

Abstract
Background:
A new approach targeting aeroallergen sensing in the early events of mucosal immunity could have greater benefit. The CSF1-CSF1R pathway has a critical role in trafficking allergens to regional lymph nodes through activating dendritic cells. Intervention in this pathway could prevent allergen sensitization and subsequent Th2 allergic inflammation.
Objective:
To examine the therapeutic effectiveness of CSF1 and CSF1R inhibition for blocking the dendritic cell function of sensing aeroallerens
Methods:
We adopted a model of chronic asthma induced by a panel of three naturally occurring allergens and novel delivery system of CSF1R inhibitor encapsulated nanoprobe
Results:
Selective depletion of CSF1 in airway epithelial cells abolished the production of allergen-reactive IgE, resulting in prevention of new asthma development as well as reversal of established allergic lung inflammation. CDPL-GW nanoprobe containing GW2580, a selective CSF1R inhibitor, showed favorable pharmacokinetics for inhalational treatment and intranasal insufflation delivery of CDPL-GW nanoprobe ameliorated asthma pathologies including allergen-specific serum IgE production, allergic lung and airway inflammation and airway hyper-responsiveness (AHR) with minimal pulmonary adverse reaction.
Conclusion:
The inhibition of the CSF1-CSF1R signaling pathway effectively suppresses sensitization to aeroallergens and consequent allergic lung inflammation in a murine model of chronic asthma. CSF1R inhibition is a promising new target for the treatment of allergic asthma.
Keywords: Airway epithelial cells, CSF1, CSF1R, chronic inflammation, dendritic cells, airway remodeling, asthma therapy
Graphical Abstract
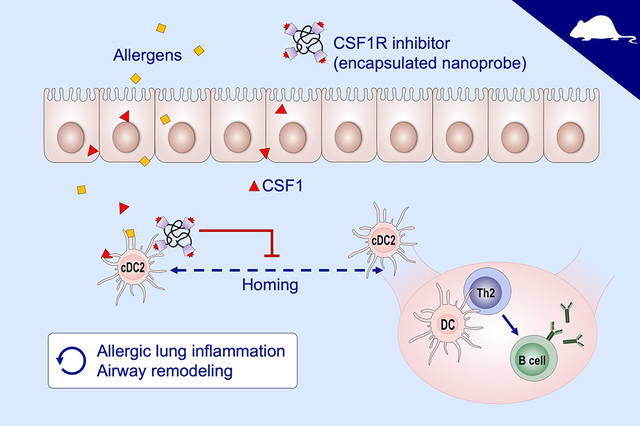
INTRODUCTION
Despite current standard medical treatment, a large number of patients with asthma remain symptomatic with long-term disabilities. There is a desperate need for developing a new treatment strategy for asthma. The events at the mucosal layer of airways are crucial to driving the development of Th2 immune responses. We now better understand the mechanisms by which Th2 immune-mediated allergic inflammation is initiated at the airway mucosa1,2. A therapeutic strategy targeting the early initial events of the mucosal immune reaction against aeroallergens could have greater therapeutic benefits because it could abolish all of the subsequent IgE- and Th2-mediated aspects of allergic inflammation in the lung.
Asthma is a chronic airway disease consisting of atopic or non-atopic asthma. In atopic asthma, people become asthmatic through the process of ‘sensitization’ against specific allergen(s) such as house dust mite and pollens. Once sensitized, individuals will develop airway inflammation and shortness of breath with re-exposure to the sensitizing allergen. Therapies targeting allergen sensitization have shown efficacy in controlling asthma3,4. The sensitization process for aeroallergens is mainly carried out by two major cell types, airway epithelial cells and dendritic cells.
Airway epithelial cells (AECs) are a gateway for sensing aeroallergens through first direct contact with inhaled particles5. Pattern recognition receptors including toll like receptors of AECs are required to recognize allergens and produce subsequent allergic inflammatory signals. Upon being activated by aeroallergens, AECs produce cytokines (TSLP, IL-1α and IL-33), chemokines (CCL2 and CCL20) and endogenous danger signals (ATP and uric acid) toward the basal side as well as luminal side and alter the micro-environmental milieu to activate innate immune cells6. AECs secrete innate cytokines which have a critical role in recruiting immune cells and skewing the immune reaction towards a predominant Th2 pattern2. On the luminal side, we and others have shown that AECs secreted chemokines into the alveolar space, recruiting monocyte-derived alveolar macrophages upon allergen challenge7,8. These findings indicate that AECs regulate the micro-environmental milieu of the airway in favor of allergic inflammation in response to aeroallergens.
During the sensitization, dendritic cells (DCs) take up and process the “invading” allergen and migrate to the regional lymph nodes (LNs), resulting in the establishment of allergen-specific Th2 memory. DCs form an essential interface between this innate and adaptive immunity, and play a key role in primary sensitization and the production of antigen-specific IgE1. DCs traffic inhaled allergens to the LNs where they launch an antigen-specific adaptive immune response involving T and B cells9. Conventional DCs (cDCs) play a key role in antigen presentation in various types of inflammation. cDCs express higher CCR7 which is involved in activation and homing of DCs to lymph nodes. In both human and mouse, two main distinct subsets of cDC, cDC1 and cDC2, possess unique characteristics and properties. cDC2 strongly depend on IRF4 and express multiple pattern recognition receptors, whereas cDC1 depend on IRF8 and activate CD8+ T cells. Especially, cDC2 have a critical role in MHC class 2-dependent antigen presentation and are more effective carriers of allergen to the regional LNs in animal models of asthma10.
However, it has not been fully elucidated how AECs and DCs interact in the process of sensing aeroallergens. We recently reported that AECs and DCs cooperate to facilitate allergen sensitization11. We found that AECs secrete CSF1 into the alveolar space in response to aeroallergen. CSF1 was markedly elevated in the BAL fluids of patients with asthma after being challenged with a sensitizing allergen via bronchoscopy. With the relevant animal models, we determined that AECs are the primary source of CSF1 in BAL fluid. We observed that the epithelial-secreted CSF1 activated DCs, particularly cDC2, and facilitated mobilization to regional LNs by regulating the DC expression of CCR7, a homing chemokine receptor to LNs11. This is the earliest event in the mucosal immune response to aeroallergen, but also occurs as a memory response that sustains asthmatic airway disease. Blocking this process could abolish sensitization to allergens and subsequent allergic inflammation upon repeated allergen exposure.
Here, we show the effectiveness of blocking CSF1 and its receptor (CSF1R) in asthma treatment. To this end, we utilized GW2580, a selective CSF1R inhibitor, which previously showed excellent pharmacokinetics11. To ensure scientific rigor, we employed a robust chronic mouse model of allergic lung and airway inflammation with naturally occurring allergens. The frequently used acute mouse asthma models have limitations because they do not recapitulate the nature of human asthma as a chronic airway inflammatory disease. To develop a model that allows study of the effects of treatment on long-lasting features of asthma, e.g. airway remodeling, while avoiding the acute effects of allergen challenge, we adopted a chronic triple allergen DRA (dust mite, ragweed and Aspergillus) asthma model as previously reported with minor modifications12. This model has many of the immunologic and pathologic features of human asthma with continued but decreased eosinophil infiltration and prominent airway remodeling.
Materials and Methods
Mice
C57BL/6, Scgb1a1-CreERT (stock# 016225), Irf4fl/fl (stock# 009380) and MAFIA (stock #005070) mice were purchased from Jackson Laboratory (Bar Harbor, ME). Scgb1a1-creERT;Csf1fl/fl and Csf1r-creERT;Irf4fl/fl were generated as described previously11. Mice were bred in a specific pathogen-free facility maintained by the University of Illinois at Chicago. All mouse experiments were approved by the Institutional Animal Care and Use Committee of University of Illinois at Chicago. Age & sex-matched 7 to 10-week-old mice were used for experiments.
DRA-induced chronic asthma model
We used the previously described DRA-induced asthma model with minor modification12. In brief, mice were subject to the DRA mixture comprised of house dust mite, ragweed and Aspergillus (5, 50, 5 μg/mouse, respectively) twice a week via intranasal insufflation for 8 weeks. The mice were then rested for 3 weeks before collecting samples at week 11. To deplete the target genes in the experiments containing Scgb1a1-creERT; Csf1f/f (CSF1ΔAEC) and Csf1r-creERT; Irf4fl/fl (IRF4ΔAPC) strains, we administered tamoxifen (Tm, 75mg/kg, Sigma-Aldrich, St. Louis, MO) via oral gavage for 5 consecutive days, according to the schedules depicted in Results.
Ex-vivo DRA antigen recall assay.
LNs were obtained from the mediastinum of the mice subjected to the DRA-induced allergic lung models. A single cell suspension was made from the LNs and a total of 4×106 cells/ml were plated in 24-well plates. For stimulation with the sensitizing allergens, the cells were treated with or without DRA (house dust mite, ragweed, and Aspergillus; 10, 100, 10 μg/ml, respectively) for 3 days. Cells were spun down and the supernatant used for ELISA.
Morphometric analysis and digital pathology
We used the Genie System (Aperio Technologies, Vista, CA, US), a tool for automated classification and quantitation of tissue inflammation in whole slide images, as described previously13. In this system, users identify the categories of tissue they want to quantify by outlining regions of interest that are examples of the tissue classes to be analyzed. We automatically quantified lung pathology over entire lung fields by categorizing them into four types: areas of normal lung parenchyma, airway, void space (such as airway lumen) and inflammation. The pathologist doing the analysis was blinded to the experimental groups.
Statistical Analysis
Student t or Mann-Whitney U test was used for two group comparisons and ANOVA was used for comparing multiple groups. Continuous variable distributions were tested for normality using Kolmogorov-Smirnov (K-S) and Shapiro–Wilk (S-W) test. SPSS 24.0 (SPSS Inc.; Chicago, IL, USA) were used for tests of normality.
Continuous variables (of which all were normally distributed) were compared between the different groups using one-way ANOVA tests in the case of homogeneity of variance between groups. If the rule of homogeneity of variance between groups was violated (K-S or S-W test p<0.05), Kruskal-Wallis test was performed. Descriptive statistics were expressed as mean value ± standard error of the mean (SEM) for continuous data and a p-value less than 0.05 was considered to be statistically significant. Each dot represents a single measurement of the parameters and the bar on the graph presents mean ± SEM. All analyses were conducted using Prism, GraphPad Software (La Jolla, CA).
RESULTS
CSF1 and CSF1R+cDCs are highly enriched in the BAL fluid of the chronic DRA-induced murine model of asthma.
To recapitulate the chronic inflammatory phenotype of human asthma, we adopted a chronic DRA-induced murine model of asthma as described in Methods (hereafter, the chronic DRA model) (Fig. 1A left panel)11,12. Unlike the acute transient asthma models, eosinophilia in lung and BAL fluid was rarely seen in the chronic DRA model. Instead, the majority of BAL cells consisted of lymphocytes and macrophages and the characteristics of chronic airway inflammation and airway remodeling are well demonstrated in this model (Fig. 2D). Since AECs secrete CSF1 into the alveolar space in response to allergen exposure11, we examined how chronic DRA exposure affects BAL CSF1 concentration. BAL CSF1 was promptly increased and peaked at week 6, whereas BAL CSF2 (GM-CSF) remained unchanged until week 6, when it began to rise (Fig.1A right panel). Conventional DCs were gated as lineage (CD3, B220, NK1.1, F4/80, CD64)−CD45+CD11c+MHCll+ and further defined either CD24high for cDC1 or CD172ahigh for cDC2 (Fig. S1A). In steady state, CSF1R positive cells were highly populated in the respiratory system, whereas their numbers were low in blood and pre-cDCs (Fig. S1B). Next, mice were subject to the chronic DRA model and the cDC2s were rapidly increased over the course of the chronic DRA model, but cDC1s were delayed until week 6. Because CSF1R+ cDC2 play critical role in allergen sensitization (10), we further analyzed CSF1R+ cDCs in the BAL fluids. Chronic DRA exposure induced an earlier and higher increase in CSF1R+ cDC2 than that of CSF1R+ cDC1 (Fig. 1B). Total and DRA-reactive serum IgE levels were also progressively increased, indicating that allergen sensitization has been intensified by repeated exposure to the allergens (Fig. 1C).
Figure 1. CSF1 and CSF1R+cDCs are highly enriched in the BAL fluid of the chronic DRA model.

(A) Left: Experimental scheme for the chronic DRA-induced murine model of asthma (the chronic DRA model). Right: The BAL fluids were obtained at the indicated time points and the level of CSF1 in the BAL fluids was measured by ELISA (n=4–6 mice per group). (B) By using the gating strategy as depicted in Figure E1, cDC and CSF1R+cDC populations were measured in the BAL fluids over the course of the chronic DRA model. (C) The concentrations of total and DRA-reactive serum IgE were measured at the indicated time points. The DRA-reactive serum IgE is expressed as fold increase, compared to the steady state IgE level. *p<0.05, **p<0.01
Figure 2. Depletion of AEC-derived CSF1 prevents allergen sensitization and abolishes chronic allergic lung inflammation.

(A) Experimental scheme for depleting AEC-derived CSF1 in the chronic DRA model using Scgb1a1-creERT;Csf1fl/fl (CSF1ΔAEC) mice. Tamoxifen was orally administered for 5 consecutive days (one cycle) every 4 weeks. All samples were collected at the end of week 11 (n=9–16 mice per group). (B-E) Tamoxifen alone has no effect on the BAL cytokines in the absence of DRA challenge (Tm_Veh), although lowering slightly the basal BAL CSF1 level (Figure 2B). DRA challenge induced marked increases in BAL CSF1, IL-4 and IL-13 (Veh_DRA), which were significantly reduced by Tm treatment (Tm_DRA) (2B & E). Total and DRA-reactive serum IgE concentrations were also suppressed by Tm treatment (2C). The DRA-reactive serum IgE are expressed as fold increase compared to that of the sham-treated control group (Veh_Veh). The increase in total BAL cells was suppressed by Tm treatment and the cells were mainly comprised of macrophages and lymphocytes (2D). (F) Morphometric digital pathologic analysis was performed over an entire single lung field as described in Methods. The color codes for the digital pathology indicate the following: green-inflammatory cells; yellow-area of the airway; brown-void space. The area of inflammation was markedly reduced by Tm treatment. The scale bars are 5 mm in middle and 500 μm in right panels. (G-I) Periodic acid Schiff (PAS) staining for goblet cells and immunofluorescence staining with anti-collagen and anti-α-smooth muscle actin antibodies were compared between the Tm-treated and sham-treated DRA groups. The signals were quantified by measurement of the signals per field. Scale bars represent 200μm. *p<0.05, **p<0.01.
AEC-derived CSF1 is necessary for development of chronic allergic lung inflammation and airway remodeling.
To test whether CSF1 secreted by AECs is required for establishing chronic allergic inflammation and airway remodeling, we used the Scgb1a1-creERT;Csf1fl/fl (hereafter CSF1ΔAEC) mice which are the inducible knock-out mice of CSF1 selectively in AECs by tamoxifen (Tm) injection11. First, we measured the duration of CSF1 depletion by one single cycle (consisting of 5 consecutive days) of Tm ingestion. The CSF1 depletion in AECs and BAL fluids continued up to 4 weeks by one cycle of oral Tm (Fig. S2). Therefore, tamoxifen was administered every 4 weeks during the chronic DRA model (Fig. 2B). CSF1 in BAL was increased about 2–3 fold in the chronic DRA group (Veh_DRA), compared to the sham-treated or only tamoxifen groups (Veh_Veh, Tm_Veh), but tamoxifen treatment abolished the increase of CSF1 (Tm_DRA) (Fig. 2B). We previously reported that CSF1 has a critical role in allergen sensitization and IgE production by facilitating DC mobilization11. In the chronic DRA model, blocking the AEC secretion of CSF1 significantly reduced total and DRA-reactive serum IgE production (Fig. 2C) and abrogated the subsequent allergic lung inflammation, inflammatory cell infiltration, Th2 cytokine production and airway remodeling, including goblet cell metaplasia, collagen deposition and smooth muscle hypertrophy (Fig. 2D–I).
CSF1 depletion reverses established chronic allergic inflammation and airway remodeling, and suppresses Th2 memory responses.
To ensure relevance to treatment of human asthma, we created a model in which CSF1 depletion took place after the allergic lung inflammation had been fully established (Fig. 3A). In this model, oral Tm ingestion was given for one cycle at week 6 of the chronic DRA model. The one cycle of oral Tm treatment resulted in significantly decreased BAL CSF1 (Fig. 3B). The late depletion of CSF1 in the chronic DRA model was able to reverse the allergen sensitization (serum IgE), inflammatory cell infiltration, allergic lung inflammation and goblet cell metaplasia (Fig. 3C–F). A single cell suspension was made from the LNs obtained from the mice subjected to the above model and used for the ex-vivo DRA antigen recall assay as described in Methods. IL-4 and IL-13 secretion were boosted by re-challenge with DRA, but blunted in the cells obtained from the CSF1-depleted mice (Fig. 3G), indicating that the Th2 memory response was significantly reduced in CSF1-depleted micewithoutaffecting IL-17 and IFN-γ secretion by DRA re-stimulation.
Figure 3. CSF1 depletion reverses established chronic allergic inflammation and airway remodeling, and suppresses Th2 memory responses in LNs.

(A) Experimental scheme for depleting AEC-derived CSF1 in the chronic DRA allergic inflammation model. Tm was administered for only one cycle, at week 6 of the chronic DRA model. (n=5–8 mice per group) (B) Single treatment with Tm (DRA/Tm) at week 6 reduced the BAL CSF1 concentration. (C-F) Tm treatment in the late phase of the model was able to reverse the BAL lymphocytosis, increased serum IgE, chronic lung inflammation and goblet cell metaplasia. The scale bars in left and right of (E) and (F) are 5 mm, 500 μm and 200μm, respectively. (G) The cells from the LNs were isolated from each group, and then re-stimulated with or without DRA for 72 hours ex-vivo in culture. The cytokines in culture supernatants were measured. The re-stimulation with DRA increased the secretion of IL-4, IL-13, IL-17 and IFN-γ in the LN cultures. However, the group treated with Tm (DRA/Tm) showed a blunted response for IL-4 and IL-13, whereas IL-17 and IFN-γ levels remained unchanged. *p<0.05, **p<0.01.
IRF4+ CSF1R+ cells are required for Th2 memory in the secondary LNs of the chronic DRA model.
We have shown that cDC2 play a critical role in CSF1-dependent allergen sensitization11. Since there is no mouse strain in which cDC2 development is selectively ablated14, we used Csf1r-creERT;Irf4fl/fl (hereafter IRF4ΔAPC) for a cDC2 targeted experiment11. Because IRF4 is required for cDC2 development and function, but not for cDC115, and Csf1r expressing macrophages are not engaged in antigen trafficking to LNs, this mouse could be used for targeting cDC2 function in the experimental model of allergen sensitization11. The mice were subjected to the chronic DRA model as depicted in Figure 4A. In these two models, the allergic inflammation was established first, and then Tm administered to deplete the CSF1R+ IRF4+ cells at either week 2 (Tm2) or 6 (Tm6). Despite the presence of sufficient CSF1 in BAL fluid (Fig. 4B), the depletion of CSF1R+IRF4+ cells resulted in reduction of inflammatory cell infiltration and total and DRA-reactive serum IgE production, and the attenuation of lung inflammation and goblet cell metaplasia, although the Tm6 group showed only modest effects (Fig. 4C–F). The ex-vivo DRA antigen recall assay showed that the depletion of CSF1R+ IRF4+ blocked the production of IL-4 and IL-13 by DRA re-stimulation in both Tm2 and Tm6 treatment groups, whereas IL-17 production remained intact in the Tm6 group, while samples from the Tm2 group showed partial reduction (Fig. 4G). Collectively, these data indicate that CSF1R+cDC2 are required for the formation of Th2 memory cells, indicating that interfering with cDC2 function should be beneficial for treating Th2-immune mediated diseases.
Figure 4. IRF4+ CSF1R+ cells are required for allergen sensitization and Th2 memory in the secondary LNs.

(A) Experimental scheme for depleting CSF1R+IRF4+ antigen presenting cells in Csf1r-creERT;Irf4fl/fl (Irf4ΔAPC) mice. Tm2 and Tm6 indicate that the Tm administration was started at weeks 2 and 6 of the chronic DRA model as depicted. (n=5–8 mice per group) (B) Depletion of CSF1R+IRF4+ in Irf4ΔAPC mice had no effect on BAL CSF1 concentration. (C-F) However, depletion of CSF1R+IRF4+ from week 2 (Tm2) resulted in a modest decrease in BAL inflammatory cells, significant reduction of total and DRA-reactive serum IgE, marked reduction of chronic lung inflammation and goblet cell metaplasia. The Tm6 group showed similar trends, but was less effective than in the Tm2 group. Left and middle scale bars in (E) are 5 mm and 500 μm, respectively. (G) Single cell suspensions of LNs were isolated from each group and re-stimulated with DRA for 72 hours. The cytokines in culture supernatants were measured. The re-stimulation with DRA boosted the secretion of IL-4, IL-13 and IL-17, compared to the sham-treated control. However, the Tm treated groups showed a blunted response for IL-4 and IL-13 production, whereas IL-17 secretion in the Tm 6 group was not affected by Tm treatment while the Tm2 group showed a mild decrease in IL-17 production. * p<0.05, **p<0.01
Nanoprobe carrying a CSF1R inhibitor blocks cDC2 migration and allergen sensitization.
Selective CSF1R inhibitors have been used in continuing clinical trials for cancer treatment with minimal adverse effects16,17. GW2580, a CSF1R inhibitor, showed excellent binding affinity to human CSF1R recombinant protein11. Simulation protein structure modeling indicated that GW2580 interacts with two critical intracellular tyrosine residues (Y546 and Y665) of the kinase domain of CSF1R (Fig. S3). Further, we have demonstrated that systemic administration of GW 2580 effectively blocked cDC2 migration to regional LNs11. The inhalational route is preferred for asthma treatment because it can deliver a compound effectively to the target organ and minimize systemic adverse effects. For an inhalational delivery, we generated a CPDL-encapsulated nanoprobe containing CSF1R inhibitor using a previously described method18. CDPL (β-, a synonym of β-cyclodextrins and amine of EPL, yclodextrins and amine of EPL) nanoprobe increase the chemical stability and bioavailability of guest molecules to ensure site-specific delivery of lipophilic GW2580 to the lung without sticking to other tissues19. Each CPDL nanoprobe carries four molecules of GW2580 as well as a ZW800–1 fluorophore (CDPL-GW) (Fig. 5A). As expected, intranasal delivery of CDPL-GW had good lung deposition (Fig. S4). Measurements by NanoSight (Solisbury, U.K.) indicated that most of the CPDL nanoprobe were less than 100nm (Fig. S5). Next, we analyzed the cellular distribution of two different doses of CDPL-GW (1ng and 100 ng/mouse) via intranasal insufflation to the lung (Fig. S6). Neutrophils, DCs and alveolar macrophages (AMs) were the major cells that took up the particles. To optimize the dose of CDPL-GW for inhibiting DC migration in lung, the mice were subjected to DRA challenge for 5 days with increasing dose of CDPL-GW (from 1pg to 1ng/mouse) and the samples were collected on day 5 (Fig. 5B). Total BAL cells were increased by DRA challenge, but suppressed by CDPL-GW in a dose-dependent manner and BAL eosinophil counts and serum IgE concentrations were also markedly reduced by 1ng/mouse of CDPL-GW (Fig. 5B–C). However, there was no significant apoptotic death of AMs or increases in BAL total protein or albumin concentration by CDPL-GW treatment (Fig. 5B & S7). To further characterize the effects of CDPL-GW on DCs, we evaluated the number of migratory DCs (CD45+lin−CD11c+MHCll+CCR7+) in regional LNs. CDPL-GW suppressed migratory cDC2 in a dose-dependent manner (Fig. 5D). From these data, we determined that 1ng of CDPL-GW/mouse (equivalent to 80,000 nanoprobe) as the therapeutic dose for the next experiment.
Figure 5. CDPL-GW nanoprobe carrying CSF1R inhibitor block cDC2 migration and allergen sensitization.

(A) Structure of the CDPL-GW nanoprobe which carries 4 molecules of GW2580, a selective CSF1R inhibitor and a ZW800–1 fluorescent dye. (B-C) The experimental scheme to optimize the dose of CDPL-GW. The total AM count and annexin V+ AMs in BAL fluids were measured by flow cytometry. AMs and eosinophils in BAL fluids were identified by CD11c+SiglecF+, CD11c−SiglecF+ respectively. DRA challenge induced increases in BAL AMs and eosinophils, which was inhibited by the CDPL-GW treatment in a dose dependent manner. However, there was no change in the proportion of annexin V+ cells up to 1ng (=1000pg) of CDPL-GW /mouse which was a sufficient dose to suppress the BAL eosinophil recruitment and total serum IgE rise (n=4–5 mice per group). (D) Migratory DCs (defined by CD45+lineage−CD11c+MHCll+CCR7+) were measured in the single cell suspensions isolated from mediastinal LNs. The number of migratory DCs in LNs gradually declined as the dose of CDPL-GW inhibitor was increased via the intranasal route (n=4–5). * p<0.05
Nanoprobe carrying CSF1R inhibitor abolish chronic allergic lung inflammation in the DRA model.
To measure the efficacy of intranasal CDPL-GW treatment for chronic allergic inflammation, we performed three different experiments as depicted in figure S8A. In these experiments, CDPL-GW intranasal treatment was started at three different time points, week 3, 5 and 7, in the chronic DRA model, during the repeated DRA exposures until week 8. After 3 weeks of rest, the mice were analyzed at week 11. Intranasal CDPL-GW treatment in all three experiments effectively attenuated asthma pathologies including airway inflammatory cell recruitment, total and DRA-reactive serum IgE, BAL Th2 cytokines, lung tissue inflammation and goblet cell metaplasia, compared to the sham treated group, although the CDPL-GW 7wk group had only modest effects (Fig. 6A–E). There was no change in the BAL concentrations of total protein and albumin (Fig. S8B). CDPL-GW treatment effectively inhibited Th2 memory cell formation in the secondary LNs, although the CDPL-GW 7wk group did not reach statistical significance (Fig. 6F). All three CDPL-GW treated groups showed reduced bronchial hyper-responsiveness, compared to the sham-treated chronic DRA group (Fig. 6G).
Figure 6. Intranasal delivery of CDPL-GW nanoprobe abolishes chronic allergenic lung inflammation in the DRA model.

Mice were subjected to the chronic DRA model, and then treated with CDPL-GW (1ng/mouse via the i.n. route, twice a week) in three different designs as depicted in Figure E8. CDPL-GW 3wk indicates that the treatment with CDPL-GW was started at week 3 of the chronic DRA model and maintained until week 8 (total of 6 weeks). CDPL-GW 5wk and 7wk were done in a similar fashion. All of the measurements were done at week 11. (n=5–10 mice per group) (A) Treatment with CDPL-GW markedly reduced the DRA-induced BAL cellular recruitment which was mainly comprised of macrophages and lymphocytes. (B-E) CDPL-GW treatment starting at 3 and 5 weeks of the chronic DRA model (CDPL-GW 3wk and 5wk) effectively blocked the increases in total and DRA-reactive serum IgE, BAL IL-4 and IL-5, and attenuated lung inflammation and goblet cell metaplasia. However, the CDPL-GW 7wk group showed similar trends, but differences failed to reach statistical significance in terms of DRA-reactive serum IgE, lung inflammation and goblet cell metaplasia. Scale bars represent 200μm. (F) Single cell suspensions of regional LNs were re-stimulated with DRA for 72 hours. DRA re-stimulation increased the secretion of IL-4, IL-13 and IL-17, but CDPL-GW treatment blocked these increases. However, cytokine levels from cells obtained from the CDPL-GW 7wk group did not reach statistical significance compared to those of the sham-treated DRA group, although they showed a declining trend. (G) Airway resistance was measured using increasing doses of methacholine in the sham and CDPL-GW treated groups after DRA challenge. All of the three CDPL-GW treated groups had significantly lower airway resistance compared to the sham treated chronic DRA group.
DISCUSSION
Our study highlights the effects of blocking or minimizing allergen sensitization on development and exacerbation of asthma pathologies, respectively. Earlier clinical studies indicate that sensitization to environmental allergens and excessive production of serum IgE precedes manifestation of allergic asthma in the early childhood20,21. In the cohort of a large group of newborns, the prevalence of asthma was significantly higher among ever-allergic sensitized children compared to never-sensitized ones22. These data strongly suggest a causal relationship between allergen sensitization and development of clinical asthma. Therapies to reduce allergen sensitization have been tried using many different approaches. Allergen immunotherapy with allergen extracts is able to lower allergen-specific IgE as well as increase specific IgG4 levels, resulting in a robust long-lasting effect on seasonal rhinitis and peanut allergy23–25. Sensitization to allergen(s) occurs not only in the development of new asthma, but also in the exacerbation of established asthma. We have shown that serum IgE is increased by re-exposure to the sensitizing allergen in the mouse DRA model of allergic asthma11. Intervening in the continuing sensitization process has been shown to prevent asthma exacerbations in the patients already sensitized. For example, IgE-targeted treatment with omalizumab, a humanized monoclonal anti-IgE antibody, showed a significant benefit in controlling asthma exacerbations in inner city children who were already sensitized to cockroach3. Pre-seasonal treatment with omalizumab was also effective in controlling seasonal exacerbations in poorly controlled asthmatics4. Here, we examined three animal models to validate the effect of intervening in the CSF1-CSF1R pathway for preventing both the development of new allergic asthma and reversing fully established allergic lung inflammation. In all three models, our data showed that blocking the CSF1-CSF1R pathway effectively abolished the sensitization process (reducing both total and allergen-reactive serum IgE) and suppressed subsequent allergic lung inflammation. These data support the concept that blocking the allergen sensitization process can effectively prevent the subsequent Th2 allergic inflammatory process regardless of the onset of allergic inflammation, and could be used as a maintenance therapy for chronic asthma.
Among DC subsets, cDC2 have been shown to play an essential role in antigen presentation in allergic lung inflammation10,11,26. Our data show a marked surge of cDC2 in BAL fluid in the DRA asthma model11. However, it is not fully understood how alveolar cDC2 are regulated in allergic inflammation. Recently, we reported that the CSF1-CSF1R pathway is critical for cDC2 activation through their homing to regional LNs and establishing subsequent Th2 immune reactions in response to inhaled allergens. We have shown that the binding of AEC-derived CSF1 to its receptor on cDC2 facilitates expression of the chemokine receptor, CCR711. Based on this scientific premise, here we rigorously examined whether intervening in this pathway would be beneficial for controlling both new and established allergic lung inflammation. To examine the therapeutic effectiveness, we adopted a mouse model of chronic asthma12. Unlike the transient acute models of allergic asthma, this model shows marked inflammatory cell infiltration and prominent airway remodeling, including structural alterations, prominent tissue fibrosis and chronic inflammation, albeit with decreased eosinophilia (Figure 2). This model is better suited for evaluating the long term effects of asthma therapeutics on well-established asthma pathologies, in this case, treatment with anti-CSF1 antibodies and nanoprobe carrying a CSF1R antagonist.
CSF1 and its receptor (CSF1R) regulate the functions of myeloid lineage cells that play key roles in innate immune responses27. CSF1R is comprised of five extracellular immunoglobulin domains, a transmembrane domain, and two intracellular domains. The CSF1R gene is evolutionally well conserved and has high sequence homology between species. Especially, the intracellular domains of human and murine CSF1R genes are identical (NCBI HomologGene). In humans, the CSF1R gene is located at the chromosomal region 5q32, which is close to genes associated with genetic susceptibility to asthma including IL-4, IL-5 and ADRB228–30. Furthermore, it has been reported that a polymorphism of the CSF1R gene is associated with an increased risk for asthma in humans31. Upon binding with its ligand, CSF1, the intracellular kinase domain of CSF1R undergoes auto-phosphorylation and activates multiple downstream pathways27. CSF1R inhibitors bind to the tyrosine residues of the intracellular kinase domain. Our structure-based simulation shows that the CSF1R inhibitor, GW2580, binds two tyrosine residues (Y546 and Y665) (Figure E3). Further studies are necessary to delineate the effect of GW2580 binding to CSF1R and modulation of downstream signaling pathways associated with allergic airway responses.
Systemic administration of CSF1R inhibitors as anti-cancer agents has been shown to have excellent clinical safety and good tolerance, even in patients with advanced-staged malignancies16,17. Here, we examined the therapeutic efficacy and potential pulmonary toxicities of locally administered CSF1R inhibitor, GW2580. Our data show a decrease in BAL eosinophil count and serum IgE level with inhalational delivery of CDPL-GW at the dose of 1 ng/mouse, which is equivalent to 8,000 nanoprobe per mouse. These data indicate that the DCs involved in allergen sensitization are highly sensitive to blockade of the CSF1-CSF1R pathway. Since not only DCs but also macrophages express CSF1R, macrophage dysfunction related to CSF1R inhibition may lead to potential adverse effects. Although not being involved in antigen presentation, AMs are a first-line of defense against inhaled foreign particles. Here, we examined the potential pulmonary toxicities of locally administered CSF1R inhibitor nanoprobe. The intranasal delivery of CDPL-GW (up to 1ng/mouse) to the airway did not increase AM cell death in terms of the number of apoptotic AMs. Various forms of pulmonary alveolar proteinosis (PAP) can develop because of impaired function of GM-CSF or its receptor (CSF2R). We examined whether CDPL-GW induced PAP in the mice, even though CSF1R is structurally different from the GM-CSF receptor and there is no known cross-reactivity between these two receptors. Our data showed that there was no increase in total protein or albumin concentrations in BAL fluid, and lung pathology showed no evidence of PAP after treatment with intranasal CDPL-GW. These data suggest that the sensitivity of the CSF1R receptor to CSF1R inhibition varies depending on the cell type. Our previous report showing variable expression of CSF1R among myeloid cells supports this hypothesis11. Of interest, blocking the CSF1-CSF1R pathway selectively inhibits Th2 memory, but Th1 and Th17 memory function remained intact, suggesting that immune defense against viral and bacterial infection may remain intact as well, although this requires further investigation. Previous reports indicate that the differentiation and survival of AMs heavily depends on GM-CSF and its receptor32,33, suggesting that CSF1R could be dispensable for macrophages in the alveolar niche. Further study is needed to clarify the biological function of the CSF1 receptor on alveolar macrophages.
In summary, our data support the efficacy of a DC centered new treatment of asthma that targets the CSF1-CSF1R pathway to block the process of allergen sensitization. Because allergen sensitization is required not only for establishing new atopic asthma, but also for exacerbation of allergic inflammation in already established asthma, the approach of blocking the sensitization process could be used as a long term maintenance therapy for the treatment of asthma.
Supplementary Material
Acknowledgments
We thank all the support from the Research Resource Center (Biophysics Core, Research Histology and Tissue Imaging Core, Flow Cytometry Core) at the University of Illinois at Chicago. This work was supported by NIH grant R01HL126852, Dr. Ralph and Marian Falk Medical Research Trust Bank of American, N.A., Trustee and Respiratory Health Association Grant (RHA2016–01-Lung cancer) (to G.Y.P.) and in part by NIH grants R01 HL137224 (to J.W.C.).
Footnotes
CONFLICT OF INTEREST
H.M. and G.Y.P. have filed a provisional patent application related to the submitted manuscript.
REFERENCES
- 1.Lambrecht BN, Hammad H. Biology of lung dendritic cells at the origin of asthma. Immunity. 2009;31(3):412–424. [DOI] [PubMed] [Google Scholar]
- 2.Hammad H, Lambrecht BN. Barrier Epithelial Cells and the Control of Type 2 Immunity. Immunity. 2015;43(1):29–40. [DOI] [PubMed] [Google Scholar]
- 3.Busse WW, Morgan WJ, Gergen PJ, et al. Randomized trial of omalizumab (anti-IgE) for asthma in inner-city children. N Engl J Med. 2011;364(11):1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teach SJ, Gill MA, Togias A, et al. Preseasonal treatment with either omalizumab or an inhaled corticosteroid boost to prevent fall asthma exacerbations. J Allergy Clin Immunol. 2015;136(6):1476–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lambrecht BN, Hammad H. Allergens and the airway epithelium response: gateway to allergic sensitization. J Allergy Clin Immunol. 2014;134(3):499–507. [DOI] [PubMed] [Google Scholar]
- 6.Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nature immunology. 2015;16(1):27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee YG, Jeong JJ, Nyenhuis S, et al. Recruited alveolar macrophages, in response to airway epithelial-derived monocyte chemoattractant protein 1/CCl2, regulate airway inflammation and remodeling in allergic asthma. Am J Respir Cell Mol Biol. 2015;52(6):772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaslona Z, Przybranowski S, Wilke C, et al. Resident alveolar macrophages suppress, whereas recruited monocytes promote, allergic lung inflammation in murine models of asthma. J Immunol. 2014;193(8):4245–4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joffre O, Nolte MA, Sporri R, Reis e Sousa C. Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol Rev. 2009;227(1):234–247. [DOI] [PubMed] [Google Scholar]
- 10.Plantinga M, Guilliams M, Vanheerswynghels M, et al. Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity. 2013;38(2):322–335. [DOI] [PubMed] [Google Scholar]
- 11.Moon H-G, Kim S-j, Jeong JJ, et al. Airway Epithelial Cell-Derived Colony Stimulating Factor-1 Promotes Allergen Sensitization. Immunity. 2018;49(2):275–287. e275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goplen N, Karim MZ, Liang Q, et al. Combined sensitization of mice to extracts of dust mite, ragweed, and Aspergillus species breaks through tolerance and establishes chronic features of asthma. Journal of Allergy and Clinical Immunology. 2009;123(4):925–932. e911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park GY, Lee YG, Berdyshev E, et al. Autotaxin production of lysophosphatidic acid mediates allergic asthmatic inflammation. Am J Respir Crit Care Med. 2013;188(8):928–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson DA 3rd, Murphy KM, Briseno CG. Development, Diversity, and Function of Dendritic Cells in Mouse and Human. Cold Spring Harb Perspect Biol. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bajana S, Turner S, Paul J, Ainsua-Enrich E, Kovats S. IRF4 and IRF8 Act in CD11c+ Cells To Regulate Terminal Differentiation of Lung Tissue Dendritic Cells. J Immunol. 2016;196(4):1666–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butowski N, Colman H, De Groot JF, et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. 2016;18(4):557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tap WD, Wainberg ZA, Anthony SP, et al. Structure-Guided Blockade of CSF1R Kinase in Tenosynovial Giant-Cell Tumor. N Engl J Med. 2015;373(5):428–437. [DOI] [PubMed] [Google Scholar]
- 18.Kang H, Gravier J, Bao K, et al. Renal Clearable Organic Nanocarriers for Bioimaging and Drug Delivery. Adv Mater. 2016;28(37):8162–8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi HS, Ashitate Y, Lee JH, et al. Rapid translocation of nanoparticles from the lung airspaces to the body. Nat Biotechnol. 2010;28(12):1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hose AJ, Depner M, Illi S, et al. Latent class analysis reveals clinically relevant atopy phenotypes in 2 birth cohorts. J Allergy Clin Immunol. 2017;139(6):1935–1945 e1912. [DOI] [PubMed] [Google Scholar]
- 21.Rhodes HL, Thomas P, Sporik R, Holgate ST, Cogswell JJ. A birth cohort study of subjects at risk of atopy: twenty-two-year follow-up of wheeze and atopic status. Am J Respir Crit Care Med. 2002;165(2):176–180. [DOI] [PubMed] [Google Scholar]
- 22.Ballardini N, Bergstrom A, Wahlgren CF, et al. IgE antibodies in relation to prevalence and multimorbidity of eczema, asthma, and rhinitis from birth to adolescence. Allergy. 2016;71(3):342–349. [DOI] [PubMed] [Google Scholar]
- 23.Durham SR, Emminger W, Kapp A, et al. SQ-standardized sublingual grass immunotherapy: confirmation of disease modification 2 years after 3 years of treatment in a randomized trial. J Allergy Clin Immunol. 2012;129(3):717–725 e715. [DOI] [PubMed] [Google Scholar]
- 24.Durham SR, Emminger W, Kapp A, et al. Long-term clinical efficacy in grass pollen-induced rhinoconjunctivitis after treatment with SQ-standardized grass allergy immunotherapy tablet. J Allergy Clin Immunol. 2010;125(1):131–138 e131–137. [DOI] [PubMed] [Google Scholar]
- 25.Du Toit G, Roberts G, Sayre PH, et al. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med. 2015;372(9):803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hammad H, Plantinga M, Deswarte K, et al. Inflammatory dendritic cells--not basophils--are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. J Exp Med. 2010;207(10):2097–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stanley ER, Chitu V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol. 2014;6(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turner S, Francis B, Vijverberg S, et al. Childhood asthma exacerbations and the Arg16 beta2-receptor polymorphism: A meta-analysis stratified by treatment. J Allergy Clin Immunol. 2016;138(1):107–113 e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu S, Chan-Yeung M, Becker AB, et al. Polymorphisms of the IL-4, TNF-alpha, and Fcepsilon RIbeta genes and the risk of allergic disorders in at-risk infants. Am J Respir Crit Care Med. 2000;161(5):1655–1659. [DOI] [PubMed] [Google Scholar]
- 30.Sordillo JE, Kelly R, Bunyavanich S, et al. Genome-wide expression profiles identify potential targets for gene-environment interactions in asthma severity. J Allergy Clin Immunol. 2015;136(4):885–892 e882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shin EK, Lee SH, Cho SH, et al. Association between colony-stimulating factor 1 receptor gene polymorphisms and asthma risk. Hum Genet. 2010;128(3):293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guilliams M, Scott CL. Does niche competition determine the origin of tissue-resident macrophages? Nat Rev Immunol. 2017;17(7):451–460. [DOI] [PubMed] [Google Scholar]
- 33.van de Laar L, Saelens W, De Prijck S, et al. Yolk Sac Macrophages, Fetal Liver, and Adult Monocytes Can Colonize an Empty Niche and Develop into Functional Tissue-Resident Macrophages. Immunity. 2016;44(4):755–768. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.