

Abstract
Genome-wide association studies (GWASs) have identified a number of genetic risk loci associated with systemic sclerosis (SSc) and Crohn’s disease (CD), some of which confer susceptibility to both diseases. In order to identify new risk loci shared between these two immune-mediated disorders, we performed a cross-disease meta-analysis including GWAS data from 5,734 SSc patients, 4,588 CD patients and 14,568 controls of European origin. We identified 4 new loci shared between SSc and CD, IL12RB2, IRF1/SLC22A5, STAT3 and an intergenic locus at 6p21.31. Pleiotropic variants within these loci showed opposite allelic effects in the two analysed diseases and all of them showed a significant effect on gene expression. In addition, an enrichment in the IL-12 family and type I interferon signaling pathways was observed among the set of SSc-CD common genetic risk loci. In conclusion, through the first cross-disease meta-analysis of SSc and CD, we identified genetic variants with pleiotropic effects on two clinically distinct immune-mediated disorders. The fact that all these pleiotropic SNPs have opposite allelic effects in SSc and CD reveals the complexity of the molecular mechanisms by which polymorphisms affect diseases.
Subject terms: Inflammatory diseases, Risk factors
Introduction
Systemic sclerosis (SSc) and Crohn’s disease (CD) are complex disorders characterized by a chronic deregulation of the immune response, in which both genetic and environmental factors are implicated in their development1,2. SSc is a chronic connective tissue disease characterized by vascular injury, excessive collagen deposition and autoantibody production1. CD is a chronic autoinflammatory disorder affecting all segments of the gastrointestinal tract, the most common being the terminal ileum and colon2.
Even though both diseases present apparently unrelated phenotypic traits, several lines of evidence support the existence of a shared genetic component between them. First of all, results from large-scale genetic studies performed in each individual disease have shown a genetic overlap between SSc and CD, with several genetic risk loci common to both conditions, such as IRF8, TYK2, STAT4, and GSDMA/IKZF33,4. In this regard, the human leukocyte antigen (HLA) region represents one of the most important shared genetic risk loci across immune-mediated diseases5, being in fact the major risk locus associated with SSc and showing a moderate effect on CD3,4. Additionally, there is an important fibrotic component in both diseases. Even when fibrosis is one of the primaries hallmarks of SSc, mainly involving skin, lungs, and gastrointestinal tract, it also appears in CD and is one of the main reasons that leads to a necessity of surgical intervention in the distal part of the small intestine6,7. In this line, it has been observed an increased risk of idiopathic pulmonary fibrosis (IPF) in individuals affected by inflammatory bowel diseases, especially in CD patients8. Fibrosis of the lungs is one of the most common complications in SSc and, indeed, both IPF and SSc lead to interstitial lung disease (ILD)9. Furthermore, the gastrointestinal tract is the internal organ most frequently involved in SSc pathogenesis, which is affected in nearly all patients, sharing this affection with CD. In most of the cases, this affection involves the upper part in SSc and the distal part in CD. However, small bowel and colorectal involvement affects 40–88% and 20–50% of SSc patients, respectively10,11, being the distal part of small bowel and colorectum the most affected areas in CD2. Thus, these observations suggest that SSc and CD are likely to share common pathogenic mechanisms of disease.
Since the advent of high-throughput genotyping platforms, including genome-wide association studies (GWASs) and the Immunochip approach, more than 15 and 140 genetic risk loci have been identified in SSc and CD, respectively3,4. However, a significant percentage of the total genetic background of both diseases remains unknown. The low prevalence of immune-mediated disorders represents an obstacle to the identification of their genetic component, making it difficult to recruit well-powered cohorts necessary to detect association signals with weak effects. Cross-phenotype meta-analyses of GWAS or Immunochip data have partially overcome this problem. In recent years, several studies have combined genotypic data from different immune-mediated phenotypes to search for shared risk alleles, either combining paired phenotypes12–17 or multiple diseases with common etiology18–20. This strategy has allowed the identification of new susceptibility loci shared among immune-mediated diseases.
Since no studies analysing the genetic overlap between SSc and CD have been performed so far, the aim of the present study was to thoroughly explore this common genetic background by combining GWAS data from both disorders.
Methods
Study population
A series of 5,734 patients diagnosed with SSc, 4,588 CD patients, and 14,568 healthy controls of European origin were enrolled in this study. Figure 1 and Supplementary Table S1 detail the cohorts included in the different stages of the study.
Figure 1.

Schema of the study design.
SSc GWAS dataset
In the discovery phase, we included GWAS data from 2,281 SSc cases and 4,410 healthy controls from Spain, USA, Germany and the Netherlands, all of them included in a previous study21 (see Supplementary Table S1).
CD GWAS dataset
The CD discovery cohort was composed of 1,988 cases and 2,978 healthy controls from the UK, included in the CD GWAS performed by the Welcome Trust Case Control Consortium (WTCCC)22 (see Supplementary Table S1).
Replication cohorts
To confirm the results obtained in the discovery phase, genotyping data of the selected polymorphisms were obtained from GWAS data from 3,453 SSc cases and 3,602 controls, and 2,600 CD cases and 3,578 controls. Specifically, the SSc replication cohort included three independent case/control sets from Spain, USA, and Italy. Regarding the CD cohort, case/control sets were recruited from Spain, USA and Germany, all of them from previously published GWASs23–25.
The control population consisted of unrelated healthy individuals that were recruited in the same geographical regions as patients. Genotyping information of each cohort is included in Supplementary Table S1.
All SSc cases were defined based on the 1980 preliminary and 2013 classification criteria of American College of Rheumatology26,27 or based on the presence of at least 3 out of 5 CREST (calcinosis, Raynaud´s phenomenon, esophageal dysmotility, sclerodactyly, telangiectasias) features typical for SSc. All CD cases were defined based on a confirmed diagnosis of CD using conventional endoscopic, radiological and histopathological criteria28.
Ethics committee approval
Approval from the Comité de Bioética del Consejo Superior de Investigaciones Científicas and the local ethical committees of the different participating centers (University of Texas Health Science Hopkins University Medical Center, Baltimore, USA; Fred Hutchinson Cancer Center-Houston, USA; The Johns Center, Seattle, USA; VU University Medical Center, Amsterdam, The Netherlands; Leiden University Medical Center, Leiden, The Netherlands; Radboud University Nijmegen Medical Centre, Nijmegen, the Netherlands; University Medical Center Utrecht, Utrecht, the Netherlands; Vall d’Hebron Hospital, Barcelona, Spain; 12 de Octubre University Hospital, Madrid, Spain; Santa Creu i Sant Pau University Hospital, Barcelona, Spain; Hospital Marqués de Valdecilla, Santander, Spain; Hospital Clínico Universitario San Cecilio, Granada, Spain; Hospital Virgen de las Nieves, Granada, Spain; Hospital Virgen de la Victoria, Málaga, Spain; Hospital Carlos Haya, Málaga, Spain; Hospital Virgen del Rocío, Sevilla, Spain; Hospital Reina Sofía, Córdoba, Spain; Hospital Clínico San Carlos, Madrid, Spain; Madrid Norte Sanchinarro Hospital, Madrid, Spain; Hospital La Princesa, Madrid, Spain; Hospital Puerta de Hierro Majadahonda, Madrid, Spain; Hospital General Universitario Gregorio Marañón, Madrid, Spain; Hospital Clinic, Barcelona, Spain; Hospital Parc Tauli, Sabadell, Spain; Hospital Del Mar, Barcelona, Spain; Hospital Universitari Mútua Terrasa, Barcelona, Spain; Hospital Universitari de Bellvitge, Barcelona, Spain; Hospital General de Granollers, Granollers, Spain; Hospital General San Jorge, Huesca, Spain; Hospital Central de Asturias, Oviedo, Spain; Hospital Xeral-Complexo Hospitalario Universitario de Vigo, Vigo, Spain; Hospital Universitario Cruces, Barakaldo, Spain; Hospital Virgen del Camino, Pamplona, Spain; Hospital Universitario Miguel Servet, Zaragoza, Spain; Hospital Universitario de Canarias, Tenerife, Spain; Hospital General Universitario de Valencia, Valencia, Spain; Hospital Universitari i Politecnic La Fe, Valencia, Spain; Hospital Universitari Doctor Peset, Valencia, Spain; Hospital Universitario A Coruña, La Coruña, Spain; Hospital Universitario La Paz, Madrid, Spain; Hospital Universitari Germans Trias i Pujol, Badalona, Spain; Hospital General de Alicante, Alicante, Spain; Hospital Clínico Universitario, Zaragoza, Spain; Hospital Clínico Universitario, Santiago de Compostela, Spain; Complejo Hospitalario de León, León, Spain; Hospital de Cabueñes, Gijón, Spain; University Hospital Cologne, Cologne, Germany; Charité University Hospital, Berlin, Germany; University of Erlangen-Nuremberg, Erlangen, Germany; University of Hannover, Hannover, Germany; Spedali Civili, Brescia, Italy; Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico di Milano, Milan, Italy; Università degli Studi di Verona, Verona, Italy; Università Politecnica delle Marche and Ospedali Riuniti, Ancona, Italy; Christian-Albrechts-University, Kiel, Germany) and informed written consent from all participants were obtained in accordance with the tenets of the Declaration of Helsinki. Genome-wide association data from Crohn’s disease patients from UK and USA were obtained from public data repositories, the Wellcome Trust Case Control Consortium (WTCCC) repository and the database of Genotypes and Phenotypes (dbGaP), respectively.
Quality control and imputation
All GWAS data were quality control (QC) filtered prior imputation. Single-nucleotide polymorphisms (SNPs) and subjects with success call rates lower than 95% were removed using PLINK V.1.9 (www.cog-genomics.org/plink/1.9/)29. SNPs showing a deviation from the Hardy–Weinberg equilibrium (P-value < 0.001) and minor allele frequencies <1% were also excluded. In addition, one subject per duplicate pair and per pair of first-degree relatives was also removed via the Genome function in PLINK V.1.9 with a Pi-HAT threshold of 0.4. Principal component analysis (PCA) was performed in order to identify and exclude outliers based on their ethnicity by using PLINK V.1.9 and the GCTA64 and R-base under GNU Public license V.2. We estimated the first five PCs using ~100.000 quality-filtered independent SNPs (r2 < 0.15). Outliers were defined as individuals who deviated more than six standard deviations from the centroid of their population. The number of SNPs before and after QC for each cohort is summarized in Supplementary Table S1.
Imputation was performed using the Michigan Imputation Server30. The software SHAPEIT31 was used in order to estimate haplotypes, and the European panel of the Haplotype Reference Consortium r1.132 was used as the reference panel for both SSc and CD genotype data in the discovery phase. Individual chunks of 50.000 Mb were used to carry out the imputation, covering whole-genome regions with a probability threshold for merging genotypes of 0.9, thus maximizing the quality of the imputed variants. Imputed data were also subjected to the above-mentioned QC filters in PLINK V.1.9. The total number of SNPs imputed for each cohort is summarized in Supplementary Table S1.
Statistical analysis
Statistical analyses were performed with PLINK V.1.9.
Discovery phase
Each GWAS case/control cohort was independently analysed by logistic regression assuming an additive model with the first five PCs as covariates, as a correcting method for population stratification. Odds ratios (ORs) and 95% confidence intervals (CIs) were calculated according to Woolf’s method. Subsequently, SSc datasets were meta-analysed by the inverse variance-weighted method. Sex chromosomes were excluded from the analysis.
In order to detect common signals for SSc and CD with the same effect, either risk or protection, we selected SNPs that showed a P-value < 1 × 10−5 in the SSc-CD meta-analysis and showed nominal significance (P-value < 0.01) with each disease separately, as well as no significant heterogeneity in the SSc meta-analysis (Cochran’s Q test > 0.05 and heterogeneity index I2 < 50%). To identify common signals for SSc and CD with opposite effect, the direction of association was flipped in the CD dataset (1/OR instead of OR). Again, we selected SNPs that showed a P-value < 1 × 10−5 in the SSc and CD meta-analysis and that were associated with each disease separately at a P-value < 0.01.
The strongest associated SNP within each locus was selected for the replication phase. Genetic variants were annotated using variant effect predictor (VEP)33 and their previous association with SSc and/or CD was explored using Immunobase (http://www.immunobase.org) and the GWAS catalog34.
Replication phase
Replication cohorts were analysed by logistic regression for the previously selected SNPs. Finally, combined analysis of the SSc and CD discovery and replication cohorts was performed using the inverse variance method. After the replication phase, we considered as statistically significant those signals that showed a P-value < 0.05 in each disease separately in the replication phase and a P-value < 5 × 10−8 in the SSc-CD cross-disease meta-analysis including both discovery and replication datasets.
The statistical power of the SSc-CD combined meta-analyses (both discovery and discovery + replication) was determined as described by Skol et al.35. In the discovery cross-disease meta-analysis, the statistical power to detect an association at a P-value of 1 × 10−5 (MAF = 20% and OR = 1.2) was 80%. In the discovery + replication meta-analysis, the statistical power to detect an association at a P-value of 5 × 10−8 (MAF = 20% and OR = 1.2) was 100%.
Independence analysis
For those SSc-CD common loci identified for which an association with any of the analysed diseases was already reported, we evaluated the independence between pleiotropic signals and genetic variants previously associated with SSc and/or CD at the genome-wide significance level according to Immunobase and the GWAS Catalog. For this purpose, we used LDlink36, a tool that provides linkage disequilibrium (LD) data between polymorphisms across a variety of ancestral populations. Only the European ancestry was taken into account for the LD analysis.
In addition, since one of the shared genetic risk loci was located close to the extended major histocompatibility complex (MHC) region, we decided to test the independence between our new common signal and the main SSc and CD HLA associations. For this, we imputed SNPs, classical HLA alleles and amino acids across the extended MHC region (29,000,000 to 34,000,000 bp in chromosome 6) using the SNP2HLA method with the Beagle software package37 and the Type 1 Diabetes Genetics Consortium reference panel, composed of 5,255 individuals of European origin38. HLA imputation of the CD discovery cohort was not possible due to the low coverage of this region included in the platform used for the genotyping of this dataset. For the SSc discovery cohort, the presence of independent effects within the extended MHC region was examined using a stepwise logistic regression by conditioning on the top independent signals.
Functional annotation
We assessed the potential regulatory function of the SSc-CD common susceptibility variants identified by means of in silico expression quantitative trait locus (eQTL) analysis using Haploreg v4.1. Haploreg v4.1 is a tool for exploring annotations at variants on haplotype blocks, providing a large collection of regulatory information, capable of the functional assignment onto any set of variants derived from GWAS or sequencing studies39. We only included eQTLs found in tissues with relevance in SSc and/or CD.
Protein-protein interaction and gene set enrichment analyses
In order to identify interactions among proteins encoded by SSc and CD common risk loci, we decided to construct a protein-protein interaction (PPI) network using the STRING database V.11.040. This software provides a critical assessment and integration of PPI, including functional (indirect) as well as physical (direct) associations.
Gene ontology (GO) was applied to perform an enrichment analysis in order to determine whether certain biological processes are overrepresented in the set of SSc-CD common genes.
Results
Meta-analysis and replication
Following QC and imputation, we performed a meta-analysis considering both diseases as a single phenotype. A total of 5,994,231 SNPs overlapped between all GWAS datasets in the discovery phase.
When we combined GWAS data from SSc and CD under the assumption that alleles had the same effect in both diseases, genetic variants at 13 loci fulfilled the replication criteria (p-value < 1 × 10−5 in the SSc-CD meta-GWAS and p-value < 0.01 in each disease-specific analysis) (Fig. 2A and Supplementary Table S2). One of these common signals was located within the IRF8 region, a known genetic risk locus shared between SSc and CD, and, therefore, it was not considered in subsequent analyses. On the other hand, we performed the analysis under the assumption that alleles had opposite directions in both diseases, identifying 12 loci that fulfilled all criteria for the replication phase (Fig. 2B and Supplementary Table S3).
Figure 2.

Manhattan plot representing the results of the cross-disease meta-analysis including systemic sclerosis and Crohn’s disease, considering same allelic effects (A) and opposite allelic effects (B). Loci selected for replication are marked in black. Significance threshold at genome-wide level is marked with a red line. Established significance threshold for the cross-disease meta-analysis (p < 1 × 10−5) is marked with a blue line.
To confirm these associations, the strongest associated SNP within each locus was selected for validation in additional sample sets. According to the criteria established for the replication analysis (genome-wide significance in the combined analysis including both discovery and replication sets, and nominal statistical significance in each disease-specific replication analysis), we identified a total of 4 genetic variants showing a pleiotropic effect in SSc and CD: two intronic variants located within IL12RB2 and STAT3, a SNP close to IRF1, and an intergenic variant at 6p21.31 located between ZBTB9 and BAK1 (Table 1). It is remarkable that an opposite allelic effect in both disorders was observed for all these new common signals.
Table 1.
Loci associated with a genome-wide significant threshold after the cross-disease meta-analysis of systemic sclerosis and Crohn’s disease.
| Discovery | Replication | Discovery + Replication | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SSc | CD | SSc-CD | SSc | CD | SSc-CD | ||||||||
| Region | Gene | SNP | Test Allele | P-value | OR | P-value | OR | P-value | P-value | OR | P-value | OR | P-value |
| 1p31.3 | IL12RB2 | rs6659932 | A | 2.47E-08 | 1.3 | 1.33E-04 | 0.79 | 1.54E-11 | 3.75E-03 | 1.13 | 3.44E-02 | 0.86 | 1.08E-11 |
| 5q31.1 | IRF1 | rs2548998 | G | 1.55E-03 | 1.27 | 3.09E-07 | 0.79 | 1.13E-08 | 2.00E-02 | 1.08 | 1.18E-03 | 0.88 | 2.18E-11 |
| 6p21.31 | ZBTB9/BAK1 | rs68191 | C | 8.15E-03 | 0.84 | 8.70E-06 | 1.39 | 8.33E-07 | 2.09E-04 | 0.82 | 1.56E-02 | 1.15 | 1.07E-10 |
| 17q21.2 | STAT3 | rs4796791 | T | 1.34E-03 | 1.13 | 1.52E-04 | 0.84 | 9.86E-07 | 3.85E-02 | 1.08 | 1.85E-02 | 0.90 | 2.52E-08 |
Results of the discovery and replication analysis for each individual disease and of the combined meta-analysis (discovery + replication) are shown.
SNP, single-nucleotide polymorphism; SSc, systemic sclerosis; CD, Crohn’s disease.
Three of these shared risk loci have been previously associated with one of the analysed diseases, IL12RB2 with SSc and IRF1 and STAT3 with CD. Shared genetic variants at the IRF1 and STAT3 loci identified in our study were linked to those polymorphisms previously associated with CD (r2 > 0.40). In the case of IL12RB2, it is an established genetic risk locus for SSc but, in addition, the IL23R gene, located within this same genomic region, is a known susceptibility gene for CD. However, LD analysis evidenced that the pleiotropic variant identified in our study (rs6659932) was independent of the IL23R SNPs previously associated with CD (Supplementary Table S4).
On the other hand, the intergenic variant at 6p21.31 (rs68191) is located close to the extended MHC region. Considering this, we decided to test the independence between our new common signal and the main HLA associations observed in the SSc and CD discovery cohorts. In the case of CD, independence between signals could not be checked due to the low coverage of the HLA region. Regarding SSc, two independent signals were observed after conditional regression analysis, HLA-DPB1*1301 (p = 1.77 × 10−19, OR = 2.79) and HLA-DRB1*1104 (p = 1.21 × 10−12, OR = 1.83). After controlling for these two classical alleles, the SSc-CD common signal remained significant in the SSc discovery cohort (p-value = 8.15 × 10−3; conditioned p-value = 2.78 × 10−2).
Functional effect on gene expression
Subsequently, we used the HaploReg database to explor wether the most strogly associated polymorphism of each shared locus acted as an eQTL. As shown in Supplementary Table S5, all the pleiotopic SNPs identified in our study appeared to affect gene expression levels. Shared genetic variants at the IL12RB2 (rs6659932) and STAT3 (rs4796791) loci affected expression levels of IL12RB2 and STAT3, respectively, whereas the pleiotropic SNP of the IRF1 locus (rs2548998) acted as an eQTL for IRF1 and SLC22A5. Interestingly, the intergenic polymorphism at the MHC extended region (rs68191) affected gene expression levels of TAPBP.
Protein-protein interaction and enrichment analysis
Finally, we also evaluated the connectivity at the protein interaction level among the genetic risk loci shared between SSc and CD, including genes whose expression levels were affected by the pleiotopic polymorphisms identified in our study, that is IRF1, SLC22A5, STAT3, IL12RB2 and TAPBP, as well as loci associated in previous studies with both SSc and CD, including STAT4, TYK2, IRF8, GSDMA and IKZF3. GSDMA and IKZF3 belong to the same LD block, however GSDMA has been set as the most probable candidate gene of this locus in SSc and IKZF3 for CD41,42. Thus, we decided to keep both genes for PPI and enrichment analyses.
The PPI network involved 9 of the 10 common proteins included in the analysis, except for SLC22A5 (Fig. 3). We observed a strongly significant PPI enrichment (p-value < 1 × 10−6), indicating that these proteins have more interactions than would be expected for a random set of proteins of similar size.
Figure 3.
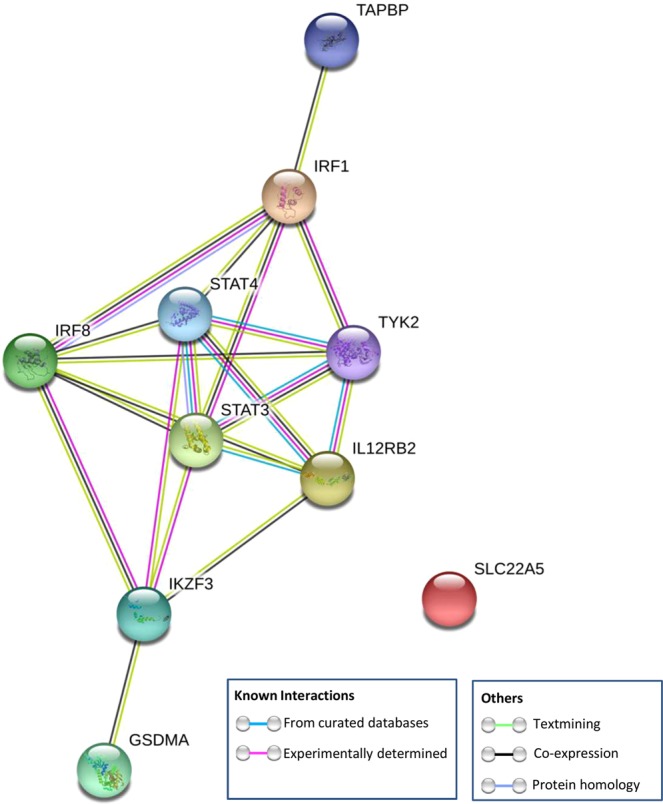
STRING protein-protein interaction network connectivity among genetic risk loci shared between systemic sclerosis and Crohn’s disease.
To further evaluate this connection, we performed a gene ontology enrichment analysis in biological processes. In this regard, we observed 29 statistically significant over-represented biological processes (p-value < 0.05). The most significantly over-represented pathways were related to interleukin-mediated signaling, especially those related with the IL-12 family and the type I interferon signaling pathway (Table 2).
Table 2.
Most significantly enriched Gene Ontology (GO)-biological processes in the set of genetic risk loci shared between systemic sclerosis and Crohn’s disease.
| Biological pathway | GO term | p-value* | Count in gene set | Shared genes involved |
|---|---|---|---|---|
| Interleukin-35-mediated signaling pathway | GO:0070757 | 1.44E-05 | 3 of 11 | STAT4, STAT3, IL12RB2 |
| Interleukin-23-mediated signaling pathway | GO:0038155 | 1.44E-05 | 3 of 9 | STAT4, STAT3, TYK2 |
| Cytokine-mediated signaling pathway | GO:0019221 | 6.16E-05 | 6 of 655 | STAT4, STAT3, IL12RB2, TYK2, IRF1, IRF8 |
| Interleukin-12-mediated signaling pathway | GO:0035722 | 3.20E-04 | 3 of 47 | STAT4, IL12RB2, TYK2 |
| Type I interferon signaling pathway | GO:0060337 | 3.60E-04 | 3 of 65 | TYK2, IRF1, IRF8 |
| Interleukin-21-mediated signaling pathway | GO:0038114 | 6.00E-04 | 2 of 8 | STAT4, STAT3 |
| Interleukin-27-mediated signaling pathway | GO:0070106 | 8.30E-04 | 2 of 11 | STAT3, TYK2 |
| Positive regulation of transcription by RNA polymerase II | GO:0045944 | 4.90E-03 | 5 of 1104 | STAT4, STAT3, IRF1, IRF8, IKZF3 |
| Positive regulation of interleukin-12 production | GO:0032735 | 5.90E-03 | 2 of 34 | IRF1, IRF8 |
| Receptor signaling pathway via JAK-STAT | GO:0007259 | 7.90E-03 | 2 of 41 | STAT4, STAT3 |
| Alpha-beta T cell differentiation | GO:0046632 | 9.70E-03 | 2 of 50 | STAT3, IRF1 |
*p-values determined by binomial statistic test and adjusted by false discovery rate correction.
New loci shared between systemic sclerosis and Crohn’s disease are in bold.
Discussion
Through the first comprehensive study of the genetic component shared between SSc and CD, we have identified four loci that contribute to suceptibility to both disorders. Of these, one had not been previously associated with any of the diseases under study (an intergenic locus at 6p21.31), whereas the remaining three represent established genetic risk loci for one but not the other condition.
Although all these pleiotropic SNPs are located in non-coding regions, functional annotation indicated that they act as regulatory variants affecting expression levels of either the gene were they mapped or close genes in cell types or tissues of relevance in the pathogenesis of SSc and/or CD. In this regard, pleiotropic variants appeared to influence expression levels of the IL12RB2, IRF1, SLC22A5, STAT3, and TAPBP genes (Supplementary Table S5). Most of these genes are key players of the immune response: IL12RB2 encodes a subunit of the IL-12 receptor complex implicated in Th1 differentiation; STAT3 encodes a transcription factor that is essential for the differentiation of Th17 cells; IRF1 encodes a transcriptional regulator of type I interferon (IFN) and IFN-inducible genes; and TAPBP is crucial for optimal peptide loading on the MHC class I molecule. In addition, the pleiotropic variant affecting IRF1 levels also regulates the expression of SLC22A5, which encodes an organic cation transporter involved in the active cellular uptake of carnitine.
Interestingly, PPI analysis evidenced a number of non-random connections among the SSc-CD common genes, including both shared risk loci previously described and comon genes identified in our study, which indicates overlap among the pathways involved in the pathogenesis of these two disorders. Specifically, the IL-12 family signaling pathways, including IL-35, IL-23, IL-12, IL-21, and IL-27-mediated signaling, were particularly compelling. This family of cytokines plays a crucial role in shaping immune responses, differentiation of naïve T cells towards different types of effector cells, as well as in the regulation of effector cell functions43. Moreover, the type I interferon signaling pathway was also enriched among the set of SSc-CD common genes. An increased expression and activation of IFN-inducible genes, known as interferon signature, has been reported in SSc44 and several interferon regulatory factors (IRFs), including IRF5, IRF4, and IRF8, have been involved in its susceptibility14,45, thus supporting the role of IRF1, previously associated with CD but not with SSc, as a new susceptibility gene for this last condition.
Considering these results, both IL-12 family and type I interferon signaling pathways could represent interesting therapeutic targets for both SSc and CD. Indeed, ustekinumab, a monoclonal antibody to the p40 subunit common to IL-12 and IL-23, has been recently approved in the EU and the USA to treat patients with CD and, therefore, this drug could be repositioned to treat SSc. However, it should be advised that all the pleiotropic variants identified in our study showed opposite allelic effects in the two analysed disorders, thus highlighting the complex effects that shared associations have on disease outcomes. This could be due to the fact that consequences of genetic variants are influenced by the cell type. For example, as previously indicated, the shared genetic variant at IL12RB2 influenced IL12RB2 gene expression levels; however, whereas the minor allele (which conferred risk to SSc in our study) correlated with an increased gene expression in whole blood, the major allele (which conferred risk to CD) had the same effect (increased IL12RB2 expression) in fibroblasts, according to GTEx data. In addition, the effect on gene expression of the pleiotropic SNP located within the 5q31.1 region was also cell type specific, influencing IRF1 expression levels in lymphoblastoid cells and SLC22A5 levels in other tissues, and, therefore, this SNP could have a different biological implication in both diseases. Indeed, higher expression levels of OCTN2, the protein encoded by SLC22A5, have been found in inflamed regions of the intestinal epithelium compared with non-inflamed areas, and a role of this protein in the intestinal homeostasis has also been reported46; whereas, given the relevance of the type 1 interferon signaling pathway in SSc, the IRF1 gene seems a more plausible candidate to be involved in SSc susceptibility. Considering this, it is possible that an effective treatment for SSc could have a detrimental effect on CD, and conversely. As previously mentioned, we observed discordant associations for variants located in genes implicated in IL-23 and Th1 differentiation pathways. In this context, IL-17-specific antibody therapy, effective in psoriasis and with promising effects on SSc47,48, has been proven to exacerbate CD49. This could be due to a deficient Th17 activation in CD owing to mutations in STAT3, which could lead to hyper-IgE syndrome, typically associated with extracellular fungal and bacterial infections50. Interestingly, according to our results, the STAT3 rs4796791 variant confers protection to CD and risk to SSc, which could lead to an exacerbate reaction in CD patients carrying this variant when treated with anti-IL17 therapy.
Interestingly, it has been reported a reduced incidence of CD in patients with SSc51,52. Although the causes of this phenomenon are not clear, our results suggest that identical genetic risk factors could have different or even opposite functional effects in both diseases. These ‘flip-flop’ associations have been extensively observed across different comparative analyses53. In this regard, a cross-disease meta-analysis including CD and type 1 diabetes54 identified two variants, such as IL27 rs4788084 and IL10 rs3024505, with opposite effects in these two conditions. Furthermore, a meta-analysis of 6 different immune-mediated disorders showed that 14% of overlapped variants were discordant regarding the risk allele across diseases55. These results suggest that predisposition to related diseases may be regulated by different dose balance of genes and genomic elements in relevant biological pathways, as well as how these differences affect a specific cell type, as previously mentioned. In this sense, differences across cell types in transcription regulation mediated by epigenetic factors such as methylation, histone modifications or long non-conding RNAs could influence these opposite effects for the same allele in different diseases56. It is, therefore, crucial to know the cell types in which genetic variants are acting to be able to elucidate their role on the pathogenesis of the disease.
Supplementary information
Acknowledgements
We thank Sofia Vargas and Gema Robledo for her excellent technical assistance and all the patients and control donors for their essential collaboration. We thank WTCCC (Welcome Trust Case Control Consortium) for the access to GWAS data of Crohn’s disease patients and healthy controls, Banco Nacional de ADN (University of Salamanca, Spain) who supplied part of the control DNA samples, and dbGap for granting access to the IBD Genetics Consortium (IBDGC) Crohn’s Disease GWAS data (phs000130.v1.p1). The IBDGC Crohn’s Disease Genome-Wide Association Study was conducted by the IBDGC Investigators and supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). This manuscript was not prepared in collaboration with Investigators of the IBDGC Crohn’s Disease Genome-Wide Association Study and does not necessarily reflect the opinions or views of the IBDGC Crohn’s Disease Genome-Wide Association Study, the NIDDK Central Repositories, or the NIDDK. This work was supported by the Spanish Ministry of Economy and Competitiveness (SAF2015-66761-P; IPT-010000-2010-36, cofunded by the European Regional Development Fund), Consejería de Innovación, Ciencia y Tecnología, Junta de Andalucía (Spain) (P12-BIO-1395) and the Cooperative Research Thematic Network (RETICS) programme (RD16/0012/0013) (RIER) from Instituto de Salud Carlos III (ISCIII, Spanish Ministry of Economy, Industry and Competitiveness). AM is recipient of a Miguel Servet fellowship (CP17/00008) from ISCIII (Spanish Ministry of Economy, Industry and Competitiveness). DGS was supported by the Spanish Ministry of Economy and Competitiveness through the FPI programme (SAF2015-66761-P). This work is part of the Doctoral Thesis “Bases Genéticas de la Esclerosis Sistémica: Integrando Genómica y Transcriptómica”.
Author contributions
A.M. and J.M. were involved in the conception and design of the study. A.M., J.M. and D.G.S. contributed in the analysis and interpretation of data. A.M. and D.G.S. drafted the manuscript. E.O., E.L.I., A.J., F.D., N.O.C., T.R.D.J.R., A.F., S.M. and M.D.M. collected samples and participated in the analysis and interpretation of data. E.O., E.L.I., A.J., F.D., N.O.C., T.R.D.J.R., A.F., S.M. and M.D.M. and J.M. revised critically the manuscript draft. The Scleroderma Genetic Consortium provided data and material and substantively revised the manuscript. All authors approved the final version of the manuscript.
Data availability
Results of the SSc-CD cross-disease meta-analysis are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
†A comprehensive list of consortium members appears at the end of the paper.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Javier Martín and Ana Márquez.
Contributor Information
Ana Márquez, Email: anamaort@ipb.csic.es.
Scleroderma Genetic Consortium:
Shervin Assassi, Xiaodong Zhou, Filemon K. Tan, Frank C. Arnett, John D. Reveille, Olga Gorlova, Wei V. Chen, Jun Ying, Peter K. Gregersen, Annette T. Lee, Alexandre E. Voskuyl, Jeska de Vries-Bouwstra, Cesar Magro-Checa, Jasper Broen, Bobby P. C. Koeleman, Carmen P. Simeón, Vicente Fonollosa, Alfredo Guillén, Patricia Carreira, Iván Castellví, Miguel A. González-Gay, Raquel Ríos, Jose Luis Callejas-Rubio, José A. Vargas-Hitos, Rosa García-Portales, María Teresa Camps, Antonio Fernández-Nebro, María F. González-Escribano, Francisco José García-Hernández, Ma. Jesús Castillo, Ma. Ángeles Aguirre, Inmaculada Gómez-Gracia, Luis Rodríguez-Rodríguez, Benjamín Fernández-Gutiérrez, Paloma García de la Peña, Esther Vicente, José Luis Andreu, Mónica Fernández de Castro, Francisco Javier López-Longo, Lina Martínez, Gerard Espinosa, Carlos Tolosa, Anna Pros, Mónica Rodríguez-Carballeira, Francisco Javier Narváez, Manel Rubio-Rivas, Vera Ortiz-Santamaría, Ana Belén Madroñero, Bernardino Díaz, Luis Trapiella, Adrián Sousa, María Victoria Egurbide, Patricia Fanlo-Mateo, Luis Sáez-Comet, Federico Díaz-González, Vanesa Hernández, Emma Beltrán, José Andrés Román-Ivorra, Elena Grau, Juan José Alegre-Sancho, Francisco J. Blanco-García, Natividad Oreiro, Mayka Freire, Alejandro Balsa, Ana M. Ortiz, Nicolas Hunzelmann, Gabriela Riemekasten, Jörg H. W. Distler, Torsten Witte, Paolo Airó, Lorenzo Beretta, Alessandro Santaniello, Chiara Bellocchi, Claudio Lunardi, Gianluca Moroncini, and Armando Gabrielli
Supplementary information
is available for this paper at 10.1038/s41598-020-58741-w.
References
- 1.Denton CP, Khanna D. Systemic sclerosis. Lancet. 2017;390:1685–1699. doi: 10.1016/S0140-6736(17)30933-9. [DOI] [PubMed] [Google Scholar]
- 2.Torres J, Mehandru S, Colombel JF, Peyrin-Biroulet L. Crohn’s disease. Lancet. 2017;389:1741–1755. doi: 10.1016/S0140-6736(16)31711-1. [DOI] [PubMed] [Google Scholar]
- 3.Angiolilli C, et al. New insights into the genetics and epigenetics of systemic sclerosis. Nat. Rev. Rheumatol. 2018;14:657–673. doi: 10.1038/s41584-018-0099-0. [DOI] [PubMed] [Google Scholar]
- 4.Wang MH, Picco MF. Crohn’s Disease: Genetics Update. Gastroenterol. Clin. North. Am. 2017;46:449–461. doi: 10.1016/j.gtc.2017.05.002. [DOI] [PubMed] [Google Scholar]
- 5.Matzaraki V, Kumar V, Wijmenga C, Zhernakova A. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol. 2017;18:76. doi: 10.1186/s13059-017-1207-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bettenworth D, et al. Assessment of Crohn’s disease-associated small bowel strictures and fibrosis on cross-sectional imaging: a systematic review. Gut. 2019;68:1115–1126. doi: 10.1136/gutjnl-2018-318081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Danese S, et al. Identification of Endpoints for Development of Antifibrosis Drugs for Treatment of Crohn’s Disease. Gastroenterology. 2018;155:76–87. doi: 10.1053/j.gastro.2018.03.032. [DOI] [PubMed] [Google Scholar]
- 8.Kim, J. et al. Increased risk of idiopathic pulmonary fibrosis in inflammatory bowel disease: A nationwide study. J Gastroenterol Hepatol, 10.1111/jgh.14838 (2019). [DOI] [PubMed]
- 9.Herzog EL, et al. Review: interstitial lung disease associated with systemic sclerosis and idiopathic pulmonary fibrosis: how similar and distinct? Arthritis Rheumatol. 2014;66:1967–1978. doi: 10.1002/art.38702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marie I, Ducrotte P, Denis P, Hellot MF, Levesque H. Outcome of small-bowel motor impairment in systemic sclerosis–a prospective manometric 5-yr follow-up. Rheumatol. 2007;46:150–153. doi: 10.1093/rheumatology/kel203. [DOI] [PubMed] [Google Scholar]
- 11.Sallam H, McNearney TA, Chen JD. Systematic review: pathophysiology and management of gastrointestinal dysmotility in systemic sclerosis (scleroderma) Aliment. Pharmacol. Ther. 2006;23:691–712. doi: 10.1111/j.1365-2036.2006.02804.x. [DOI] [PubMed] [Google Scholar]
- 12.Ellinghaus D, et al. Combined analysis of genome-wide association studies for Crohn disease and psoriasis identifies seven shared susceptibility loci. Am. J. Hum. Genet. 2012;90:636–647. doi: 10.1016/j.ajhg.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Festen EA, et al. A meta-analysis of genome-wide association scans identifies IL18RAP, PTPN2, TAGAP, and PUS10 as shared risk loci for Crohn’s disease and celiac disease. PLoS Genet. 2011;7:e1001283. doi: 10.1371/journal.pgen.1001283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopez-Isac E, et al. Brief Report: IRF4 Newly Identified as a Common Susceptibility Locus for Systemic Sclerosis and Rheumatoid Arthritis in a Cross-Disease Meta-Analysis of Genome-Wide Association Studies. Arthritis Rheumatol. 2016;68:2338–2344. doi: 10.1002/art.39730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marquez A, et al. A combined large-scale meta-analysis identifies COG6 as a novel shared risk locus for rheumatoid arthritis and systemic lupus erythematosus. Ann. Rheum. Dis. 2017;76:286–294. doi: 10.1136/annrheumdis-2016-209436. [DOI] [PubMed] [Google Scholar]
- 16.Martin JE, et al. A systemic sclerosis and systemic lupus erythematosus pan-meta-GWAS reveals new shared susceptibility loci. Hum. Mol. Genet. 2013;22:4021–4029. doi: 10.1093/hmg/ddt248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhernakova A, et al. Meta-analysis of genome-wide association studies in celiac disease and rheumatoid arthritis identifies fourteen non-HLA shared loci. PLoS Genet. 2011;7:e1002004. doi: 10.1371/journal.pgen.1002004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Acosta-Herrera M, et al. Genome-wide meta-analysis reveals shared new loci in systemic seropositive rheumatic diseases. Ann. Rheum. Dis. 2019;78:311–319. doi: 10.1136/annrheumdis-2018-214127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellinghaus D, et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat. Genet. 2016;48:510–518. doi: 10.1038/ng.3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marquez A, et al. Meta-analysis of Immunochip data of four autoimmune diseases reveals novel single-disease and cross-phenotype associations. Genome Med. 2018;10:97. doi: 10.1186/s13073-018-0604-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Radstake TR, et al. Genome-wide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat. Genet. 2010;42:426–429. doi: 10.1038/ng.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wellcome Trust Case Control, C Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nat. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franke A, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Julia A, et al. A genome-wide association study on a southern European population identifies a new Crohn’s disease susceptibility locus at RBX1-EP300. Gut. 2013;62:1440–1445. doi: 10.1136/gutjnl-2012-302865. [DOI] [PubMed] [Google Scholar]
- 25.Rioux JD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Preliminary criteria for the classification of systemic sclerosis (scleroderma) Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis Rheum. 1980;23:581–590. doi: 10.1002/art.1780230510. [DOI] [PubMed] [Google Scholar]
- 27.van den Hoogen F, et al. Classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013;65:2737–2747. doi: 10.1002/art.38098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lennard-Jones J. E. Classification of Inflammatory Bowel Disease. Scandinavian Journal of Gastroenterology. 1989;24(sup170):2–6. doi: 10.3109/00365528909091339. [DOI] [PubMed] [Google Scholar]
- 29.Chang CC, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das S, et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016;48:1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat. Methods. 2011;9:179–181. doi: 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- 32.McCarthy S, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016;48:1279–1283. doi: 10.1038/ng.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McLaren W, et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinforma. 2010;26:2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacArthur J, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog) Nucleic Acids Res. 2017;45:D896–D901. doi: 10.1093/nar/gkw1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat. Genet. 2006;38:209–213. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- 36.Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants: Fig. 1. Bioinforma. 2015;31:3555–3557. doi: 10.1093/bioinformatics/btv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jia X, et al. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS One. 2013;8:e64683. doi: 10.1371/journal.pone.0064683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown WM, et al. Overview of the MHC fine mapping data. Diabetes Obes. Metab. 2009;11(Suppl 1):2–7. doi: 10.1111/j.1463-1326.2008.00997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szklarczyk D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu JZ, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015;47:979–986. doi: 10.1038/ng.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terao C, et al. Transethnic meta-analysis identifies GSDMA and PRDM1 as susceptibility genes to systemic sclerosis. Ann. Rheum. Dis. 2017;76:1150–1158. doi: 10.1136/annrheumdis-2016-210645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun L, He C, Nair L, Yeung J, Egwuagu CE. Interleukin 12 (IL-12) family cytokines: Role in immune pathogenesis and treatment of CNS autoimmune disease. Cytokine. 2015;75:249–255. doi: 10.1016/j.cyto.2015.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu M, Assassi S. The role of type 1 interferon in systemic sclerosis. Front. Immunol. 2013;4:266. doi: 10.3389/fimmu.2013.00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bossini-Castillo L, Lopez-Isac E, Martin J. Immunogenetics of systemic sclerosis: Defining heritability, functional variants and shared-autoimmunity pathways. J. Autoimmun. 2015;64:53–65. doi: 10.1016/j.jaut.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 46.Fujiya M, et al. Cytokine regulation of OCTN2 expression and activity in small and large intestine. Inflamm. Bowel Dis. 2011;17:907–916. doi: 10.1002/ibd.21444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gaowa S, et al. Effect of Th17 and Treg axis disorder on outcomes of pulmonary arterial hypertension in connective tissue diseases. Mediators Inflamm. 2014;2014:247372. doi: 10.1155/2014/247372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park MJ, et al. IL-1-IL-17 Signaling Axis Contributes to Fibrosis and Inflammation in Two Different Murine Models of Systemic Sclerosis. Front. Immunol. 2018;9:1611. doi: 10.3389/fimmu.2018.01611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hueber W, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693–1700. doi: 10.1136/gutjnl-2011-301668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Minegishi Y, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nat. 2007;448:1058–1062. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 51.Koumakis E, Dieude P, Avouac J, Kahan A, Allanore Y. Familial autoimmunity in systemic sclerosis–results of a French-based case-control family study. J. Rheumatol. 2012;39:532–538. doi: 10.3899/jrheum.111104. [DOI] [PubMed] [Google Scholar]
- 52.Tseng CC, et al. Reduced incidence of Crohn’s disease in systemic sclerosis: a nationwide population study. BMC Musculoskelet. Disord. 2015;16:251. doi: 10.1186/s12891-015-0693-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin PI, Vance JM, Pericak-Vance MA, Martin ER. No gene is an island: the flip-flop phenomenon. Am. J. Hum. Genet. 2007;80:531–538. doi: 10.1086/512133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang K, et al. Comparative genetic analysis of inflammatory bowel disease and type 1 diabetes implicates multiple loci with opposite effects. Hum. Mol. Genet. 2010;19:2059–2067. doi: 10.1093/hmg/ddq078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat. Rev. Genet. 2013;14:661–673. doi: 10.1038/nrg3502. [DOI] [PubMed] [Google Scholar]
- 56.Jonkers IH, Wijmenga C. Context-specific effects of genetic variants associated with autoimmune disease. Hum. Mol. Genet. 2017;26:R185–R192. doi: 10.1093/hmg/ddx254. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Results of the SSc-CD cross-disease meta-analysis are available from the corresponding author on reasonable request.