

Abstract
Congenital fibrosis of the extraocular muscles (CFEOM) is one of the congenital cranial dysinnervation disorders (CCDDs). This review discusses the characteristics of the CFEOM phenotypes and the CCDDs, the fibrosis associated with these disorders and the processes, and genes involved in the embryological development of cranial neuromuscular units. In particular, it focuses on the genetics of neural crest identity, axon guidance, and axon construction in relation to the CFEOMs and some consideration of treatment strategies.
Subject terms: Neurological disorders, Eye diseases
摘要
先天性眼外肌纤维化 (CFEOM) 是一种先天性颅神经功能障碍 (CCDDs) 。本文综述了CFEOM与CCDDs的临床表型的特点, 纤维化与与这两种疾病的相关性以及在颅神经肌肉单元的胚胎发育中的相关过程及与其相关的基因。我们特别着重描述了与CFEOMs相关的神经嵴识别的基因、轴突引导和轴突构建及其针对性的治疗策略。
Introduction
Congenital fibrosis of the extra-ocular muscles (CFEOM) has had a number of names over the years including congenital ophthalmoplegia, generalised fibrosis, ophthalmoplegia imperfecta, and abiotrophic ophthalmoplegia externa. This is testimony to the fact that the underlying cause was unknown. Similar to Duane syndrome, it was assumed to be a developmental abnormality of the extra-ocular muscles for well over a century. It was only relatively recently that it was found to be a developmental abnormality of the oculomotor nucleus and nerve (Cranial Nerve III) [1]. It is one of a group of disorders now known as Congenital Cranial Dysinnervation Disorders (CCDD) which all involve the abnormal development of cranial nerves and/or their nuclei [2]. CFEOM is rare with a minimum prevalence of 1/250,000 [3].
Characteristics of CFEOM
Phenotypically, CFEOM presents in three forms
CFEOM 1 is dominantly inherited and fully penetrant and typically presents with relatively symmetrical bilateral ptosis with severe restriction of ocular motility with the eyes most commonly fixed in a depressed and exotropic position. Patients tend to adopt a chin-up abnormal head posture because of the tight inferior recti and bilateral ptosis (Fig. 1). It is not generally associated with systemic abnormalities.
Fig. 1.

A father and daughter with CFEOM type 1, both with ptosis and restriction of elevation of the eyes and a chin-up abnormal head posture
CFEOM 2 is rare and recessively inherited. It is characterised by bilateral IIIrd and IVth nerve paresis, often the pupils are small and sluggishly reactive and occasionally is associated with retinal dystrophy.
CFEOM 3 is also dominantly inherited but with variable penetrance and expressivity. Individual family members may present very differently. The ptosis and oculomotility restrictions may be unilateral or asymmetrical. CFEOM 3 is often associated with other neurological developmental abnormalities and intellectual or social disability. Another rare form, recessively inherited Tukel syndrome, is associated with limb developmental abnormalities [4].
The other CCDDs
Many of the other cranial nerves may develop abnormally and it seemed sensible to group these conditions into a collective assembly [2]. The CCDDs share a number of features. They are congenital, nonprogressive developmental abnormalities of one or more cranial nerve nuclei. They may be associated with other developmental abnormalities, either neurological or systemic, including developmental limb abnormalities.
The cranial nerve abnormality is a primary innervation failure with an absence of normal innervation of the affected muscles where neurons fail to develop or are misguided. There may be a secondary aberrant innervation of the affected muscles by neurons of one of the other cranial nerves. As a consequence of reduced innervation, the affected muscles become atrophic and the unopposed action of other muscles results in fibrosis causing severe restriction of movement. The group includes congenital facial diplegia (CN VII), Moebius syndrome (CN VII and VI and associated horizontal gaze palsy), Duane syndrome (CN VI), congenital IVth nerve paresis and some cases of Brown syndrome (superior oblique tendon sheath syndrome) [5], Monocular elevation deficiency (superior division CN III), CFEOM (CNIII and occasionally CNIV) Marcus–Gunn jaw wink ptosis (CN III and CN V). Another related condition is horizontal gaze palsy and progressive scoliosis, a failure of crossing internuclear neuron development [6].
Fibrosis associated with the CCDDs
Little is known about the exact mechanism of fibrosis in these conditions. The initial innervation failure is early in the embryological time-frame (maybe as early as 9 weeks gestation). Foetal eye movements have been identified by ultrasound at 14 weeks of gestation [7], although these movements are rudimentary. More sustained lateral movements of the eyes are seen by 23 weeks gestation and Rapid Eye Movements at 24 weeks [8]. This means that there must be complex agonist/antagonist relationships developing between the extraocular muscles ontogenetically early in the second trimester. Interrupting the innervation at this early stage is going to cause unique changes in muscle development.
Histological investigations in CFEOM have shown significant fibrous tissue in the restricted muscles (usually the inferior rectus and lateral rectus). Harley et al. [9] performed histology on fourteen patients with dominantly inherited CFEOM. They found that, in most of the restricted muscles biopsied, muscle tissue was replaced with fibrous tissue and collagen, confirmed with electron microscopy. Many of the other muscles were replaced with fibrous tissue as well, with few myofibrils present. There was an accumulation of glycogen and lipid. They noticed fibrous bands restricting eye movement at surgery.
Histological examination of the superior rectus muscles shows them to be thin and membranous. Engle et al. [1] were unable to find the superior rectus at all in one patient and a biopsy from another patient showed that the superior rectus was composed of fat, connective tissue, and a few myofibres. Fat seems to be a common component of the atrophic muscles. An increase in myocyte nuclei has been reported [10, 11] but the significance of this remains unclear.
Although the weak muscles are described as “atrophic” this may not be an appropriate term as this assumes that they were once normal in size and have diminished. It is more likely that the muscles do not develop appropriately because they have not been innervated appropriately. The antagonist muscles, being unopposed, also develop abnormally from an early stage. These muscles are unlikely to have normal innervation as there are changes in all divisions of the nerve [1] but asymmetrically with the superior division being more affected.
Some of the mechanisms involved in muscle atrophy and fibrosis after denervation have been studied by transecting nerves in various animal studies. Rebelledo [12] transected the sciatic nerve in mice. They showed that there was an accumulation of extracellular matrix (ECM) proteins such as collagen and fibronectin replacing functional myofibres. There was an increase in levels of the pro-fibrotic factors transforming growth factor β (TGFβ) and connective tissue growth factor (CTGF-CCN2) suggesting that both were important in the fibrotic response to denervation. Extraocular muscle atrophy has been studied by transecting cranial nerves in primates and looking at the subsequent changes in the extraocular muscles. Demer et al. [13] transected the trochlear nerve in five juvenile macaque monkeys. This resulted in a 40–50% reduction in cross-sectional area of the muscle. Giant muscle fibres developed in one animal but not the others. Although some of these processes may be involved in the fibrosis and accumulation of collagen in CFEOM, these models study atrophy of developed muscles after denervation and so are not ideal models for a condition that is characterised by the embryological failure of innervation and the consequent failure of extraocular muscle development.
Processes involved in developing cranial neuromotor units
All of the CCDDs are caused by a failure somewhere in the complex developmental processes involved in creating cranial neuromotor units. A number of genetic abnormalities have been identified which have helped to elucidate some of these processes. However, the genetic and molecular basis for many of the CCDDs remains obscure.
Neural crest identity
Neural crest cells are a transient population of cells that migrate during CNS development as the precursors of a wide range of derivative cells. There is a complex interplay between signals secreted by embryonic signalling centres and transcription genes which transfer this pre-programming to the transcriptional apparatus [14]. These transcription factors are specific for embryological time period, anatomical regional area, and cell type. They control the development of specific neuronal cell identity in complex combinations and sequences [15]. During hindbrain development HOX (homeobox) transcription genes play a particularly important role in determining the segmentation of the hindbrain into rhombomeres (segments from which the various cranial nerves arise). Each specific region has a “HOX-code” depending on the expression of various members of the HOX gene family. It is unsurprising, therefore, that HOX gene mutations have been associated with CCDDs (HOX A1 mutations cause a syndromic form of Duane syndrome [16], HOXB1 mutations cause congenital bilateral facial paresis [17]). Other important transcription factors associated with hindbrain differentiation are from the PHOX family (paired-like homeodomain protein). PHOX 2a and PHOX 2b are both essential for the development of the rostral cranial nerve sites. If both copies of Phox2a are knocked out in mice there is failure to develop both the IIIrd and IVth cranial nerve nuclei [18]. PHOX 2a mutations in humans cause CFEOM type 2 which is characterised by dysinnervation of the IIIrd and IVth cranial nerve [19].
Axon guidance and construction
Once the cranial nuclei have been determined, axons have to make their way to their respective extraocular muscles. This requires guidance of the developing axons and construction processes to elongate the axons.
There is a complex arrangement of chemotactic signals both attracting and repelling axons to ensure they are guided to the appropriate place. A number of gene mutations have been identified in this process which cause aberrant guidance and result in CCDDs. The protein α2-chimaerin is integral to the orchestration of guidance signals for both the IIIrd and the VIth cranial nerve development by a largely inhibitory influence on various semiphorins [20]. α2-chimaerin is encoded by the CHN1 gene and mutations in this gene cause migration failure of the VIth cranial nerve and are responsible for some cases of syndromic Duane Syndrome [21].
CFEOM 1 and 3 are phenotypic categories but more recent identification of some of the genetic aberrations associated with CFEOMs has shown that there is considerable overlap between the two conditions.
A majority of cases of CFEOM 1, the dominantly inherited condition which is usually not associated with other neurological conditions, are caused by mutations in KIF 21A [22]. KIF 21A encodes a kinesin motor protein that is involved in transportation along the growing axon and patients with KIF21 A CFEOM show MRI evidence of significant oculomotor nerve hypoplasia and some evidence of abducens nerve developmental abnormality [23]. Studies of mice models of CFEOM 1 have shown that the mechanism by which Kif21A mutations cause oculomotor developmental abnormality are by interrupting the association of the kinesin motor protein with the microtubules along which it is travelling. Almost all of the different mutations result in an up-regulation of Kif21A inhibitory influence on microtubule associated protein 1b (Map 1b), preventing it disengaging from the microtubule and restricting movement [24].
Microtubules are a principle component of axon structural development. Abnormalities of the tubulins from which microtubules are constructed are associated with a number of neurological conditions. Elisabeth Engle and her co-workers have systematically investigated all forms of CFEOM and the genetic abnormalities underlying the conditions. They discovered that many families with CFEOM 3 had mutations in a gene (TUBB 3) that encodes a tubulin protein that is specific to neuron development (β-tubulin isotope III) [25]. Other brain abnormalities in the affected families include dysgenesis of the corpus callosum, anterior commissure, and corticospinal tract. They have subsequently shown quite specific genotype–phenotype correlations, with some families having cortical abnormalities but no ocular signs, and some predominantly the ocular phenotype [26] which would phenotypically be categorised as CFEOM type 1. Mutations in another tubulin gene TUBB 2B cause CFEOM 3 with a specific brain abnormality, polymicrogyria [27].
Treatment strategies
The ptosis and severe oculomotor restrictions require the adoption of a chin-up abnormal head posture (AHP) that, in the long term, can cause significant neck problems (Fig. 1). Binocularity is rare and so amblyopia management is required from an early age. Getting glasses to fit effectively is technically difficult because of the AHP. Refractive errors can, however, be effectively managed with contact lenses.
Developing an area of binocular vision is not a realistic objective and the main indication for surgical intervention is correction of the AHP. This requires addressing the ptosis and the extraocular muscle restriction. Levator function is usually minimal and correction of the ptosis requires brow suspension. Although Bell’s phenomenon is limited because of the upgaze restriction, eyelid closure is often unaffected. The risk of corneal exposure can be reduced by performing simultaneous brow suspension and inferior rectus recession. It is wise to use a temporary material such as prolene to perform the brow suspension in the first instance to ensure that it can be tolerated, later converting to a more permanent solution such as autologous fascia lata. Children are more tolerant of brow suspension, one factor favouring early intervention.
Extraocular muscle surgery is challenging, and the achievable outcomes are limited. There may be horizontal and vertical mis-alignment, usually exotropia and hypotropia. The fundamental problem is that there may be virtually no superior rectus or medial rectus function and so relieving the restrictive element (by inferior rectus and/or lateral rectus recession) only allows the eyes to achieve a fixed position close to the midline at best. The innervation abnormality affects the whole orbit and even when the tight muscles are detached from the eye, forced duction tests may be restricted. At surgery, fibrous bands may be seen between the muscles and the surrounding connective tissues of the orbit (Fig. 2).
Fig. 2.
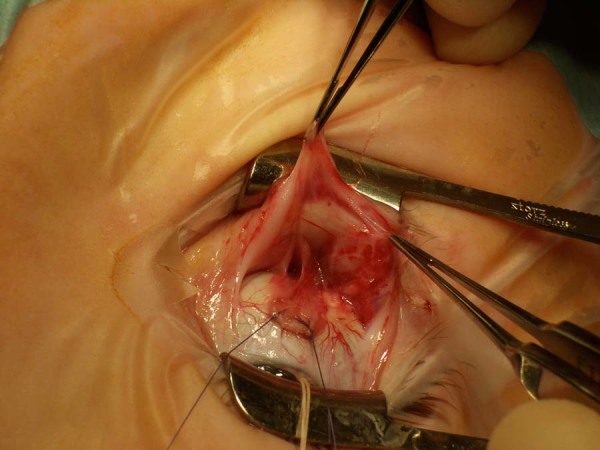
A disinserted inferior rectus muscle with fibrotic bands between the muscle and connective tissues of the orbit
Conclusion
Constructing a cranial neuromuscular unit involves complex cascades of gene interactions early in the embryological process. Failure at various points in the pathway results in dysinnervation phenotypes characterised by extraocular muscle paresis and fibrosis. The exact mechanism of fibrosis remains unclear. It may simply be secondary to shortening of the muscles caused by unopposed action from early in pregnancy, but it seems more likely that disordered innervation has widespread effects on the development of many of the orbital tissues.
Compliance with ethical standards
Conflict of interest
The author declares no conflicts of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Engle EC, Goumnerov BC, McKeown CA, Schatz M, Johns DR, Porter JD, et al. Oculomotor nerve and muscle abnormalities in congenital fibrosis of the extraocular muscles. Ann Neurol. 1997;41:314–25. doi: 10.1002/ana.410410306. [DOI] [PubMed] [Google Scholar]
- 2.Gutowski NJ, Bosley TM, Engle EC. 110th ENMC International Workshop: the congenital cranial dysinnervation disorders (CCDDs). Naarden, The Netherlands, 25–27 October, 2002. Vol. 13, Neuromuscular disorders: NMD. England; 2003. p. 573–8. [DOI] [PubMed]
- 3.Reck AC, Manners R, Hatchwell E. Phenotypic heterogeneity may occur in congenital fibrosis of the extraocular muscles. Br J Ophthalmol. 1998;82:676–9. doi: 10.1136/bjo.82.6.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tukel T, Uzumcu A, Gezer A, Kayserili H, Apak MY, Uyguner O, et al. A new syndrome, congenital extraocular muscle fibrosis with ulnar hand anomalies, maps to chromosome 21qter. J Med Genet. 2005;42:408–15. doi: 10.1136/jmg.2004.026138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaeser P-F, Brodsky MC. Fourth cranial nerve palsy and Brown syndrome: two interrelated congenital cranial dysinnervation disorders? Curr Neurol Neurosci Rep. 2013;13:352. doi: 10.1007/s11910-013-0352-5. [DOI] [PubMed] [Google Scholar]
- 6.Jen JC, Chan W-M, Bosley TM, Wan J, Carr JR, Rüb U, et al. Mutations in a human ROBO gene disrupt hindbrain axon pathway crossing and morphogenesis. Science. 2004;304:1509–13. doi: 10.1126/science.1096437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horimoto N, Hepper PG, Shahidullah S, Koyanagi T. Fetal eye movements. Ultrasound Obstet Gynecol. 1993;3:362–9. doi: 10.1046/j.1469-0705.1993.03050362.x. [DOI] [PubMed] [Google Scholar]
- 8.Okawa H, Morokuma S, Maehara K, Arata A, Ohmura Y, Horinouchi T, et al. Eye movement activity in normal human fetuses between 24 and 39 weeks of gestation. PLoS ONE. 2017;12:e0178722. doi: 10.1371/journal.pone.0178722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harley RD, Rodrigues MM, Crawford JS. Congenital fibrosis of the extraocular muscles. Trans Am Ophthalmol Soc. 1978;LXXVI:197–226. [PMC free article] [PubMed]
- 10.Engle EC, Goumnerov BC, McKeown C, Schatz M, Johns DR, Porter J, et al. Ocularmotor nerve and muscle abnormalities in congenital fibrosis of the extraocular muscles. Ann Neurol. 1997;41:314–25. doi: 10.1002/ana.410410306. [DOI] [PubMed] [Google Scholar]
- 11.Chen H, Liu T, Zeng Z, Wang Y, Lin Y, Cheng L, et al. Clinical characteristics of a KIF21A mutation in a Chinese family with congenital fibrosis of the extraocular muscles type 1. Medicine. 2017;96:e8068. doi: 10.1097/MD.0000000000008068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rebolledo DL, González D, Faundez-Contreras J, Contreras O, Vio CP, Murphy-Ullrich JE, et al. Denervation-induced skeletal muscle fibrosis is mediated by CTGF/CCN2 independently of TGF-β. Matrix Biol. 2019;82:20–37. doi: 10.1016/j.matbio.2019.01.002. [DOI] [PubMed] [Google Scholar]
- 13.Demer JL, Poukens V, Ying H, Shan X, Tian J, Zee DS. Effects of intracranial trochlear neurectomy on the structure of the primate superior oblique muscle. Investig Ophthalmol Vis Sci. 2010;51:3485–95. doi: 10.1167/iovs.09-5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parker HJ, Pushel I, Krumlauf R. Coupling the roles of Hox genes to regulatory networks patterning cranial neural crest. Dev Biol. 2018;444:S67–78. doi: 10.1016/j.ydbio.2018.03.016. [DOI] [PubMed] [Google Scholar]
- 15.Pattyn A, Hirsch M, Goridis C, Brunet JF. Control of hindbrain motor neuron differentiation by the homeobox gene Phox2b. Development. 2000;127:1349 LP–1358. doi: 10.1242/dev.127.7.1349. [DOI] [PubMed] [Google Scholar]
- 16.Bosley TM, Alorainy IA, Salih MA, Aldhalaan HM, Abu-Amero KK, Oystreck DT, et al. The clinical spectrum of homozygous HOXA1 mutations. Am J Med Genet A. 2008;146A:1235–40. doi: 10.1002/ajmg.a.32262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vogel M, Velleuer E, Schmidt-Jimenez LF, Mayatepek E, Borkhardt A, Alawi M, et al. Homozygous HOXB1 loss-of-function mutation in a large family with hereditary congenital facial paresis. Am J Med Genet A. 2016;170:1813–9. doi: 10.1002/ajmg.a.37682. [DOI] [PubMed] [Google Scholar]
- 18.Pattyn A, Morin X, Cremer H, Goridis C, Brunet JF. Expression and interactions of the two closely related homeobox genes Phox2a and Phox2b during neurogenesis. Development. 1997;124:4065 LP–4075. doi: 10.1242/dev.124.20.4065. [DOI] [PubMed] [Google Scholar]
- 19.Nakano M, Yamada K, Fain J, Sener EC, Selleck CJ, Awad AH, et al. Homozygous mutations in ARIX(PHOX2A) result in congenital fibrosis of the extraocular muscles type 2. Nat Genet. 2001;29:315–20. doi: 10.1038/ng744. [DOI] [PubMed] [Google Scholar]
- 20.Ferrario JE, Baskaran P, Clark C, Hendry A, Lerner O, Hintze M, et al. Axon guidance in the developing ocular motor system and Duane retraction syndrome depends on Semaphorin signaling via alpha2-chimaerin. Proc Natl Acad Sci USA. 2012;109:14669–74. doi: 10.1073/pnas.1116481109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyake N, Chilton J, Psatha M, Cheng L, Andrews C, Chan W-M, et al. Human CHN1 mutations hyperactivate alpha2-chimaerin and cause Duane’s retraction syndrome. Science. 2008;321:839–43. doi: 10.1126/science.1156121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamada K, Andrews C, Chan W-M, McKeown CA, Magli A, de Berardinis T, et al. Heterozygous mutations of the kinesin KIF21A in congenital fibrosis of the extraocular muscles type 1 (CFEOM1) Nat Genet. 2003;35:318–21. doi: 10.1038/ng1261. [DOI] [PubMed] [Google Scholar]
- 23.Demer JL, Clark RA, Engle EC. Magnetic resonance imaging evidence for widespread orbital dysinnervation in congenital fibrosis of extraocular muscles due to mutations in KIF21A. Investig Ophthalmol Vis Sci. 2005;46:530–9. doi: 10.1167/iovs.04-1125. [DOI] [PubMed] [Google Scholar]
- 24.Cheng L, Desai J, Miranda CJ, Duncan JS, Qiu W, Nugent AA, et al. Human CFEOM1 mutations attenuate KIF21A autoinhibition and cause oculomotor axon stalling. Neuron. 2014;82:334–49. doi: 10.1016/j.neuron.2014.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tischfield MA, Baris HN, Wu C, Rudolph G, Van Maldergem L, He W, et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell. 2010;140:74–87. doi: 10.1016/j.cell.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whitman MC, Engle EC. Ocular congenital cranial dysinnervation disorders (CCDDs): insights into axon growth and guidance. Hum Mol Genet. 2017;26(R1):R37–44. doi: 10.1093/hmg/ddx168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cederquist GY, Luchniak A, Tischfield MA, Peeva M, Song Y, Menezes MP, et al. An inherited TUBB2B mutation alters a kinesin-binding site and causes polymicrogyria, CFEOM and axon dysinnervation. Hum Mol Genet. 2012;21:5484–99. doi: 10.1093/hmg/dds393. [DOI] [PMC free article] [PubMed] [Google Scholar]