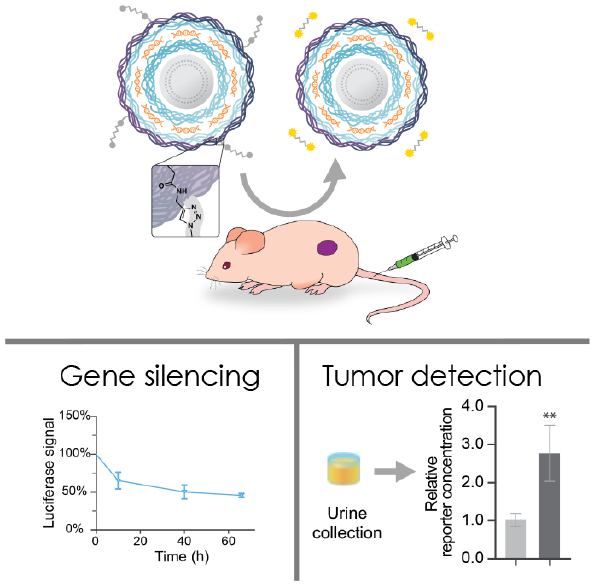
A new class of nanotheranostics was created by combining multicomponent features and tumor targeting capabilities of layer-by-layer assembly with functional biosensing peptides. siRNA-loaded nanoparticles functionalized with a synthetic biosensing peptide able to report back on MMP9 overexpression in the tumor microenvironment mediated noninvasive urinary-based diagnostics and simultaneous gene silencing.
INTRODUCTION
The term nanotheranostics refers to multifunctional nanotechnology capable of interweaving therapeutic and diagnostic utilities.[1] In the context of cancer, nanotheranostics have been applied to bulk tumor imaging, but emerging versions now aim to provide molecular and cellular information about the tumor that can allow for patient stratification and treatment personalization. This is typically achieved by combining both an imaging probe and a therapy in a single drug delivery vehicle.[2] Such an ability is particularly of interest in cancer types for which monitoring of disease can be relatively difficult, including ovarian and pancreatic cancer, and metastatic cancers of all types. In such systems, the ability to target the tumor cells and tissues of interest can greatly enhance the efficiency and efficacy of such theranostic systems.
Although imaging provides local information of the disease sites, obtaining information on precise pathological changes requires the use of more dynamic molecular imaging probes capable of identifying signature features of disease. Recently, urinary-based detection methods have been developed using biosensing peptides to distinguish pathological extracellular matrix protein signatures when attached to nanoparticles (NPs).[3] These peptides are designed to be activated by particular proteases overexpressed in the cancer microenvironment. When cleaved by target proteases, a synthetic biomarker is released from the NP, which can then be detected in the urine. Such an approach noninvasively provides both tumor detection and information about the protease environment, which is known to correlate with patient prognosis.[4]
Integrating this biosensor peptide technology into the layer-by-layer (LbL) platform marks a significant development towards more sophisticated nanotheranostics. LbL assembly provides a natural route towards multifunctional nanotheranostics by introducing functional nanoscale polymer films onto colloidal substrates that serve to modulate drug release and extend plasma half-life.[5] In addition to easily interchanging the components that comprise the substrate and polyelectrolyte films, the LbL platform is distinctly capable of incorporating small molecule drugs,[6] therapeutic nucleic acids,[7] imaging modalities,[8] and molecular probes all within a discrete NP.[3b, 8–9] Therefore, the modularity of this approach also lends itself towards personalizing such NPs to specific clinical contexts.
To achieve a theranostic LbL NP capable of dynamic monitoring of disease state, we opted to use copper(I)-catalyzed click conjugation chemistry to attach cleavable peptide linkers to the surfaces of LbL-coated liposomes modified with alkyne-functional macromolecules as outer layers (Scheme 1). While click chemistry techniques have been used previously to build multilayered assemblies on planar[10] and microparticle[11] substrates, and to cross-link hydrogen-bonded assembled LbL films,[12] limited efforts have been made to introduce this key technique to LbL NPs to add additional functionality. By employing click conjugation to modify LbL NPs, we provide a means to introduce multiple functional macromolecules that are otherwise inaccessible via traditional LbL assembly, including biosensing peptides and tumor penetrating peptides such as iRGD. We chose to incorporate iRGD as we have previously demonstrated its ability to enhance biosensor functionality due to its tumor targeting and penetrating capabilities.[13] Using LbL NPs constructed from liposomal cores, small interfering RNA (siRNA), and polypeptides, we demonstrate both the therapeutic and diagnostic utilities of this approach in vivo, using siRNA to silence a model gene and biosensing peptides to detect the MMP9 protease signature in a variety of metastatic tumor models.
Scheme 1.

Biosensor peptides can be conjugated onto siRNA-containing LbL liposomes via copper(I)-catalyzed click chemistry to create theranostic LbL NPs. In the tumor microenvironment, biosensor peptides are cleaved, and the resulting fragments can be non-invasively detected in the urine to monitor disease progression. PLR = poly-l-arginine, pPLD = propargyl-modified poly-l-aspartic acid..
RESULTS AND DISCUSSION
To produce biosensor LbL NPs, we first synthesized click-compatible LbL NPs through the sequential adsorption of poly-l-arginine (PLR), siRNA, and propargyl-modified poly-l-aspartate (pPLD) onto an anionic liposomal core using previously described conditions.[14] We chose to utilize liposomes as the colloidal substrate due to their clinical precedence, ability to encapsulate both hydrophobic and hydrophilic drugs, tunable surface charge density, and compatibility with extrusion techniques that can yield monodisperse particles of specific sizes.[15] The PLR coatings were used due to their well-documented ability to mediate transfection while avoiding nonspecific cytotoxicity.[16] A commercially available locked nucleic acid analog to siRNA was utilized in these particles due to its stability to degradation by nucleases. The PLD outer layer was chosen to provide a strongly anionic surface that promotes colloidal stability and repels negatively charged serum proteins. Additionally, approximately 12% of the carboxylic side chains of the PLD were converted to propargyl groups to yield pPLD to allow for the downstream click conjugation with biosensor peptides and other neutral macromolecules (Figure S1). The NP size, uniformity and charge were tracked during LbL synthesis (Figure S2a–b), and the collective data demonstrated the controlled growth (from Z-average diameter of 95±1 nm to 124±3 nm, polydispersity index (PDI) below 0.2 throughout) and sequential charge reversal indicative of successful LbL assembly.
The therapeutic capabilities of this nanotheranostic hinge on efficient loading of siRNA to potently mediate gene silencing in vivo. We quantified the purification kinetics in order to determine the encapsulation efficiency of siRNA (Figure S2c–d). After siRNA deposition, permeate fractions were collected during tangential flow filtration and analyzed using a UV-spectrophotometer, indicating an encapsulation efficiency of 37 % and a notably high weight percent loading of 41%. This corresponds to approximately 1000 siRNA molecules per particle.
Having confirmed that the LbL NPs possessed acceptable size, charge, and siRNA loading, we used modified conjugation conditions to click on various combinations of the MMP9 biosensor peptide (+S), iRGD targeting peptide (+R), and PEG pendants (+P) (Scheme 1, Figure 1). We used copper-catalyzed azide alkyne cycloaddition to conjugate azide-functionalized substrates onto pPLD coated LbL NPs.[17] Because LbL NPs are easily aggregated in the presence of salts,[18] this method has advantages over traditional amide bond formation reactions in that it requires minimal buffering and exhibits high reactivity at low reagent and catalyst concentrations, all of which reduce the ionic strength of the reaction medium. For the click conjugation reaction, a matrix of conditions was tested to determine optimal conditions for NP stability by varying absolute and relative concentrations of LbL NPs and azide functionalized ligands while keeping constant concentrations of remaining reagents. Of the tested conditions, we found that high NP concentration (1 mg/mL, 0.027 μM liposome core) with low azide-functionalized ligand concentration (0.28 μM) consistently resulted in stable NPs with acceptable size (Table S1) and PDI (Table S2). This corresponds to a molar ratio of approximately 10:1 peptide to NP (liposome core). While peptide loading is limited by the number of propargyl groups present on pPLD, the sensitivity of MMP9-responsive biosensors has been established to be very high, with detection of average tumor sizes of 36 mm3 possible in murine ovarian cancer models. This is due to the catalytic activity of the proteolytic enzyme that provides signal amplification, and kidney filtration and renal concentration of the cleaved fragments.[13] LbL NPs tolerated the click conjugation of the biosensor, iRGD, and PEG, yielding NPs with mean Z-average hydrodynamic diameters ranging from 121 to 184 nm, PDIs ranging from 0.14 to 0.23, and zeta potentials ranging from −24 to −43 mV (Figure 1). As expected, conjugation of PEG caused an increase in the hydrodynamic diameter (from 124±3 nm to 184±15 nm for NP+SP and 175±4 nm for NP +SRP) and a reduction in zeta potential (from −50±6 mV to −30±1 mV for NP+SP and −24±1 mV for NP+SRP, p<0.0001 one-way ANOVA with the Dunnett post-hoc test). The observed increase in hydrodynamic diameter is in agreement with previously published reports,[19] though we note that interactions of PEG with the LbL film that may cause some small amount of particle aggregation could also contribute to size increase. Additionally, the change in zeta potential was expected due to the incorporation of a neutral polymer (PEG) capable of shielding coulombic interactions on the negative nanoparticle surface. We had initially chosen to investigate whether the addition of PEG pendant chains would be required to guard against colloidal destabilization due to the addition of the biosensor peptides, which are relatively hydrophobic; however, size and zeta characterization of the various formulations ultimately indicated PEG was not necessary for stability.
Figure 1. Click-functionalized LbL NPs remain stable and exhibit protease-responsive behavior.

(a) Diameter, polydispersity index, and (b) zeta potential of click-modified LbL NPs. (c) In vitro cleavage of biosensor LbL NPs in the presence of MMP9. Nanoparticle modifications are abbreviated as follows: biosensor peptide (+S), iRGD targeting peptide (+R), and PEG pendants (+P). Error bars represent standard deviation, and statistical analysis of panel b uses one-way ANOVA with a Dunnett post-hoc test and alpha of 0.05.
To confirm that diagnostic utility was preserved, LbL NPs were modified with biosensor and PEG or biosensor, iRGD, and PEG to yield NP+SP and NP+SRP, respectively. These NPs were then exposed to recombinant MMP9 (12.5 nM) to verify protease-induced cleavage of the reporter fragment from the NP.[20] The MMP9-digested NPs were pelleted via centrifugation and the supernatant was analyzed for the peptide reporter fragment (PRF) using enzyme-linked immunosorbent assay (ELISA) (Figure 1c). The results indicated that the biosensor on the surface of the LbL NP is still accessible to the MMP9 protein, providing detectable peptide signal in the supernatant for both NP+S and NP+SR.
After initial validation of stability and function, the diagnostic capabilities of the biosensor LbL NPs were assessed across mouse models of colorectal (subcutaneous flank tumor), pancreatic (metastatic model) and ovarian cancer (flank and metastatic models). We systemically tail-vein injected NP+SP at an injection concentration of 0.5–1 mg/mL NP to provide a dosage of biosensor peptide equivalent to 0.03 mg/kg (based on an average mouse weight of 25 g). This concentration was chosen to match the NP concentration used for subsequent gene silencing experiments. One hour after injection, urine from individual mice was collected and analyzed using ELISA. Tumor-free mice were also injected with the same particles as a control, and their urine was used to determine baseline PRF concentrations in the urine. We detected significantly enriched PRF concentration in the urine of tumor-bearing mice relative to tumor-free mice across all three tested tumor models, demonstrating the broad applicability of our theranostic LbL NP platform. (Figure 2a–c). These results indicate that in vivo diagnostic capabilities of the peptide biosensor are preserved after conjugation to LbL NPs. Click conjugation of urinary-based reporter peptides can therefore facilitate the development of advanced LbL nanotheranostics capable of sensing protein signatures in the tumor microenvironment, all without the need of cumbersome imaging infrastructure.
Figure 2. Addition of the biosensing peptide onto siRNA-loaded LbL NPs provides a NP capable of simultaneous tumor detection and gene silencing.

Analysis of the urine of the mice one hour after injection indicates that theranostic LbL NPs yield significantly elevated levels of the peptide reporter fragment in the urine in a (a) pancreatic cancer flank model, (b) a metastatic model of colorectal cancer, and (c) two models of ovarian cancer. (d) Theranostic LbL NPs were intravenously administered via tail vein injection to the subcutaneous flank ovarian cancer model to provide an siRNA dosage of 0.5 mg/kg to silence the model gene luciferase in vivo. (e) Bioluminescence levels were monitored over three days using an IVIS imaging device, also used to generate the images shown in a–c. Error bars represent SEM, and statistical analysis of panels a and b uses students t-tests, while panel c uses a one-way ANOVA with a Dunnett post-hoc test and alpha of 0.05. Panel e uses a one-way ANOVA with a Tukey post-hoc test and alpha of 0.05.
Having confirmed the diagnostic capabilities of the biosensor LbL NP across three different models of cancer, we decided to focus further platform development efforts on ovarian cancer due to its high mortality rate, which stems from poor diagnostic capabilities and resistance to traditional cancer treatments, such as platinum-based therapies.[21] We investigated the ability to concurrently deliver siRNA in vivo in a flank xenograft model of ovarian cancer transfected to stably express firefly luciferase (Figure 2d–e). For this experiment, biosensor LbL NPs were prepared using firefly luciferase siRNA, and the outer pPLD layer was click-conjugated with biosensor, iRGD, and PEG to yield NP+SP and NP+SRP. These particles were then systemically administered to mice to provide a one-time siRNA dosage of 0.5 mg/kg. We found that unconjugated LbL NPs were able to mediate knockdown of the luciferase gene to 68±6% signal, with a peak effect at 3 days, compared to the control group (5% dextrose) which showed increasing luciferase expression levels over time corresponding to increasing tumor burden. This sustained reduction in luciferase signal demonstrated that our theranostic LbL NP platform is able to effectively traffic to the tumor site and deliver its cargo. We noted that addition of the biosensor and PEG to the NP surface (NP+SP) mediated a more substantial knockdown, decreasing luciferase signal to 51±2% at day 2 (Figure S3). However, these silencing effects dissipated quickly, with expression rebounding by the third day (88±17% luciferase signal, relative to pre-treatment). This was in contrast to the sustained silencing observed with the unconjugated LbL NPs. We found that the addition of tumor targeting motifs (NP+SRP) increased both the extent and duration of gene silencing compared to NP+SP. Dosing with NP+SRP mediated a consistent and durable knockdown, decreasing luciferase expression by 50±9% and 46±3% at two and three days post treatment, respectively.
To study how NP surface modification impacts particle association with ovarian cancer cells, we performed additional in vitro experiments to probe NP-cell binding and uptake (Figure 3). We chose to utilize bilayer LbL NPs without siRNA, which consisted of 100 nm carboxylate-modified latex (CML) cores labeled with an infrared fluorescent dye and coated with PLR and pPLD layers to probe the impact of the NP surface chemistry interactions with ovarian cancer cells via flow cytometry. To this end, we synthesized the following bilayer particles: NP+S, NP+SP, NP+SR, and NP+SRP. The patient-derived high grade serous ovarian cancer cell line OVCAR8 was utilized for these experiments. Cells were incubated with NPs for 4 or 24 hours prior to analysis via flow cytometry, where shifts in the NP-associated fluorescence channel were quantified (Figure 3a–d, Tables S3–S4). As expected, NP association increased over time, with only 18–30% of cells being NP-positive at 4 hours and 94–98% of cells being NP-positive at 24 hours. Functionalization of the NPs with iRGD (NP+SR, NP+SRP) led to moderate increases in mean NP-associated fluorescence intensity at both 4 and 24 hours, with the most potent trends occurring in the non-PEGylated formulation (NP+SR). This result is expected as the presence of PEG on the NP surface may interfere with iRGD-receptor interactions. Interestingly, the biosensor also appeared to improve uptake of NPs compared to unconjugated LbL NP. Further, while we noted that PEGylation of the NPs increased the total number of NP-positive cells, it depressed the amounts of NPs per cell at both 4 and 24 hours compared to the other LbL NP formulations (Figure 3c–d). Additionally, this trend was only statistically significant at 24 hours for NP+SP, and addition of iRGD (NP+SRP) mitigated this effect. Overall, these results indicated moderate improvement in NP binding to ovarian cancer cells after conjugation of either the biosensor or iRGD peptides as well as a negative effect on binding after conjugation of PEG.
Figure 3. Addition of targeting ligands and biosensor peptides improves LbL NP binding to ovarian cancer cells.

Flow cytometry was used to assess NP-associated fluorescence of OVCAR8 cells after incubating with NPs for (a) 4 and (b) 24 hours. Analysis of the mean NP-associated fluorescence intensity of the NP-positive cell population at (c) 4 and (d) 24 hours. Error bars represent SEM, and statistical analysis was carried out using a one-way ANOVA with Tukey’s post-hoc test and an alpha of 0.05. (e) Super resolution microscopy images of OVCAR8 cells incubated with sulfoCy-3 labeled LbL NPs for 24 hours. Wheat germ agglutinin, shown in red, was used to stain endosomal membranes. NPs are shown in green and nuclei in cyan. Scale bar = 10 μm.
To further probe how surface functionalization with PEG, iRGD, and biosensor impacts uptake and intracellular trafficking in ovarian cancer cells, we utilized super resolution microscopy to image OVCAR8 cells incubated for 24 hours with bilayer NP, NP+SR, or NP+SP wherein the NP core consisted of sulfoCy3-labeled liposomes (Figures 3e–g, S4). We observed that for both NP and NP+SR, a portion of particles reside on the outside of the cell while the remainder was internalized. However, the way in which the particles were internalized appeared to be quite distinct. For the cells incubated with NP, individual particles could be resolved within small vesicles (Figure 3e). For the cells incubated with NP+SR (Figure 3g), particles appeared to co-localize with perinuclear vesicles that seem enlarged relative to those found in Figures 3e–f. This is in contrast to NP+SP, where the majority of the NPs were found on the cell surface with little internalization. We postulate that this difference could possibly be explained by PEG blocking the interactions between the cell and nanoparticle surface that mediate the uptake observed for NP and NP+SR.
Having gained information about the effects of LbL surface functionalization with biosensor, PEG, and iRGD on interactions with OVCAR8 cells, we then assessed the biodistribution profiles of the biosensor LbL NPs after both intraperitoneal (IP) and intravenous (IV) (tail vein) administration in an orthotopic model of metastatic ovarian cancer 72 hours after administration (Figure 4a). We evaluated the IP administration route specifically for its emerging relevance in the management of ovarian cancer patients in the clinic.[22] The LbL NPs utilized in this study were constructed from Cy7-labeled liposomal cores coated with PLR/siRNA/PLR/pPLD, and then functionalized further with biosensor (+S) or biosensor/iRGD (+SR). PEGylation was omitted for the reasons discussed previously. Mice were sacrificed 72 hours post injection to determine relative NP levels in the tumor, liver and spleen. We observed more significant penetration of LbL NPs into tumor tissue for IP-administered NPs relative to IV-administered NPs, likely due to IP-administered particles having a higher concentration in the intraperitoneal space and extended interactions with tumor tissue compared to the IV-administered NPs (Figures 4b, S5–6). When comparing tumor accumulation of IP-administered NP+S and NP+SR, there is a significant increase of NP signal observed in the tumor tissue for NP+SR, highlighting potential increased tumor targeting and penetration due to iRGD functionalization, though we note that the method of quantification used was based on tissue surface area, and could be skewed by differences in tumor burden between the two treatment groups. In comparison to IP administration, NP+SR administered IV (at the same dosing and analysis time points as noted for IP above) did not traffic to the tumor as consistently, resulting in reduced NP accumulation there overall. It is important to note that the IV-administered NPs did not appear in the liver or spleen at this time point, which we propose is due to degradation and clearance from these organs, consistent with the biodegradable nature of these NPs. Further analysis of NP accumulation in tissues of interest via cryohistology of the tumors revealed a consistent distribution of NPs throughout the outer surface of the tumors for the IP-injected mice (Figures 4d–e, S7a–b), but less NP signal in the tumors from IV-injected mice (Figures 4f,S7c). Overall, these results demonstrate the tumor-homing ability of the biosensor LbL NPs, especially when administered IP, and that these NPs are likely biodegradable and easily cleared from filtration organs.
Figure 4. Theranostic LbL NP biodistribution in a metastatic model of ovarian cancer indicates tumor targeting capabilities, especially using intraperitoneal (IP) administration.

(a) A metastatic model of ovarian cancer was used to evaluate whether IP-administration of nanoparticles improved biodistribution and sensistivity of the diagnostic function of the theranostic NPs. (b) NP accumulation in liver, spleen, and tumors was measured 72 hours post-administration using a fluorescent flatbed scanner (Li-COR Odyssey). Tissue autofluorescence was used to generate a region-of-interest (indicated by the white outline) in which to quantify signal from the Cy7-labeled NPs (Figure S6). (c) Urine was collected 1-hour post-IP administration of NP+SR and analyzed for the MMP9-sensitive biomarker. (d–f) Cryohistology was performed on tumor tissue and imaged using a fluorescent slide scanner to detect tissue autofluorescence (pseudo-colored magenta) and NP signal (pseudo-colored green). These representative images of LbL NP distribution through tumors when administered (d–e) IP and (f) intravenously. Scale bar = 100 μm. Error bars represent SEM, and statistical analysis was carried out using an unpaired student’s t-test.
Because of the improved NP accumulation in tumor tissue of the metastatic ovarian cancer mouse model upon IP injection, we hypothesized that this administration method would result in improved biosensor readout sensitivity compared to the systemic (IV) administration reported in Figure 2. To this end, we collected and analyzed urine from mice injected IP with NP+SR for PRF concentration and observed a significant signal increase in the tumor bearing mice (p = 0.0159) compared to the control group (Figure 4c). These data suggest that matching the administration method to the disease model is a way to improve diagnostic sensitivity and utility.
CONCLUSION
Nanotheranostics provide the capability to monitor and characterize tumors and their response to therapies. This approach has tremendous potential for nanomedicine, as it can be used to identify patients who may benefit the most from particular treatments. Here, we described a method for incorporating advanced functionality into the LbL NP platform using click chemistry conjugation techniques. Combining RNA interference with biosensing and tumor-homing peptides, we demonstrate 54±3% knockdown of a model gene and sensitive detection of tumor burden across three models of cancer, including colorectal, pancreatic, and ovarian. This approach provides a highly modular system in which biosensing molecules, antifouling agents, and targeting ligands can be readily incorporated onto a highly-sophisticated drug carrier. The versatility of the LbL NP provides further levels of modularity in this system, such as the ability to use different RNAs for particular applications as well as the option to include drugs or imaging modalities in the liposomal core. Moreover, biosensor functionalization of these LbL NPs provides the ability to monitor enzymatic signatures with prognostic relevance, a strategy that could be employed to detect changes in specific protease levels and correlate them with disease progress and treatment response over time. Continued development of this platform will offer a route towards dynamic, noninvasive monitoring of disease while simultaneously delivering next-generation therapeutics.
Supplementary Material
Acknowledgements
N.B., S.C., and L.H. contributed equally to this work. N.B. and S.C. wrote the manuscript. N.B., S.C., L.H., W.W., and J.P.S. edited the manuscript. W.W. synthesized the pPLD utilized in this work, and W.W. and J.P.S. optimized click conjugation conditions. S.N.B. and P.T.H. advised on experimental design, scope, and editing of the final manuscript. All authors have given approval to the final version of the manuscript.
The authors would like to thank Elad Deiss-Yehiely for helpful discussion and editorial feedback during the preparation of this work and Chris Straehla for his help on figure design.
This work was supported by the Department of Defense’s Ovarian Cancer Research Program (W81XWH-13-1-0151) and a grant from the Koch Institute’s Marble Center for Cancer Nanomedicine. The authors also thank the MIT Koch Institute Swanson Biotechnology Center, which is supported by the Koch Institute Core Grant P30-CA14051 from the NCI, for the use of facilities, specifically: the Peterson (1957) Nanotechnology Core Facility, The Hope Babette Tang (1983) Histology Facility, the Flow Cytometry Facility, the Biopolymers & Proteomics Core Facility, the Microscopy Core Facility, the High Throughput Sciences Facility, and the Animal Imaging & Preclinical Testing Core Facility.
This material is based on work made possible by a fellowship from the Koch Institute’s Marble Center for Cancer Nanomedicine for N.B., Sloan UCEM and Siebel Scholars fellowships for S.C., a MIT Ludwig Center for Molecular Oncology fellowship for L.H., and a Research Training in Pediatric Oncology fellowship for J.P.S. (T32 CA136432–08). S.N.B. is an HHMI investigator.
Footnotes
Publisher's Disclaimer: This manuscript has been accepted after peer review and appears as an Accepted Article online prior to editing, proofing, and formal publication of the final Version of Record (VoR). This work is currently citable by using the Digital Object Identifier (DOI) given below. The VoR will be published online in Early View as soon as possible and may be different to this Accepted Article as a result of editing. Readers should obtain the VoR from the journal website shown below when it is published to ensure accuracy of information. The authors are responsible for the content of this Accepted Article.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest:
The authors declare no conflict of interest.
Contributor Information
Natalie Boehnke, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139.
Santiago Correa, Department of Bioengineering, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139.
Liangliang Hao, Institute for Medical Engineering and Science, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139.
Wade Wang, Department of Chemistry, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139.
Joelle P. Straehla, Department of Pediatric Oncology, Dana-Farber/Boston Children’s Cancer and Blood Disorders Center, Boston, MA 02115 Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139.
Sangeeta N. Bhatia, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology Institute for Medical Engineering and Science, Massachusetts Institute of Technology, Cambridge, MA 02139; Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA 02115; Broad Institute of Massachusetts Institute of Technology and Harvard, Cambridge, Massachusetts 02139; Howard Hughes Medical Institute, Cambridge, MA 02139.
Paula T. Hammond, Department of Chemical Engineering, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139
REFERENCES:
- [1].Chen H, Zhang W, Zhu G, Xie J, Chen X, Nat. Rev. Mater 2017, 2, 17024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chen HM, Zhang WZ, Zhu GZ, Xie J, Chen XY, Nat. Rev. Mater 2017, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Warren AD, Kwong GA, Wood DK, Lin KY, Bhatia SN, Proc. Natl. Acad. Sci. USA 2014, 111, 3671; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kim JS, Rieter WJ, Taylor KM, An H, Lin W, Lin W, J. Am. Chem. Soc 2007, 129, 8962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Pellikainen JM, Ropponen KM, Kataja VV, Kellokoski JK, Eskelinen MJ, Kosma V-M, Clin. Cancer Res 2004, 10, 7621; [DOI] [PubMed] [Google Scholar]; b) Wu ZS, Wu Q, Yang JH, Wang HQ, Ding XD, Yang F, Xu XC, Int. J. Cancer 2008, 122, 2050; [DOI] [PubMed] [Google Scholar]; c) Sier CFM, Kubben FJGM, Ganesh S, Heerding MM, Griffioen G, Hanemaaijer R, vanKrieken JHJM, Lamers CBHW, Verspaget HW, Br. J. Cancer 1996, 74, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Morton SW, Poon ZY, Hammond PT, Biomaterials 2013, 34, 5328; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Choi KY, Correa S, Min J, Li JH, Roy S, Laccetti KH, Dreaden E, Kong S, Heo R, Roh YH, Lawson EC, Palmer PA, Hammond PT, Adv. Funct. Mater 2019, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dreaden EC, Kong YW, Morton SW, Correa S, Choi KY, Shopsowitz KE, Renggli K, Drapkin R, Yaffe MB, Hammond PT, Clin. Cancer Res 2015, 21, 4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gu L, Deng ZJ, Roy S, Hammond PT, Clin. Cancer Res 2017, 23, 7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dang X, Gu L, Qi J, Correa S, Zhang G, Belcher AM, Hammond PT, Proc. Natl. Acad. Sci. USA 2016, 113, 5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Correa S, Dreaden EC, Gu L, Hammond PT, J. Controlled Release 2016, 240, 364; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shi XY, Wang SH, Swanson SD, Ge S, Cao ZY, Van Antwerp ME, Landmark KJ, Baker JR, Adv. Mater 2008, 20, 1671. [Google Scholar]
- [10].a) Bergbreiter DE, Chance BS, Macromolecules 2007, 40, 5337; [Google Scholar]; b) Such GK, Quinn JF, Quinn A, Tjipto E, Caruso F, J. Am. Chem. Soc 2006, 128, 9318; [DOI] [PubMed] [Google Scholar]; c) De Geest BG, Van Camp W, Du Prez FE, De Smedt SC, Demeester J, Hennink WE, Macromol. Rapid Commun 2008, 29, 1111. [Google Scholar]
- [11].Such GK, Tjipto E, Postma A, Johnston AP, Caruso F, Nano Lett. 2007, 7, 1706. [DOI] [PubMed] [Google Scholar]
- [12].a) Kinnane CR, Such GK, Antequera-Garcia G, Yan Y, Dodds SJ, Liz-Marzan LM, Caruso F, Biomacromolecules 2009, 10, 2839; [DOI] [PubMed] [Google Scholar]; b) Leung MK, Such GK, Johnston AP, Biswas DP, Zhu Z, Yan Y, Lutz JF, Caruso F, Small 2011, 7, 1075. [DOI] [PubMed] [Google Scholar]
- [13].Kwon EJ, Dudani JS, Bhatia SN, Nat. Biomed. Eng 2017, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Correa S, Boehnke N, Deiss-Yehiely E, Hammond PT, ACS Nano 2019, 13, 5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Torchilin VP, Nat. Rev. Drug Discov 2005, 4, 145; [DOI] [PubMed] [Google Scholar]; b) Akbarzadeh A, Rezaei-Sadabady R, Davaran S, Joo SW, Zarghami N, Hanifehpour Y, Samiei M, Kouhi M, Nejati-Koshki K, Nanoscale Res. Lett 2013, 8, 102; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Levchenko TS, Rammohan R, Lukyanov AN, Whiteman KR, Torchilin VP, Int. J. Pharm 2002, 240, 95; [DOI] [PubMed] [Google Scholar]; d) Miller CR, Bondurant B, McLean SD, McGovern KA, O’Brien DF, Biochemistry 1998, 37, 12875. [DOI] [PubMed] [Google Scholar]
- [16].a) Deng ZJ, Morton SW, Bonner DK, Gu L, Ow H, Hammond PT, Biomaterials 2015, 51, 250; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang C, Tang N, Liu X, Liang W, Xu W, Torchilin VP, J. Controlled Release 2006, 112, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Presolski SI, Hong VP, Finn MG, Curr. Protoc. in Chem. Biol 2011, 3, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schneider G, Decher G, Langmuir 2008, 24, 1778. [DOI] [PubMed] [Google Scholar]
- [19].a) Gref R, Luck M, Quellec P, Marchand M, Dellacherie E, Harnisch S, Blunk T, Muller RH, Colloids Surf 2000, 18, 301; [DOI] [PubMed] [Google Scholar]; b) Suk JS, Xu QG, Kim N, Hanes J, Ensign LM, Adv. Drug Deliver. Rev 2016, 99, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dudani JS, Ibrahim M, Kirkpatrick J, Warren AD, Bhatia SN, Proc. Natl. Acad. Sci. USA 2018, 115, 8954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].a) Luvero D, Milani A, Ledermann JA, Ther. Adv. Med. Oncol 2014, 6, 229; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bowtell DD, Bohm S, Ahmed AA, Aspuria PJ, Bast RC, Beral V, Berek JS, Birrer MJ, Blagden S, Bookman MA, Brenton JD, Chiappinelli KB, Martins FC, Coukos G, Drapkin R, Edmondson R, Fotopoulou C, Gabra H, Galon J, Gourley C, Heong V, Huntsman DG, Iwanicki M, Karlan BY, Kaye A, Lengyel E, Levine DA, Lu KH, McNeish IA, Menon U, Narod SA, Nelson BH, Nephew KP, Pharoah P, Powell DJ, Ramos P, Romero IL, Scott CL, Sood AK, Stronach EA, Balkwill FR, Nat. Rev. Cancer 2015, 15, 668; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, Gaudet MM, Jemal A, Siegel RL, CA: Cancer J. Clin 2018, 68, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) Wright AA, Cronin A, Milne DE, Bookman MA, Burger RA, Cohn DE, Cristea MC, Griggs JJ, Keating NL, Levenback CF, Mantia-Smaldone G, Matulonis UA, Meyer LA, Niland JC, Weeks JC, O’Malley DM, J. Clin. Oncol 2015, 33, 2841; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) van Driel WJ, Koole SN, Sikorska K, Schagen van Leeuwen JH, Schreuder HWR, Hermans RHM, de Hingh I, van der Velden J, Arts HJ, Massuger L, Aalbers AGJ, Verwaal VJ, Kieffer JM, Van de Vijver KK, van Tinteren H, Aaronson NK, Sonke GS, N. Engl. J. Med 2018, 378, 230; [DOI] [PubMed] [Google Scholar]; c) Narod S, Nat. Rev. Clin. Oncol 2016, 13, 255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.