

Abstract
Patients with ulcerative colitis (UC) have an increased risk of developing colitis-associated colon cancer (CACC). Changes in glycosylation of the oncoprotein MUC1 commonly occur in chronic inflammation, including UC, and this abnormally glycosylated MUC1 promotes cancer development and progression. It is not known what causes changes in glycosylation of MUC1. Gene expression profiling of myeloid cells in inflamed and malignant colon tissues showed increased expression levels of inflammatory macrophage-associated cytokines compared to normal tissues. We analyzed the involvement of macrophage-associated cytokines in the induction of aberrant MUC1 glycoforms. A co-culture system was used to examine the effects of M1 and M2 macrophages on glycosylation-related enzymes in colon cancer cells. M2-like macrophages induced the expression of the glycosyltransferase ST6GALNAC1, an enzyme that adds sialic acid to O-linked GalNAc residues, promoting the formation of tumor-associated sialyl-Tn (sTn) O-glycans. Immunostaining of UC and CACC tissue samples confirmed the elevated number of M2-like macrophages as well as high expression of ST6GALNAC1 and the altered MUC1-sTn glycoform on colon cells. Cytokine arrays and blocking antibody experiments indicated that the macrophage-dependent ST6GALNAC1 activation was mediated by IL-13 and CCL17. We demonstrated that IL-13 promoted phosphorylation of STAT6 to activate transcription of ST6GALNAC1. A computational model of signaling pathways was assembled and used to test IL-13 inhibition as a possible therapy. Our findings revealed a novel cellular cross-talk between colon cells and macrophages within the inflamed and malignant colon that contributes to the pathogenesis of UC and CACC.
Keywords: IL-13, CCL17, ST6GALNAC1, MUC1-sTn, Macrophages
Introduction
Approximately 1.4 and 2.2 million people are affected with inflammatory bowel diseases (IBD) in the US and Europe, respectively (1,2). IBD, including ulcerative colitis (UC) and Crohn’s disease (CD), are chronic intestinal inflammation disorders that affect the gastrointestinal tract; namely the colon in UC and anywhere in the gastrointestinal tract in CD. Patients with UC often present with bloody diarrhea while those with CD tend to have weight loss and abdominal pain. Histologically, there is a disruption of crypt architecture and inflammatory cell infiltration (3). Patients with UC and CD colitis have an increased risk of developing colitis-associated colon cancer (CACC). This risk is proportional to the duration and extent of disease, with a cumulative incidence as high as 30% in individuals with longstanding UC and widespread colonic involvement (4).
During tumor initiation and progression, epithelial cells acquire new capabilities that allow them to become tumorigenic and ultimately malignant. Glycosylation changes are one of the most common post-translational modifications that occur during malignant transformation (5). Among all glycoproteins, mucin-type O-glycans are frequently altered during UC and progression to colon cancer (6-8). The mucin MUC1, is expressed at low levels on the apical surface of normal epithelial cells and is heavily glycosylated in the tandem repeat region of the extracellular domain rich in proline, serine, and threonine residues (9,10). During colonic malignant transformation, MUC1 is over-expressed and it loses its apical polarity and displays an altered glycosylation profile, becoming predominantly hypoglycosylated (6-8). Aberrant and tumor-associated mucin glycoforms expose long stretches of naked peptide backbone decorated with sialyl-LewisX (SLeX), prematurely terminated monosaccharides (Tn antigens) or disaccharides (T antigens) and their sialylated forms sTn and sT to the immune system (9). We demonstrated that the presence of human MUC1 exacerbates chronic inflammation and induces tumorigenesis in the azoxymethane/dextran sodium sulfate (AOM/DSS) mouse model of CACC (11). Chronic inflammation promotes the expression of aberrant MUC1 glycosylation in colon epithelial cells whereas no or very low expression of hypoglycosylated MUC1 is detected in colon tissues of healthy mice (11).
Altered O-glycans on mucins can result either from mutations in the cell chaperone COSMC or changes in ST6GALNAC1, enzymes involved in the biosynthesis of Tn/sTn glycans (12). An inflammatory microenvironment can also induce changes in the glycan composition of cells via modulation of glycosyltransferases (13,14).
Tumor-associated macrophages (TAMs), by secreting inflammatory cytokines and chemokines, play a key role in tumor initiation, promotion, invasion and metastasis. During intestinal inflammation, such as UC, macrophages acquire a typical inflammatory phenotype, present antigens, undergo phagocytic activities and secrete inflammatory cytokines and chemokines thus contributing to UC pathogenesis and progression to CACC (15-17).
Differentiated macrophages are classified into two subpopulations: the classically activated macrophages (M1 phenotype) and the alternatively activated macrophages (M2 phenotype) (18). In vitro studies have shown that upon stimulation with inflammatory factors, interferon-γ (IFN-γ) and lipopolysaccharides (LPSs), macrophages polarize to a M1 state and secrete pro-inflammatory cytokines IL-6, IL-1β, and TNF-α. By contrast, in response to anti-inflammatory signals IL-4 and IL-13, macrophages polarize to a M2 state, secrete tumor promoting cytokines such as arginase (Arg)-1, and express the mannose receptor (MR), IL-10, and Fizz1 (18). Within tumors, both pro- and anti-inflammatory signals are simultaneously present, resulting in a more complex spectrum of macrophage polarization states (19).
Here, we showed that markers associated with both types of macrophages were highly expressed in UC and CACC tissues. The expression of TAM markers, glycosylation-associated enzymes and tumor-associated MUC1 glycoforms were assessed in human tissues and in a co-culture model system. Computational modeling was carried out to evaluate the involvement of macrophage-induced cytokines in aberrant glycosylation, in particular in the regulation of glycosyltransferase ST6GALNAC1. A novel regulatory mechanism was discovered involving macrophage-derived CCL17 and macrophage-enhanced IL-13 in the induction of ST6GALNAC1 expression in colon cancer cells. Chromatin immunoprecipitation assays of human UC and CACC samples indicated that IL-13 via STAT6 directly promoted the transcriptional activity of the ST6GALNAC1 gene. Thus, increased expression of ST6GALNAC1 resulted in the production of the MUC1-sTn glycoform, which is associated with colonic inflammation and cancer. A computational model demonstrated the signaling, cross-talk and dynamics involved in regulating gene expression, and identified a potential therapeutic intervention.
Material and Methods
Cell culture and Reagents.
SW480 (ATCC® CCL-228) and HT-29 (ATCC® HTB-38) cell lines were purchased from American Type Culture Collection in 2016 and frozen upon initial expansion (<5 passages). Cells were cultured for a maximum of 15 passages in RPMI-1640 (Royal Park Memorial Institute Medium, Cellgro, Mediatech, Inc.) medium supplemented with 10% heat-inactivated fetal bovine serum, 100 units/ mL penicillin, 100 μg/mL streptomycin and 2 mmol/L L-glutamine. Both cell lines were regularly tested for Mycoplasma contamination. Cells were not reauthenticated. For specific experiments IL-13 and CCL17 (R&D Systems) were used at 20, 50 100 ng/mL for 24h. IL-13 neutralizing antibody (JES10-5A2, Thermo Scientific) and CCL17 (AF364, R&D Systems) were used at 1:200 dilution.
Patients and tissue samples.
Archived paraffin sections of colonic biopsies of UC patients in remission (non-inflamed), with active disease, and those with colitis associated colon cancer were selected and collected in the Department of Gastroenterology, University of Pittsburgh. The study was approved by the institutional review board of the University of Pittsburgh (PRO16090194). Chart review to identify patients who had biopsy samples during colonoscopy and/or surgery was performed. In order to perform IHC staining, a waiver of consent and HIPAA was requested.
Fresh colon tissues were obtained under the approved IRB PRO19070174. Patients had previously provided a signed informed consent at the time their tissue was collected. Tissues were processed and stored in under standard operating procedures of the Pittsburgh Biospecimen Core.
Plasmids and Transfection.
The cDNA for ST6 N-Acetylgalactosaminide Alpha-2,6-Sialyltransferase 1 (ST6GALNAC1) was subcloned into the pcDNA3 plasmid vector (Invitrogen) (20). Empty vector was used as the negative control. Transfections were performed with Lipofectamine 3000 (cat. L3000008, Thermo Fisher Scientific,) according to manufacturer’s instruction.
Antibodies.
The following primary antibodies were used: anti-p65 (sc-8008), anti-pp65 (sc-166748) anti-IκBα (sc-1643), anti-MUC1 VU-4H5 (sc-7313, Santa Cruz), anti-ST6GALNAC1 (PA5-31200 and 15363-1-AP), anti-IL-13 (AHC0132), anti-actin (MA5-11869, Thermo Fisher Scientific), anti-MUC1 5E5 (TAB-418MZ, Creative Biolabs), anti-CD163 (NBP2-36494, Novus Biologicals), anti-CCL17 (ab182793), anti-sialyl Tn, clone sTn 219 (ab115957), anti-CD68 (ab955), anti-CD86 (ab53004, Abcam), anti-AKT (C67E7, #4691), anti-p-AKT (D9E, #4060), p-STAT1 (D4A7, #7649), p-STAT3 (D3A7, #9145), p-STAT6 (D8S9Y, #56554, Cell Signaling).
Western Blotting.
Total proteins from human colon tissues and cancer cell lines were extracted using RIPA buffer (150 mM NaCl, 0.5 sodium deoxycholate, 0.1% SDS, 1% NP-40 and 50 mM Tris-HCl) with commercial protease inhibitors (Complete Protease Inhibitor Cocktail, Roche) and phosphatase inhibitors (Phosphatase Inhibitor Cocktail II, Sigma- Aldrich). Proteins were separated by SDS-PAGE and transferred to PVDF membranes. Blots were incubated with the primary antibodies indicated above. Immunoblots were developed with m-IgGκ BP-HRP (sc-516102) or mouse anti-rabbit IgG-HRP (sc-2357, Santa Cruz) and chemiluminescence reagents (SuperSignal West Pico Substrate, cat. #34580, Pierce). Intensity of signals was determined by densitometric scanning (Kodak, Image Station 4000MM). Densitometry of western immunoblotting results was performed using Image J software.
ELISA.
IL-13 concentrations in the conditioned medium of HT-29 or SW480 cancer cells co-cultured with M1 or M2 macrophages were measured using a human ELISA kit (DY213, BD Biosciences) following manufacturer’s protocol. Mono-cultures of HT-29 or SW480, M1 and M2 cells were used as control. IL-13 concentrations detected were within the range of the standard curve. All points were done in triplicate, and the experiments were repeated three times. Samples were read in a microplate reader (Infinite 200 Pro, Tekan).
Immunofluorescence confocal microscopy.
Cultured cells were fixed in 4% paraformaldehyde for 20 min and permeabilized in 0.5% Triton-X100 for 20 min. The fixed tissues or cells were incubated with primary Abs for 1h at RT followed by secondary anti-mouse Alexa-488 or Cy3 antibody (Invitrogen Life Technologies) for 1h at RT. Abs were diluted in 1% bovine serum albumin (BSA). Nuclei were stained with mounting medium containing DAPI (VectorLab). Confocal images were captured on an Olympus Fluoview 1000 confocal microscope.
Immunohistochemistry (IHC).
Slides were deparaffinized by baking overnight at 59°C. Endogenous peroxidase activity was eliminated by treatment with 30% H2O2 for 15 min at room temperature. Antigen retrieval was performed by microwave heating in 0.1% citrate buffer for 10 min at 850V. Nonspecific binding sites were blocked with 2% BSA. Reaction with anti-CD163, anti-CD68, anti-CD86, anti-sTn, anti-ST6GALNAC1 (listed above) was for 16h at 4C. Staining was performed by the avidin-biotin-peroxidase complex method with a commercial kit (PK4000, Vectastain ABC HRP kit; Vector Laboratories) according to the manufacturer’s protocol. Positive signals were visualized by a DAB Substrate Kit (cat. #550880, BD Pharmingen) according to the manufacturer’s protocol. The total inflammation score for each sample was determined as previously described [8]. For double staining IHC, ImmPRESS duet staining HRP/AP polymer kits, anti-rabbit IgG-brown and anti-mouse IgG red was used (MP-7714, Vector Laboratories) according the manufacturer’s protocol. Histology sections were observed using an Olympus BX40 microscope. Images were acquired using a Leica DFC420 camera and Leica Application Suite version 2.7.1 R1.
Migration and invasion assay.
Migration studies were conducted using 24-well Transwells, 8 μm pore size (Costar Transwell; Corning Inc.). In each well, 4 X 104 HT-29 or SW480 cells were plated into the upper chamber in serum free medium whereas 700 μl of 5% FBS containing medium was placed in the lower chamber. After 16 h, cells remaining in the upper chamber were removed using cotton swabs. Cells adhering to the lower surface were fixed with 4% paraformaldehyde for 20 min and stained with Christal Violet 1%. Cells were then counted in 5 different fields using a microscope at 40X magnification.
Peripheral blood monocyte isolation and macrophage differentiation.
Peripheral blood mononuclear cells (PBMCs) were freshly isolated by density gradient centrifugation using Ficoll Paque Plus (Sigma-Aldrich) for 50 min at 400g. At least 20 de-identified human Buffy Coat samples purchased from the Pittsburgh Central Blood Bank (or Vitalant) fulfilling the basic exempt criteria 45 CFR 46.101(b)(4) in accordance with the University of Pittsburgh IRB guidelines. Monocytes were then isolated with CD14+ microbeads (MACS Miltenyi) and incubated for 5 days in RPMI/10%FCS and 1% penicillin/streptomycin solution (Sigma) supplemented with 25 ng/ml human M-CSF (R&D Systems) to stimulate macrophage differentiation. Macrophages were then washed and primed by incubating with RPMI media supplemented with 100 ng/ml IFN-γ (R&D Systems) or 50 ng/ml IL4 and IL13 (R&D Systems) for 24h to drive M1 or M2 polarization respectively (21). Unprimed macrophages were incubated with non-supplemented RPMI media. By adding 20 ng/ml LPS to media containing priming stimuli for another 24h, M1 and M2 macrophages were activated. Cells were thoroughly washed in PBS before being transferred to co-culture with colon cancer cells.
Indirect co-culture assay.
CD14+ monocytes were differentiated in Transwell insert dishes (Corning) as described above. After differentiation into M1 and M2, macrophages were washed three times with RPMI and then placed in 6-well plates where HT-29 or SW480 cells (2 X 105 cells/wells) were pre-plated the day before in RPMI media. Macrophages and colon cancer cells were co-cultured for 48 hours in RPMI media.
Cytokine and chemokine expression detection.
Bead-based multiplex assay panel.
The supernatants of HT29 and SW480 cells, M1 and M2 macrophages, and the conditioned medium of HT-29 cells co-cultured with macrophages were tested for cytokines and chemokines using the Legendplex™ human inflammation 10-plex panel (Biolegend LEGENDplex Cat. 740508) for IL-1β, IL-1RA, TNFα, IP-10, IL-6, IL-10, IL-12p70, IL-12p40, CCL17 and IL-23. Samples were treated following the manufacturer’s instructions, measured with a FACS LSRFortessa (BD Biosciences) and analyzed with FlowJo v.10 software. Human cytokine array. Supernatants and condition medium were also analyzed using a cytokines array (Raybiotech, AAH-CYT-5-2). The procedure was performed according to the manufacturer’s instructions. Membranes were developed, and the dots were quantified using Image J plug-in protein array analyzer according to the developer’s instructions (http://image.bio.methods.free.fr/ImageJ/Protein-A).
Gene expression profiling.
Total RNA from colon tissues of patients with no inflammation (control, n=3), active and severe ulcerative colitis (inflamed, n=3), colitis-associated colon cancer (CACC, n=3) was isolated using RNeasy Micro kit (Qiagen) following the manufacturer’s instructions. The myeloid innate immune response was examined using the nCounter Human Myeloid Innate Immunity Panel v2 (NanoString Technologies). The protocol was carried out at the University of Pittsburgh NanoString facility using 80 ng of total RNA from each sample following their commercial protocol. Data were analysed using the NanoStringDiff R-package, following the procedure described in the package’s instructions (22). Normalization of mRNA content, for heatmap visualization purposes, was performed by using the NanoString Data Normalization function, which adjusts for positive control size factors, background noise and housekeeping genes size factors. Differentially expressed genes were detected by using the glm.LRT function. P-values were adjusted for multiple comparison using the procedure of Benjamini and Hochberg. A gene was considered significantly over-expressed if associated with an adjusted p < 0.01 and a logFC > 1.
Chromatin immunoprecipitation assay.
The chromatin immunoprecipitation assay was performed on frozen human colon tissues, utilizing the commercially available ChromaFlash High Sensitivity ChIP Kit (cat. P-2027, Epigentek), according to the manufacturer’s instructions. Cells were fixed with 1% formaldehyde for 10 min at 37°C. Chromatin was precipitated with 4 μg of anti-pSTAT6 (Thermo Fisher Scientific) or anti-p-p65 (Santa Cruz) at 4°C overnight. The presence of ST6GALNAC1 gene promoter sequences in immunoprecipitated DNA was identified by RT-PCR using the following primer sequences: F1: AGTTGGATCTGGACCCCAAG, R1: CACGTATGAGGGCTCACTCT; F2: CCCTCATACGTGCTGGTCAT, R2: AACCCATCTGCCGCCATATAA. In control samples, primary antibody was replaced with non-immune IgG. All experiments were repeated at least three times.
Quantitative real-time PCR.
Total RNA was extracted from colon cancer cell lines, murine and human colon tissues using QIAshredder (cat. 79654) and RNeasy mini kit (cat. 74104, Qiagen) according to the manufacturer’s instructions. RNA was measured using a Gen5 microplate reader (Biotek). A total of 2 μg of RNA was reverse-transcribed using the RT2 HT First Strand Kit (cat. 330411, Qiagen ). A total of 4 μl of RT products was used to amplify ST6GALNAC1 and GAPDH as an internal control. Real-time PCR was performed using a SYBR Green PCR kit (Qiagen) and a StepOnePlus realtime PCR system (Applied Biosystems). Gene expression was determined using the 2-ΔCt. All experiments were repeated three times in triplicate.
Gene expression profiling of glycosylation associated enzymes.
Gene expression profiling of human glycosylation was performed using the RT2 Profiler PCR Array (PAHS-046Z, Qiagen). This PCR array is a 96-well plate containing the RT2 Profiler PCR Primer Assays for a set of 84 related genes, plus five housekeeping genes and three controls. Data analyses were performed using the web-based analysis software (http://www.sabiosciences.com/pcrarraydataanalysis.php) and described in detail before (11).
Statistical analysis.
Differences between two conditions were analyzed by the student’s t test or one-way ANOVA with Tukey post-tests for multiple pairwise comparisons. In all cases, p < 0.05 was considered statistically significant. Statistics were calculated with Prism software (Graphpad).
Computational modeling and simulation.
The protein interactions involved in signaling pathways investigated in this study were collected from the literature and entered using the BioRECIPES tabular format [23], a model representation format that includes the name, type (protein, gene or a chemical), cellular location, number of possible discrete states, and formatted list of regulators for each model element. Three levels were used to represent activation or inhibition of elements. Level 0 was if the element has low activity, level 1 was if the element has moderate activity and 2 was if the element has high activity. All elements were initialized to 0 except input cytokines that were set to match experimental conditions (levels indicated in Fig. 6B). From the tabular representation an executable discrete model were created using the element update functions generated from the formatted regulator lists. Simulations of the model were performed using the publicly available stochastic simulator, DiSH [24]. Different experimental conditions, scenarios, were defined by assigning initial values to all model elements, and a set of inputs for scenarios of normal, UC, CACC and IL-13 inhibitor. Many independent runs of the scenario represent multiple cells in an experiment that have the same starting point but traverse through time steps differently. From these individual simulation runs, we computed average trajectories to plot and visualize element behavior over time.
Figure 6. Computational model of signaling pathways in UC and CACC.

A) Interaction map of the UC and CACC model. Pointed arrows represent activation, blunted arrows represent inhibition. The cytokines (triangles) were selected from experiments and represented as inputs, these ligands bound to the receptors (orange shape) at the plasma membrane, and the signal was transduced across the membrane by activating the receptors. Signaling cascades then relayed the signal through the cytosol to the transcription factors, STAT1, STAT3, STAT6 and AKT. The latter were translocated into the nucleus to regulate the ST6GALNAC1 gene (rectangle) to influence the amount of enzyme in the Golgi and ultimately the glycosylation of extracellular sTn form of MUC1. B) Table showing cytokine input for UC and CACC. C. Simulation results showing the average behavior from 200 runs over 1000 time steps for MUC1 sTn for three different scenarios (Normal, UC and CACC). D) Simulation results showing the average behavior from 200 runs over 1000 time steps for MUC1 sTn for two different scenarios (UC + IL-13 inhibitor and CACC + IL-13 inhibitor).
Results
Increased macrophage-associated markers in colonic inflammation and colon cancer
To investigate changes in the innate immune-related genes during progression from UC to CACC, 770 genes involved in the innate immune response were profiled using the NanoString nCounter Human Myeloid Innate Immunity Panel. Tissue samples of non-inflamed colon of UC patients (Control) were compared to severe active UC and CACC (Supplemental Fig. S1A). Increased expression of 7 genes was unique to UC and of 221 genes was unique to CACC, with 45 genes up-regulated in both UC and CACC (Fig. 1A). Up-regulated genes were defined as having a log fold change of expression >1 as compared to those in control tissues.
Figure 1. Abundance of macrophage-associated cytokines and markers in UC and CACC patients.

Colon tissues collected from three patients with non-inflamed colon tissues (Control, n=4), ulcerative colitis (UC, n=4) or colitis- associated colon cancer (CACC, n=4) were assessed on the NanoString platform. A) Venn diagram representing genes up-regulated in CACC and/or UC compared to control samples. B-C) Relative gene expression of indicated genes from NanoString analysis. Statistical analyses were performed using a two-way ANOVA with Tukey’s multiple comparison, comparing UC vs Control or CACC vs Control. D) Representative image of CD68 immunostaining in paraffin-embedded human colon tissues sections from patients with uninflamed colon tissue (Control, n=10), ulcerative colitis (UC, n=10) and colitis-associated colon cancer (CACC, n=8). Scale bars, 20 μm. E, F, G) Number of CD68+, CD86+ and CD163+ positive cells in tissues sections from 6 high power fields. H and I) Ratio of double positive CD163+ and CD68+ in UC and CACC vs Control samples. P values were calculated by one-way ANOVA with Tukey post-tests. Error bars represent the SEM. *p<0.05 **p<0.01 ***p<0.001.
Cytokines and chemokines involved in macrophage chemotaxis were significantly upregulated in UC and CACC samples. In particular, S100A8, S100A9, and VEGF-A, known to mediate the migration of macrophages to the tumor site (23), were up-regulated both in UC and CACC (Fig. 1B). Expression levels of macrophage-attracting cytokines and chemokines CCL2, CCL19 and CCL21 were significantly higher in CACC (Fig. 1B). These data suggest that macrophages were recruited and continued to accumulate during inflammation and tumor progression. Genes associated with the a pro-inflammatory (M1-like) phenotype, including NOS2, TNFA, IL-1A and IL-23 were significantly up-regulated in UC samples when compared to control and CACC samples (Fig. 1C). The expression of genes linked with an anti-inflammatory (M2-like) phenotype, such as IL-6, IL-1R1, IL-13, CD163, CCL17 and TGF-β were up-regulated in both inflamed tissue and tumor samples (Fig. 1C). Notably, some of the M2-associated markers, including CD163, IL-6, and IL-13 were higher in CACC samples compared to UC samples suggesting a potential role of M2-like cells in CACC promotion.
To further examine the involvement of inflammatory macrophages in inflammation leading to colon cancer, the abundance of infiltrating macrophages in human UC (n=10) and CACC (n=8) tissues was evaluated. Tissues collected from UC patients in remission with no evidence of disease (n=10) were used as controls. Biopsies were taken from left colon and rectum regions. Immunohistochemistry (IHC) was performed using anti-CD68 antibody that recognizes all macrophages, anti-CD86 antibody that recognizes M1-like macrophages, and anti-CD163, a well-known marker of M2-like macrophages (18). The number of CD68+, CD86+ and CD163+ cells increased in UC and CACC samples compared to control (Fig. 1D-G). Importantly, the ratio of CD163+/CD68+ was significantly higher in both UC and CACC than in controls (Fig. 1I and Supplemental Data S1B).
Exposure to M2 macrophages induced epithelial ST6GALNAC1 and sTn-MUC1 expression
Having observed that tissue-infiltrating macrophages in UC and CACC are located adjacent to epithelial cells (Fig 1D), we postulated that these macrophages and epithelial cells may communicate. Using changes in MUC1 as a potential biomarker of this interaction, we investigated in a trans-well co-culture system whether macrophage-secreted factors could influence the status of MUC1 glycosylation in human colon tumor cells. Freshly isolated human peripheral blood monocytes cultured in trans-well inserts were differentiated and polarized into M1 (IFN-γ + LPS) or M2 (IL-4 + IL-13) macrophages. After 24 hours, the inserts containing macrophages were added to trans-wells containing HT-29, a colon tumor cell line that has been used to study mature intestinal cells, IBD, CACC and colon cancer (24-26).
Aberrant glycosylation of MUC1 can result from aberrant glycosyltransferase activity and therefore, we first studied the ability of macrophages to modulate the expression of glycosyltransferases. An RT2 Profiler PCR array of 84 glycosylation-associated genes was performed in HT-29 cells co-cultured with macrophages. HT-29 cells cultured alone were used as a control. The gene array revealed a distinct glycosyl/fucosyl/sialyl-transferase expression profile in HT29 cells after co-culture with macrophages (Fig. 2A). Exposure to M1 or M2 macrophages induced up- or down-regulation of 24 and 17 genes, respectively, when compared to control HT-29 cells (Fig. 2B). M1 macrophages promoted the expression of B3GNT4, B3GNT8, EDEM1, GALNT9, GALNT13, GALNT16, MAN2A2, POMT1, ST8SIA2, and ST8SIA3 whereas M2 induced the expression GCNT1, GALNT1, MAN2B1, ST6GALNAC1, ST6GAL1, MAN2A1 and MGAT4A. Both M1 and M2 modulated GALNT11, NEU1, MGAT2 and ST3GAL1. We focused on genes that encoded enzymes involved in the initial steps of O-glycan chain elongation, whose activity would lead to shorter and more tumor-associated MUC1 forms (9). One of these enzymes is ST6GALNAC1, a glycosyltransferase that catalyzes the transfer of a sialic acid residue in alpha-2,6-linkage to O-linked GalNAc residues on mucins to form the cancer-associated sialyl-Tn (sTn) antigen (Fig. 2C) (9).
Figure 2. M2 polarized macrophages induced ST6GALNAC1 in colon cancer cells.

A) Heatmap and B) Venn diagram of eighty-four genes related to glycosylation differentially expressed in HT-29 colon cancer cells co-cultured with polarized M1 and M2 macrophages or cultured alone. C) Schematic model of Tn and s-Tn antigens. The addition of α2,6-linked sialic acid on the Tn antigen is mediated by ST6GALNAC1 and results in the biosynthesis of sTn antigen. D) Proteins isolated from HT-29 and SW-480 colon cancer cells co-cultured with M1 and M2 macrophages were immunoblotted with anti-ST6GALNAC1 Ab. Actin was used as loading control. Graph represents the relative protein expression measured by densitometry scanning of western blots. Results are representative of at least four independent experiments. E) Representative image of ST6GALNAC1 (brown) and CD163 (magenta) double staining in paraffin-embedded human colon tissues sections from non-inflamed (Control, n=9), ulcerative colitis (UC, n=10), and colitis- associated colon cancer (CACC, n=7). Scale bars, 20 μm. F) Quantification of ST6GALNAC1-positive cells in tissues sections from 6 high power fields. G) Representative image of low ST6GALNAC1 and CD163 expression in a selected area of a CACC sample. Error bars represent the SEM (D and F). P values were calculated using one-way ANOVA test. ***p<0.001 (D and F).
ST6GALNAC1 is overexpressed in gastric, colon, breast, ovarian and pancreatic cancers (27-31). Western blot analysis confirmed that M2-secreted factors induced a significant up-regulation of ST6GALNAC1 protein in HT29 and SW480 colon cancer cells (Fig. 2D).
High expression of ST6GALNAC1 have been associated with tumorigenicity, increased cell proliferation and migration in different types of cancer cells (32,33). To evaluate the effect of ST6GALNAC1 over-expression on cell migration capacity, HT-29 and SW-480 cells were transfected with a plasmid for high level constitutive expression of ST6GALNAC1 (Supplemental Fig. S2A) and analyzed after 48 hours. Then a cell migration assay was performed using a Boyden chamber. The migration rate of ST6GALNAC1-overexpressing HT-29 and SW480 cells was significantly increased compared with cells transfected with the empty vector control (Supplemental Fig. S2B and C).
Next, to assess the clinical relevance of M2-like macrophages and ST6GALNAC1 expression in UC and CACC, we performed double IHC staining with anti-CD163 (magenta) and anti-ST6GALNAC1 (brown) antibodies (Abs) on tissue sections. Representative images are shown in Fig. 2E. ST6GALNAC1 showed a homogenous cytoplasmic staining mainly in the Golgi of glandular cells, it was over-expressed in both inflamed and tumor tissues and was absent or weakly stained in non-inflamed tissues (Fig. 2E and F). Tissues characterized by high accumulation of CD163+ macrophages over-expressed ST6GALNAC1 (Fig. 2E) whereas small areas of inflamed or cancer tissues with low abundance of CD163+ cells presented low expression of ST6GALNAC1 (Fig. 2G).
To determine whether M2 macrophages induced ST6GALNAC1 in HT-29 cells results in the induction of the MUC1-sTn glycoform, confocal immunofluorescence was performed using anti-MUC1 5E5 that recognizes MUC1-Tn/sTn structures, and anti-MUC1 CT2 that recognizes the cytoplasmic tail of MUC1. Our results showed that MUC1-Tn/sTn expression was increased upon co-culture with M2 macrophages when compared to M1 or HT-29 alone (Fig. 3A). To detect the amount of M2-induced MUC1-sTn in HT-29 cells, we performed a co-immunoprecipitation (Co-IP) assay. Specifically, protein lysates, extracted from HT-29 cells co-cultured with M1 and M2 macrophages, were immunoprecipitated with anti-MUC1 5E5 and then immunoblotted with anti-sTn (sTn 219), which displayed a strong band around 150 kDa (Fig. 3B). This assay confirmed that M2 macrophage exposure specifically induced higher expression MUC1-sTn in HT-29 cells compared to M1 and no macrophage samples.
Figure 3. M2 polarized macrophages induced MUC1-sTn tumor forms in inflamed and tumor colon tissues.
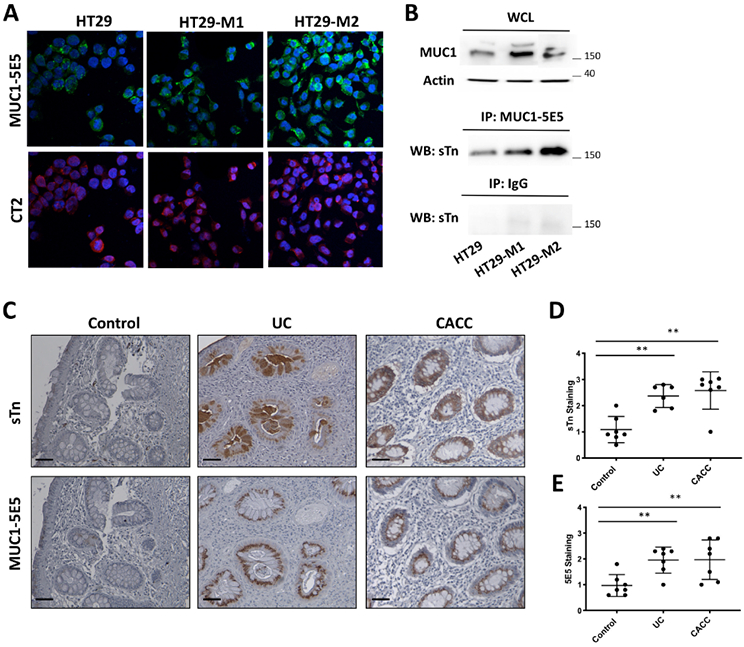
A) Double color immunofluorescence staining for MUC1 5E5 and MUC1 CT2 was performed on HT-29 cells co-cultured with M1 or M2 macrophages or left alone. DAPI stained nuclei. Results are representative of three independent experiments. B) Cell lysates from indicated cells were immunoprecipitated with anti-MUC1 VU-5E5 or IgG Abs. Immunoprecipitated proteins were immunoblotted with anti-sialyl Tn (sTn) Ab. Levels of MUC1 and actin were detected in the whole cell lysates (WCL) as control. Results are representative of three independent experiments. C) Representative sTn and MUC1-5E5 immunohistochemical staining of colon samples from patients with non-inflamed colon (Control, n= 7), ulcerative colitis (UC, n=6), and colitis- associated colon cancer (CACC, n=7). Scale bars, 20 μm. Right panel. Quantification of ST6GALNAC1-positive cells in tissues sections from 6 high power fields. P values were calculated by one-way ANOVA with Tukey post-tests. Error bars represent the SEM **p<0.01.
The expression of MUC1-sTn was also evaluated in inflamed UC and CACC tissues by performing an IHC staining with anti-MUC1 5E5 and anti-sTn (sTn 219). All samples expressing the sTn antigen also showed positive staining with MUC1 5E5 (Fig. 3C-E). Altogether, our results demonstrate over-expression of MUC1-sTn in human UC and CACC samples.
M2-induced ST6GALNAC1 expression is mediated by IL-13 and CCL17.
To explore the mechanism by which M2 macrophages increased the expression of glycosylation-associated enzymes, we used a human cytokine array containing antibodies against 80 cytokines. Cytokine profiling was conducted on the conditioned medium (CM) of HT-29 colon cancer cells co-cultured with M1 or M2 macrophages and compared to HT-29, M1 and M2 macrophages cultured alone (Fig. 4A). IL-13 and CCL17 emerged as the most up-regulated cytokines in the CM of HT-29 co-cultured with M2 macrophages (Fig. 4A). M1/M2 multiplex cytometric bead and ELISA assays further confirmed the increase of IL-13 and CCL17 in the CM from HT-29 colorectal cancer cells co-cultured with M2 polarized macrophages (Fig. 4B and C). The CM of M2 and SW480 colon cancer cell line, showed a similar cytokine profile (Supplemental Fig. S3A and B). IL-13 was higher in the CM of the co-culture compared to M2 alone indicating that M2-exposed colon cancer cells secreted IL-13 or stimulated increased macrophage secretion of IL-13. As previously reported (34,35), M2 macrophages secreted the chemokine CCL17 whereas colon cancer cells did not (Fig. 4A and B). Levels of CCL17 were slightly lower in HT29-M2 co-culture compared to monocultures. As HT-29 express CCR4 (36), the receptor of CCL17, this result could suggest that cells might consume this cytokine.
Figure 4. IL-13 and CCL17 modulated the expression of ST6GALNAC1 in colon cancer cells.

A) Upper panel. Human cytokine arrays were conducted using conditioned media of HT-29 co-cultured with M1(HT-M1) or M2 macrophages (HT-M2) and media of HT-29 or M2 cultured alone. Each sample was performed in duplicate. Lower panel. List summarizing relative intensities determined by Image J of the most increased cytokines. B) HT-29, M1 and M2 macrophages supernatants and conditioned medium of HT-29 co-cultured M1 or M2 were harvested for cytokine and chemokine analysis using the LegendPlex chemokine array. C) IL-13 cytokine expression was determined using ELISA assay. D-E) Real-time PCR and western blotting analyses of ST6GALNAC1 in HT-29 cells alone or co-cultured with M2 macrophages with or without IL-13 neutralizing Ab (D) or CCL17 neutralizing Ab (E). F) ST6GALNAC1 and sTn expression in IL-13- and CCL17-treated HT-29 cells was detected by immunoblotting analysis. Actin was used as loading control. G) The expression of mRNA of ST6GALNAC1 in IL-13- and CCL17-treated cells were detected using real-time PCR. GAPDH was used as housekeeping gene. P values were calculated using one-way (C,G) or two-way (B,D,E) ANOVA with Tukey post-tests for multiple comparisons. Error bars represent the SEM (B-E and G). *p<0.05, **p<0.01, ***p<0.001. All results are representative of three independent experiments.
To confirm IL-13 and CCL17 mediated the increased expression of ST6GALNAC1, HT-29 and SW480 colon cancer cells were incubated with neutralizing antibodies against IL-13 or CCL17 during co-culture with M2 macrophages. Inhibition of IL-13 or CCL17 significantly decreased ST6GALNAC1 mRNA levels almost to baseline in both cell lines (Fig. 4D and E). This result was confirmed by Western blotting (Fig. 4D and E). To further substantiate the role of these cytokines in the control of ST6GALNAC1 expression, 20, 50 or 100 ng/mL of exogenous recombinant IL-13 or CCL17 were added for 16 hours to the culture medium of HT-29 and SW480 cells. Both IL-13 and CCL17 increased the expression of sTn form as well as ST6GALNAC1 protein and mRNA expression in HT-29 (Fig. 4F and G) and SW480 cells (Supplemental Fig. S3C-E).
IL-13 increased ST6GALNAC1 expression in colon cancer cells through p-STAT6.
Given our observation that IL-13 and CCL17 were both involved in the increased expression of ST6GALNAC1 in response to M2-like macrophages, we investigated the intracellular signaling that could mediate this effect. First, using real-time PCR, we analyzed IL-13 expression in colon tissues of UC and CACC patients. IL-13 gene expression was significantly increased in highly inflamed and tumor tissues (Fig. 5A). IL-13 is known to activate transcriptional activity of target genes via phosphorylation of STAT6 (37,38). P-STAT6 was highly expressed in HT-29 cells co-cultured with M2 macrophages whereas it was not detected in HT-29 co-cultured with M1 macrophages or HT-29 cells cultured alone (Fig. 5B, Upper panel). Furthermore, IL-13 addition to HT-29 cells significantly increased p-STAT6 (Fig. 5B, Lower panel). Next, we assessed if IL-13-pSTAT6 mediated ST6GALNAC1 transcriptional activity in UC and CACC. The Motif Map system was used to identify consensus binding motifs for p-STAT6 (39). Two consensus elements were detected: NNYTTCYH (AC(T)TTCT(C)) and NDDTTCNN (TA(T)T(A)TTCC) (Fig. 5C). These regulatory elements were found in the ST6GALNAC1 promoter at – 444 (TATTCC) and −17 (CCTTCCT) of the transcription start site. To confirm the association of p-STAT6 with the ST6GALNAC1 promoter, we performed a chromatin immunoprecipitation (ChIP) assay. Briefly, chromatin was isolated from colon tissues from UC (n=4) and CACC (n=4) patients and immunoprecipitated with an anti-p-STAT6 Ab. The immunoprecipitated chromatin was then amplified using two sets of primers spanning from −175 to +3 and from −540 to −352. Our results showed that p-STAT6 associated with ST6GALNAC1 promoter with highest affinity toward the CCTTCCT element (Fig. 5D).
Figure 5. IL-13 and CCL17 induced the transcription activity of ST6GALNAC1 in colon cells of UC and CACC samples.

A and E) The mRNA expression of IL-13 (A) and CCL17 (E) from colon tissues of UC (n=4) and CACC patients (n=4) was detected via qPCR. Non-inflamed colon tissues were used as control (n=4). P values were calculated using a two-tailed t-test. **p<0.01. B) Western blotting analysis of STAT6 and p-STAT6 in HT-29 cells alone or co-cultured with M1 and M2 macrophages (upper panel) or in IL-13-treated HT-29 cells (lower panel). Actin was used as loading control. C and H) The human ST6GALNAC1 promoter region contains putative STAT6-binding sites (C) and p-p65 consensus site (H) detected by MotifMap. D and I) ChIP assays were performed with anti-p-STAT6 (D) and p-p65 (I) Abs followed by Real-Time PCR to measure ST6GALNAC1 promoter in UC, CACC and non-inflamed colon tissues (Control). F and G) Western blotting analysis of indicated proteins in HT-29 cells alone or in CCL17-treated HT-29 cells. P values are calculated using one-way Anova. Error bars represent the SEM (A, D and I) ***p<0.001. All results are representative of three independent experiments.
CCL17 activated ST6GALNAC1 expression in a p65-dependent manner.
Real-time PCR indicated up-regulation of CCL17 in UC and CACC samples compared to the control tissues (Fig. 5A). In order to test whether CCL17 signaling pathway and IL-13 signaling pathway both mediate their effects via p-STAT6 we checked the state of STAT6 activation in CCL17-treated HT-29 cells. Unlike in IL-13 treated HT-29, we saw no activation of pSTAT6 (Fig. 5E). As AKT and STAT3 were abundantly phosphorylated in HT-29 co-cultured with M2 macrophages (Fig. 5F), we analyzed the expression of p-STAT3 and p-AKT in CCL17-treated cells and found that CCL17 induced only p-AKT (Fig 5G). The PI3K-AKT signaling pathway can activate NF-κB, p65 through activation of IKK and p-IκB-α (40). We investigated whether CCL17 stimulation induced the expression of p-IκB-α and p-p65 in HT-29 cancer cells. Our results revealed that CCL17 induced phosphorylation and activation of IκB and p65 (Fig. 5G). One κb consensus motif (GGRNKTYCCCHN) (Fig. 5H) was found on the ST6GALNAC1 promoter from −33 to −21 (GGAGTTTCCCTT). ChIP assay confirmed the association of p-p65 with the ST6GALNAC1 promoter in human UC and CACC samples (Fig. 5I).
Computational model of activated signaling pathways and ST6GALNAC1 in UC and CACC.
We assembled a computational model that incorporated our experimental data and what has been reported in the literature to simulate signaling pathways potentially involved in the development of UC and CACC (Supplemental Table S1). The granular modeling framework utilized a standardized tabular framework (41) and the DiSH simulator (42). In this modeling approach, discrete elements change state depending on the influences of positive or negative regulators. The inputs were ligands present in the tumor microenvironment (Fig. 1C) that stimulated signaling cascades to influence the ST6GALNAC1 gene and MUC1 protein of colon cells (Fig. 6A). The stochastic simulation results reflected the experimental results for the normal, UC and CACC scenarios, when microenvironment signaling ligands were used as inputs (Fig. 6B and C). Higher IL13 and CCL17 observed in M2 co-culture with HT-29 cells and CACC resulted in increased transcription of ST6GALNAC1 and MUC1 sTn mediated by p-STAT6 and p65. Introduction of an IL-13 inhibitor resulted in decreased sTn only when introduced to UC and not CACC, suggesting early intervention as a therapeutic strategy (Fig. 6D). The model had IL-6 and IL-13 activating STAT6, however IL-6 was present in CACC and therefore the IL-13 inhibitor alone did not prevent ST6GALNAC1 transcription as the model still predicted its expression would occur downstream of IL-6. The computational model also indicated the IKK/IKB/P65 pathway was active in the presence of IL13 and CCL17 regulating the PI3K/AKT pathway through inhibition of PTEN. Further development of this model will allow for a more detailed investigation of the regulation of the signaling pathways.
Discussion
In this study, we have uncovered novel regulatory axes between macrophages and colon cells involved in UC and CACC. Our results indicated that CCL17 and IL-13, present in UC and CACC and produced by M2 macrophages co-cultured with colon cells, induced activation of multiple oncogenic pathways including AKT and STAT6. CCL17 and IL-13 also induced aberrant over-expression of the ST6GALNAC1 glycosyltransferase that lead to an increase in expression of the tumor glycoform MUC1-sTn.
An abundance of infiltrating anti-inflammatory (M2-like) macrophages in the tumor microenvironment is generally associated with poor prognosis and lymph node metastasis of laryngeal squamous cell carcinoma, breast, gastric, ovarian and colon cancer (43-46) suggesting involvement in tumor progression. Macrophages are highly plastic and able to change their phenotype and function according to the surrounding environment. In normal epithelium, macrophages are located just beneath the intestinal epithelial layer to perform surveillance activities (47). Tissue-infiltrating macrophages are also involved in the pathogenesis and the chronicity of UC and progression to CACC (47-49). However, many open questions remain regarding the specific mechanisms that are involved with aggravating inflammation and promoting tumorigenesis.
As healthy tissues evolve progressively to a neoplastic state, they acquire new tumorigenic and ultimately malignant potential. A noticeable event during the malignant transformation of epithelial cells is the alteration in glycosylation state of cell surface and secreted glycoproteins (7,9,12). We have previously reported that MUC1’s glycosylation pattern is markedly changed during chronic inflammation and CACC (6,9,11). In this study we have investigated the role of microenvironment factors in regulating the glycosylation state of MUC1 on inflamed and tumor colon cells.
Our gene expression profiling of myeloid-associated immune response indicated that chemokines and cytokines involved in the recruitment and migratory activities of macrophages are elevated in inflamed and malignant colon tissues. Moreover, analyses of infiltrating macrophages showed that CD163+ cells are significantly increased in both UC and CACC. These data suggest that M2-like macrophages might be involved in UC inflammation and CACC tumor progression.
IL-13 is a cytokine markedly elevated in UC patients produced by natural killer T (NKT) cells, T helper cells (Th) 2 and macrophages (50). A previous study reported that, in a murine model of UC, blockade of IL-13 prevented development of colitis (51). IL-13 modulates epithelial barrier permeability via binding IL-13Rα1 and activation of the expression of Claudin-2, a pore forming tight junction. Here, we expanded the knowledge of the mechanistic role of IL-13 in colitis and described its potential role in CACC as well. We showed that IL-13 was capable of inducing ST6GALNAC1 expression via phosphorylation of STAT6 in colonic cells. This agrees with other studies showing that the activity of IL-13 in colon cancer cells is STAT6 dependent (37,52) and these findings together with proteins involved in other signaling pathways were incorporated into the computational model.
Considering the role of IL-13 in the pathogenesis of UC, various clinical trials have been implemented to target IL-13 as a treatment strategy. We can speculate that inhibition of IL-13 would result not only in the reduction of colitis but could also prevent tumor progression as shown by the modeling if the inhibitor is implemented during UC.
We also demonstrated that another cytokine is involved in the control of the ST6GALNAC1 of colonic epithelial cells, CCL17, a chemokine produced by macrophages and dendritic cells (34,35,53). The receptor for CCL17 in colon cancer cells is CCR4 (36). Our data and computational model also indicated that CCL17 stimulation involved the activation of IκBα and p65 via the AKT pathway. The computational model provided indications for the involvement of NF-κB and also suggested future studies involving VEGF-A signaling. The computational model will allow for rapid, inexpensive testing of multiple scenarios to replace difficult and expensive experiments. Scenarios involving therapeutic interventions, such as inhibiting IL-13, and their timing can be performed using our computational model to advance knowledge of complex signaling networks involved in the transition from UC and CACC.
In this report, we also demonstrated that up-regulation of ST6GALNAC1 correlated with MUC1-sTn form expression on colon cells of UC and CACC. This is in agreement with previous studies showing that ST6GALNAC1 promotes the synthesis of the tumor-associated MUC1-sTn glycoform in breast and gastrointestinal cancer (54). MUC1-sTn has been associated with tumor progression and cancer (55). In addition, MUC1-sTn glycans might influence immune suppression through inhibition of dendritic cell maturation (56), activity of NK cells (57) and via interaction with macrophage galactose C-type lectin (MGL) (54) which is expressed on immature monocytes and M2-like macrophages. All these events would establish a positive feedback between MUC1-sTn and immune cells that could result in the perpetuation of chronic inflammation of UC and CACC.
Supplementary Material
Acknowledgements
This study was supported by the grants Fondazione Ri.MED (S.C.), NCI 1R35CA210039 (OJF) and DARPA W911NF-17-1-0135 (NMZ). The authors thank the Center for Biological Imaging (CBI) of the University of Pittsburgh (NIH grant number 1S10OD019973-01).
Footnotes
Conflict of Interest: The authors declare no potential conflicts of interest.
References
- 1.Economou M, Pappas G. New global map of Crohn’s disease: Genetic, environmental, and socioeconomic correlations. Inflamm Bowel Dis 2008;14:709–20 [DOI] [PubMed] [Google Scholar]
- 2.Shanahan F, Bernstein CN. The evolving epidemiology of inflammatory bowel disease. Curr Opin Gastroenterol 2009;25:301–5 [DOI] [PubMed] [Google Scholar]
- 3.Xia B, Crusius J, Meuwissen S, Pe?a A. Inflammatory bowel disease: definition, epidemiology, etiologic aspects, and immunogenetic studies. World J Gastroenterol 1998;4:446–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ekbom A, Helmick C, Zack M, Adami HO. Increased risk of large-bowel cancer in Crohn’s disease with colonic involvement. Lancet 1990;336:357–9 [DOI] [PubMed] [Google Scholar]
- 5.Stowell SR, Ju T, Cummings RD. Protein glycosylation in cancer. Annu Rev Pathol 2015;10:473–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beatty PL, Plevy SE, Sepulveda AR, Finn OJ. Cutting edge: transgenic expression of human MUC1 in IL-10−/− mice accelerates inflammatory bowel disease and progression to colon cancer. J Immunol 2007;179:735–9 [DOI] [PubMed] [Google Scholar]
- 7.Brockhausen I. Mucin-type O-glycans in human colon and breast cancer: glycodynamics and functions. EMBO Rep 2006;7:599–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krishn SR, Kaur S, Smith LM, Johansson SL, Jain M, Patel A, et al. Mucins and associated glycan signatures in colon adenoma-carcinoma sequence: Prospective pathological implication(s) for early diagnosis of colon cancer. Cancer Lett 2016;374:304–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cascio S, Finn OJ. Intra- and Extra-Cellular Events Related to Altered Glycosylation of MUC1 Promote Chronic Inflammation, Tumor Progression, Invasion, and Metastasis. Biomolecules 2016;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nath S, Mukherjee P. MUC1: a multifaceted oncoprotein with a key role in cancer progression. Trends Mol Med 2014;20:332–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cascio S, Faylo JL, Sciurba JC, Xue J, Ranganathan S, Lohmueller JJ, et al. Abnormally glycosylated MUC1 establishes a positive feedback circuit of inflammatory cytokines, mediated by NF-kappaB p65 and EzH2, in colitis-associated cancer. Oncotarget 2017;8:105284–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Varki A, Kannagi R, Toole B, Stanley P. Glycosylation Changes in Cancer In: rd, Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, et al. , editors. Essentials of Glycobiology. Cold Spring Harbor (NY)2015. p 597–609. [Google Scholar]
- 13.Bassaganas S, Allende H, Cobler L, Ortiz MR, Llop E, de Bolos C, et al. Inflammatory cytokines regulate the expression of glycosyltransferases involved in the biosynthesis of tumor-associated sialylated glycans in pancreatic cancer cell lines. Cytokine 2015;75:197–206 [DOI] [PubMed] [Google Scholar]
- 14.Padro M, Mejias-Luque R, Cobler L, Garrido M, Perez-Garay M, Puig S, et al. Regulation of glycosyltransferases and Lewis antigens expression by IL-1beta and IL-6 in human gastric cancer cells. Glycoconj J 2011;28:99–110 [DOI] [PubMed] [Google Scholar]
- 15.Erreni M, Mantovani A, Allavena P. Tumor-associated Macrophages (TAM) and Inflammation in Colorectal Cancer. Cancer Microenviron 2011;4:141–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steinbach EC, Plevy SE. The role of macrophages and dendritic cells in the initiation of inflammation in IBD. Inflamm Bowel Dis 2014;20:166–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Subramaniam R, Mizoguchi A, Mizoguchi E. Mechanistic roles of epithelial and immune cell signaling during the development of colitis-associated cancer. Cancer Res Front 2016;2:1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 2014;41:14–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kratochvill F, Neale G, Haverkamp JM, Van de Velde LA, Smith AM, Kawauchi D, et al. TNF Counterbalances the Emergence of M2 Tumor Macrophages. Cell Rep 2015;12:1902–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kinlough CL, Poland PA, Gendler SJ, Mattila PE, Mo D, Weisz OA, et al. Core-glycosylated mucin-like repeats from MUC1 are an apical targeting signal. J Biol Chem 2011;286:39072–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith MP, Sanchez-Laorden B, O’Brien K, Brunton H, Ferguson J, Young H, et al. The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFalpha. Cancer Discov 2014;4:1214–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H, Horbinski C, Wu H, Liu Y, Sheng S, Liu J, et al. NanoStringDiff: a novel statistical method for differential expression analysis based on NanoString nCounter data. Nucleic Acids Res 2016;44:e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xia C, Braunstein Z, Toomey AC, Zhong J, Rao X. S100 Proteins As an Important Regulator of Macrophage Inflammation. Front Immunol 2017;8:1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al-Ghadban S, Kaissi S, Homaidan FR, Naim HY, El-Sabban ME. Cross-talk between intestinal epithelial cells and immune cells in inflammatory bowel disease. Sci Rep 2016;6:29783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dosh RH, Jordan-Mahy N, Sammon C, Le Maitre CL. Long-term in vitro 3D hydrogel co-culture model of inflammatory bowel disease. Sci Rep 2019;9:1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noben M, Vanhove W, Arnauts K, Santo Ramalho A, Van Assche G, Vermeire S, et al. Human intestinal epithelium in a dish: Current models for research into gastrointestinal pathophysiology. United European Gastroenterol J 2017;5:1073–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baldus SE, Hanisch FG. Biochemistry and pathological importance of mucin-associated antigens in gastrointestinal neoplasia. Adv Cancer Res 2000;79:201–48 [DOI] [PubMed] [Google Scholar]
- 28.Itzkowitz SH, Bloom EJ, Kokal WA, Modin G, Hakomori S, Kim YS. Sialosyl-Tn. A novel mucin antigen associated with prognosis in colorectal cancer patients. Cancer 1990;66:1960–6 [DOI] [PubMed] [Google Scholar]
- 29.Kobayashi H, Terao T, Kawashima Y. Serum sialyl Tn as an independent predictor of poor prognosis in patients with epithelial ovarian cancer. J Clin Oncol 1992;10:95–101 [DOI] [PubMed] [Google Scholar]
- 30.Marcos NT, Bennett EP, Gomes J, Magalhaes A, Gomes C, David L, et al. ST6GalNAc-I controls expression of sialyl-Tn antigen in gastrointestinal tissues. Front Biosci (Elite Ed) 2011;3:1443–55 [DOI] [PubMed] [Google Scholar]
- 31.Yonezawa S, Tachikawa T, Shin S, Sato E. Sialosyl-Tn antigen. Its distribution in normal human tissues and expression in adenocarcinomas. Am J Clin Pathol 1992;98:167–74 [DOI] [PubMed] [Google Scholar]
- 32.Wang WY, Cao YX, Zhou X, Wei B, Zhan L, Sun SY. Stimulative role of ST6GALNAC1 in proliferation, migration and invasion of ovarian cancer stem cells via the Akt signaling pathway. Cancer Cell Int 2019;19:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu X, Wu Q, Wang L, Zhao Y, Zhang Q, Meng Q, et al. Silencing of ST6GalNAc I suppresses the proliferation, migration and invasion of hepatocarcinoma cells through PI3K/AKT/NF-kappaB pathway. Tumour Biol 2016;37:12213–21 [DOI] [PubMed] [Google Scholar]
- 34.Achuthan A, Cook AD, Lee MC, Saleh R, Khiew HW, Chang MW, et al. Granulocyte macrophage colony-stimulating factor induces CCL17 production via IRF4 to mediate inflammation. J Clin Invest 2016;126:3453–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katakura T, Miyazaki M, Kobayashi M, Herndon DN, Suzuki F. CCL17 and IL-10 as effectors that enable alternatively activated macrophages to inhibit the generation of classically activated macrophages. J Immunol 2004;172:1407–13 [DOI] [PubMed] [Google Scholar]
- 36.Al-haidari AA, Syk I, Jirstrom K, Thorlacius H. CCR4 mediates CCL17 (TARC)-induced migration of human colon cancer cells via RhoA/Rho-kinase signaling. Int J Colorectal Dis 2013;28:1479–87 [DOI] [PubMed] [Google Scholar]
- 37.Cao H, Zhang J, Liu H, Wan L, Zhang H, Huang Q, et al. IL-13/STAT6 signaling plays a critical role in the epithelial-mesenchymal transition of colorectal cancer cells. Oncotarget 2016;7:61183–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kraus J, Borner C, Hollt V. Distinct palindromic extensions of the 5’-TTC…GAA-3’ motif allow STAT6 binding in vivo. FASEB J 2003;17:304–6 [DOI] [PubMed] [Google Scholar]
- 39.Daily K, Patel VR, Rigor P, Xie X, Baldi P. MotifMap: integrative genome-wide maps of regulatory motif sites for model species. BMC Bioinformatics 2011;12:495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bai D, Ueno L, Vogt PK. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int J Cancer 2009;125:2863–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sayed CAT K, Butchy AA, and Miskov-Zivanov N. Recipes for translating big data machine reading to executable cellular signaling models Lect Notes Comput Sci (including Subser Lect Notes Artif Intell Lect Notes Bioinformatics). Volume 10710 LNCS: DiSH simulator: Capturing dynamics of cellular signaling with heterogeneous knowledge; 2018. [Google Scholar]
- 42.Sayed YHK K, Kulkarni A, and Miskov-Zivanov N. DiSH simulator: Capturing dynamics of cellular signaling with heterogeneous knowledge. Proc - Winter Simul Conf 2018. [Google Scholar]
- 43.Forssell J, Oberg A, Henriksson ML, Stenling R, Jung A, Palmqvist R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin Cancer Res 2007;13:1472–9 [DOI] [PubMed] [Google Scholar]
- 44.Sun W, Wei FQ, Li WJ, Wei JW, Zhong H, Wen YH, et al. A positive-feedback loop between tumour infiltrating activated Treg cells and type 2-skewed macrophages is essential for progression of laryngeal squamous cell carcinoma. Br J Cancer 2017;117:1631–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang C, Wei C, Wang S, Shi D, Zhang C, Lin X, et al. Elevated CD163(+)/CD68(+) Ratio at Tumor Invasive Front is Closely Associated with Aggressive Phenotype and Poor Prognosis in Colorectal Cancer. Int J Biol Sci 2019;15:984–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan X, Zhang J, Li D, Mao Y, Mo F, Du W, et al. Prognostic significance of tumor-associated macrophages in ovarian cancer: A meta-analysis. Gynecol Oncol 2017;147:181–7 [DOI] [PubMed] [Google Scholar]
- 47.Bain CC, Mowat AM. Macrophages in intestinal homeostasis and inflammation. Immunol Rev 2014;260:102–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest 2008;118:560–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu T, Dai Y, Wang W, Teng G, Jiao H, Shuai X, et al. Macrophage targeting contributes to the inhibitory effects of embelin on colitis-associated cancer. Oncotarget 2016;7:19548–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Endo Y, Marusawa H, Kou T, Nakase H, Fujii S, Fujimori T, et al. Activation-induced cytidine deaminase links between inflammation and the development of colitis-associated colorectal cancers. Gastroenterology 2008;135:889–98, 98 e1-3 [DOI] [PubMed] [Google Scholar]
- 51.Heller F, Fuss IJ, Nieuwenhuis EE, Blumberg RS, Strober W. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity 2002;17:629–38 [DOI] [PubMed] [Google Scholar]
- 52.Junttila IS. Tuning the Cytokine Responses: An Update on Interleukin (IL)-4 and IL-13 Receptor Complexes. Front Immunol 2018;9:888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stutte S, Quast T, Gerbitzki N, Savinko T, Novak N, Reifenberger J, et al. Requirement of CCL17 for CCR7- and CXCR4-dependent migration of cutaneous dendritic cells. Proc Natl Acad Sci U S A 2010;107:8736–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beatson R, Maurstad G, Picco G, Arulappu A, Coleman J, Wandell HH, et al. The Breast Cancer-Associated Glycoforms of MUC1, MUC1-Tn and sialyl-Tn, Are Expressed in COSMC Wild-Type Cells and Bind the C-Type Lectin MGL. PLoS One 2015;10:e0125994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van Elssen CH, Frings PW, Bot FJ, Van de Vijver KK, Huls MB, Meek B, et al. Expression of aberrantly glycosylated Mucin-1 in ovarian cancer. Histopathology 2010;57:597–606 [DOI] [PubMed] [Google Scholar]
- 56.Carrascal MA, Severino PF, Guadalupe Cabral M, Silva M, Ferreira JA, Calais F, et al. Sialyl Tn-expressing bladder cancer cells induce a tolerogenic phenotype in innate and adaptive immune cells. Mol Oncol 2014;8:753–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ogata S, Maimonis PJ, Itzkowitz SH. Mucins bearing the cancer-associated sialosyl-Tn antigen mediate inhibition of natural killer cell cytotoxicity. Cancer Res 1992;52:4741–6 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.