

Abstract
Glycogen storage disease type Ia (GSD Ia) is caused by autosomal mutations in glucose-6-phosphatase α catalytic subunit (G6PC) and can present with severe hypoglycemia, lactic acidosis and hypertriglyceridemia. In both children and adults with GSD Ia, there is over-accumulation of hepatic glycogen and triglycerides that can lead to steatohepatitis and a risk for hepatocellular adenoma or carcinoma. Here, we examined the effects of the commonly used peroxisomal proliferated activated receptor α agonist, fenofibrate, on liver and kidney autophagy and lipid metabolism in 5-day-old G6pc −/− mice serving as a model of neonatal GSD Ia. Five-day administration of fenofibrate decreased the elevated hepatic and renal triglyceride and hepatic glycogen levels found in control G6pc −/− mice. Fenofibrate also induced autophagy and promoted β-oxidation of fatty acids and stimulated gene expression of acyl-CoA dehydrogenases in the liver. These findings show that fenofibrate can rapidly decrease hepatic glycogen and triglyceride levels and renal triglyceride levels in neonatal G6pc −/− mice. Moreover, since fenofibrate is an FDA-approved drug that has an excellent safety profile, our findings suggest that fenofibrate could be a potential pharmacological therapy for GSD Ia in neonatal and pediatric patients as well as for adults. These findings may also apply to non-alcoholic fatty liver disease, which shares similar pathological and metabolic changes with GSD Ia.
Introduction
Glycogen storage disease type Ia (GSD Ia), also known as von Gierke’s disease, is caused by mutations in the G6PC gene (1–4). G6PC encodes glucose-6-phosphatase (G6Pase), which dephosphorylates glucose-6-phospate (G6P) to glucose in the last step of gluconeogenesis (5). G6Pase is primarily expressed in the liver and kidneys and is important for maintaining glucose homeostasis in the body. In GSD Ia, pathogenic variants in G6PC decrease G6Pase enzyme activity, which leads to intracellular accumulation of G6P, and increased glycogen synthesis as well as fatty acid and triglyceride production. These metabolic changes result in build-up of glycogen and triglycerides in the liver and kidneys (6). Besides these changes in the liver and kidney, GSD Ia is characterized by severe hypoglycemia, lactic acidosis and hypertriglyceridemia (7). Severe hypoglycemia due to the interruption of feeding has been associated with seizures and death (8). However, the acute clinical manifestations of these biochemical abnormalities can largely be controlled by frequent oral supplementation of glucose in infants and an adherence to a strict schedule of uncooked cornstarch supplementation in children and adults (9, 10). Dietary therapy can achieve metabolic control as demonstrated by decreased blood lactate and triglyceride concentrations, and improved metabolic control has correlated with decreased frequency of long-term complications including hepatic adenoma formation and proteinuria (9–11). Unfortunately, dietary therapy does not successfully maintain metabolic control in all patients, including those with identical genotypes, which implicates unidentified variables including environmental and genetic factors that determine the phenotype of individual patients (12). Dietary therapy does not reliably prevent long-term complications in all patients with GSD Ia, including the development of hepatocellular adenomas and hepatocarcinoma (13, 14). It also appears that GSD Ia patients are especially susceptible to developing hepatic adenomas as three-quarters of patients over 25 years of age have at least one hepatocellular adenoma (13). This disease progression resembles the histological, metabolic and pathological changes that occur in non-alcoholic fatty liver disease (NAFLD), a condition that affects approximately 40% adult Americans (15, 16). Additionally, there are non-hepatic long-term complications such as chronic renal failure, hyperlipidemia with a potential risk for atherosclerosis, growth retardation and rarely pancreatitis or pulmonary hypertension (7).
We and others previously showed that there is impaired macroautophagy, hereafter referred to as autophagy, in cell culture, murine and canine models of GSD Ia (17). Autophagy in the liver is critical for β-oxidation of fatty acids and mitochondrial turnover, functions that are critical for normal lipid metabolism, aerobic glycolysis and oxidative phosphorylation (18). We previously showed that induction of autophagy with mammalian target of rapamycin inhibitor rapamycin, or the thyroid hormone agonist, VK2809, increased β-oxidation of fatty acids and reduced hepatosteatosis in G6pc −/− mice (19). We also showed that the pan-peroxisomal proliferator activated receptor (PPAR) agonist, bezafibrate, reduced glycogen and triglyceride in the livers of GSD Ia mice. Bezafibrate is approved for the treatment of hypertriglyceridemia in Europe, but restricted in the U.S. PPARs are members of the nuclear hormone receptor superfamily. The PPARα isoform is highly expressed in the liver and kidneys (20), the same tissues that are primarily affected in GSD Ia. As a ligand-inducible transcription factor, PPARα regulates the expression of key genes involved in peroxisomal and mitochondrial β-oxidation, fatty acid catabolism, energy homeostasis and lipogenesis (21–26).
Chronic treatment with fenofibrate, an FDA-approved PPARα-selective agonist, led to decreased hepatic triglyceride and glycogen in an adult GSD Ia mouse model featuring inducible liver- and kidney-specific knockout of G6Pase (16). Currently, little is known about the hepatic metabolic effects of fenofibrate in neonatal mice, which have undergone chronic intrahepatic and intrarenal accumulation of G6P, glycogen and triglycerides in utero and postnatally. In the present study, we assess the ability of the PPARa agonist, fenofibrate, to rapidly decrease hepatic triglycerides and glycogen in neonatal GSD1a mice.
Results
Fenofibrate lowers lipid and glycogen levels in the liver of G6pc −/− mice
Neonatal G6pc −/− mice were treated with fenofibrate at five days of age for 5 days to examine the effect of fenofibrate administration on the liver and kidney in this GSD Ia model. Control and knockout mice were sacrificed 24 hours after administration of the last dose. Liver mass and body mass were measured and were found to be decreased in G6pc −/− mice treated with fenofibrate (Figs. 1A and S1A). However, the liver weight was unchanged by fenofibrate administration when normalized to body weight (Fig. 1B). Serum glucose levels of G6pc −/− mice were below the level of detection and did not change with fenofibrate administration (Fig. S1B). When liver histology was examined by hematoxylin and eosin (H&E) stain, hepatosteatosis was almost completely reversed by fenofibrate administration (Fig. 1C). Hepatic triglycerides and glycogen were higher in G6pc −/− mice treated with vehicle, when compared to wild-type (WT) mice treated with vehicle (Fig. 1D–H) as reported previously (19). When fenofibrate was administered, there was decreased intrahepatic liver triglyceride levels by biochemical assay and by oil red O staining (Fig. 1D–F). Fenofibrate also significantly decreased hepatic glycogen levels in in G6pc −/− mice as determined by biochemical assay and by periodic acid Schiff (PAS) glycogen staining (Fig. 1G and H).
Figure 1.
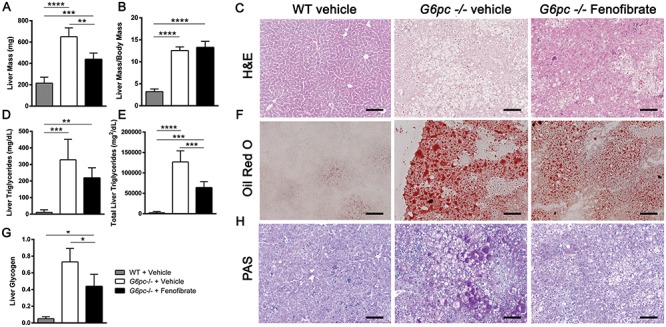
Liver triglycerides and glycogen are reduced by fenofibrate treatment in G6pc −/− mice. (A) Liver mass (mg) (n = 4–5). (B) Liver mass proportional to body mass (n = 4–5). (C) Representative liver H&E stain from three mice. Scale bar = 100 μm. (D) Liver triglycerides (mg/dL) measured by colorimetric assay (n = 4–5). (E) Total liver triglycerides (mg2/dL) calculated from liver weight and liver triglyceride assay (n = 4–5). (F) Representative liver Oil Red O stain from three mice. Scale bar = 100 μm. (G) Liver glycogen content measured by biochemical assay (n = 4). (H) Representative liver PAS glycogen stain from three mice. Scale bar = 100 μm. One-way ANOVA with Tukey’s multiple comparison test was performed. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Fenofibrate induces autophagic flux in liver of G6pc −/− mice
We previously showed that autophagy flux was impaired in G6pc −/− mice. Induction of autophagy by rapamycin and bezafibrate reduced the hepatic triglyceride and glycogen over-accumulation of these mice (17, 19). Fenofibrate treatment increased hepatic microtubule-associated protein 1A/1B-light chain 3 (LC3)-II protein expression (Fig. 2A and B), but had no significant effect on Lc3 mRNA expression (Fig. 2C). This suggests that increased LC3-II is related to autophagosome formation, rather than increased Lc3 transcription. In vehicle-treated G6pc −/− mice, P62 was increased when compared with control WT mice suggesting a block in autophagosome degradation (Fig. 2A, D and E). P62 protein was significantly decreased in the livers of fenofibrate-treated G6pc −/− mice when compared to vehicle-treated G6pc −/− mice, and was not significantly different from the level observed in WT mice (Fig. 2A and D). Of note, hepatic p62 transcripts were significantly decreased in both fenofibrate and vehicle-treated G6pc −/− mice, suggesting the decrease in p62 protein caused by fenofibrate in G6pc −/− mice occurred post-transcriptionally (Fig. 2E). Beclin1, a joint regulator of autophagy and apoptosis, was decreased in vehicle-treated G6pc −/− liver; however, it was increased by fenofibrate (Fig. S2). Taken together, these data suggested that fenofibrate restored autophagic flux in G6pc −/− mice. Considering the results of Beclin1 and since apoptosis has previously been shown to be increased in G6pc −/− mouse liver, we evaluated the expression of other apoptosis-related genes to assess the effect of fenofibrate (27). We found these genes were unchanged following fenofibrate administration and that apoptosis genes were significantly downregulated between WT and G6pc −/− vehicle treated mice (Fig. S2) (27).
Figure 2.
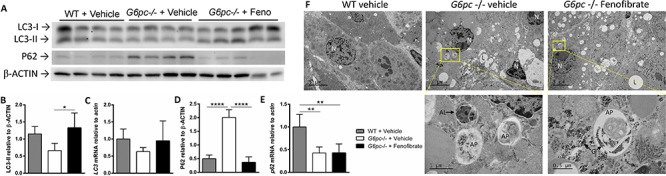
Autophagy is induced by fenofibrate in G6pc −/− mice. (A) Western blot of LC3 and P62 with β-ACTIN loading control. Each lane represents a biological replicate (n = 4–5). (B) Quantification of LC3-II protein from A relative to β-ACTIN. (C) LC3 transcripts relative to actin. (D) P62 protein from A relative to β-ACTIN. (E) p62 transcripts relative to actin. (F) TEM images of the liver (n = 2). (Top) 5000× images for overall morphology. Scale bar = 5 μm. (Bottom) Higher magnification of autophagosomes is shown in the lower panels, including autophagosomes containing intracellular cargo and autolysosomes from the boxed area of the top panel. The autophagosome in the liver of G6pc −/− fenofibrate-treated mice also contained electron dense glycogen granules. Scale bar = 1 μm or 0.5 μm. N: nucleus, L: lipid, G: glycogen granules, AP: autophagosome, AL: autolysosome. One-way ANOVA with Tukey’s multiple comparison test was performed. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
TEM demonstrated accumulation of lipids and autophagosomes in G6pc −/− liver (Fig. 2F). Autophagosomes in G6pc −/− fenofibrate treated mice contained glycogen, in contrast to G6pc −/− vehicle treated mice. Autophagosomes can deliver glycogen to the lysosomes that can be degraded by GAA (28), which provides a mechanism for autophagy induction to decrease accumulated glycogen in GSD Ia.
Fenofibrate induces expression PPARα targets and promotes β-oxidation
Plasma acylcarnitines in several essential metabolic pathways were significantly increased in G6pc −/− liver, and significantly normalized following fenofibrate administration (Fig. 3). PPAR agonists might enhance fatty acid β-oxidation (FAO), and therefore, the expression of genes related to FAO were analyzed. The mitochondrial acylcarnitine dehydrogenases Acadm, Acadl and Acadvl are significantly decreased in vehicle-treated G6pc −/− liver, in comparison with WT (Fig. 3A); however, palmitoyl-CoA oxidase gene, Acox1, appeared lower without significantly decreasing. Fenofibrate administration significantly increased Acox1 transcripts in G6pc −/− liver (Fig. 3A). Lower Acadvl expression corresponds with decreased very long-chain acylCoA dehydrogenase (VLCAD) protein, which correlates with increased plasma acylcarnitines including C12, C12:1, C14, C14:1 and C14:2 species (29). Fenofibrate administration restored normal C12, C12:1, C14, C14:1 and C14:2 concentrations to the normal range (Fig. 3B). Similarly, Acadl and Acadvl transcripts normalized in the liver following fenofibrate administration (Fig. 3A).
Figure 3.
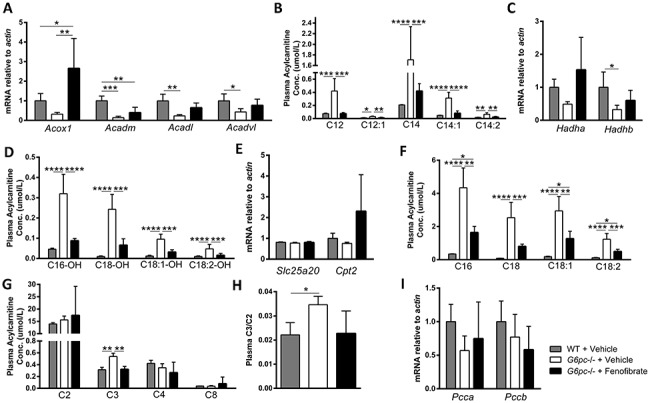
Fenofibrate normalizes plasma acylcarnitines and restores acyl-CoA dehydrogenase transcripts. (A) Acyl-CoA dehydrogenase transcripts (n = 4–5). (B) Plasma acylcarnitines related to acyl-CoA dehydrogenases (n = 4–5). (C) MTFP subunit transcripts, Hadha and Hadhb (n = 4–5). (D) Plasma acylcarnitines associated with LCHAD and MTFP (n = 4–5). (E) Transcripts of Cpt2 and Slc25a20 implicated in CPTII/CACT deficiencies (n = 4–5). (F) Plasma acylcarnitine species implicated in CPTII/CACT deficiencies (n = 4–5). (G) Short chain plasma acylcarnitines (n = 4–5). (H) Ratio of C2/C3 plasma acylcarnitines (n = 4–5). (I) Succionate metabolism transcripts, Pcca and Pccb (n = 4–5). One-way ANOVA with Tukey’s multiple comparison test was performed. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Since Acox1, Acadm, Acadl and Acadvl are all known targets of PPARα regulation, we assessed PPARα transcript and protein levels (30–32). No significant differences in Pparα, Pparβ/δ or Pparγ transcripts were observed between groups (Fig. S3A). PPARα was significantly decreased in vehicle-treated G6pc −/− liver, but not significantly affected by fenofibrate administration (Fig. S3B and C).
The expression of genes implicated in long-chain hydroxylacylcarnitine dehydrogenase (LCHAD) and mitochondrial trifunctional protein (MTFP) deficiencies, Hadha and Hadhb, were decreased in vehicle-treated G6pc −/− liver (Fig. 3C). Plasma acylcarnitines associated with LCHAD and MTFP C16-OH, C18-OH, C18:1-OH and C18-OH are significantly increased between WT and G6pc −/− mice (Fig. 3D) (33). Despite the lack of a significant effect on transcription of Hadha and Hadhb, fenofibrate decreased these accumulated species of acylcarnitines associated with LCHAD and MTFP deficiencies in G6pc −/− mice, such that they were not significantly different from WT levels (Fig. 3D).
The expression in liver for Cpt2, the gene responsible for carnitine palmitoyltransferase II (CPTII) deficiency, and for Slc25a20, the gene causing carnitine-acylcarnitine translocase (CACT) deficiency, was not significantly changed in G6pc −/− mice or by fenofibrate administration. (Fig. 3E). However, plasma acylcarnitine species implicated in CPTII/CACT deficiencies, C16, C18, C18:1 and C18:2, were significantly increased in G6pc −/− mice (Fig. 3F) (33). Fenofibrate administration decreased concentrations of these CPTII/CACT deficiency-implicated acylcarnitines (Fig. 3F), although the fluctuations in these species might also be attributable to changes in Acadl and Acadvl transcripts that normalized in the liver following fenofibrate administration (Fig. 3A).
Short-chain plasma acylcarnitines were not significantly different between groups with the exception of C3. Plasma C3 was increased in G6pc −/− mice, decreased following fenofibrate exposure to concentrations equivalent to those observed in WT mice (Fig. 3G). The C2/C3 ratio, which reflects succinate metabolism, followed the same pattern (Fig. 3H). Genes for propionyl-CoA carboxylase Pcca and Pccb were not significantly changed by genotype or fenofibrate administration (Fig. 3I). All measured plasma acylcarnitines are reported in Table S3, along with relevant statistics.
Fenofibrate affects kidney of G6pc −/− mice
Kidney mass was not significantly altered between WT and G6pc −/− mice or by fenofibrate administration, but kidney mass normalized to body weight was significantly increased in groups of G6pc −/− mice (Fig. 4A and B). Kidney histology was unchanged by fenofibrate administration (Fig. 4C) with the exception that renal lipids visualized by ORO staining were decreased (Fig. 4D). Furthermore, renal glycogen detected by PAS staining was unchanged (Fig. 4E). Significant changes in LC3-II and P62 protein and transcripts were not detected in G6pc −/− kidney, suggesting that experimental conditions did not allow detection of known alterations in renal autophagy related to GSD Ia (Fig. S4). Fenofibrate exposure had no significant effects on metabolic genes in the kidney, in contrast with conditions in the liver (Fig. S5). Similarly to the liver, there were no significant changes in Pparα, Pparβ/δ or Pparγ transcripts with genotype or fenofibrate administration (Fig. S6A). PPARα was significantly decreased in G6pc −/− kidney, but unchanged by fenofibrate exposure (Fig. S6B and C).
Figure 4.
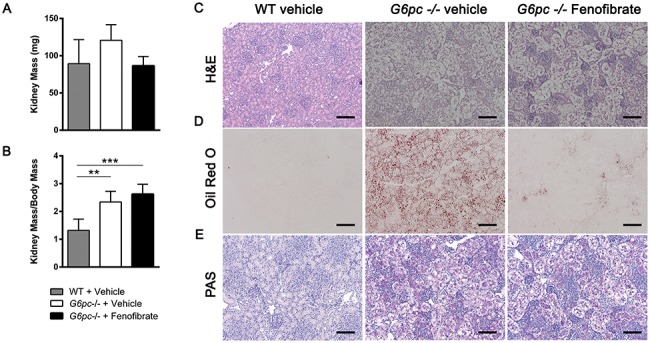
Fenofibrate decreases renal triglycerides. (A) Kidney mass (mg) (n = 4–5). (B) Kidney mass proportional to body mass (n = 4–5). (C) Representative kidney H&E stain from three mice. (D) Representative kidney Oil Red O stain from three mice. (E) Representative liver PAS glycogen stain from three mice. One-way ANOVA with Tukey’s multiple comparison test was performed. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Discussion
GSD Ia is the most common liver glycogen storage disease and is characterized by increased hepatic triglycerides and glycogen, and impaired hepatic autophagy (17, 34). Although nutritional management has enabled survival beyond childhood, most patients develop severe and progressive NAFLD that leads to cirrhosis and increased risk for hepatomas by their 40s. Liver transplantation is the only available treatment for late-stage disease. Currently, there are no pharmacological treatments for GSD Ia, so the identification of approved drugs or development of new drugs are urgently needed. We previously found that neonatal G6pc −/− mice treated with rapamycin, bezafibrate and VK2809 induced autophagy and reduced intrahepatic triglyceride and glycogen content (17, 19, 35). Although these agents highlight the key role of impaired autophagy in the pathogenesis of GSD Ia, their clinical application may be limited since chronic treatment with rapamycin has been associated with insulin resistance and increased inflammation (36, 37), and bezafibrate and VK2809 have not been approved for clinical use in the USA, although bezafibrate is widely used in Europe (38).
Fenofibrate currently is one of the most widely prescribed fibrate drugs for treating hypertriglyceridemia, and has received marketing approval from the FDA. Thus, it can be repurposed and prescribed off-label to treat patients with the metabolic and hepatic/renal complications of GSD Ia (38, 39). Fenofibrate also is a selective agonist for PPARα, the major PPAR isoform likely mediating the beneficial biochemical and physiological effects observed previously with bezafibrate. This selectivity gives fenofibrate the theoretical advantages of having more PPARα-selective effects and less cross-signaling side effects from other PPAR isoforms such as the pan-PPAR agonist bezafibrate would have. In this connection, a recent study showed that chronic administration of fenofibrabe was effective in reducing hepatic triglycerides and glycogen in adult tissue-specific mouse models of GSD Ia (15). However, little is known about the efficacy and mechanism of fenofibrate action on hepatic metabolism in neonates with GSD Ia.
Our study examined the effects of short-term (5 day) administration of fenofibrate on hepatic and renal autophagy and metabolism in neonatal, universal G6pc −/− mice. First, we demonstrated the ability of fenofibrate to quickly decrease hepatic triglyceride and glycogen content by stimulating autophagy in young G6pc −/− mice. Next, we confirmed our previous findings that liver autophagy deficiency was impaired in G6pc −/− mice (17). These findings are particularly relevant to GSD Ia since this condition typically is diagnosed early in children, and early intervention may be important to prevent or delay disease sequelae.
As we observed previously, the restoration of autophagic flux in GSD Ia is likely the mechanism for enhanced degradation of lipids and glycogen in the lysosomes (19). We observed that fenofibrate significantly decreases hepatic triglycerides and glycogen and renal triglycerides (Figs. 1 and 4). G6pc −/− mice have impaired β-oxidation of fatty acids; however, our metabolomic analysis of acylcarnitines strongly suggested that fenofibrate increases fatty acid β-oxidation in the mitochondria since plasma acylcarnitines and expression of genes involved in hepatic β-oxidation in fenofibrate-treated G6pc −/− mice returned to levels not significantly different from those seen in untreated WT controls (Fig. 3). It is noteworthy that the decreases in liver glycogen and triglycerides in G6pc −/− mice following fenofibrate treatment were consistent with our previously reported findings with bezafibrate. Although bezafibrate and fenofibrate can decrease lipid and glycogen accumulations in the liver, gene therapy has been necessary to achieve long-term survival of G6pc −/− mice (40, 41).
Limitations of the current study include the lack of a renal effect. Although triglycerides appeared to be decreased based upon kidney histology, glycogen accumulations and gene expression related to autophagy were not affected by fenofibrate treatment in contrast to an earlier study of chronic fenofibrate administration in adult, kidney-specific G6pc −/− mice with GSD Ia (15). Similarly, a short-term treatment with rapamycin reversed abnormalities of autophagy and decreased glycogen accumulations in the kidney of G6pc −/− mice (42). Despite these limitations, the current study demonstrated important benefits in the liver following short-term treatment with fenofibrate in the severe, congenital G6pc −/− mouse model.
In summary, our study demonstrated the efficacy of fenofibrate to induce autophagy, decrease excessive triglyceride and glycogen accumulation in the liver, and normalize hepatic and plasma acylcarnitines in a neonatal G6pc −/− model of GSD Ia. Since it is already approved by the FDA to lower high cholesterol and triglyceride levels, fenofibrate can potentially be repurposed for the treatment of metabolic complications in adult and young patients with GSD Ia. Clinical studies in GSD Ia patients are needed in order to verify the efficacy of fenofibrate for the treatment of hepatic, renal and metabolic derangements associated with this condition. Finally, since GSD Ia shares similar clinical features with NAFLD, fenofibrate may be a candidate for drug therapy in patients with NAFLD.
Materials and Methods
Study approval
All animals received human care and animal studies were approved by the Duke University Institutional Animal Care and Use Committee under the protocol A038–17-02.
Mouse experiments
Carrier G6pc −/+ mice were bred to produce homozygous G6pc −/− offspring. By day three, G6pc −/− neonates are easily distinguishable from unaffected (+/+ and +/−) littermates based on distended abdomens and size. G6pc −/− mice are treated with 0.1–0.2 mL of 10% dextrose once identified and throughout the course of the experiment. Fenofibrate (Sigma Chemical Co, St. Louis MO, PHR1246) was dissolved in 1:10 DMSO (Sigma, D2650) and 15% kolliphor HS 15 (Sigma, 42 966). At day 5, G6pc −/− mice were treated with either fenofibrate or vehicle by intraperitoneal injection. Administration continued for 5 days with daily injection. Twenty-four hours after the last drug administration, mice were sacrificed according to Duke University IACUC protocol. Blood glucose was measured using AlphaTrak glucometer and test strips (ADW Diabetes, Pompano Beach, FL, 32107). Liver and kidney were weighed and collected by freezing at −80°C, flash freezing in OCT Tissue-Tek (Sakura Finetek USA, Inc., Torrance, CA 4583) for cryostat sectioning, fixed in 10% neutral buffered formalin (VWR, 89370–094) for paraffin embedding and sectioning and fixed in glutaraldehyde 3% sodium phosphate buffer (Electron Microscopy Sciences, Hatfield, PA, 16539) for transmission electron microscopy.
Western blotting
Frozen liver tissue was homogenized in RadioImmuno-Precipitation assay lysis buffer (Thermo-Fisher, Waltham MA, 89900) with a cocktail of protease inhibitors, and protein concentration was determined via Pierce BCA Assay Kit (Thermo-Fisher, 23 225). Samples were diluted to 2.5 mg/μL with water and 4X SDS sample buffer, then boiled for 10 min and stored at −20°C until ready to use. Samples were electrophoresed on housemade or premade SDS-polyacrylamide gels (Bio-Rad Laboratories, Hercules CA, 4568096) and transferred with Bio-Rad Trans-Blot SD cell to polyvinylidene difluoride (PVDF) membrane (Bio-Rad, 1 620 177). Ponceau (Armesco, Framingham MA, 0860) was used to check transfer efficiency. PVDF membrane was blocked in 5% skim milk in PBST, incubated with primary antibody (Table S1) diluted in 5% milk in PBST overnight at 4°C, rinsed in PBST and incubated in HRP secondary antibody for 1 hour at room temperature. Blots were developed with SuperSignal® Chemiluminescent substrate (Thermo-Fisher, 34 080, 34 095) and visualized on Bio-Rad ChemiDoc™ Imaging System. Band intensity was quantified using Bio-Rad Image Lab.
Quantitative PCR
RNA was isolated from frozen liver and kidney tissue using Trizol Reagent (Thermo-Fisher, 15 596 026) and chloroform extraction. cDNA was reverse transcribed from this RNA with RevertAid first strand cDNA synthesis kit (Thermo-Fisher, K1622) and stored at −20°C. Master mix with iTaq™ Universal SYBR® Green Supermix (Bio-Rad, 1 725 124), forward and reverse primers (Table S2), and H2O was added to a 384-well plates (Thermo-Fisher, 4 309 849). Genes were quantified in triplicate with 1 μL of cDNA on the QuantStudio 6 Flex (Applied Biosystems from Thermo-Fischer). Graphs represent the average ΔΔCt for each experimental group (n = 3–5, indicated on each graph).
Biochemical assays
Glycogen quantification was performed on liver homogenate as previously described (43). Triglyceride concentration in the liver was determined using a colorimetric assay kit (Cayman Chemical Company, Ann Arbor MI, 10010303) following manufacturer’s instructions.
Histology
Oil Red O: Oil Red O (ORO, 00625, Sigma) staining was performed on OCT flash frozen liver and kidney samples (8 μm thick) stored at −80°C. ORO stock solution was prepared (2.5 g of ORO in 400 mL of 99% (vol/vol) isopropyl alcohol) and dissolved by magnetic stirring for 2 hours at room temperature. Before using, ORO stock solution was diluted with distilled water (18 mL of ORO stock solution to 12 mL of water) and the diluted solution was stirred for 10 min at 4°C and filtered with filter paper to remove precipitates. The sections were incubated with ORO dilute solution for 5–10 min at room temperature and washed under running tap water for up to 30 min. The sections were mounted with a water-soluble mounting medium. Images were taken using a Keyence BZ-X710 (Keyence America, Itasca IL).
H&E staining. Formalin-fixed, paraffin-embedded tissues sections (4 μm thick) were stained with hematoxylin (Sigma, MHS16) for 2 min, reacted with bluing reagent (Sigma, S5134) and stained with eosin-Y (Sigma, 318 906) for 2 min. The slides were mounted and images were taken using a Keyence BZ-X710 (Keyence America).
PAS staining for glycogen. Formalin-fixed tissues were embedded in paraffin and sectioned (thickness 6 μm). The sections were deparaffinized and hydrated to water and oxidized in 0.5% Periodic acid (Sigma, P7875) solution for 5 min. The slides were rinsed in distilled water and stained in Schiff reagent (Millipore from Sigma, 2818–71) for 15 min, followed by counterstaining with H&E. Slides were mounted and images were taken using a Keyence BZ-X710 microscope (Keyence America).
Transmission electron microscopy (TEM)
Glutaraldehyde-fixed samples were washed in three 20 min changes of cold 67 mM Sorensen’s buffer, pH 7.2, post-fixed in 2% OsO4 in the same buffer for 1 hour, cold and in the dark, followed by three additional buffer washes as above. Samples were then dehydrated in a graded ethanol series (30%, 50%, 70%, 95% and 3 X 100%) and infiltrated and embedded in Spurr’s resin (Ladd Research, Williston, VT) at 70oC.
Cured blocks were hand-trimmed with a single-edge razor blade and thin-sectioned at 80 nm using a Leica EM UCT6rt Ultramicrotome (Leica Microsystems Inc., Buffalo Grove, IL) and a diamond knife (Diatome, USA, Hatfield, PA). Sections were double-stained with 4% aqueous uranyl acetate for 1 hour in the dark at room temperature, followed by 4 min in Reynold’s lead citrate.
Grids were viewed in a JEOL JEM-1200EX TEM (JEOL USA, Peabody, MA) at 80 kV. Images were digitally acquired using a Gatan Model 795 ES 1000 W Erlangshen CCD camera, and minimally processed in Photoshop CS6.
Acylcarnitine profiling
Standards included the following: Acetyl-L-carnitine-(N-methyl-d3) HCl (d3-C2), tetradecanoyl-L-carnitine-(N-methyl-d3) HCl (d3-C14), palmitoyl-L-carnitine-(N-methyl-d3) HCl (d3-C16) (Sigma Chemical Co, St. Louis, MO); propionyl-L-carnitine-(N-methyl-d3) HCl ((d3-C3) and L-glutarylcarnitine-d6 chloride (d6-C5-DC) (Toronto Research Co, North York, ON, Canada); butyryl-L-carnitine-(N-methyl-d3) HCl, octanoyl-L-carnitine-(N-methyl-d3) HCl, d6-glutarylcarnitine (C5-DC) and unlabelled acylcarnitines (NSK-B and NSK-B-G1 kits) (Cambridge Isotopes Labs, Tewksbury, MA); 3 M methanolic HCl (Supelco, Bellefonte, PA); Seracon II CD 1 L stripped, delipidated plasma (SeraCare Life Sciences, Inc. Milford, MA); all other reagents were purchased from VWR (Radnor, PA).
Plasma acylcarnitines were analyzed by tandem mass spectrometry based on previously published method (49) with modifications. Briefly, 5 or 10 L of ethylenediaminetetraacetic acid treated plasma was combined with a deuterated internal standard mixture containing 12.5,6.3, 2.5, 2.5, 2.5, 2.5 and 5 pmols of d3-C2, d3-C3, d3-C4, d3-C8, d6-C5DC, d3-C14, d3-C16 acylcarnitine species in 50:50 (v/v) methanol: diH2O, respectively. Calibrators were prepared using delipidated plasma spiked with unlabelled acylcarnitine species. Calibrators were prepared for C2, C3, C4, C5, C5-DC, 3-hydroxyisovalerylcarnitine (C5-OH), C8, C12, C14, C16 and C18 with concentrations from 1.1 to 37 μmol/L (C2), 0.2 to 7.5 μmol/L (C3, C4, C5, C5-DC, C8, C12, C14) and 0.4 to 15 μmol/L (C16, C18). Samples were deproteinized using methanol, and an aliquot of the supernatant liquid was dried under nitrogen and derivatized by incubation with 50 μL 3 M methanolic HCl at 50oC for 15 min. The reagent was removed by drying under nitrogen and samples were reconstituted in 85:15 (v/v) methanol:diH2O.
Acylcarnitines were analyzed on an Acquity Xevo-TQ (Waters Corp, Milford, MA) LC-MS/MS system with electrospray ionization and flow injection analysis. About, 5 μL sample was injected onto the instrument and 80:20 (v/v) methanol:diH2O was used as the mobile phase. Methylated acylcarnitine species were detected using a precursor ion of m/z 99 from 200 to 500 amu. Acylcarnitine species were quantified using the ion intensity ratio of analyte to the assigned deuterated internal standard for that analyte. Concentrations were determined using response factors generated by the calibration curves for the analyte, or for the analyte closest in mass to the derivatized acylcarnitine species.
Statistics
Statistical analysis was performed in GraphPad Prism 6 (GraphPad Software, La Jolla, CA). Samples were analyzed for normality by the Shapiro–Wilks normality test, and none showed abnormal distribution. Statistical significance was determined using a one-way ANOVA with Tukey’s multiple comparison test. The statistical significance of comparisons is indicated as *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Author Contributions
ZAY designed and conducted experiments, analyzed data, wrote the paper and edited the manuscript. HRK performed histology and analyzed the data. LRW performed the mouse experiment and analyzed data. BHB provided interpretation of electron microscopic data. PMY provided critical editing of the manuscript. SPY supervised plasma acylcarnitine analysis. DDK supervised the studies and wrote the paper.
Funding
Singapore NMRC (NMRC/CIRG/1457/2016; NRMC/CSA/0054/2013 to P.M.Y.) and National Institute of Diabetes and Digestive and Kidney Diseases (R01DK105434), as well as by support from the Alice and Y.T. Chen Center for Genetics and Genomics to D.D.K. H.-R.K. was funded by a Pfizer NC Biotechnology Gene Therapy Fellowship (Agreement GTF-A-4026). D.D.K. has received a grant from Viking Therapeutics. He received an honorarium and grant support in the past from Genzyme Sanofi.
Supplementary Material
Acknowledgements
We would like to thank Seth Allen for assistance in preparing western blot samples and running biochemical assays. We are grateful to Valerie Lapham at North Carolina State University for her contribution to TEM and Songtao Li for mouse management. We thank Dr Janice Chou of the National Institute of Health and Human Development for providing G6pc -/- mice. We would also like to acknowledge inspiration and support from Dr Emory and Mrs Mary Chapman and their son Christopher, and from Dr John and Mrs Michelle Kelly.
Conflicts of Interest statement. D.D.K. served on a data and safety monitoring board for Baxter International. None of the other authors had a conflict of interest to declare.
References
- 1. von Gierke E. (1929) Liver and kidney in glycogen storage disease. Beitr. Pathol. Anat., 82, 497–513. [Google Scholar]
- 2. Chou J.Y. and Mansfield B.C. (2008) Mutations in the glucose-6-phosphatase-α (G6PC) gene that cause type Ia glycogen storage disease. Hum. Mut., 29, 921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bruni N., Rajas F., Montano S., Chevalier-Porst F., Maire I. and Mithieux G. (1999) Enzymatic characterization of four new mutations in the glucose-6 phosphatase (G6PC) gene which cause glycogen storage disease type 1a. Ann. Hum. Genet., 63, 141–146. [DOI] [PubMed] [Google Scholar]
- 4. Takahashi K., Akanuma J., Matsubara Y., Fujii K., Kure S., Suzuki Y., Wataya K., Sakamoto O., Aoki Y. and Ogasawara M. (2000) Heterogeneous mutations in the glucose-6-phosphatase gene in Japanese patients with glycogen storage disease type Ia. Am. J. Med. Genet., 92, 90–94. [PubMed] [Google Scholar]
- 5. Lei K.-J., Shelly L.L., Pan C.-J., Sidbury J.B. and Chou J.Y. (1993) Mutations in the glucose-6-phosphatase gene that cause glycogen storage disease type 1a. Science, 262, 580–583. [DOI] [PubMed] [Google Scholar]
- 6. Greene H.L., Swift L.L. and Knapp H.R. (1991) Hyperlipidemia and fatty acid composition in patients treated for type IA glycogen storage disease. J. Pediatr., 119, 398–403. [DOI] [PubMed] [Google Scholar]
- 7. Kishnani P.S., Austin S.L., Abdenur J.E., Arn P., Bali D.S., Boney A., Chung W.K., Dagli A.I., Dale D., Koeberl D. et al. (2014) Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genet. Med., 16, e1. [DOI] [PubMed] [Google Scholar]
- 8. Leonard J.V. and Dunger D.B. (1978) Hypoglycaemia complicating feeding regimens for glycogen-storage disease. Lancet, 2, 1203–1204. [DOI] [PubMed] [Google Scholar]
- 9. Wang D.Q., Fiske L.M., Carreras C.T. and Weinstein D.A. (2011) Natural history of hepatocellular adenoma formation in glycogen storage disease type I. J. Pediatr., 159, 442–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dambska M., Labrador E.B., Kuo C.L. and Weinstein D.A. (2017) Prevention of complications in glycogen storage disease type Ia with optimization of metabolic control. Pediatr. Diabetes, 18, 327–331. [DOI] [PubMed] [Google Scholar]
- 11. Okechuku G.O., Shoemaker L.R., Dambska M., Brown L.M., Mathew J. and Weinstein D.A. (2017) Tight metabolic control plus ACE inhibitor therapy improves GSD I nephropathy. J. Inherit. Metab. Dis., 40, 703–708. [DOI] [PubMed] [Google Scholar]
- 12. Peeks F., Steunenberg T.A.H., de Boer F., Rubio-Gozalbo M.E., Williams M., Burghard R., Rajas F., Oosterveer M.H., Weinstein D.A. and Derks T.G.J. (2017) Clinical and biochemical heterogeneity between patients with glycogen storage disease type IA: the added value of CUSUM for metabolic control. J. Inherit. Metab. Dis., 40, 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bianchi L. (1993) Glycogen storage disease I and hepatocellular tumours. Eur. J. Pediatr., 152(Suppl 1), S63–S70. [DOI] [PubMed] [Google Scholar]
- 14. Franco L.M., Krishnamurthy V., Bali D., Weinstein D.A., Arn P., Clary B., Boney A., Sullivan J., Frush D.P., Chen Y.T. et al. (2005) Hepatocellular carcinoma in glycogen storage disease type Ia: a case series. J. Inherit. Metab. Dis., 28, 153–162. [DOI] [PubMed] [Google Scholar]
- 15. Browning J.D., Szczepaniak L.S., Dobbins R., Nuremberg P., Horton J.D., Cohen J.C., Grundy S.M. and Hobbs H.H. (2004) Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology, 40, 1387–1395. [DOI] [PubMed] [Google Scholar]
- 16. Monteillet L., Gjorgjieva M., Silva M., Verzieux V., Imikirene L., Duchampt A., Guillou H., Mithieux G. and Rajas F. (2018) Intracellular lipids are an independent cause of liver injury and chronic kidney disease in non alcoholic fatty liver disease-like context. Mol. Metab., 16, 100–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Farah B.L., Landau D.J., Sinha R.A., Brooks E.D., Wu Y., Fung S.Y.S., Tanaka T., Hirayama M., Bay B.H., Koeberl D.D. et al. (2016) Induction of autophagy improves hepatic lipid metabolism in glucose-6-phosphatase deficiency. J. Hepatol., 64, 370–379. [DOI] [PubMed] [Google Scholar]
- 18. Singh R., Kaushik S., Wang Y., Xiang Y., Novak I., Komatsu M., Tanaka K., Cuervo A.M. and Czaja M.J. (2009) Autophagy regulates lipid metabolism. Nature, 458, 1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Waskowicz L.R., Zhou J., Landau D.J., Brooks E.D., Lim A., Yavarow Z.A., Kudo T., Zhang H., Wu Y., Grant S. et al. (2019) Bezafibrate induces autophagy and improves hepatic lipid metabolism in glycogen storage disease type Ia. Hum. Mol. Genet., 28, 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Braissant O., Foufelle F., Scotto C., Dauca M. and Wahli W. (1996) Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, −beta, and -gamma in the adult rat. Endocrinology, 137, 354–366. [DOI] [PubMed] [Google Scholar]
- 21. Hashimoto T., Cook W.S., Qi C., Yeldandi A.V., Reddy J.K. and Rao M.S. (2000) Defect in peroxisome proliferator-activated receptor alpha-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J. Biol. Chem., 275, 28918–28928. [DOI] [PubMed] [Google Scholar]
- 22. Xu J., Xiao G., Trujillo C., Chang V., Blanco L., Joseph S.B., Bassilian S., Saad M.F., Tontonoz P., Lee W.N. et al. (2002) Peroxisome proliferator-activated receptor alpha (PPARalpha) influences substrate utilization for hepatic glucose production. J. Biol. Chem., 277, 50237–50244. [DOI] [PubMed] [Google Scholar]
- 23. Desvergne B. and Wahli W. (1999) Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr. Rev., 20, 649–688. [DOI] [PubMed] [Google Scholar]
- 24. Issemann I. and Green S. (1990) Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature, 347, 645–650. [DOI] [PubMed] [Google Scholar]
- 25. Linden D., Alsterholm M., Wennbo H. and Oscarsson J. (2001) PPARalpha deficiency increases secretion and serum levels of apolipoprotein B-containing lipoproteins. J. Lipid. Res., 42, 1831–1840. [PubMed] [Google Scholar]
- 26. Schoonjans K., Staels B. and Auwerx J. (1996) The peroxisome proliferator activated receptors (PPARS) and their effects on lipid metabolism and adipocyte differentiation. Biochim. Biophys. Acta., 1302, 93–109. [DOI] [PubMed] [Google Scholar]
- 27. Sun B., Li S., Yang L., Damodaran T., Desai D., Diehl A.M., Alzate O. and Koeberl D.D. (2009) Activation of glycolysis and apoptosis in glycogen storage disease type Ia. Mol. Genet. Metab., 97, 267–271. [DOI] [PubMed] [Google Scholar]
- 28. Kim S.Y. and Bae Y.S. (2009) Cell death and stress signaling in glycogen storage disease type I. Mol. Cells, 28, 139–148. [DOI] [PubMed] [Google Scholar]
- 29. Rinaldo P., Cowan T.M. and Matern D. (2008) Acylcarnitine profile analysis. Genet. Med., 10, 151–156. [DOI] [PubMed] [Google Scholar]
- 30. Gulick T., Cresci S., Caira T., Moore D.D. and Kelly D.P. (1994) The peroxisome proliferator-activated receptor regulates mitochondrial fatty acid oxidative enzyme gene expression. Proc. Natl. Acad. Sci. U. S. A., 91, 11012–11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aoyama T., Peters J.M., Iritani N., Nakajima T., Furihata K., Hashimoto T. and Gonzalez F.J. (1998) Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha). J. Biol. Chem., 273, 5678–5684. [DOI] [PubMed] [Google Scholar]
- 32. Rakhshandehroo M., Hooiveld G., Muller M. and Kersten S. (2009) Comparative analysis of gene regulation by the transcription factor PPARalpha between mouse and human. PLoS One, 4, e6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McCoin C.S., Knotts T.A. and Adams S.H. (2015) Acylcarnitines--old actors auditioning for new roles in metabolic physiology. Nat. Rev. Endocrinol., 11, 617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Froissart R., Piraud M., Boudjemline A.M., Vianey-Saban C., Petit F., Hubert-Buron A., Eberschweiler P.T., Gajdos V. and Labrune P. (2011) Glucose-6-phosphatase deficiency. Orphanet J. Rare. Dis., 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou J., Waskowicz L.R., Lim A., Liao X.H., Lian B., Masamune H., Refetoff S., Tran B., Koeberl D.D. and Yen P.M. (2019) A liver-specific thyromimetic, VK2809, decreases hepatosteatosis in glycogen storage disease type Ia (GSD Ia). Thyroid, 29, 1158–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schindler C.E., Partap U., Patchen B.K. and Swoap S.J. (2014) Chronic rapamycin treatment causes diabetes in male mice. Am. J. Physiol. Regul. Integr. Comp. Physiol., 307, R434–R443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shameem R., Lacouture M. and Wu S. (2015) Incidence and risk of high-grade stomatitis with mTOR inhibitors in cancer patients. Cancer Invest., 33, 70–77. [DOI] [PubMed] [Google Scholar]
- 38. Ashburn T.T. and Thor K.B. (2004) Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov., 3, 673–683. [DOI] [PubMed] [Google Scholar]
- 39. Perkins K.A., Karelitz J.L., Michael V.C., Fromuth M., Conklin C.A., Chengappa K.N., Hope C. and Lerman C. (2016) Initial evaluation of Fenofibrate for efficacy in aiding smoking abstinence. Nicotine Tob. Res., 18, 74–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koeberl D.D., Sun B.D., Damodaran T.V., Brown T., Millington D.S., Benjamin D.K. Jr., Bird A., Schneider A., Hillman S., Jackson M. et al. (2006) Early, sustained efficacy of adeno-associated virus vector-mediated gene therapy in glycogen storage disease type Ia. Gene Ther., 13, 1281–1289. [DOI] [PubMed] [Google Scholar]
- 41. Koeberl D.D., Pinto C., Sun B., Li S., Kozink D.M., Benjamin D.K. Jr., Demaster A.K., Kruse M.A., Vaughn V., Hillman S. et al. (2008) AAV vector-mediated reversal of hypoglycemia in canine and murine glycogen storage disease type Ia. Mol. Ther., 16, 665–672. [DOI] [PubMed] [Google Scholar]
- 42. Farah B.L., Landau D.J., Wu Y., Sinha R.A., Loh A., Bay B.H., Koeberl D.D. and Yen P.M. (2017) Renal endoplasmic reticulum stress is coupled to impaired autophagy in a mouse model of GSD Ia. Mol. Genet. Metab., 122, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang G., Young S.P., Bali D., Hutt J., Li S., Benson J. and Koeberl D.D. (2014) Assessment of toxicity and biodistribution of recombinant AAV8 vector-mediated immunomodulatory gene therapy in mice with Pompe disease. Mol. Ther. Methods. Clin. Dev., 1, 14018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.