

Abstract
Endophytic microorganisms absorb nutrients and prevent pathogen damage, supporting healthy plant growth. However, the relationship between endophytic bacteria and berberine synthesis in the medicinal plant Coptis teeta Wall. remains unclear. Herein, we explored the community composition of endophytic bacteria related to berberine in roots, stems, and leaves of wild-type and cultivated C. teeta. Endophytic bacterial communities were analyzed by 16S rRNA sequencing, and berberine content in roots was analyzed by high-performance liquid chromatography. Proteobacteria, Actinobacteria, and Bacteroidetes were the major phyla, and Mycobacterium, Salmonella, Nocardioides, Burkholderia-Paraburkholderia, and Rhizobium were the dominant genera in root, stem, and leaf tissues. Root berberine content was positively correlated with total N, total P, total K, and available K in rhizosphere soil. In addition, root berberine content was positively correlated with Microbacterium and norank_f_7B-8, whereas soil total K was positively correlated with Microbacterium and Burkholderia-Paraburkholderia in roots. Our results demonstrated a clear correlation between dominant endophytic bacteria and berberine synthesis in C. teeta. The findings are useful for the promotion of berberine production in C. teeta via manipulation of endophytic bacteria.
Electronic supplementary material
The online version of this article (10.1007/s13205-020-2084-y) contains supplementary material, which is available to authorized users.
Keywords: Coptis teeta Wall., Berberine, Endophytic bacteria, 16S rDNA sequencing, Metabonomics
Introduction
The genus Coptis belongs to the family Ranunculaceae. In Chinese herbal medicine, Coptidis rhizome refers to the dried rhizome of Coptis chinensis Franch., C. deltoidea CY Cheng et Hsiao, and C. teeta Wall. (Committee 2015). Coptidis rhizome is often used for clearing heat, eliminating dampness, and detoxification (Wang et al. 2019). It can also be used to treat vomiting, diarrhea, jaundice, high fever, fainting, heartburn, upset stomach, toothache, and various other ailments (Moon et al. 2017; Goswami and Gogoi 2019). In addition to use in traditional Chinese medicine, Coptidis rhizome is a source of compounds used in proprietary Chinese pharmaceutical products such as Huanglian 9 and Yinglian tablet. Although statistics are incomplete, there are numerous pharmaceutical preparations containing Coptidis rhizome as a raw ingredient. Botanical and pharmacological studies have shown that Coptidis rhizome contains a variety of alkaloids such as palmatine and the more famous berberine (Committee, 2015; Moon et al. 2017; Goswami et al. 2019). The root of C. teeta contains 6–7% berberine, while the stem contains 2–3% and the leaf contains 1–1.97% (Committee, 2015; Wang et al. 2019).
Coptis teeta is a shade-tolerant plant that grows at high altitudes, in cold mountainous and rainy regions such as the Gaoligong Mountain, northwestern Yunnan, China. As long ago as the Ming Dynasty, Lan Mao recorded in the Diannan Materia Medica that Coptidis rhizome grown in Yunnan, namely C. teeta, was more efficacious than that grown in Sichuan, China. Indeed, C. teeta is widely considered to produce the best quality Coptidis rhizome. Due to slow growth, it takes 6–7 years after sowing before products can be harvested. The slow growth is often affected by a variety of diseases and pests, such as anthracnose (Tan et al. 2016). The yield of C. teeta is very low, because the rhizome is small and it produces unusual ‘breeding branches’ that consume substantial nutrients (Cui et al. 2017). Furthermore, due to long-term over-harvesting and continuous habitat destruction, the wild population is on the verge of extinction, and the species is listed as a second-class endangered plant in China.
As the growth of C. teeta is dependent on various environmental factors including nutrients, moisture, light, and temperature, these must be taken into full consideration when attempting to maximize the yield and quality of medicinal materials (Bi and Zhao 2018). N, P, and K play important roles in plant vegetative growth, indicating that their combined application may be the key to achieving a high yield. Although fertilizer application can increase the growth and development of C. teeta (Zhou 2011), the rhizome quality may decline under cultivated conditions. Artificial cultivation of C. teeta is the only method capable of meeting demand, hence the need for an improved, standardized approach. Understanding the effects of fertilizer application on the rhizosphere soil of medicinal plants is vital for improving the yield and quality of medicinal materials (Veach et al. 2019).
In recent years, plant microbiomes have received much attention from researchers (Kwak et al. 2018; Rodriguez et al. 2019), with knowledge gradually being converted into field applications (Beckers et al. 2017; Cordovez et al. 2019). Leaf endophytes, stem endophytes, and rhizosphere soil microorganisms form a planet-wide ecosystem that plays a crucial role in many processes on a global scale (Zhu et al. 2018). A better understanding of the relationships between plant microbiomes, crop growth and development, and soil environment could help us to improve fertilizer use efficiency (Bulgarelli et al. 2012). In addition, microorganisms are closely related to plant health; some beneficial bacteria such as Bacillus and Pseudomonas can reduce the incidence of scab (Cernava et al. 2019). Relevant knowledge could allow us to reduce the use of chemical fertilizers and pesticides whilst improving the yield and quality of plant products (Muller et al. 2016; Lu et al. 2018). With the development of molecular biological technology, it is possible to study interactions between microbial communities and functional components or stress resistance in plants (Yang et al. 2017). However, previous studies have mainly focused on food crops such as rice and wheat (Cernava et al. 2019; Jiang et al. 2019; Zhang et al. 2019), whereas less research is available on the relationships between the soil environment, endophytic bacteria, and medicinal components of medicinal plants (Cicatelli et al. 2019; Wolfgang et al. 2019).
The present work expands on recent research on C. teeta by investigating the endophytic bacterial community composition in the roots, stems, and leaves of wild-type (WT) and cultivated plants. We hypothesized that berberine synthesis in C. chinensis is related to the endophytic bacterial community shaped by soil environmental factors. WT and cultivated C. teeta samples were collected from the country of origin in Yunnan, China, and endophytic bacterial communities of root, stem, and leaf tissues were analyzed by 16S rRNA sequencing. Rhizosphere soil samples were collected to determine the content of total P (TP), total N (TN), total K (TK), and available K (AK). In addition, berberine content in roots was analyzed by high-performance liquid chromatography (HPLC).
Materials and methods
Sample collection
A total of 10 C. teeta plants (5 WT and 5 cultivated, aged 5 years) were collected from Tengchong (98° 49′ E, 25° 03′ N), Yunnan, China. The surface of plant samples was cleaned with sterile water, disinfected with 95% ethanol, and washed again with sterile water. Root, stem, and leaf tissues were separated and stored at − 80 °C until used. Rhizosphere soil samples were collected at the same time as plants and thoroughly air-dried before testing. A flowchart of the experimental process is presented in Fig. 1.
Fig. 1.

Flowchart of experimental processes
16S rRNA gene sequencing of endophytic bacteria
DNA extraction and PCR amplification
Extraction of total genomic DNA from plant samples was performed using E.Z.N.A.® soil DNA Kit (Omega Bio-tek, Norcross, GA, USA) according to the manufacturer’s protocols. The final DNA concentration and purity were determined using a NanoDrop 2000 UV–Vis spectrophotometer (Thermo Scientific, Wilmington, DE, USA), and DNA quality was checked by 1% agarose gel electrophoresis. The V3-V4 hypervariable regions of the bacteria 16S rRNA gene were amplified with barcoded primers 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) (Cregger et al. 2018).
PCR was performed on an ABI GeneAmp 9700 thermocycler (Applied Biosystems, Foster City, CA, USA). All reactions were performed in triplicate 20-μL mixture containing 4 μL of 5 × FastPfu Buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.4 μL of FastPfu Polymerase, and 10 ng of template DNA. The PCR program was as follows: 3 min of denaturation at 95 °C, 27 cycles of 30 s at 95 °C, 30 s for annealing at 55 °C, and 45 s for elongation at 72 °C, and a final extension at 72 °C for 10 min. The resulted PCR products were extracted from a 2% agarose gel, followed by purification using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) and quantification using QuantiFluor™-ST (Promega, Fitchburg, WI, USA) according to the manufacturers’ protocol.
Illumina Miseq library construction and sequencing
The Illumina adaptor sequence was added to the outer end of the target region by PCR. Then PCR products were recovered using a gel extraction kit, eluted with Tris–HCl buffer, analyzed by 2% agarose electrophoresis, and hydrolyzed with sodium hydroxide. A single-stranded DNA fragment was purified using the TruSeq DNA Sample Prep Kit (Illumina, San Diego, CA, USA). Purified amplicons were pooled in equimolar and paired-end sequenced (2 × 300 bp) on an Illumina Miseq® platform (Illumina, San Diego, CA, USA) according to the standard protocols by Majorbio Bio-pharm Technology Co., Ltd. (Shanghai, China).
Data processing and bioinformatics analysis
Raw sequencing data were analyzed using the free online Majorbio I-Sanger Cloud Platform (https://www.i-sanger.com/). Raw fastq files were quality-filtered by Trimmomatic and merged by FLASH with the following criteria: (1) The reads were truncated at any site receiving an average quality score < 20 over a 50 bp sliding window. (2) Sequences with overlap > 10 bp were merged according to their overlap with mismatch ≤ 2 bp. (3) Sequences of each sample were separated according to barcodes (exactly matching) and primers (allowing 2 nucleotide mismatching), and reads containing ambiguous bases were removed.
Sequences were clustered into operational taxonomic unit (OTU) at 97% similarity using UPARSE v7.1 (https://drive5.com/uparse/) with a novel ‘greedy’ algorithm that performs chimera filtering and OTU clustering simultaneously. Taxonomic classification was performed by RDP Classifier algorithm (https://rdp.cme.msu.edu/) against the Silva (SSU123) 16S rRNA database using confidence threshold of 70% (Sonnenburg et al. 2016). Sequencing depth analysis was performed on the OTU dataset. Bacterial alpha-diversity was analyzed using different indices that reflect the richness and diversity of microbial communities. The Chao index reflects community richness, and the Shannon diversity and evenness indices indicate community diversity (Caporaso et al. 2010).
Two taxonomic levels (OTU and genus) were selected to calculate the beta-diversity of endophytic bacterial communities. Principal component analysis (PCoA) plots of 30 samples at the OTU level were generated based on the unweighted UniFrac distance matrix. Hierarchical clustering (Hcluster) was performed to reveal the distances of sample branches, and a hierarchical clustering tree was generated based on the unweighted pair group method with arithmetic mean (UPGMA). In addition, an analysis of similarities (ANOSIM) was performed using the Bray–Curtis algorithm to calculate differences between pairs of samples, and to test whether differences between pairs of groups were significantly greater than intra-group differences.
Determination of soil nutrients
N analysis
Soil TN content was determined using the Kjeldahl method (Rukun 2000). In a sealed diffuser, soil samples were hydrolyzed with a 1.8 M NaOH solution. The nitrogen converted to an ammonia adduct at constant temperature (40 °C) was continuously diffused and absorbed by boric acid (H3BO3). The content of soil hydrolysable nitrogen was determined by titration with standard hydrochloric acid.
P analysis
Soil TP content was determined by spectrophotometry (Rukun 2000). Briefly, 1 g of soil sample was digested with 5 mL of H2SO4 solution. The digested solution was brought to a constant volume of 50 mL with water after filtration or clarification. A 10.00-mL aliquot (phosphorus 0.05–0.75 mg) was transferred into a 50-mL volumetric flask, followed by the addition of two drops of dinitrophenol. The indicator was neutralized to a yellow color by dropwise addition of 6 M NaOH, and 10.00 mL of ammonium vanadium molybdate reagent was added to react with phosphorus. The solution volume was adjusted to a constant volume with water. After 15 min, the absorbance of the sample was measured at a wavelength of 440 mm. The instrument was calibrated with a blank digestion solution prepared as described above. A standard curve was established and its linear regression equation was obtained. The P concentration in solution was estimated using the regression equation.
K analysis
Soil TK content was determined by flame photometry (Rukun 2000). Briefly, 5 g of soil sample was digested with 100 mL of NaHCO3 solution. The digested solution (5.00–10.00 mL) in a 50-mL volumetric flask was diluted to the required volume using water and directly analyzed for K by flame photometry at a wavelength of 700 nm. A linear regression equation for the standard curve was established and used to estimate the K concentration in solution. Soil AK was extracted using a 1 M NH4OAc solution (Rukun 2000). The K concentration in solution was directly determined by flame photometry.
Berberine analysis by HPLC
Instruments and reagents
HPLC analysis was performed on an Agilent 1260 HPLC system (Agilent Technologies, Santa Clara, CA, USA), with a diode-array detector coupled to an Agilent Zorbax SB C18 column (250 × 4.6 mm, 5 µm internal diameter). An appropriate amount of berberine reference standard (purity ≥ 99%; Puxin, Beijing, China) was added to methanol to prepare a reference test solution containing 20% per mL.
Sample preparation
Root samples were completely air dried and prepared into a powder, then passed through a No. 4 sieve. Approximately 0.l g of powder was added to 50 mL of methanol, sealed, weighed, and sonicated (power = 250 W, frequency = 40 kHz) for 1 h. After cooling, the mixture was weighed again, and lost weight was supplemented with methanol. The mixture was shaken and filtered, and the filtrate was used to prepare test solutions.
Chromatographic analysis
For chromatography, octadecylsilane-bonded silica gel (Agilent SB C18 column) served as a filler. The mobile phase consisted of acetonitrile and 0.22 M potassium dihydrogen phosphate solution (25:75, v/v). The detection wavelength was 265 nm and the sample size was 10 µL. Berberine content was determined from the HPLC chromatograms.
Statistical analysis
All data are expressed as mean ± standard deviation. Significant differences between groups were determined by t tests. Canonical correspondence analysis (CCA) was used to explore the relationship between berberine content in roots, major nutrients in rhizosphere soil, and root endophytes at the genus level. Spearman correlation was used to assess the relationship between selected bacteria and soil environment. p values less than 0.05 was considered to indicate statistical significance.
Results
Quality metrics of sequencing analysis
The average number of sequences cross all 30 samples was 903,568, the average number of nucleotides was 356,700,253 bp, and the average read length was 394 bp (Supplementary file 1). The sequences were classified into 887 OTUs. In the WT plants, 54, 40, and 9 unique QTUs were identified in roots, stems, and leaves, respectively. In the artificial cultivar, 230, 59, and 35 unique QTUs were detected in roots, stems, and leaves, respectively. A total of 406 common OTUs were found in the six groups (Fig. 2). The total number of OTUs was the highest in cultivated root samples (1413) and lowest in WT leaf samples (816). Generally, cultivated samples yielded a larger number of OTUs than the corresponding WT samples, and the number of OTUs in various plant tissues was ordered roots > stems > leaves (Supplementary file 2). The quality metrics of sequencing analysis indicated uniform, high-quality sequencing data suitable for subsequent analysis.
Fig. 2.

Venn diagram of bacterial operational taxonomic units (OTUs) identified in different parts of wild-type and cultivated C. teeta plants. WL, WS, and WR represent roots, stems, and leaves of wild-type plants, respectively. AL, AS, and AR represent roots, stems, and leaves of artificial cultivar, respectively. Different colors represent various groupings, overlapping parts represent OTUs shared among multiple groups, non-overlapping parts represent OTUs unique to groupings, and numbers indicate the number of corresponding OTUs
Alpha-diversity endophytic bacteria
Alpha-diversity analysis (Fig. 3) revealed that the Chao index of C. teeta root endophytes was largest (p < 0.01) and the value was greater in cultivated samples than WT samples. Regarding the Shannon index, the value of stem endophytes for WT plants was largest, while the value of root endophytes in the artificial cultivar was largest. In addition, the Shannon index value of root endophytes was greater in the artificial cultivar than WT plants (p < 0.01). According to the Shannon evenness index, the value of stem endophytes was largest for WT plants, while the value of root endophytes was largest for the artificial cultivar. The Shannon evenness index value of root endophytes in the cultivar was greater than that of WT plants (p < 0.01).
Fig. 3.

Alpha-diversity estimates of bacterial communities in different parts of wild-type (WT) and cultivated C. teeta plants. a OTU richness estimates (Chao index). b OTU diversity estimates (Shannon diversity index). c OTU evenness estimates (Shannon evenness index). WL, WS, and WR represent roots, stems, and leaves of wild-type plants, respectively. AL, AS, and AR represent roots, stems, and leaves of artificial cultivar, respectively. The chart shows significant differences between groups (*p < 0.05, **p < 0.01, and ***p < 0.001). The abscissa is the group name and the ordinate is the exponential average of index value for each group
Beta-diversity of endophytic bacteria
PCoA analysis based on the unweighted UniFrac distances (Fig. 4a) showed that endophytic bacteria in different parts of WT and cultivated plants varied considerably. PC1 explained 27.72% of total variation at the OTU level, and PC2 accounted for 13.46%. Bacteria in roots, stems, and leaves could be clearly distinguished. Based on sample-level clustering, samples could be divided into six distinct groups according to OTUs (Fig. 4b), illustrating differences between WT and artificial cultivars. ANOSIM results at different phylogenetic levels (OTU and genus) also revealed significant compositional differences in endophytic bacterial communities between various parts of WT and cultivated plants (Table 1). The difference was most pronounced between the leaves (or roots) and stems of WT plants.
Fig. 4.
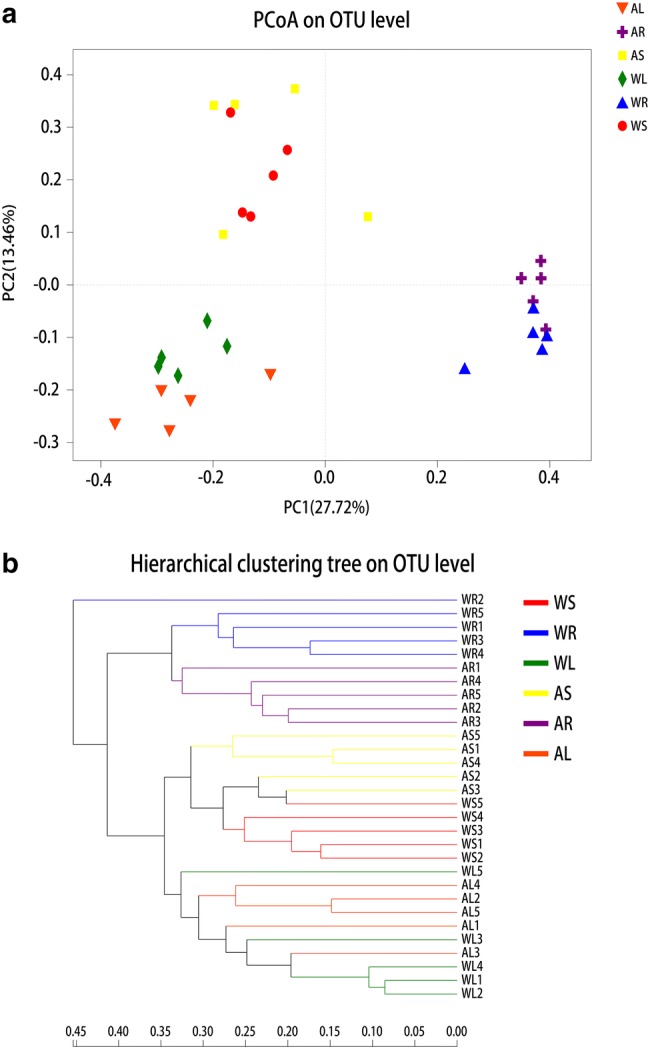
Beta-diversity estimates of bacterial communities in different parts of wild-type (WT) and cultivated C. teeta plants. a Principal component analysis at the OTU level. WL, WS, and WR represent roots, stems, and leaves of wild-type plants, respectively. AL, AS, and AR represent roots, stems, and leaves of artificial cultivar, respectively. Horizontal and vertical coordinates represent two selected principal coordinate components, and the percentage represents the contribution of the principal coordinate component to community composition differences. Different colored points and shapes represent samples from various groups, and the closer two sample points are, the more similar the composition of the two sample species. b Hierarchical clustering tree at the OTU level. The length of branches represents the distance between samples, and different groups are shown in different colors
Table 1.
Analysis of similarities (ANOSIM) of endophytic bacterial community composition using the Bray–Curtis algorithm
| Taxonomic level | OTU | Genus | ||
|---|---|---|---|---|
| ANOSIM output | R | P | R | P |
| WL vs AL | 0.396 | 0.024 | 0.42 | 0.024 |
| WS vs AS | 0.468 | 0.032 | 0.368 | 0.037 |
| WR vs AR | 0.58 | 0.012 | 0.524 | 0.01 |
| WR vs WL | 0.924 | 0.011 | 0.98 | 0.014 |
| WL vs WS | 0.648 | 0.009 | 0.624 | 0.008 |
| WR vs WS | 0.812 | 0.005 | 0.76 | 0.007 |
| AR vs AL | 0.992 | 0.011 | 0.996 | 0.007 |
| AR vs AS | 0.888 | 0.008 | 0.856 | 0.013 |
| AL vs AS | 0.8 | 0.01 | 0.82 | 0.012 |
WL, WS, and WR represent roots, stems, and leaves of wild-type plants, respectively. AL, AS, and AR represent roots, stems, and leaves of artificial cultivar, respectively. The closer the R value is to 1, the greater the intra-group differences; the smaller the R value, the less significant the differences between and within groups
Dominant members of the endophytic bacterial community
At the phylum level (Fig. 5a), Actinobacteria (51.9%), Proteobacteria (43.8%), Firmicutes (2.3%), and Bacteroidetes (1.4%) were identified in stem samples of WT plants. Similar phyla were detected in root and leaf samples of WT plants, despite their proportions in the total community varied. For example, the percentages of Proteobacteria (76.0%) and Bacteroidetes (11.1%) increased, while the percentages of Actinobacteria (11.1%) and Firmicutes (1.2%) deccreased in root samples compared with leaf samples of WT plants. Moreover, Proteobacteria (62.4%), Actinobacteria (32.3%), Bacteroidetes (3.3%), and Firmicutes (0.8%) were identified in stem samples of artificial cultivar. These phyla also occurred in the root and leaf samples of artificial cultivar, accounting for different proportions of the total community. In summary, bacterial phyla making the largest contributions were Proteobacteria, Actinobacteria, and Bacteroidetes, while Firmicutes and Acidobacteria made minor contributors. The relative abundances of Proteobacteria, Actinobacteria, and Bacteroidetes varied widely among the six groups (p values < 0.05; Fig. 5b).
Fig. 5.

Distribution of bacterial OTUs at phylum and genus levels. a Percentage abundance at the phylum level. Different colored columns represent various species, and the length of columns represents the proportion of identified phyla. b The top 10 dominant phyla. The vertical axis represents the phylum name, and the column length corresponding to the phylum represents its average relative abundance in each sample group, with different colors indicating various groupings. The p-value is shown in the right (*p < 0.05, **p < 0.01, and ***p < 0.001). c Percentage abundance at the genus level. Different colored columns represent various genera, and the length of columns represents the proportion of identified genera. d The top 10 dominant genera. The vertical axis represents the genus name, and the column length corresponding to the genus represents its average relative abundance in each sample group, with different colors indicating various groupings. The p-value is shown on the right (*p < 0.05, **p < 0.01, and ***p < 0.001)
At the genus level (Fig. 5c), Mycobacterium, Salmonella, Nocardioides, Burkholderia-Paraburkholderia, and Rhizobium were the dominant bacteria. In addition, the relative abundances of Mycobacterium, Salmonella, and Nocardioides differed substantially among the six groups (p values < 0.05; Fig. 5d).
Major nutrients in rhizosphere soil and berberine content in roots
Generally, TN, TP, TK, and AK contents were higher in the rhizosphere soil of WT plants compared with that of cultivated plants. In addition, root berberine content was significantly higher in WT plants than cultivated plants (Table 2).
Table 2.
Major nutrient contents in rhizosphere soil and berberine content in roots of wild-type (WT) and cultivated C. teeta plants (mean ± SD; n = 5; mg/kg)
| WR | AR | |
|---|---|---|
| Total N | 355 ± 34.6 | 346 ± 37.4 |
| Total P | 1098 ± 111.7 | 972 ± 266.5 |
| Total K | 7342 ± 1546.7 | 5042 ± 1749.4 |
| Available K | 1196 ± 284.2 | 987 ± 131.6 |
| Berberine | 2,009,987 ± 489,034 | 1,489,243 ± 354,791* |
*p < 0.05
Relationship of berberine synthesis, endophytic bacteria, and soil environment
The CCA results showed that berberine content in roots had a significant positive correlation with TK, TP, TN, and AK contents in rhizosphere soil (Fig. 6a, b). Further, correlation analysis based on 50 genera selected from root samples revealed that root berberine was positively correlated with Microbacterium and norank_f_7B-8 (p < 0.05), whereas soil TK was positively correlated with Microbacterium (p < 0.01). In addition, soil TN and AK were positively correlated with g_Burkholderia-Paraburkholderia (p < 0.05; Fig. 6c). Based on the correlation results and the community composition characteristics of endophytic bacteria in WT and cultivated plants, 10 genera (Bradyrhizobium, Serratia, Microbacterium, Aquincola, Acidibacter, Herbaspirillum, Cupriavidus, Acidothermus, norank_f_7B-8, and Granulicella) were identified as key bacteria related to berberine synthesis in C. teeta (Fig. 6d).
Fig. 6.

Correlations between berberine synthesis, endophytic bacteria, and soil environment. a Canonical correspondence analysis (CCA) at the genus level (1). b CCA at the genus level (2). Different colored dots and shapes represent sample groups, green arrows represent bacterial genera, red arrows represent environmental factors, and the length of environmental factors arrows represents the extent to which these factors affect the genus data. The angle between s environmental factors arrows represents the correlation (sharp angle = positive correlation, obtuse angle = negative correlation, and right angle = no correlation). c Spearman correlation heatmap. The x- and y-axes are environmental factors and bacterial genera, respectively. R values are shown in different colors, p values < 0.05 are labeled with an asterisk (*), and the legend on the right shows color intervals for different R values (*p < 0.05, **p < 0.01, and ***p < 0.001). d The top 10 related genera. The vertical axis represents the genus name, and the column length corresponding to the genus represents its average relative abundance of the genus in each sample group, with different colors indicating various groupings. The p value is shown on the right (*p < 0.05, **p < 0.01, and ***p < 0.001)
Discussion
In this study, we characterized the endophytic bacterial communities in C. teeta to explore their relationship with berberine synthesis. We found that the number of OTUs identified in cultivated and WT plants was always higher in roots than stems and leafs. Soils are among the most abundant microbial ecosystems on earth. Generally, plant tissues closer to soil yield a greater number of bacterial OTUs. In addition, we observed a larger number of OTUs in each tissue of artificial cultivar compared with WT plants. This result indicates that artificially cultivated C. teeta plants could harbor more endophytic bacteria in a relatively stable environment.
According to the results of alpha-diversity indices (Chao, Shannon diversity, and evenness indices), we found that the richness, diversity, and evenness of endophytic bacteria were all greater in cultivated plants than in WT plants. This may be related to the fact that C. teeta is a shade-tolerant plant that grows at high altitudes. It is often found in cold mountainous and rainy areas with harsh environmental conditions, which might reduce the alpha-diversity of endophytic bacteria in WT C. teeta compared with cultivated plants.
Differences in endophytic bacteria may be explained from another aspect. The results of beta-diversity analysis based on PCoA, Hcluster, and ANOSIM revealed significant differences in endophytic bacterial communities between WT and cultivated plants, and among roots, stems, and leaves. The plant root system is the interface between these multicellular eukaryotes and soil (Hu et al. 2018; Sasse et al. 2018). The interleaf and associated microorganisms constitute a large and dynamic ecosystem that is important at a global scale (Flues et al. 2017).
Based on the OTU dataset, we found that the dominant phyla were Proteobacteria, Actinobacteria, and Bacteroidetes, and the most abundant genera were Mycobacterium, Salmonella, Nocardioides, Burkholderia-Paraburkholderia, and Rhizobium. These results revealed details of the endophytic bacterial community composition in the medicinal plant C. teeta. Compositional differences at phylum and genus levels could potentially be used to distinguish between WT and artificial cultivars. In addition, the distinct dominant bacteria may be related to the absorption of nutrients from soil and the biosynthesis of pharmaceutical compounds in the plant.
To explore the influence of soil environment, we determined TP, TN, TK, and AK contents in the rhizosphere soil of C. teeta. All these major nutrients occurred at higher levels in rhizosphere soil samples of WT plants than artificial cultivar. Endophytic bacteria can enhance the adsorption of N, P, K, and other nutrient elements by host plants (Jacoby et al. 2017). N is an indispensable component of all organisms, and it is also necessary for the synthesis of key cellular compounds such as proteins and nucleic acids (Filannino et al. 2018). However, N in the atmosphere is the largest stock of freely available N, and few bacteria can fix this inert gas (Lammel et al. 2018). Soil pH is an important factor determining the diversity and structure of N-fixing bacterial communities (Lammel et al. 2018). Taking into account the lower OTU number and diversity indices for WT plants, we considered that soil nutrient content is not the only environmental factor affecting endophytic bacteria in C. teeta. Furthermore, these environmental conditions may be related to the synthesis of medicinal components.
Berberine is an important medicinal component present in C. teeta (Meng et al. 2018). Herein, we analyzed the berberine content in the roots of WT and cultivated plants by HPLC. We found that berberine content in WT roots were higher than in cultivated samples, indicating a potential decline in the rhizome quality of artificial cultivar. WT plants of C. teeta are well adapted to the natural environment and can freely absorb nutrients from the soil, which may result in a superior synthesis of medicinal components such as berberine. Our results revealed that root berberine content was positively correlated with soil TK, TP, TN, and AK contents in rhizosphere soil, indicating the relevance of these major nutrients to berberine synthesis in C. teeta. In addition, root berberine was positively correlated with Microbacterium and norank_f_7B-8, whereas particular soil nutrients were positively correlated with Microbacterium and g_Burkholderia-Paraburkholderia. These findings indicate that some dominant endophytes are related to berberine synthesis in C. teeta and affected by the soil environment.
Based on the correlation results and the community characteristics of endophytic bacteria in WT and cultivated C. teeta, 10 genera were identified as key members related to berberine synthesis. Some of these genera may be positively linked to berberine synthesis, and others may interfere with berberine synthesis. For example, Bradyrhizobium species are believed to utilize sugars and organic acids, and studies have found that this genus is resistant to antibiotics (Filannino et al. 2018). Serratia are found in soil, water, and on the surfaces of plants, and some members of this genus are conditional pathogens of humans (Konecka et al. 2019). In addition, members of Microbacterium can produce the plant growth hormone indoleacetic acid and solubilize phosphate (Madhaiyan et al. 2010). Microbacterium strain EC8 was found to enhance root and shoot biomass formation in lettuce and tomato (Cordovez et al. 2018), whereas a Microbacterium strain isolated from soil could suppress Rhizoctonia root rot infection and enhance seedling growth in wheat (Barnett et al. 2006). In the present work, Microbacterium was strongly related to berberine synthesis in C. teeta. The higher abundance of this genus in WT C. teeta may explain why WT C. teeta produced more berberine in roots than cultivated plants. However, further verification is needed to explore the function of Microbacterium in berberine synthesis by this medicinal plant.
Herein, we identified endophytic bacteria related to berberine synthesis in the Chinese medicinal plant C. teeta using 16S rDNA sequencing. The bacterial community composition of WT and cultivated plants differed, and endophytes in the roots, stems, and leaves varied. Furthermore, our results demonstrated a clear correlation between dominant endophytic bacteria and berberine synthesis in C. teeta. The findings provide evidence for the potential role of endophytic bacteria in berberine production and make it possible to improve berberine yield of C. teeta through manipulation of endophytic bacteria.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary file 1: Sequence information table (XLS 9 kb)
Supplementary file 2: OUT information table (XLS 126 kb)
Author contribution
LgC, JlY, and SzT participated in study design, searched databases, and helped to draft the manuscript. ThL and XmZ carried out the statistical analysis of data.
Funding
The work was supported by the National Natural Science Foundation of China (Nos: 31600018, 21620248) and Yunnan Basic Applied Research Project (Nos: 2016FD049).
Data availability
The data used to support this study can be made freely available. The 16S rDNA gene sequencing data were analysed using the free online Majorbio I-Sanger Cloud Platform (https://www.i-sanger.com). The sequence data are available at the NIH Sequence Read Archive (https://submit.ncbi.nlm.nih.gov/subs/bioproject/.) under the Bioproject accession number PRJNA556939.
Compliance with ethical standards
Conflict of interest
All authors declared that there are no conflicts of interest.
References
- Barnett SJ, Roget DK, et al. Suppression of Rhizoctonia solani AG-8 induced disease on wheat by the interaction between Pantoea, Exiguobacterium, and Microbacteria. Aust J Soil Res. 2006;44(4):331–342. doi: 10.1071/SR05113. [DOI] [Google Scholar]
- Beckers B, Op DBM, et al. Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees. Microbiome. 2017;5(1):25. doi: 10.1186/s40168-017-0241-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi YQ, Zhao ZL. identification and control measures of anthrax pathogen of Coptis yunhuanglian. Yunnan Agric Sci Technol. 2018;S1:87–88. [Google Scholar]
- Bulgarelli D, Rott M, et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature. 2012;488(7409):91–95. doi: 10.1038/nature11336. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cernava T, Erlacher A, et al. Enterobacteriaceae dominate the core microbiome and contribute to the resistome of arugula (Eruca sativa Mill.) Microbiome. 2019;7(1):13. doi: 10.1186/s40168-019-0624-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicatelli A, Ferrol N, et al. Editorial: effects of plant-microbiome interactions on phyto- and bio-remediation capacity. Front Plant Sci. 2019;10:533. doi: 10.3389/fpls.2019.00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Committee TNP. Pharmacopoeia of the people's Republic of China. Beijing: China Medical Science and Technology Press; 2015. [Google Scholar]
- Cordovez V, Schop S, et al. Priming of plant growth promotion by volatiles of root-associated Microbacterium spp. Appl Environ Microbiol. 2018 doi: 10.1128/AEM.01865-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordovez V, Dini-Andreote F, et al. Ecology and evolution of plant microbiomes. Annu Rev Microbiol. 2019;73:69–88. doi: 10.1146/annurev-micro-090817-062524. [DOI] [PubMed] [Google Scholar]
- Cregger MA, Veach AM, et al. The Populus holobiont: dissecting the effects of plant niches and genotype on the microbiome. Microbiome. 2018;6(1):31. doi: 10.1186/s40168-018-0413-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui R, Liang YL, et al. Effects of application of phosphorus and compound fertilizer on growth, yield and quality of Coptis yunhuanglian. J Yunnan Agric Univ (Nat Sci) 2017;32(04):697–702. [Google Scholar]
- Filannino P, Di Cagno R, et al. Metabolic and functional paths of lactic acid bacteria in plant foods: get out of the labyrinth. Curr Opin Biotechnol. 2018;49:64–72. doi: 10.1016/j.copbio.2017.07.016. [DOI] [PubMed] [Google Scholar]
- Flues S, Bass D, et al. Grazing of leaf-associated Cercomonads (Protists: Rhizaria: Cercozoa) structures bacterial community composition and function. Environ Microbiol. 2017;19(8):3297–3309. doi: 10.1111/1462-2920.13824. [DOI] [PubMed] [Google Scholar]
- Goswami AK, Gogoi N, et al. Development and validation of high-performance thin-layer chromatographic method for quantification of berberine in rhizomes of Coptis teeta Wall, an endangered species collected from Arunachal Pradesh, India. J Chromatogr Sci. 2019;57(5):411–417. doi: 10.1093/chromsci/bmz009. [DOI] [PubMed] [Google Scholar]
- Hu L, Robert C, et al. Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nat Commun. 2018;9(1):2738. doi: 10.1038/s41467-018-05122-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby R, Peukert M, Succurro A, et al. The role of soil microorganisms in plant mineral nutrition-current knowledge and future directions. Front Plant Sci. 2017;8:1617. doi: 10.3389/fpls.2017.01617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Cao S, et al. An expanded subfamily of G-protein-coupled receptor genes in Fusarium graminearum required for wheat infection. Nat Microbiol. 2019;4(9):1582–1591. doi: 10.1038/s41564-019-0468-8. [DOI] [PubMed] [Google Scholar]
- Konecka E, Mokracka J, et al. Evaluation of the pathogenic potential of insecticidal Serratia marcescens strains to humans. Polish J Microbiol. 2019;68(2):185–191. doi: 10.33073/pjm-2019-018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak MJ, Kong HG, et al. Author Correction: Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat Biotechnol. 2018;36(11):1117. doi: 10.1038/nbt1118-1117. [DOI] [PubMed] [Google Scholar]
- Lammel DR, Barth G et al (2018) Direct and indirect effects of a pH gradient bring insights into the mechanisms driving prokaryotic community structures. Microbiome 6(106) [DOI] [PMC free article] [PubMed]
- Lu T, Ke M, et al. Investigation of rhizospheric microbial communities in wheat, barley, and two rice varieties at the seedling stage. J Agric Food Chem. 2018;66(11):2645–2653. doi: 10.1021/acs.jafc.7b06155. [DOI] [PubMed] [Google Scholar]
- Madhaiyan M, Poonguzhali S, et al. Microbacterium Azadirachtae sp. nov., a plant-growth-promoting actinobacterium isolated from the rhizoplane of neem seedlings. Int J Syst Evol Microbiol. 2010;60(Pt 7):1687–1692. doi: 10.1099/ijs.0.015800-0. [DOI] [PubMed] [Google Scholar]
- Meng F, Wu Z et al (2018) Coptidis rhizoma and its main bioactive components: recent advances in chemical investigation, quality evaluation and pharmacological activity. Chin Med 13(13) [DOI] [PMC free article] [PubMed]
- Moon M, Huh E, et al. Coptidis rhizoma prevents heat stress-induced brain damage and cognitive impairment in mice. Nutrients. 2017;9(10):1057–1075. doi: 10.3390/nu9101057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller DB, Vogel C, et al. The plant microbiota: systems-level insights and perspectives. Annu Rev Genet. 2016;50:211–234. doi: 10.1146/annurev-genet-120215-034952. [DOI] [PubMed] [Google Scholar]
- Rodriguez PA, Rothballer M, et al. Systems biology of plant-microbiome interactions. Mol Plant. 2019;12(6):804–821. doi: 10.1016/j.molp.2019.05.006. [DOI] [PubMed] [Google Scholar]
- Rukun L. Chemical analysis method of soil agriculture. Beijing: China Agricultural Press; 2000. [Google Scholar]
- Sasse J, Martinoia E, et al. Feed your friends: do plant exudates shape the root microbiome? Trends Plant Sci. 2018;23(1):25–41. doi: 10.1016/j.tplants.2017.09.003. [DOI] [PubMed] [Google Scholar]
- Sonnenburg ED, Smits SA, et al. Diet-induced extinctions in the gut microbiota compound over generations. Nature. 2016;529(7585):212–215. doi: 10.1038/nature16504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HL, Chan KG, et al. Rhizoma coptidis: a potential cardiovascular protective agent. Front Pharmacol. 2016;7:362. doi: 10.3389/fphar.2016.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veach AM, Morris R, et al. Rhizosphere microbiomes diverge among Populus trichocarpa plant-host genotypes and chemotypes, but it depends on soil origin. Microbiome. 2019;7(1):76. doi: 10.1186/s40168-019-0668-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wang L, et al. Coptidis rhizoma: a comprehensive review of its traditional uses, botany, phytochemistry, pharmacology and toxicology. Pharm Biol. 2019;57(1):193–225. doi: 10.1080/13880209.2019.1577466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfgang A, Taffner J, et al. Novel strategies for soil-borne diseases: exploiting the microbiome and volatile-based mechanisms toward controlling meloidogyne-based disease complexes. Front Microbiol. 2019;10:1296. doi: 10.3389/fmicb.2019.01296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Danzberger J, et al. Dominant groups of potentially active bacteria shared by barley seeds become less abundant in root associated microbiome. Front Plant Sci. 2017;8:1005. doi: 10.3389/fpls.2017.01005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Liu YX, et al. NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat Biotechnol. 2019;37(6):676–684. doi: 10.1038/s41587-019-0104-4. [DOI] [PubMed] [Google Scholar]
- Zhou B (2011) The effect of main mineral elements on the growth and development of Coptis chinensis in the early stage of growth, Huazhong Agricultural University. Master's degree 64
- Zhu H, Li S, et al. Molecular characterization of eukaryotic algal communities in the tropical phyllosphere based on real-time sequencing of the 18S rDNA gene. BMC Plant Biol. 2018;18(1):365. doi: 10.1186/s12870-018-1588-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file 1: Sequence information table (XLS 9 kb)
Supplementary file 2: OUT information table (XLS 126 kb)
Data Availability Statement
The data used to support this study can be made freely available. The 16S rDNA gene sequencing data were analysed using the free online Majorbio I-Sanger Cloud Platform (https://www.i-sanger.com). The sequence data are available at the NIH Sequence Read Archive (https://submit.ncbi.nlm.nih.gov/subs/bioproject/.) under the Bioproject accession number PRJNA556939.