

Close to half of a collection of 27 clinical a/α isolates of Candida albicans underwent white-to-opaque switching. Complementation experiments revealed that while approximately half of the a/α switchers were due to EFG1 mutations, the remaining half were due to mutations in other genes. In addition, the results of competition experiments in a mouse GI tract colonization model support previous observations that efg1/efg1 cells rapidly outcompete EFG1/EFG1 strains, but direct microscopic analysis reveals that the major colonizing cells were opaque, not gray.
KEYWORDS: Candida albicans, EFG1 mutation, clinical isolates, switching to opaque
ABSTRACT
The transcription factor EFG1 functions as a suppressor of white-to-opaque and white-to-gray switching in a/α strains of Candida albicans. In a collection of 27 clinical isolates, 4 of the 17 EFG1/EFG1 strains, 1 of the 2 EFG1/efg1 strains, and all 8 of the efg1/efg1 strains underwent white-to-opaque switching. The four EFG1/EFG1 strains, the one EFG1/efg1 strain, and one of the eight efg1/efg1 strains that underwent switching to opaque did not switch to gray and could not be complemented with a copy of EFG1. Competition experiments in a mouse model for gastrointestinal (GI) colonization confirmed that efg1/efg1 cells rapidly outcompete EFG1/EFG1 cells, and in plating experiments, formed colonies containing both gray and opaque cells. Direct microscopic analysis of live cells in the feces, however, revealed that the great majority of cells were opaque, suggesting opaque, not gray, may be the dominant phenotype at the site of colonization.
IMPORTANCE Close to half of a collection of 27 clinical a/α isolates of Candida albicans underwent white-to-opaque switching. Complementation experiments revealed that while approximately half of the a/α switchers were due to EFG1 mutations, the remaining half were due to mutations in other genes. In addition, the results of competition experiments in a mouse GI tract colonization model support previous observations that efg1/efg1 cells rapidly outcompete EFG1/EFG1 strains, but direct microscopic analysis reveals that the major colonizing cells were opaque, not gray.
INTRODUCTION
Candida albicans remains a pervasive opportunistic fungal pathogen, colonizing a majority of humans as a commensal (1). It was initially believed that its success as an opportunistic pathogen was due in part to its capacity to invade tissue through a single developmental program, the transition from a budding yeast to a filamentous hypha (1, 2). However, in 1987, a second reversible phenotypic transition, the white-opaque switching system, was identified in a strain of C. albicans, WO-1, isolated from a bone marrow transplant patient (3, 4). Soon after, it was reported that only select strains of C. albicans underwent the white-opaque transition (5). Switching affected not only colony morphology but also impacted most aspects of cell morphology and cell wall architecture (3, 6, 7). In the 15 years after the discovery of white-opaque switching, unique patterns of differential gene expression were described (8–14), and differences in virulence were demonstrated in mouse models of skin colonization (15) and systemic colonization (16). However, the role of switching remained elusive. That appeared to end in 2002, when Miller and Johnson (17) discovered that the configuration of the mating type locus (MTL) regulated switching and switching in turn regulated mating. They discovered that hemizygous MTL derivatives of a heterozygous MTL (a/α) strain, the latter representing the predominant MTL genotype among clinical isolates (18, 19), could switch. This led to the conclusion that minority a/a and α/α strains could switch, but majority a/α strains could not (19). This perception, however, ended when Xie et al. (20) reported that approximately one third of a collection of clinical a/α isolates could be induced to switch on agar medium containing N-acetylglucosamine (GlcNAc) at 25°C in 5% CO2. Furthermore, Xie et al. (20) demonstrated that individually deleting any one of the genes for the transcription factors (TFs) Efg1, Rfg1, or Brg1, derepressed switching to opaque in a/α cells, and Park et al. (21) subsequently demonstrated that deletion of the TF gene SFL2 also derepressed switching in a/α strains. Tao et al. (22) subsequently demonstrated that select a/α clinical isolates that underwent white-opaque switching could also switch to a “gray” phenotype, and this was confirmed by Park et al. (21), suggesting the existence of a triphasic switching system. Liang et al. (23) further presented evidence that clinical a/α strains that underwent switching to gray harbored a mutation in EFG1, and that, as previously demonstrated by Pierre and Kumamoto (24), a/α EFG1 mutants outcompeted wild-type cells in a mouse gastrointestinal (GI) colonization model. Using a CHROMagar plating assay, Liang et al. (23) further presented evidence indicating that the colonizing EFG1 mutant cells expressed the gray phenotype, although Park et al. (21) presented evidence that the gray phenotypes did not appear to be expressed by a/α EFG1 mutant cells at physiological temperature (37°C).
Here, we have explored the role of EFG1 in white-to-opaque or white-to-gray switching, and GI colonization, in a collection of 27 clinical a/α isolates. We found that 13 of the 27 isolates could be induced to switch to opaque. Four of the 17 EFG1/EFG1 strains, 1 of the 2 EFG1/efg1 strains, and all 8 of the efg1/efg1 strains underwent switching from white to opaque. However, only 7 of the 13 a/α isolates that switched were complemented by site-specific integration of a copy of EFG1, and all 7 were efg1/efg1 mutants. In all cases in which complementation with EFG1 reestablished repression of white-opaque switching, it also reestablished repression of white-gray switching. These results demonstrate that mutants of genes that repress white-to-opaque switching other than EFG1 may be just as prevalent as EFG1 mutants in clinical a/α strains that switch. Finally, results from competition experiments in a mouse gastrointestinal colonization model confirmed previous results (23, 24) that efg1Δ/efg1Δ cells or efg1/efg1 cells outcompete EFG1/EFG1 cells. When we plated the cells that colonized the GI tract, the results suggested that they formed gray and opaque cells. However, direct microscopic observations of live colonizing cells in the feces revealed that a/α strains with EFG1 null mutations expressed almost exclusively the opaque, not gray, phenotype, suggesting plating experiments may not accurately assess the cell phenotypes expressed at the site of GI colonization. Therefore, the latter results suggest that a/α efg1/efg1 strains colonizing the GI tract may express the opaque, not gray, phenotype.
RESULTS
EFG1 deletion mutants of strains SC5314 and P37039.
To assess the role of EFG1 in switching in a/α strains, we previously generated homozygous EFG1 deletion mutants of two a/α EFG1/EFG1 strains that did not switch, SC5314 and P37009 (see Table S1 in the supplemental material), and analyzed them for switching from white to opaque and white to gray on agar media under eight sets of environmental conditions (21). The conditions included all combinatorial permutations of the sugar source (agar with 1.25% glucose [Gluc-agar] versus 2.0% N-acetylglucosamine [GlcNAc-agar]), temperature (25°C versus 37°C) and atmosphere (air [0.04% CO2] versus 5% CO2) (21) in supplemented Lee’s medium (25). In our previous study (21), the four cellular phenotypes that were distinguished included the original white cell phenotype (Fig. 1A), the gray phenotype, which included the tiny elongate phenotype (Fig. 1B) and the transition phenotype (Fig. 1C), and the elongate, pimpled opaque cell phenotype (Fig. 1D). On both Gluc-agar and GlcNAc-agar at 25°C or 37°C, in air or 5% CO2, white cells of the SC3514 and P37039 a/α parent strains, which were EFG1/EFG1, did not switch. On Gluc-agar at 25°C or 37°C, in air or 5% CO2, white cells of both efg1Δ/efg1Δ derivatives of strains SC5314 and P37039 also did not switch to the gray or opaque phenotype (21). However, on GlcNAc-agar under all four sets of environmental conditions, the two efg1Δ/efg1Δ strains switched to either gray, opaque and gray, or opaque. On GlcNAc agar at 25°C in air, both efg1Δ/efg1Δ strains formed colonies containing a majority of gray cells and a minority of white cells, but no opaque cells (Fig. 1E and 2A). On GlcNAc-agar at 25°C in 5% CO2, both efg1Δ/efg1Δ strains formed colonies containing a majority of opaque cells and a minority of white and gray cells (Fig. 1E and 2A). On GlcNAc-agar at 37°C in air, efg1Δ/efg1Δ strains formed colonies containing a majority of opaque cells and a minority of white cells but no gray cells (Fig. 1E and 2A). On GlcNAc-agar at 37°C in 5% CO2, efg1Δ/efg1Δ strains formed opaque colonies containing exclusively opaque cells (Fig. 1E and 2A).
FIG 1.
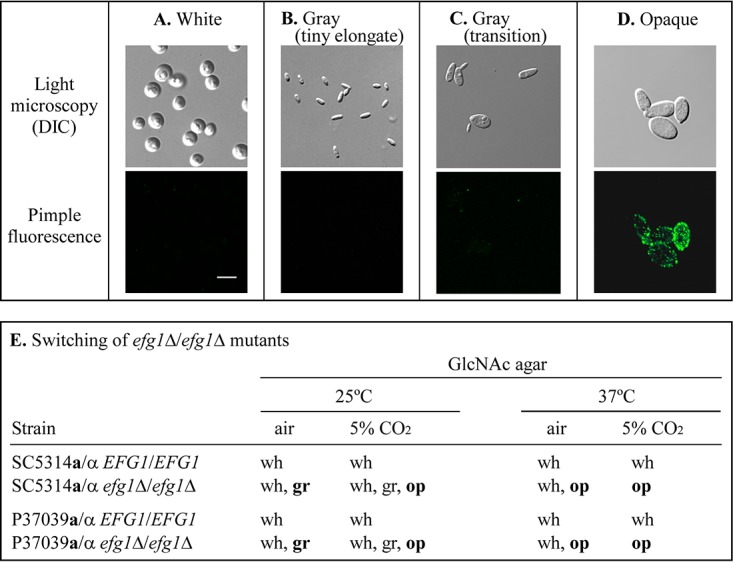
Examples of a/α cellular phenotype assessed in the switching studies and a summary of switching by the two strains SC5314 and P37039 and their deletion derivatives, previously analyzed in detail (21). Note that “tiny elongate” and “transition” represent two subgroups of the gray phenotype. (A) White phenotype; (B) gray tiny elongate phenotype; (C) gray transition phenotype; (D) opaque phenotype. In panels A to D, a differential interference contrast (DIC) image is presented in the top panel, and the corresponding immunostained image, with anti-opaque-specific pimple antibody, is shown in the bottom panel. (E) The phenotypes of the two parental a/α strains and deletion derivatives are presented under the four tested sets of environmental conditions on GlcNAc-agar. Cell phenotypes in boldface type in panel E are the dominant phenotypes formed in mixtures. wh, white; gr, gray; op, opaque. Bar in panel A, 5 μm.
FIG 2.
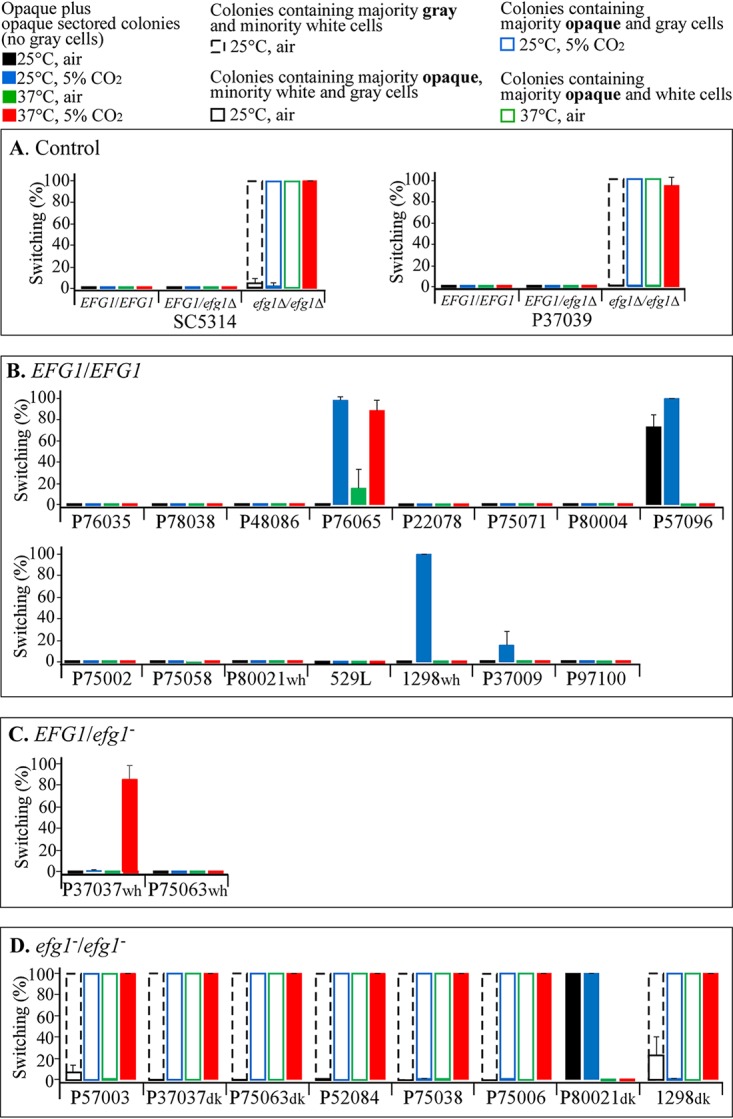
Switching to opaque and gray by the 27 clinical isolates and the generated deletion derivatives of C. albicans strains SC5314 and P37039 on GlcNAc-agar under the four sets of environmental conditions (25°C and air, 25°C and 5% CO2, 37°C and air, and 37°C and 5% CO2). The bars are explained in the key at the top of the figure. The majority cell phenotypes in the mixtures in the key are shown in boldface type. (A) Parent a/α SC5314 and P37039 EFG1/EFG1 strains and their heterozygous EFG1/efg1Δ and homozygous efg1Δ/efg1Δ mutant derivatives. (B) Fifteen additional clinical EFG1/EFG1 strains. (C) Two clinical EFG1/efg1 strains. (D) Eight clinical efg1/efg1 strains.
Strains used in this study. Download Table S1, DOCX file, 0.02 MB (25.5KB, docx) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Collection of clinical isolates.
The 27 clinical a/α isolates in the collection (Table S1) have been analyzed by restriction fragment length polymorphisms (RFLPs) probed with the mid-repeat sequence (Ca3 and by multilocus sequence typing [MLST]) as explained in the legend for Table S2. Clades I, SA, E, and III, determined by Ca3 fingerprinting, correlate with clades 1, 4, 3, and 11, determined by MLST (Table S2). The collection, based on the consensus of the clade analyses, grouped 8 in clade SA (MLST 4), 1 in clade III (MLST 3), 5 in clade E (MLST 11), and 10 in clade I (MLST 1) (Table S2). Four isolates were not tested by probed RFLPs (Table S2). One proved to be in MLST clade 3, two were in MLST 18, and one remained untested. Eight of the 27 isolates represented four pairs, each pair isolated from one of four individuals (P37037wh and P37037dk, P75063wh and P75063dk, P80021wh and P80021dk, and 1298wh and 1298dk). Using multilocus sequence typing (10, 11), we found that two strains in each of three pairs (P37037wh and P37037dk, P75063wh and P75063dk, and 1298wh and 1298dk) were highly related. The coisolates in the fourth pair (P80021wh and P80021dk) were not related (Table S2). The coisolates in each of the four pairs were treated individually in this study.
Ca3/MLST clade assignments for the 27 isolates used in this study. Ca3 fingerprinting was performed in the Soll lab. The MLST analysis and clade assignment of 10 strains were from collaboration (M.-E. Bougnoux, C. Pujol, D. Diogo, C. Bouchier, et al., Fungal Genet Biol 45:221–231, 2008, https://doi.org/10.1016/j.fgb.2007.10.008) or from different groups (F. C. Odds, M.-E. Bougnoux, D. J. Shaw, J. M. Bain, et al., Eukaryot Cell 6:1041–1052, 2007, https://doi.org/10.1128/EC.00041-07; M. P. Hirakawa, D. A. Martinez, S. Sakthikumar, M. Z. Anderson, et al., Genome Res 5:413–425, 2015, https://doi.org/10.1101/gr.174623.114). MLST typing of isolates P75063wh, P75063dk, P80021wh, P80021dk, 1298wh, 1298dk, P37037wh, and P37037dk was performed in our lab according to the consensus MLST scheme described by Bougnoux et al. (M.-E. Bougnoux, A. Tavanti, C. Bouchier, N. A. R. Gow, et al., J Clin Microbiol 41:5265–5266, 2003, https://doi.org/10.1128/JCM.41.11.5265-5266.2003) which is commonly used worldwide. The MLST clade assignment for these isolates was performed by generating a dendrogram, including MLST data retrieved from the Candida albicans MLST website (https://pubmlst.org/calbicans/) for 24 control isolates and strain 529L, which does not appear to have been previously assigned to a clade. The control isolates were selected, three per clade, from the seven most frequent clades (MLST clades 1, 2, 3, 4, 8, 9, and 11) found among a collection of 1,391 isolates (F. C. Odds, M.-E. Bougnoux, D. J. Shaw, J. M. Bain, et al., Eukaryot Cell 6:1041–1052, 2007, https://doi.org/10.1128/EC.00041-07). Three isolates from clade 18, a major clade in Asia (J. E. Shin, M.-E. Bougnoux, C. d’Enfert, S. H. Kim, et al., J Clin Microbiol 49:2572–2577, 2011, https://doi.org/10.1128/JCM.02153-10), were also used as controls. 529L did not cluster with any other strain. The Ca3 fingerprinting clades I, II, III, SA, and E have been repeatedly shown to correspond nearly perfectly to MLST clades 1, 2, 3, 4, and 11, respectively (A. Tavanti, A. D. Davidson, M. J. Fordyce, N. A. R. Gow, et al., J Clin Microbiol 43:5601–5613, 2005, https://doi.org/10.1128/JCM.43.11.5601-5613.2005; M.-E. Bougnoux, C. Pujol, D. Diogo, C. Bouchier, et al., Fungal Genet Biol 45:221–231, 2008, https://doi.org/10.1016/j.fgb.2007.10.008; F. C. Odds, M.-E. Bougnoux, D. J. Shaw, J. M. Bain, et al., Eukaryot Cell 6:1041–1052, 2007, https://doi.org/10.1128/EC.00041-07; B. A. McManus and D. C. Coleman, Infect Genet Evol 21:166–178, 2014, https://doi.org/10.1016/j.meegid.2013.11.008). It should be noted that clade assignments are no longer displayed on the C. albicans MLST website. Download Table S2, DOCX file, 0.01 MB (13.7KB, docx) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
EFG1 mutations among the collection.
EFG1 was sequenced in all 27 clinical isolates. EFG1 alleles harboring nonsense mutations or a glycine-to-aspartic acid substitution, will be indicated as “efg1,” whereas the deletion alleles we generated in strains SC5314 and P37039 will be indicated as “efg1Δ.” Of the 27 clinical isolates in the collection, which includes the parent strains SC5314 and P37039, 17 were EFG1/EFG1, 2 were EFG1/efg1, and 8 were efg1/efg1 (Table 1). In the key at the top of Fig. 3, the sequenced EFG1 alleles and the deduced proteins are diagrammed. In the EFG1/efg1 strain P37037wh, the efg1 allele harbored a base substitution of aspartic acid for glycine at genomic position 755, resulting in a missense mutation G252D within the APSES core domain (Table 1 and Fig. 3B). In the EFG1/efg1 strain P35063wh, the efg1 allele had an insertion of 22 nucleotides at genomic position 849, resulting in a frameshift causing a nonsense mutation at position 289 (Table 1 and Fig. 3B). Seven of the eight efg1/efg1 strains (P57003, P75063dk, P52084, P75038, P75006, P80021dk, and 1298dk) harbored nonsense mutations caused by point mutations or indel frameshifts, in both EFG1 alleles (Table 1 and Fig. 3C). A G252D missense mutation in both EFG1 alleles and partial loss of heterozygosity in strain P37037dk, indicated that it was a derivative of the heterozygous EFG1/efg1 partner isolate P37037wh (Fig. 3B). The results from sequence analyses supported that conclusion (Fig. 3B and C). Sequence analysis also revealed that P75063dk was derived from P75063wh (Fig. 3B and C).
TABLE 1.
Origins, clades, and EFG1 genotypes of the 27 clinical a/α isolates and derived deletion mutants analyzed in this studya
|
EFG1
genotypeb |
Isolate no. |
Strainc | Body location(s)d |
Country of origine |
Cladef
Ca3 (MLST) |
EFG1 allelesg |
|---|---|---|---|---|---|---|
| EFG1/EFG1 | 1 | SC5314 | NI | USA | I (1) | EFG1/EFG1 |
| 2 | P37039 | HI, Sp | USA | I (1) | EFG1/EFG1 | |
| 3 | P76035 | BSI | USA | I (1) | EFG1/EFG1 | |
| 4 | P78038 | BSI | USA | I | EFG1/EFG1 | |
| 5 | P48086 | BSI | USA | I | EFG1/EFG1 | |
| 6 | P76065 | BSI | USA | I | EFG1/EFG1 | |
| 7 | P22078 | BSI | UK | SA | EFG1/EFG1 | |
| 8 | P75071 | BSI | Italy | SA (4) | EFG1/EFG1 | |
| 9 | P80004 | BSI | USA | SA (4) | EFG1/EFG1 | |
| 10 | P57096 | BSI | Brazil | E (11) | EFG1/EFG1 | |
| 11 | P75002 | BSI | Spain | E | EFG1/EFG1 | |
| 12 | P75058 | BSI | Switz. | E (11) | EFG1/EFG1 | |
| 13 | P80021wh | BSI | Italy | NG (3) | EFG1/EFG1 | |
| 14 | 529L | OC | UK | NG | EFG1/EFG1 | |
| 15 | 1298wh | NI | Uganda | NG (18) | EFG1/EFG1 | |
| 16 | P37009 | HI, Sp | Unknown | NG | EFG1/EFG1 | |
| 17 | P97100 | BSI | Czech. R | NG (NG) | EFG1/EFG1 | |
| EFG1/efg1Δ | SC5314 | USA | I (1) | EFG1/efg1Δ | ||
| P37039 | USA | I (1) | EFG1/efg1Δ | |||
| EFG1/efg1− | 18 | P37037wh | HI, Sp | USA | I (1) | EFG1/EFG1(G252D) |
| 19 | P75063wh | BSI | France | SA (4) | EFG1/EFG1g.849ins22nt (289*) | |
| efg1Δ/efg1Δ | SC5314 | USA | I (1) | efg1Δ/efg1Δ | ||
| P37039 | USA | I (1) | efg1Δ/efg1Δ | |||
| efg1−/efg1− | 20 | P57003 | BSI | USA | I (1) | EFG1g.93-298del(52*)/EFG1g.93-289del(52*) |
| 21 | P37037dk | HI, Sp | USA | I (1) | EFG1(G252D)/EFG1(G252D) | |
| 22 | P75063dk | BSI | France | SA (4) | EFG1g.849ins22nt(289*)/EFG1g.849ins22nt(289*) | |
| 23 | P52084 | BSI | Canada | SA (4) | EFG1(Q34*)/EFG1(Q34*) | |
| 24 | P75038 | BSI | Turkey | SA | EFG1(Q199*)/EFG1(Q199*) | |
| 25 | P75006 | BSI | Spain | E (11) | EFG1(Q93*)/EFG1(Q93*) | |
| 26 | P80021dk | BSI | Italy | E (11) | EFG1g.1361ins1nt(459*)/EFG1g.1361ins1nt(459*) | |
| 27 | 1298dk | NI | Uganda | NG (18) | EFG1(Y220*)/EFG1(Y220*) | |
See Table S1 for the origins of strains noted in references.
Deletion derivatives generated for strains SC5314 and P37039 (21) are shown in boldface type and were not considered or numbered as part of the basic collection of clinical a/α isolates. The wild-type parent strains SC5314 and P37039 were considered members of the collection of the 27 clinical a/α isolates. Deletion derivatives are therefore not numbered and are distinguished from natural EFG1 mutants by a delta symbol.
Each of four pairs of isolates (P37037wh and P37037dk, P75063wh and P75063dk, P80021wh and P80021dk, and 1298wh and 1298dk) was obtained from one of four individuals.
NI, not identified: HI, healthy individual; Sp, sputum; BSI, bloodstream; OC, oral cavity.
USA, United States of America; UK, United Kingdom; Switz., Switzerland; Czech. R, Czech Republic.
Clades were determined by RFLPs identified by the mid-repeat sequence CA3 (42, 43). See explanation for MLST clades in Table S2 in the supplemental material.
The numbers after EFG1 and in parentheses refer to nucleotide positions and amino acid positions, respectively. g, genomic position in EFG1 gene; del, deletion; ins, insertion; nt, nucleotide; D, aspartic acid; G, glycine; Q, glutamine; *, stop codon; Y, tyrosine; A, adenine; G, guanine; C, cytosine; T, thymine.
FIG 3.

EFG1 allelic sequences of the 27 a/α clinical isolates and the deduced Efg1 proteins. The names of the isolates that did not switch to opaque are black, and those that did switch are brown. (A) Allelic sequences and the deduced proteins of EFG1/EFG1 isolates. (B) Allelic sequences and the deduced proteins of EFG1/efg1− isolates. (C) Allelic sequences and deduced proteins of efg1−/efg1− isolates. The keys for color coding and abbreviations are presented at the top of figure. SNPs, single nucleotide polymorphisms; stop, stop codon; base exchange, base exchange resulting in an amino acid (a.a.) exchange compared to the reference sequence (http://www.candidagenome.org/cgi-bin/seqTools); indel, insertion or deletion of base(s). PolyQ stretch, regions rich in glutamine (Q); APSES core domain, DNA binding helix-loop-helix domain which is essential for the function of Efg1; *, protein termination due to stop codon; G252D, amino acid exchange from glycine to aspartic acid.
EFG1 polymorphisms of the collection were used to generate a dendrogram of relatedness (see Fig. S1 in the supplemental material), using the neighbor-joining method (26). The EFG1 sequences of 25 of the 27 isolates separated into three clades, consistent with clades SA (MLST 4), clade E (MLST 11), and I (MLST 1) (Fig. S1). It should be noted that the efg1/efg1 mutants were distributed among the three major clades (Fig. S1). It should also be noted that the dendrogram generated by the EFG1 sequences is highly consistent with the groupings of Ca3 fingerprinting and MLST analyses (Fig. S1).
A dendrogram of EFG1 relatedness based on EFG1 polymorphisms. Clade assignments are presented immediately to the right, based on RFLPs probed with the mid-repeat sequence CA3 and, in parentheses, MLST when available. See Table S2 for explanations about the Ca3 and MLST analysis. The EFG1 genotypes of the clinical isolates are presented to the far right. The efg1− alleles in the isolates are shown in red. Download FIG S1, PDF file, 0.2 MB (172.8KB, pdf) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Switching under the eight sets of conditions.
None of the 27 strains in the collection formed either gray or opaque colonies on Gluc-agar (1.25% glucose) under any of the four sets of environmental conditions (25°C and air, 25°C and 5% CO2, 37°C and air, and 37°C and 5% CO2). However, on GlcNAc-agar, 13 of the 27 strains in the collection (48%) formed opaque colonies under one or more sets of conditions (Fig. 2). Seven of the 13 clinical isolates that switched to opaque formed gray cells, but only at 25°C, as was the case for the efg1Δ/efg1Δ derivatives of SC5314 and P37039 (21) (Fig. 1E and 2A). Switching by isolates in each of the three EFG1 genotypes is dealt with separately in the following sections.
Switching by EFG1/EFG1 isolates.
Of the 17 clinical isolates that were EFG1/EFG1, four (P76065, P57096, 1298wh, and P37009) underwent switching from white to opaque on GlcNAc-agar under one or more sets of environmental conditions (Fig. 2B and Table S3). P76065 switched to opaque at 25°C in 5% CO2 and 37°C in air and 5% CO2 (Fig. 2B and Table S3). P57096 switched to opaque in 25°C in air and 5% CO2 (Fig. 2B and Table S3). 1298wh and P37009 switched to opaque only in air at 5% CO2. All three switching patterns differed from those exhibited by the two efg1Δ/efg1Δ derivatives of strains SC5314 and P37039 (Fig. 2A). None of the 17 EFG1/EFG1 strains, including those that switched to opaque, formed gray cells under any of the four sets of conditions on GlcNAc-agar (Fig. 2B and Table 2). These results suggest that mutations in genes other than EFG1 resulted in the capacity to switch to opaque and the fact that these mutations did not derepress switching to gray reinforces that suggestion.
TABLE 2.
Summary of the phenotypic switching characteristics and complementation results for the 27 clinical a/α isolates and deletion derivatives of SC5314 and P37039a
| EFG1 genotype | Isolate no. | Strain | Phenotypic transitionb
|
Complementation by scEFG1c
|
||||
|---|---|---|---|---|---|---|---|---|
| Wh to Op | Wh to Gr | Hy | Wh to Op | Wh to Gr | Hy | |||
| EFG1/EFG1 | 1 | SC5314 | − | − | + | |||
| 2 | P37039 | − | − | + | ||||
| 3 | P76035 | − | − | + | ||||
| 4 | P78038 | − | − | − | nt | |||
| 5 | P48086 | − | − | + | ||||
| 6 | P76065 | + | − | + | No | |||
| 7 | P22078 | − | − | + | ||||
| 8 | P75071 | − | − | + | ||||
| 9 | P80004 | − | − | + | ||||
| 10 | P57096 | + | − | + | No | |||
| 11 | P75002 | − | − | − | nt | |||
| 12 | P75058 | − | − | − | nt | |||
| 13 | P80021wh | − | − | + | ||||
| 14 | 529L | − | − | − | nt | |||
| 15 | 1298wh | + | − | + | No | |||
| 16 | P37009 | + | − | + | No | |||
| 17 | P97100 | − | − | + | ||||
| EFG1/efg1Δ | SC5314 | − | − | + | ||||
| P37039 | − | − | + | |||||
| EFG1/efg1− | 18 | P37037wh | + | − | + | No | ||
| 19 | P75063wh | − | − | + | ||||
| efg1Δ/efg1Δ | SC5314 | + | + | − | Yes | Yes | Yes | |
| P37039 | + | + | − | Yes | Yes | Yes | ||
| efg1−/efg1− | 20 | P57003 | + | + | − | Yes | Yes | Yes |
| 21 | P37037dk | + | + | − | Yes | Yes | Yes | |
| 22 | P75063dk | + | + | − | Yes | Yes | Yes | |
| 23 | P52084 | + | + | − | Yes | Yes | Yes | |
| 24 | P75038 | + | + | − | Yes | Yes | Yes | |
| 25 | P75006 | + | + | − | Yes | Yes | Yes | |
| 26 | P80021dk | + | − | − | No | na | No | |
| 27 | 1298dk | + | + | − | Yes | Yes | Yes | |
Summary of the phenotypic switching characteristics, including gray and hypha formation, and complementation results for the collection of 27 clinical a/α isolates and the deletion derivatives of strains SC5314 and P37039. Wh, white; Op, opaque; Gr, gray; Hy, hyphae.
The white-to-opaque transition (Wh to Op) and the white-to-gray transition (Wh to Gr) was tested on GlcNAc-agar under four sets of environmental conditions (data presented in Fig. 2). The yeast-to-hypha formation transition was tested in suspension and on agar plates, both in the presence of 10% serum.
Complementation by scEFG1 represents reestablishment of repression of the white-to-opaque transition, reestablishment of repression of the white-to-gray transition and reestablishment of hypha induction by serum. nt, not tested; na, not applicable; yes, complemented; no, not complemented.
Switching frequency of the four EFG1/EFG1 clinical isolates that switched to opaque and their EFG1/scEFG1 derivatives generated by site-specific integration at one of the EFG1 alleles. Switching was assessed on GlcNAc-agar under the four sets of environmental conditions. The switching data represent the means ± standard deviations for three independently performed experiments. Switching includes data for homogenous opaque colonies and opaque sectored colonies in which the sectors contained only opaque cells. These strains did not form gray cells under any of the conditions tested. Download Table S3, DOCX file, 0.02 MB (19KB, docx) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Switching by EFG1/efg1 isolates.
There were only two EFG1/efg1 strains in the collection, P37037wh and P75063wh. One of these two strains, P37037wh, underwent switching to opaque, but only on GlcNAc-agar at 37°C in 5% CO2 (Fig. 2C and Table S4), a response pattern different from those of the 12 other isolates that switched (Fig. 2A, B, and D). This mutant did not form gray cells under any of the four conditions (Table 2). P37037wh harbored one efg1 allele with an aspartic acid substitution for glycine (Table 1 and Fig. 3). These results suggest that just as in the case of the EFG1/EFG1 strains that switched to opaque, repression was due to a mutation in one or more genes other than EFG1. This conclusion was supported by the fact that the two generated EFG1/efg1Δ derivatives of SC5314 and P37039 did not switch (Fig. 2A and Table S4).
Switching frequency of white to opaque for the efg1Δ/efg1Δ mutants, efg1−/efg1− natural strains, and their wild type and efg1−/scEFG1 complemented derivatives generated by site-specific integration of one of the EFG1 alleles. The switching was assessed on GlcNAc-agar under the four sets of environmental conditions. The switching data represent the means ± standard deviations for three independently performed experiments. These data include only switching from white to opaque (homogenous opaque colonies and colonies with homogenous opaque sectors); they do not include switching from white to gray. Download Table S4, DOCX file, 0.02 MB (23.3KB, docx) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Switching by efg1/efg1 isolates.
All eight of the clinical isolates that were efg1/efg1 underwent switching from white to opaque on GlcNAc-agar under more than one of the four sets of conditions on GlcNAc-agar (Fig. 2D and Table S4). Seven of the eight underwent switching to gray and opaque in response to the four sets of environmental conditions (Fig. 2D and Table S4) in a fashion highly similar to that of the efg1Δ/efg1Δ deletion derivatives of SC5314 and P37039 (Fig. 2A and Table S4). Only one of the eight clinical efg1/efg1 strains, P80021dk, that did not switch to gray (Fig. 2D and Table 2) exhibited a switching pattern under the four sets of conditions (Fig. 2D and Table S4) that differed from that of the two efg1Δ/efg1Δ derivatives (Fig. 2A), but it was highly similar to that of the EFG1/EFG1 strain P57096 (Fig. 2B), suggesting that the mutation was in the same gene in the two clinical isolates, even though the former was EFG1/EFG1 and the latter was efg1/efg1. Another explanation is that the deduced domains remaining in the truncated P80021dk Efg1 protein resulted in partial function.
Complementation of EFG1 mutants.
To test whether the repression of switching by a/α clinical isolates could be reinstated (i.e., complemented) by reintroducing a functional copy of EFG1, we performed site-specific integration at the native EFG1 locus of one copy of EFG1 from strain SC5314 (“scEFG1”), under the control of the native promoter (21). As we previously demonstrated (21), reintroducing a copy of scEFG1 into the SC5314 efg1Δ/efg1Δ and P37039 efg1Δ/efg1Δ derivatives, under the control of the wild-type EFG1 promoter, reinstated switching repression. Site-specific integration of one copy of scEFG1 in the four EFG1/EFG1 strains that switched to opaque, generating strains P76065 EFG1/scEFG1, P57096 EFG1/scEFG1, 1298wh EFG1/scEFG1, and P37009 EFG1/scEFG1 (Table S1) did not reinstate repression of switching (i.e., did not result in complementation) in any of the four strains (Tables 2, 3, and S3). These results support the conclusion that derepression of switching in these four clinical EFG1/EFG1 isolates is due to mutations in one or more repressor genes other than EFG1. We next tested whether integration of scEFG1 complemented the one clinical EFG1/efg1 mutant, P37037wh, which switched to opaque. It did not (Tables 2, 3, and S4), again indicating that a mutation in a gene other than EFG1 derepressed switching to opaque. Finally, we tested whether the addition of one copy of scEFG1 complemented the seven clinical efg1/efg1 strains that switched to gray and opaque and the one efg1/efg1 strain (P80021dk) that switched to opaque but not gray. The addition of scEFG1 reestablished the repression of switching (i.e., complementation) in the seven strains that switched to opaque and gray (Fig. 2D and Tables 2 and 3). However, the site-specific integration of scEFG1 into P37037dk, generating P37037dk efg1/scEFG1, did not repress switching to opaque and repressed switching to gray (Tables 2, 3, and S4), as was the case for P37037wh (Fig. 2 and Table S4). Site-specific integration of scEFG1 in P80021dk did not restore repression of switching, indicating a mutation in a repressor gene other than EFG1. These results support the conclusion that seven of the eight efg1/efg1 strains, which represent 30% of the entire collection of clinical isolates, switched to gray and opaque due to efg1 mutations and that only one efg1/efg1 strain carried a second mutation that derepressed switching to opaque.
TABLE 3.
Results of complementation experimentsa
| Category | Strain | Switchingb
|
|||
|---|---|---|---|---|---|
| 25°C |
37°C |
||||
| Air | 5% CO2 | Air | 5% CO2 | ||
| Controls | SC5314 efg1Δ/efg1Δ | (+) | (++++) | (++++) | ++++ |
| SC5314 efg1Δ/scEFG1 | − | − | − | − | |
| P37039 efg1Δ/efg1Δ | − | (++++) | (++++) | ++++ | |
| P37039 efg1Δ/scEFG1 | − | − | − | − | |
| EFG1/EFG1 | P76065 EFG1/EFG1 | − | ++++ | ++ | ++++ |
| P76065 EFG1/scEFG1 | − | ++++ | ++ | +++ | |
| P57096 EFG1/EFG1 | +++ | ++++ | − | − | |
| P57096 EFG1/scEFG1 | ++++ | ++++ | − | − | |
| 1298wh EFG1/EFG1 | − | ++++ | − | − | |
| 1298wh EFG1/scEFG1 | − | ++++ | − | − | |
| P37009 EFG1/EFG1 | − | ++ | − | − | |
| P37009 EFG1/ scEFG1 | − | ++ | − | − | |
| EFG1/efg1− | P37037wh EFG1/efg1− | − | − | − | +++ |
| P37037wh EFG1/scEFG1 | − | − | − | ++++ | |
| efg1−/efg1− | P57003 efg1−/efg1− | − | (++++) | (++++) | ++++ |
| P57003 efg1−/scEFG1 | − | − | − | − | |
| P37037dk efg1−/efg1− | (+) | (++++) | (++++) | ++++ | |
| P37037dk efg1−/scEFG1 | − | − | − | ++++ | |
| P75063dk efg1−/efg1− | − | (++++) | (++++) | ++++ | |
| P75063dk efg1−/scEFG1 | − | − | − | − | |
| P52084 efg1−/efg1− | − | (++++) | (++++) | ++++ | |
| P52084 efg1−/scEFG1 | − | − | − | − | |
| P75038 efg1−/efg1− | − | (++++) | (++++) | ++++ | |
| P75038 efg1−/scEFG1 | − | − | − | − | |
| P75006 efg1−/efg1− | − | (++++) | (++++) | ++++ | |
| P75006 efg1−/scEFG1 | − | − | − | − | |
| P80021dk efg1−/efg1− | ++++ | ++++ | − | − | |
| P80021dk efg1−/scEFG1 | ++++ | ++++ | − | − | |
| 1298dk efg1−/efg1− | (++) | (++++) | (++++) | ++++ | |
| 1298dk efg1−/scEFG1 | − | − | − | − | |
Results of complementation experiments, in which one copy of SC5314 EFG1 (scEFG1) was inserted into the native locus at one allele, under the control of native promoter, by site-specific integration in strains that switched to opaque. See Tables S3 and S4 for switching frequencies. Switching analyses were performed on GlcNAc-agar.
++++, 80 to 100% opaque colonies; +++, 21 to 80% opaque colonies; ++, 11 to 20% opaque colonies; −, no opaque or mixed colonies. Parentheses indicates colonies with mixed cellular phenotypes. At 25°C in air or 5% CO2, the mixtures include a majority of opaque cells and a minority of white and gray cells; at 37°C in air, the mixtures include a majority of opaque cells and a minority of white cells (no gray cells). (++++), 80 to 100%; (++), 11 to 20%; (+), 1 to 10%. Each assessment represents the results of pooled data for three independent experiments. The total number of colonies ranged between 425 and 2,059.
Hypha formation.
EFG1 not only plays a negative regulatory role in white-opaque switching (10, 27), but it also plays a positive regulatory role in hypha formation (28, 29). Serum induced hypha formation in the EFG1/EFG1 control strains SC5314 and P37039, but not in the deletion derivatives SC5314 efg1Δ/efg1Δ and P37039 efg1Δ/efg1Δ (Fig. 4A and Table 2). Site-specific integration of a copy of scEFG1, generating strains SC5314 efg1Δ/scEFG1 and P37039 efg1Δ/scEFG1, reestablished hypha induction (i.e., caused complementation) (Fig. 4A and Table 2). Serum induced hypha formation in 13 of the 17 EFG1/EFG1 isolates (Table 2). Most notably, serum induced hypha formation in all four of the EFG1/EFG1 strains that underwent white-opaque switching (Table 2), supporting the conclusion that mutations in genes other than EFG1 were responsible for the derepression of switching in those strains. The four EFG1/EFG1 strains that did not form hyphae also did not undergo the white-to-opaque transition (Table 2), suggesting that a mutation in a gene other than EFG1 was responsible for the lack of hypha induction. Both EFG1/efg1 clinical isolates formed hyphae, as was the case for the EFG1/efg1Δ derivatives of strains SC5314 and P37039 (Table 2). Seven of the eight efg1−/efg1 clinical isolates, which switched to opaque and gray, did not form hypha, which was the case for the efg1Δ/efg1Δ derivatives of SC5314 and P37039 (Table 2). Representative micrographs of four of these isolates (P37037dk, P75063dk, P52084, and P75038) are presented in Fig. 4B. In seven of the eight efg1/efg1 isolates, for which site-specific integration of scEFG1 reestablished the repression of switching, the addition of scEFG1 also reestablished serum-induced hypha formation (Table 2 and Fig. 4B). The one efg1/efg1 isolate, P80021dk, which switched to opaque but not gray, and for which the addition of scEFG1 does not complement switching, was also not complemented for hypha formation by the addition of scEFG1 (Table 2). These results support the conclusion that, in the case of seven of the eight efg1/efg1 isolates, the mutation in EFG1 was solely responsible for the derepression of switching and the repression of hypha formation. In the case of one efg1/efg1 isolate, P80021dk, a mutation in a gene in addition to the mutation in EFG1 repressed switching and the induction of hypha formation.
FIG 4.
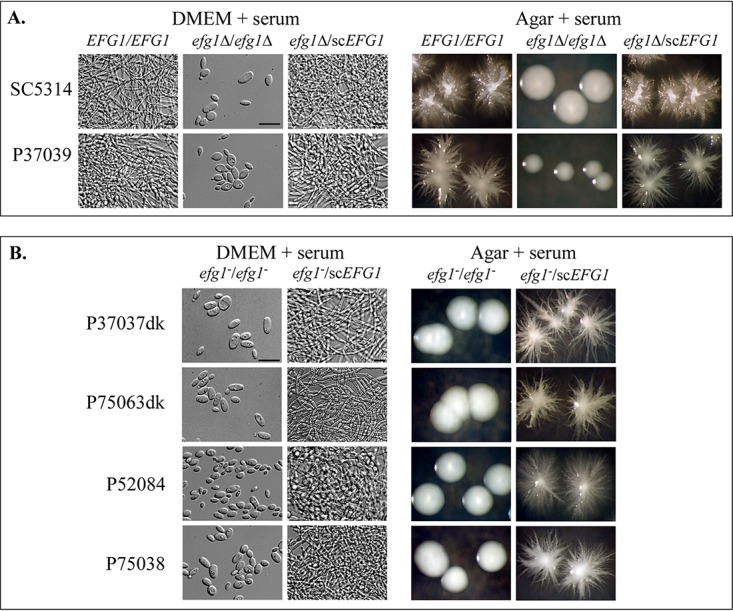
Induction of hypha formation in clinical a/α strains and their complemented derivatives. Hypha induction was tested in suspension cultures in DMEM medium plus 10% serum (DMEM + serum), and on nonnutrient agar containing 10% serum (Agar + serum) at 37°C in 5% CO2. Complementation was tested by the addition of a single copy of scEFG1 through site-specific integration. (A) Cellular phenotypes and colony phenotypes under hypha-inducing conditions for SC5314 and P37039 EFG1/EFG1, their efg1Δ/efg1Δ derivatives, and the complemented efg1Δ/scEFG1 derivatives. (B) Examples of the phenotypes of efg1/efg1 strains and the complemented efg1−/scEFG1 derivatives. Bars, 10 μm.
Intestinal colonization.
Previous studies have demonstrated that efg1/efg1 strains outcompete EFG1/EFG1 strains in mouse gastrointestinal (GI) colonization models (23, 24, 30, 31). Liang et al. (23) further demonstrated that in competition experiments in which fecal samples were plated on CHROMagar over time and incubated in air at 22°C for 5 days, the colonizing efg1Δ/efg1Δ or efg1/efg1 cells formed colonies containing almost exclusively gray cells after 2 days postingestion. However, in an in vitro study, we found that cells of efg1Δ/efg1Δ derivatives of strains SC5314 and P37039 that were plated on GlcNAc-agar and incubated at 25°C formed gray cells, but not opaque cells, and at 37°C, they formed opaque cells, but not gray cells (21). Hence, ingested cells in the GI tract may not be capable of expressing the gray phenotype at the site of colonization because of the physiological temperature (37°C). Hence, plating assays performed at 25°C or 22°C may induce opaque cells to switch back to gray. We therefore considered the possibility that the proportion of colony phenotypes assessed after 5 days on CHROMagar Candida, a glucose-based agar containing a “chromogenis mix” (32), in air at 22°C (23), may not have accurately reflected the actual phenotypes of progenitor cells at the site of colonization. We performed similar competition experiments between SC5314 EFG1/EFG1 versus SC5314 efg1Δ/efg1Δ cells (50:50) and between P57003 efg1/scEFG1 versus P57003 efg1/efg1 cells (50:50) in which we plated fecal samples over time on GlcNAc-agar and incubated the plates for 5 days at 25°C in air, conditions we previously found stabilized both the opaque and gray phenotype but did not induce the opaque phenotype (21). The genotypes and cell phenotypes of the initial strains in the four competition experiments are presented in Fig. 5A, the experimental protocol is shown in Fig. 5B, the methods for computing the proportions of initial strains over time are shown in Fig. 5C, and the results are shown in Fig. 5D. The efg1Δ/efg1Δ strain outcompeted the EFG1/EFG1 strain in combinations 1, 2, and 3, and the efg1/efg1 strain outcompeted the efg1/scEFG1 strain in combination 4. For all four combinations, the homozygous mutant strains represented more than 90% of the colonizing populations after 2 days, and close to 100% after 3 days postingestion (Fig. 5D), results similar to those of Liang et al. (23). For all four combinations, the gray phenotype dominated after 2 days postingestion and remained dominant through 8 days postingestion, again consistent with the results of Liang et al. (23). However, the proportion of opaque cells differed between our study and that of Liang et al. (23) as time progressed. Whereas Liang et al. (23) found that in competition experiments between a/α EFG1/EFG1 and efg1/efg1 cells, the colonizing populations did not form a/α opaque colonies 10 or more days postingestion, we found here a significant proportion of opaque colonies, approximately 25% of the colonies from feces in combinations 1, 2, and 3 and 50% in combination 4 after 8 days (Fig. 5E). After 15 days, the proportions increased to 30, 30, 30, and 80% (Fig. 5E).
FIG 5.

Competition experiments between SC5314 EFG1/EFG1 strain and efg1Δ/efg1Δ derivatives and between the complemented P57003 efg1/scEFG1 efg1/efg1 strain in the mouse model of gastrointestinal colonization. (A) Cell phenotype and genotypes of the pairs of strains in each of the four tested combinations. (B) Experimental protocol used to analyze colonizing populations. (C) Calculations for assessing the proportions of the two strains in each combination. (D) Proportion of each of the two strains and the proportion of the colony phenotypes in the feces over a 15-day period following ingestion.
We considered the possibility that the differences in the proportions of opaque cells observed in our study versus those of Liang et al. (23) may be due to differences in the agar media. Agar based on supplemented Lee’s medium (25) and containing GlcNAc was employed in our plating study, whereas glucose-based CHROMagar Candida medium (32) was used in the study by Liang et al. (23). Our major concern was that the incubation conditions for assessing phenotype in both of the studies may have in fact influenced phenotype, and therefore, may not have accurately produced results that reflected the actual proportions of cellular phenotypes at the site of colonization. We therefore examined the phenotypes of live cells directly in feces in combination 1 (Fig. 5A) after 8 days postingestion. Feces were simply diluted, and the live C. albicans cells were viewed microscopically. Since the colonizing SC5314 efg1Δ/efg1Δ cells harbored the gene for mCherry under the control of the promoter of the opaque-specific gene OP4 (12, 33) (Table S1), the live cells were examined by phase-contrast microscopy for morphology and by fluorescence microscopy for opaque-specific mCherry expression. White and gray cells of SC5314 efg1Δ/efg1Δ do not express OP4 and therefore do not fluoresce, whereas opaque cells express OP4 and therefore fluoresce throughout their cytoplasm (Fig. 6A). Of 100 cells examined directly in fecal samples, more than 90% exhibited the elongate opaque, not gray, cell morphology, and more than 70% exhibited mCherry fluorescence (Fig. 6B). These results indicate that the majority of efg1Δ/efg1Δ cells colonizing the feces after 8 days postingestion express the opaque, not gray, phenotype, and therefore, that the results of the plating experiments performed here (Fig. 5E) and previously (23) may not accurately reflect the actual cellular phenotype of efg1/efg1 cells at the site of GI colonization.
FIG 6.
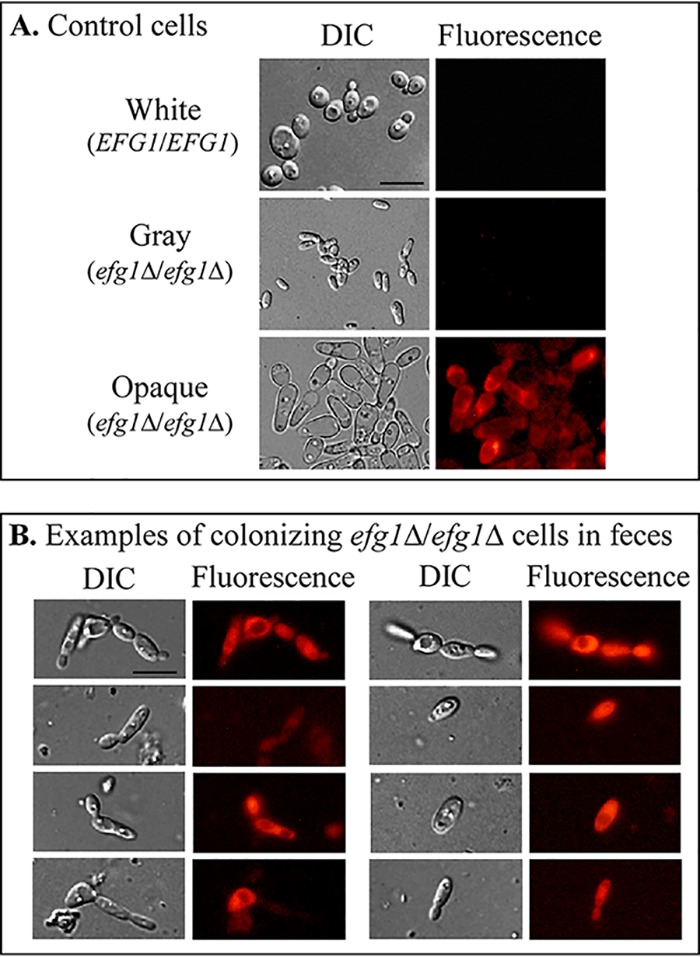
The majority of live cells in 8 day fecal samples of mice that ingested a combination of 50% SC5314 EFG1/EFG1 white cells and 50% SC5314 efg1Δ/efg1Δ, OP4p-mCh white cells exhibited almost exclusively opaque cell morphologies and expressed mCherry, which was under the regulation of the native opaque-specific OP4 promoter, indicating that opaque was the dominating phenotype in the colonizing cell population. Cell morphology was assessed by DIC microscopy and expression of mCherry by fluorescence microscopy. (A) Cell morphologies and mCherry expression of EFG1/EFG1 white cells, efg1Δ/efg1Δ gray cells, and efg1Δ/efg1Δ opaque cells generated in vitro. (B) Cell morphologies and mCherry expression by cells colonizing the feces 8 days after ingestion of the 50:50 mixture of SC5314 EFG1/EFG1 white cells and efg1Δ/efg1Δ OP4p-mCh white cells (combination 1 in Fig. 5A). Of 100 cells analyzed, more than 90% exhibited the distinct opaque phase phenotype, and more than 70% expressed mCherry throughout their cytoplasm. Bars, 10 μm.
DISCUSSION
Xie et al. (20) first reported that approximately one third of clinical isolates, which are predominately a/α (18, 19), can be induced to switch from white to opaque by plating white cells on GlcNAc-agar and incubating the plates at 25°C in 5% CO2. We have found here, using a broader screen that included eight combinations of environmental conditions, that close to half of a collection of 27 clinical a/α isolates could be induced to switch. Xie et al. (20) further demonstrated that deletion of any one of three transcription factor genes, RFG1, BRG1, or EFG1, derepressed white-to-opaque switching in an a/α strain that did not switch. Park et al. (21) subsequently reported the same for deletion of SFL2. Liang et al. (23) further reported that 6 of 31 clinical isolates (19%) harbored hemizygous or homozygous EFG1 mutations and that homozygous EFG1 mutants switched to the gray phenotype, which was previously shown to exhibit unique characteristics and to be part of a triphasic switching system (22). We report here that 8 of the 27 clinical a/α isolates (30%) were efg1/efg1 deletion mutants and that 7 of the 8 could switch to gray and all 8 could switch to opaque (21). However, we also found that four EFG1/EFG1 isolates and one EFG1/efg1 isolate (19%) could switch, but as a result of a mutation in a gene other than EFG1.
EFG1 and white-to-opaque switching.
It has been almost 20 years since Sonneborn et al. (10) first demonstrated that Efg1 suppresses switching in MTL homozygous strains of C. albicans. Subsequent studies then revealed that EFG1 null mutants outcompeted EFG1/EFG1 cells in mouse models of gastrointestinal (GI) colonization (24, 34). The results of Tao et al. (22) and Liang et al. (23), and the results presented here, further support these previous studies that suggest that EFG1 plays a role in GI colonization. The results reported here further demonstrate that 30% of a collection of 27 clinical a/α isolates were efg1/efg1. All eight efg1/efg1 strains in our study underwent white-to-opaque switching, and seven of the eight were complemented for white-opaque switching by reintroducing a functional copy of EFG1. Surprisingly, we also found that 4 of the 17 EFG1/EFG1 a/α strains and 1 of the 2 EFG1/efg1 a/α strains in the tested collection underwent white-to-opaque switching and that switching in none of these strains was complemented by the addition of a functional copy of EFG1. Together, these results suggest that while half of the a/α strains that switch do so as a result of mutations in both EFG1 alleles, the other half that switch do so due to mutations in genes other than EFG1.
EFG1 and white-to-gray switching.
Tao et al. (22) and Liang et al. (23) have provided strong evidence that the gray cell phenotype is distinct from the white and opaque cell phenotypes, in morphology, gene expression, and virulence. However, we demonstrated (21) that switching to gray cells could be induced in vitro at 25°C, but not at 37°C, and at the single cell level, that individual tiny elongate, gray cells could be induced to morph phenotypically to the larger opaque, pimpled cell morphology prior to cell division. In doing so, they pass smoothly through a morphology that is intermediate between the tiny elongate morphology and the final opaque morphology (21). In support of the contention that gray does represent a unique phenotype (22, 23), we found that the four EFG1/EFG1 clinical a/α isolates, the one EFG1/efg1 clinical a/α isolate, and the one efg1/efg1 clinical a/α isolate that switched to opaque but could not be complemented for white-opaque switching by site-specific integration of scEFG1 could also not be induced to form gray cells, suggesting that there are genes other than EFG1 that, when mutated, derepress opaque cell formation, but not gray cell formation.
EFG1, switching, and hypha formation.
Opaque cells share several characteristics with hyphae, most notably an elongate shape, an enlarged vacuole, surface antigens (4, 6), a number of regulatory genes (10, 35, 36), and downregulation of the white-specific gene (WH11) (37). There are, however, a number of differences, including budding patterns (4), wall ultrastructure, most notably the formation of opaque cell wall pimples (7), the release of extracellular membrane-bound vesicles (7), and the expression of opaque-specific genes, like OP4 (12, 13, 38). Here, we have presented evidence that a majority of a/α EFG1/EFG1 clinical isolates that can be induced to form hyphae cannot be induced to switch, and a majority of the efg1/efg1 clinical isolates that cannot be induced to form hyphae, can be induced to switch. However, there are exceptions to this yin-yang pattern, indicating the existence of additional repressor genes, other than EFG1, specific for switching and additional activator genes, other than EFG1, specific for hypha formation.
Gastrointestinal colonization.
Earlier studies demonstrated that EFG1 deletion mutants outcompete wild-type EFG1/EFG1 cells in competition experiments in a mouse GI model for colonization (24, 30, 31). Liang et al. (23) recently reported that in such competition experiments, the colonizing efg1/efg1 cells express almost exclusively the gray phenotype. Liang et al. (23) employed a plating assay on CHROMagar, which contains glucose as the sugar source and a proprietary chromogenic mix (32). We performed similar competition experiments, also using a plating assay to assess the phenotype of colonizing cells, employing GlcNAc rather than glucose as the sugar source. In both studies, the temperature (22°C and 25°C, respectively) was far below the physiological temperature. We previously demonstrated (21) that this agar at 25°C in air stabilized the opaque phenotype of a/α cells induced in vitro. Here, we found that regardless of the initial phenotype of the efg1Δ/efg1Δ cells (white, gray, or opaque) in combination with EFG1/EFG1 white cells, the efg1Δ/efg1Δ cells rapidly dominated colonization in the mouse GI colonization model. Also, in the case of the combination of efg1/scEFG1 white cells and efg1/efg1 gray cells, the efg1/efg1 cells dominated. These results were consistent with those of Liang et al. (23). However, Liang et al. (23) found almost exclusively gray colony formation by the colonizing cells, whereas we found that by 15 days postingestion, a third of the colonizing cells were opaque in combinations 1, 2, and 3, and more than three quarters were opaque in combination 4. This difference between the two studies, combined with our earlier observation (21) that the gray phenotype is not expressed at physiological temperature in vitro (21), led us to question the accuracy of both the plating procedure of Liang et al. (23) and that of our study in assessing the phenotypes of the cells in feces. We considered the possibility that both assay procedures, which include 5 days of incubation on an agar substrate in air at temperatures well below the physiological temperature may influence phenotype. We therefore directly examined microscopically the phenotype(s) of live cells in fresh feces. We found that more than 90% of the cells in feces exhibited the opaque cell morphology and more than 70% expressed mCherry under the regulation of an opaque-specific promoter. Our results suggest, therefore, that plating experiments may not accurately reflect the phenotype of cells colonizing the GI tract, and that opaque, not gray, may be the dominant phenotype at the site of colonization. The results of Pande et al. (31) are also pertinent to our finding. They found that overexpressing WOR1 (WOR1OE) in a/α cells causes a competitive advantage over wild-type a/α cells in the mouse GI colonization model and that the colonizing WOR1OE cells formed opaque-like cells without wall pimples that were unstable on glucose-based agar at 25°C (31). These results support the general conclusion that a/α strains that can switch, regardless of the switching repressor or a switching activator gene that is mutated, outcompete cells that cannot switch in the mouse GI colonization model, and add weight to the conclusion that an opaque or opaque-like phenotype is expressed at the site of GI colonization.
Final comments.
Our results indicate that, as previously reported, a third or more of clinical isolates, which are mainly a/α, can undergo the white-to-opaque transition. Half of these strains switch due to mutations in the repressor EFG1, while half switch due to mutations in other repressor genes. Since the efg1/efg1 a/α strains outcompete wild-type EFG1 strains in the mouse GI model, they appear to have an advantage in GI colonization, and this advantage appears to be accompanied by expression of the opaque phenotype. On the other hand, the efg1/efg1 strains may lose invasiveness since they do not appear to form hyphae. If true, one might consider the possibility that opaque efg1/efg1 cells may serve to colonize the GI tract as a commensal but not function as an opportunistic pathogen. However, seven of the eight efg1/efg1 isolates that can switch from white to opaque and that cannot be induced to form hyphae were isolated from the bloodstream, suggesting invasiveness and opportunism. This apparent contradiction warrants further investigation.
MATERIALS AND METHODS
Strains and media.
The C. albicans strains used in this study are described in Table 1 and Table S1 in the supplemental material. The strains were maintained at room temperature on agar containing YPD medium (1% yeast extract, 2% peptone, 2% glucose). Escherichia coli strain XL1-Blue (Agilent Technologies, TX, USA), used to generate and maintain plasmids, was grown in LB medium (1% tryptone, 0.5% yeast extract, 1% NaCl) plus 100 μg/ml ampicillin.
Plasmids and construction of C. albicans strains.
The plasmid pEFG1C (21), which contains a wild-type copy of EFG1 obtained from strain SC5314 (scEFG1), was used to test for complementation by site-specific integration at the EFG1 locus. A pmCherry-HygB plasmid (21) was employed to create strains harboring the mCherry gene under the regulation of the opaque-specific OP4 promoter. For selection of cells transformed with the gene insertion cassettes from the plasmids above, YPD agar containing 200 μg/ml of nourseothricin or 1 mg/ml of hygromycin B was used, depending on the selection marker. The C. albicans SAT1 (CaSAT1) gene was used to confer resistance to nourseothricin, and the CaHygB gene was used to confer resistance to hygromycin B. Integration into proper loci was verified by PCR. The genotypes of the constructed strains are provided in Table S1.
Sequence analysis.
To sequence EFG1 in the 27 clinical isolates, we prepared genomic DNA from strains grown in 5 ml of a YPD suspension culture as described previously (39). Genomic DNA was used as a template for PCR amplification with the primer pairs EFG1-5´F/-3´R (Table S5). Phusion high-fidelity DNA polymerase (New England Biolabs) was employed to amplify the genes by PCR. The amplified DNA fragments of EFG1 were sequenced by Sanger sequencing with the primers EFG1-seq1 through EFG1-seq5 (Table S5).
Primers used in this study. Download Table S5, DOCX file, 0.02 MB (16KB, docx) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Dendrogram for EFG1 relatedness.
Polymorphisms along the EFG1 sequences were translated into allelic frequencies, with 1.0 for the presence of a homozygous polymorphism, 0.5 for a heterozygous polymorphism, and 0.0 for the absence of a polymorphism. Allelic frequencies were used to generate Nei’s pairwise genetic distances (40) with the GENDIST program of the PHYLIP package, version 3.695 (http://evolution.genetics.washington.edu/phylip.html). The unrooted dendrogram (see Fig. S1 in the supplemental material) was generated by the neighbor-joining method (26) implemented in the NEIGHBOR program of the PHYLIP package.
Switching assay.
Switching was tested on agar containing 1.25% glucose (Gluc-agar) or 2.00% GlcNAc (GlcNAc-agar), with the agar containing supplemented Lee’s medium (sLee’s) (25, 41) and 5 μg/liter of phloxine B, which stains opaque colonies or opaque sectors pink to red (6). Eight sets of environmental conditions were tested, which included all combinatorial permutations of sugar source (1.25% glucose versus 2% GlcNAc), temperature (25°C versus 37°C), and atmosphere (air [0.04% CO2] versus 5% CO2). Cells from YPD suspension cultures were grown overnight (∼20 h) at 25°C and then plated on Gluc-agar or GlcNAc-agar plates. Colony phenotypes were analyzed after 5 days of incubation. Homogeneous opaque colonies containing exclusively opaque cells and white colonies with opaque sectors with the latter containing exclusively opaque cells were counted as opaque. Representative colonies were analyzed for cellular phenotypes as described in a previous study (21). In all assays, more than 10 colonies were assessed microscopically for white, opaque, and gray cells. Switching experiments were repeated at least three times, and in some cases, the data were pooled. A total of at least 500 colonies were analyzed for each strain under each condition.
Hypha formation assays.
Hypha formation was assessed in two ways, in suspension cultures containing Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS) in 24-well plates and incubated at 37°C in 5% CO2 for 24 h or by plating on nonnutrient agar containing 10% serum and incubating at 37°C in 5% CO2 for 3 days.
MTL genotypes of a/α opaque cells.
Cells from at least six individual opaque colonies of each strain of the clinical a/α collection were genotyped for the MTL configuration by PCR with the two primer pairs, MTLa1F/R and MTLα2F/R (Table S5).
Immunolocalization of the opaque-specific pimple marker.
Rabbit-derived polyclonal antipimple antiserum against an opaque pimple antigen (7) was used to visualize the formation of opaque-specific pimples by immunocytochemistry. Opaque cells were heat killed in a 65°C water bath for 1 h, pelleted, and resuspended in phosphate-buffered solution (PBS) supplemented with 10% normal goat serum to block nonspecific binding. A 1:50 dilution of rabbit serum was preabsorbed five times with heat-killed homozygous MTL white cells to remove antibodies to surface antigens common to white and opaque cells (7). After staining with the primary antiserum, cells were washed with PBS and treated with Alexa Fluor 488-tagged goat anti-rabbit secondary antibody (Jackson ImmunoResearch, West Grove, PA). Fluorescent images were captured using a Leica TCS SP8 confocal microscope, and all images were similarly processed with Image J software.
Imaging colonies and cells.
Colonies grown on agar plates were imaged through a stereo microscope equipped with a Nikon E990 digital camera. Differential interference contrast (DIC) and fluorescence microscopic images of cells expressing mCherry were obtained with a Canon Rebel T3i digital camera attached to a Nikon-PE2000 inverted microscope through a 60× plan water immersion objective. To image cells directly in fecal samples, fecal pellets were mixed in distilled H2O at a dilution of 1:10. Cells were imaged by phase-contrast and fluorescence microscopy.
Mouse gastrointestinal tract colonization.
All procedures complied with regulatory guidelines defined by the Iowa University IACUC committee. C57BL/6J (Jackson Laboratory) female mice 6 to 7 weeks old were treated with 1 mg/ml of penicillin G and 2 mg/ml of streptomycin in their drinking water for 4 days. Mice were orally inoculated with 0.2 ml (3 × 107 cells) of a cell combination (Fig. 5A) in PBS. Six mice were tested for each cell mixture. To assess the levels of colonization, competition, and phenotype switching, fresh fecal samples were collected postingestion at time intervals. Each fecal sample was homogenized in 0.2 ml of water, and dilutions of the fecal samples were plated on YPD agar plates, YPD+nourseothricin agar (YPD+clonNAT), YPD+hygromycin B agar plates (YPD+HygB), and GlcNAc-agar plates (Fig. 5B). All agar media contained 50 μg/ml of chloramphenicol. Cultures were incubated at 25°C in air for 3 to 6 days, and colony phenotypes were assessed (Fig. 5B). GlcNAc-agar plates were used to determine phenotypic switching to opaque, since this agar medium maintains the cellular phenotype of efg1Δ/efg1Δ and efg1/efg1 mutants and does not induce switching to opaque or gray when incubation is performed at 25°C in air (21). The data from six mice were pooled for each combination of cells at each time point.
ACKNOWLEDGMENTS
We thank Julian R. Naglik for strain 529L.
These studies were supported by the Developmental Studies Hybridoma Bank (DSHB), a National Resource created by NIH and housed in the Biology Department at The University of Iowa.
REFERENCES
- 1.Odds FC. 1988. Candida and candidosis: a review and bibliography, 2nd ed Baillière Tindall, London, United Kingdom. [Google Scholar]
- 2.Cutler JE. 1991. Putative virulence factors of Candida albicans. Annu Rev Microbiol 45:187–218. doi: 10.1146/annurev.mi.45.100191.001155. [DOI] [PubMed] [Google Scholar]
- 3.Soll DR. 1992. High-frequency switching in Candida albicans. Clin Microbiol Rev 5:183–203. doi: 10.1128/cmr.5.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Slutsky B, Staebell M, Anderson J, Risen L, Pfaller M, Soll DR. 1987. White-opaque transition“: a second high-frequency switching system in Candida albicans. J Bacteriol 169:189–197. doi: 10.1128/jb.169.1.189-197.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soll DR, Langtimm CJ, McDowell J, Hicks J, Galask R. 1987. High-frequency switching in Candida strains isolated from vaginitis patients. J Clin Microbiol 25:1611–1622. doi: 10.1128/JCM.25.9.1611-1622.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson JM, Soll DR. 1987. Unique phenotype of opaque cells in the white-opaque transition of Candida albicans. J Bacteriol 169:5579–5588. doi: 10.1128/jb.169.12.5579-5588.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson J, Mihalik R, Soll DR. 1990. Ultrastructure and antigenicity of the unique cell wall pimple of the Candida opaque phenotype. J Bacteriol 172:224–235. doi: 10.1128/jb.172.1.224-235.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White TC, Miyasaki SH, Agabian N. 1993. Three distinct secreted aspartyl proteinases in Candida albicans. J Bacteriol 175:6126–6133. doi: 10.1128/jb.175.19.6126-6133.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Srikantha T, Soll DR. 1993. A white-specific gene in the white-opaque switching system of Candida albicans. Gene 131:53–60. doi: 10.1016/0378-1119(93)90668-s. [DOI] [PubMed] [Google Scholar]
- 10.Sonneborn A, Tebarth B, Ernst JF. 1999. Control of white-opaque phenotypic switching in Candida albicans by the Efg1p morphogenetic regulator. Infect Immun 67:4655–4660. doi: 10.1128/IAI.67.9.4655-4660.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morrow B, Srikantha T, Soll DR. 1992. Transcription of the gene for a pepsinogen, PEP1, is regulated by white-opaque switching in Candida albicans. Mol Cell Biol 12:2997–3005. doi: 10.1128/mcb.12.7.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morrow B, Srikantha T, Anderson J, Soll DR. 1993. Coordinate regulation of two opaque-phase-specific genes during white-opaque switching in Candida albicans. Infect Immun 61:1823–1828. doi: 10.1128/IAI.61.5.1823-1828.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lan CY, Newport G, Murillo LA, Jones T, Scherer S, Davis RW, Agabian N. 2002. Metabolic specialization associated with phenotypic switching in Candida albicans. Proc Natl Acad Sci U S A 99:14907–14912. doi: 10.1073/pnas.232566499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hube B, Monod M, Schofield DA, Brown AJ, Gow NA. 1994. Expression of seven members of the gene family encoding secretory aspartyl proteinases in Candida albicans. Mol Microbiol 14:87–99. doi: 10.1111/j.1365-2958.1994.tb01269.x. [DOI] [PubMed] [Google Scholar]
- 15.Kvaal C, Lachke SA, Srikantha T, Daniels K, McCoy J, Soll DR. 1999. Misexpression of the opaque-phase-specific gene PEP1 (SAP1) in the white phase of Candida albicans confers increased virulence in a mouse model of cutaneous infection. Infect Immun 67:6652–6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kvaal CA, Srikantha T, Soll DR. 1997. Misexpression of the white-phase-specific gene WH11 in the opaque phase of Candida albicans affects switching and virulence. Infect Immun 65:4468–4475. doi: 10.1128/IAI.65.11.4468-4475.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller MG, Johnson AD. 2002. White-opaque switching in Candida albicans is controlled by mating-type locus homeodomain proteins and allows efficient mating. Cell 110:293–302. doi: 10.1016/s0092-8674(02)00837-1. [DOI] [PubMed] [Google Scholar]
- 18.Odds FC, Bougnoux M-E, Shaw DJ, Bain JM, Davidson AD, Diogo D, Jacobsen MD, Lecomte M, Li S-Y, Tavanti A, Maiden MCJ, Gow NAR, d’Enfert C. 2007. Molecular phylogenetics of Candida albicans. Eukaryot Cell 6:1041–1052. doi: 10.1128/EC.00041-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lockhart SR, Pujol C, Daniels KJ, Miller MG, Johnson AD, Pfaller MA, Soll DR. 2002. In Candida albicans, white-opaque switchers are homozygous for mating type. Genetics 162:737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie J, Tao L, Nobile CJ, Tong Y, Guan G, Sun Y, Cao C, Hernday AD, Johnson AD, Zhang L, Bai FY, Huang G. 2013. White-opaque switching in natural MTLa/alpha isolates of Candida albicans: evolutionary implications for roles in host adaptation, pathogenesis, and sex. PLoS Biol 11:e1001525. doi: 10.1371/journal.pbio.1001525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park YN, Conway K, Conway TP, Daniels KJ, Soll DR. 2019. Roles of the transcription factors Sfl2 and Efg1 in white-opaque switching in a/alpha strains of Candida albicans. mSphere 4:e00703-18. doi: 10.1128/mSphere.00703-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tao L, Du H, Guan G, Dai Y, Nobile CJ, Liang W, Cao C, Zhang Q, Zhong J, Huang G. 2014. Discovery of a “white-gray-opaque” tristable phenotypic switching system in Candida albicans: roles of non-genetic diversity in host adaptation. PLoS Biol 12:e1001830. doi: 10.1371/journal.pbio.1001830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang SH, Anderson MZ, Hirakawa MP, Wang JM, Frazer C, Alaalm LM, Thomson GJ, Ene IV, Bennett RJ. 2019. Hemizygosity enables a mutational transition governing fungal virulence and commensalism. Cell Host Microbe 25:418–431.e6. doi: 10.1016/j.chom.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pierce JV, Kumamoto CA. 2012. Variation in Candida albicans EFG1 expression enables host-dependent changes in colonizing fungal populations. mBio 3:e00117-12. doi: 10.1128/mBio.00117-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bedell GW, Soll DR. 1979. Effects of low concentrations of zinc on the growth and dimorphism of Candida albicans: evidence for zinc-resistant and -sensitive pathways for mycelium formation. Infect Immun 26:348–354. doi: 10.1128/IAI.26.1.348-354.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saitou N, Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 27.Srikantha T, Tsai LK, Daniels K, Soll DR. 2000. EFG1 null mutants of Candida albicans switch but cannot express the complete phenotype of white-phase budding cells. J Bacteriol 182:1580–1591. doi: 10.1128/jb.182.6.1580-1591.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stoldt VR, Sonneborn A, Leuker CE, Ernst JF. 1997. Efg1p, an essential regulator of morphogenesis of the human pathogen Candida albicans, is a member of a conserved class of bHLH proteins regulating morphogenetic processes in fungi. EMBO J 16:1982–1991. doi: 10.1093/emboj/16.8.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown AJ, Gow NA. 1999. Regulatory networks controlling Candida albicans morphogenesis. Trends Microbiol 7:333–338. doi: 10.1016/s0966-842x(99)01556-5. [DOI] [PubMed] [Google Scholar]
- 30.Tyc KM, Herwald SE, Hogan JA, Pierce JV, Klipp E, Kumamoto CA. 2016. The game theory of Candida albicans colonization dynamics reveals host status-responsive gene expression. BMC Syst Biol 10:20. doi: 10.1186/s12918-016-0268-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pande K, Chen C, Noble SM. 2013. Passage through the mammalian gut triggers a phenotypic switch that promotes Candida albicans commensalism. Nat Genet 45:1088–1091. doi: 10.1038/ng.2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Odds FC, Bernaerts R. 1994. CHROMagar Candida, a new differential isolation medium for presumptive identification of clinically important Candida species. J Clin Microbiol 32:1923–1929. doi: 10.1128/JCM.32.8.1923-1929.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lockhart SR, Nguyen M, Srikantha T, Soll DR. 1998. A MADS box protein consensus binding site is necessary and sufficient for activation of the opaque-phase-specific gene OP4 of Candida albicans. J Bacteriol 180:6607–6616. doi: 10.1128/JB.180.24.6607-6616.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pierce JV, Dignard D, Whiteway M, Kumamoto CA. 2013. Normal adaptation of Candida albicans to the murine gastrointestinal tract requires Efg1p-dependent regulation of metabolic and host defense genes. Eukaryot Cell 12:37–49. doi: 10.1128/EC.00236-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noble SM, Gianetti BA, Witchley JN. 2017. Candida albicans cell-type switching and functional plasticity in the mammalian host. Nat Rev Microbiol 15:96–108. doi: 10.1038/nrmicro.2016.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu H. 2002. Co-regulation of pathogenesis with dimorphism and phenotypic switching in Candida albicans, a commensal and a pathogen. Int J Med Microbiol 292:299–311. doi: 10.1078/1438-4221-00215. [DOI] [PubMed] [Google Scholar]
- 37.Srikantha T, Tsai LK, Soll DR. 1997. The WH11 gene of Candida albicans is regulated in two distinct developmental programs through the same transcription activation sequences. J Bacteriol 179:3837–3844. doi: 10.1128/jb.179.12.3837-3844.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tuch BB, Mitrovich QM, Homann OR, Hernday AD, Monighetti CK, De La Vega FM, Johnson AD. 2010. The transcriptomes of two heritable cell types illuminate the circuit governing their differentiation. PLoS Genet 6:e1001070. doi: 10.1371/journal.pgen.1001070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park YN, Morschhauser J. 2005. Tetracycline-inducible gene expression and gene deletion in Candida albicans. Eukaryot Cell 4:1328–1342. doi: 10.1128/EC.4.8.1328-1342.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nei M. 1972. Genetic distance between populations. Am Nat 106:283–292. doi: 10.1086/282771. [DOI] [Google Scholar]
- 41.Lee KL, Buckley HR, Campbell CC. 1975. An amino acid liquid synthetic medium for the development of mycelial and yeast forms of Candida albicans. Sabouraudia 13:148–153. doi: 10.1080/00362177585190271. [DOI] [PubMed] [Google Scholar]
- 42.Pujol C, Pfaller M, Soll DR. 2002. Ca3 fingerprinting of Candida albicans bloodstream isolates from the United States, Canada, South America, and Europe reveals a European clade. J Clin Microbiol 40:2729–2740. doi: 10.1128/jcm.40.8.2729-2740.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pujol C, Joly S, Lockhart SR, Noel S, Tibayrenc M, Soll DR. 1997. Parity among the randomly amplified polymorphic DNA method, multilocus enzyme electrophoresis, and Southern blot hybridization with the moderately repetitive DNA probe Ca3 for fingerprinting Candida albicans. J Clin Microbiol 35:2348–2358. doi: 10.1128/JCM.35.9.2348-2358.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Strains used in this study. Download Table S1, DOCX file, 0.02 MB (25.5KB, docx) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Ca3/MLST clade assignments for the 27 isolates used in this study. Ca3 fingerprinting was performed in the Soll lab. The MLST analysis and clade assignment of 10 strains were from collaboration (M.-E. Bougnoux, C. Pujol, D. Diogo, C. Bouchier, et al., Fungal Genet Biol 45:221–231, 2008, https://doi.org/10.1016/j.fgb.2007.10.008) or from different groups (F. C. Odds, M.-E. Bougnoux, D. J. Shaw, J. M. Bain, et al., Eukaryot Cell 6:1041–1052, 2007, https://doi.org/10.1128/EC.00041-07; M. P. Hirakawa, D. A. Martinez, S. Sakthikumar, M. Z. Anderson, et al., Genome Res 5:413–425, 2015, https://doi.org/10.1101/gr.174623.114). MLST typing of isolates P75063wh, P75063dk, P80021wh, P80021dk, 1298wh, 1298dk, P37037wh, and P37037dk was performed in our lab according to the consensus MLST scheme described by Bougnoux et al. (M.-E. Bougnoux, A. Tavanti, C. Bouchier, N. A. R. Gow, et al., J Clin Microbiol 41:5265–5266, 2003, https://doi.org/10.1128/JCM.41.11.5265-5266.2003) which is commonly used worldwide. The MLST clade assignment for these isolates was performed by generating a dendrogram, including MLST data retrieved from the Candida albicans MLST website (https://pubmlst.org/calbicans/) for 24 control isolates and strain 529L, which does not appear to have been previously assigned to a clade. The control isolates were selected, three per clade, from the seven most frequent clades (MLST clades 1, 2, 3, 4, 8, 9, and 11) found among a collection of 1,391 isolates (F. C. Odds, M.-E. Bougnoux, D. J. Shaw, J. M. Bain, et al., Eukaryot Cell 6:1041–1052, 2007, https://doi.org/10.1128/EC.00041-07). Three isolates from clade 18, a major clade in Asia (J. E. Shin, M.-E. Bougnoux, C. d’Enfert, S. H. Kim, et al., J Clin Microbiol 49:2572–2577, 2011, https://doi.org/10.1128/JCM.02153-10), were also used as controls. 529L did not cluster with any other strain. The Ca3 fingerprinting clades I, II, III, SA, and E have been repeatedly shown to correspond nearly perfectly to MLST clades 1, 2, 3, 4, and 11, respectively (A. Tavanti, A. D. Davidson, M. J. Fordyce, N. A. R. Gow, et al., J Clin Microbiol 43:5601–5613, 2005, https://doi.org/10.1128/JCM.43.11.5601-5613.2005; M.-E. Bougnoux, C. Pujol, D. Diogo, C. Bouchier, et al., Fungal Genet Biol 45:221–231, 2008, https://doi.org/10.1016/j.fgb.2007.10.008; F. C. Odds, M.-E. Bougnoux, D. J. Shaw, J. M. Bain, et al., Eukaryot Cell 6:1041–1052, 2007, https://doi.org/10.1128/EC.00041-07; B. A. McManus and D. C. Coleman, Infect Genet Evol 21:166–178, 2014, https://doi.org/10.1016/j.meegid.2013.11.008). It should be noted that clade assignments are no longer displayed on the C. albicans MLST website. Download Table S2, DOCX file, 0.01 MB (13.7KB, docx) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
A dendrogram of EFG1 relatedness based on EFG1 polymorphisms. Clade assignments are presented immediately to the right, based on RFLPs probed with the mid-repeat sequence CA3 and, in parentheses, MLST when available. See Table S2 for explanations about the Ca3 and MLST analysis. The EFG1 genotypes of the clinical isolates are presented to the far right. The efg1− alleles in the isolates are shown in red. Download FIG S1, PDF file, 0.2 MB (172.8KB, pdf) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Switching frequency of the four EFG1/EFG1 clinical isolates that switched to opaque and their EFG1/scEFG1 derivatives generated by site-specific integration at one of the EFG1 alleles. Switching was assessed on GlcNAc-agar under the four sets of environmental conditions. The switching data represent the means ± standard deviations for three independently performed experiments. Switching includes data for homogenous opaque colonies and opaque sectored colonies in which the sectors contained only opaque cells. These strains did not form gray cells under any of the conditions tested. Download Table S3, DOCX file, 0.02 MB (19KB, docx) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Switching frequency of white to opaque for the efg1Δ/efg1Δ mutants, efg1−/efg1− natural strains, and their wild type and efg1−/scEFG1 complemented derivatives generated by site-specific integration of one of the EFG1 alleles. The switching was assessed on GlcNAc-agar under the four sets of environmental conditions. The switching data represent the means ± standard deviations for three independently performed experiments. These data include only switching from white to opaque (homogenous opaque colonies and colonies with homogenous opaque sectors); they do not include switching from white to gray. Download Table S4, DOCX file, 0.02 MB (23.3KB, docx) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Primers used in this study. Download Table S5, DOCX file, 0.02 MB (16KB, docx) .
Copyright © 2020 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.