

Abstract
Kabuki syndrome is an autosomal dominant developmental disorder with high similarities to CHARGE syndrome. It is characterized by a typical facial gestalt in combination with short stature, intellectual disability, skeletal findings and additional features like cardiac and urogenital malformations, cleft palate, hearing loss and ophthalmological anomalies. The major cause of Kabuki syndrome are mutations in KMT2D, a gene encoding a histone H3 lysine 4 (H3K4) methyltransferase belonging to the group of chromatin modifiers. Here we provide evidence that Kabuki syndrome is a neurocrestopathy, by showing that Kmt2d loss-of-function inhibits specific steps of neural crest (NC) development. Using the Xenopus model system, we find that Kmt2d loss-of-function recapitulates major features of Kabuki syndrome including severe craniofacial malformations. A detailed marker analysis revealed defects in NC formation as well as migration. Transplantation experiments confirm that Kmt2d function is required in NC cells. Furthermore, analyzing in vivo and in vitro NC migration behavior demonstrates that Kmt2d is necessary for cell dispersion but not protrusion formation of migrating NC cells. Importantly, Kmt2d knockdown correlates with a decrease in H3K4 monomethylation and H3K27 acetylation supporting a role of Kmt2d in the transcriptional activation of target genes. Consistently, using a candidate approach, we find that Kmt2d loss-of-function inhibits Xenopus Sema3F expression, and overexpression of Sema3F can partially rescue Kmt2d loss-of-function defects. Taken together, our data reveal novel functions of Kmt2d in multiple steps of NC development and support the hypothesis that major features of Kabuki syndrome are caused by defects in NC development.
Introduction
Neural crest (NC) cells form a migratory cell population that is unique to vertebrates and contributes to a large number of different organ systems. Various human syndromes or congenital diseases have been linked to defects in NC development and subsumed under the term neurocrestopathies (1). These conditions can be caused by defects at any step of NC development including specification, migration and differentiation. For example, CHARGE syndrome, a sporadic, autosomal dominant malformation disorder that encompasses symptoms like coloboma, heart defects, atresia of the choanae, retarded growth and development, genital hypoplasia, ear anomalies and deafness (2), has been linked to defects in NC development (3–6). Through molecular and functional analyses of CHD7, the gene mutated in CHARGE patients, it was shown in Xenopus that all major CHARGE symptoms can be attributed to defects in NC development (4). Kabuki syndrome (OMIM 147920), another developmental disorder characterized by the combination of a typical facial gestalt, short stature, intellectual disability, skeletal findings, dermatoglyphic anomalies and variable additional features (7,8), shows a striking phenotypic overlap to CHARGE syndrome. Especially in young children, the clinical distinction between CHARGE and Kabuki syndrome can be challenging, because a multitude of organ malformations might fit to the spectrum of both syndromes, and the characteristic facial gestalt of Kabuki syndrome is often not fully evident in newborn patients. Recently, we and others found further evidence supporting the link between CHARGE and Kabuki syndrome (9–12) suggesting that Kabuki syndrome—like CHARGE syndrome—might belong to the group of neurocrestopathies.
The major genetic cause of Kabuki syndrome are heterozygous mutations in the KMT2D gene (13). In humans, KMT2D maps to chromosome 12q13.12 and consists of 54 coding exons (MIM 602113), encoding a 600 kDa large protein (human: 5262 amino acids). KMT2D is a chromatin modifier expressed widely during embryonic development (14), and homozygous knockout in mouse embryos causes lethality at embryonic day 9.5 (15). KMT2D belongs to the SET1 family of histone methyltransferases, which are responsible for transferring up to three methyl groups from a cofactor (AdoMet) to lysine 4 on histone H3 (16,17). SET1 family enzymes exert their function through the catalytic SET domain (18,19). H3K4 methylation occurs at enhancers and promoters as well as in gene bodies and has been associated with active transcription (20–23). Differential methylation states of H3K4 are related to particular cellular functions (17). Several studies in different model systems, including Drosophila, MEFs, MESCs and human cells, find evidence that KMT2D primarily functions as a monomethyltransferase at enhancers, acting as a coactivator of genes crucial for embryonic development and differentiation (15,18,24–28). Recent findings also suggest that KMT2D can facilitate p300 H3K27 acetylation activity as well as Pol II binding to enhancers, further promoting a fully active enhancer environment to boost the transcription of target genes (26,28).
The pathomechanism of Kabuki syndrome as well as the impact of various cellular signaling pathways are still largely undiscovered. Consistent with the heterogeneous clinical symptoms of Kabuki syndrome, a range of KMT2D target genes and related pathways—including HOX genes as well as members of the MAPK, Notch, canonical Wnt and retinoic acid signaling pathways—have been identified, pointing to a role of KMT2D in multiple signaling events during embryonic development (16,27,29–33). Some of the most characteristic Kabuki syndrome features have been analyzed in mouse and zebrafish models, providing evidence that KMT2D is crucial for the formation of craniofacial structures (34,35), heart development (35–37) and brain formation (34,35). Moreover, KMT2D knockout mice displayed a shorter body axis as well as defects in adipocyte and myocyte differentiation (15,34). Previously, we have shown that Xenopus Kmt2d is required for the formation and differentiation of cardiac tissue, which is reminiscent of the congenital heart defects frequently observed in Kabuki patients (37). However, the effect of KMT2D loss-of-function on NC cell development has not been investigated in more detail.
In this study, we used loss-of-function approaches to analyze the role of Kmt2d during Xenopus NC development. Our results demonstrate that major clinical symptoms of Kabuki syndrome can be recapitulated using the Xenopus model system. Furthermore, we provide evidence that Kmt2d is required for NC formation and migration, supporting the hypothesis that Kabuki syndrome belongs to the neurocrestopathies.
Results
Kabuki-like craniofacial malformations can be reproduced in Xenopus laevis embryos
To investigate a potential NC contribution to the Kabuki syndrome phenotype, we asked if we can recapitulate the craniofacial malformations, typically observed in patients, in KMT2D-deprived embryos. Therefore, Xenopus embryos were injected with an antisense Kmt2d morpholino oligonucleotide (Kmt2d MO) in one blastomere at the two-cell stage and phenotypically analyzed for craniofacial defects at tadpole stages. Indeed, knockdown of Kmt2d caused a severe reduction of craniofacial structures—characterized by frontal protrusion, reduced facial width or microcephaly—on the injected side (Fig. 1A and B). In addition, eye formation was impaired in the majority of morphant embryos, as indicated by smaller or absent eyes (Fig. 1A). In contrast, embryos injected with a five base-pair mismatch MO (mmMO) did not display significant defects. Alcian Blue staining of the cranial cartilage supports this observation. Kmt2d loss-of-function led to a marked reduction of the mandibular, hyoid and branchial cartilage, while injection of the mmMO did not affect cartilage formation (Fig. 1C). To confirm the specificity of the observed craniofacial defects, we used a splice-blocking MO in addition to the translation-blocking MO. The splice MO targets the Kmt2d-SET domain and efficiently blocks splicing of the kmt2d RNA (we have previously demonstrated this in 37). In this study, Xenopus embryos injected with the splice MO exhibited the same craniofacial phenotype as embryos treated with the translation-blocking MO (Fig. 1A and B), providing evidence for a specific effect of the Kmt2d loss-of-function on Xenopus craniofacial development. Taken together with our previous observation that Kmt2d loss-of-function causes cardiac defects (37), these results demonstrate that major Kabuki syndrome symptoms can be reproduced in Xenopus.
Figure 1.
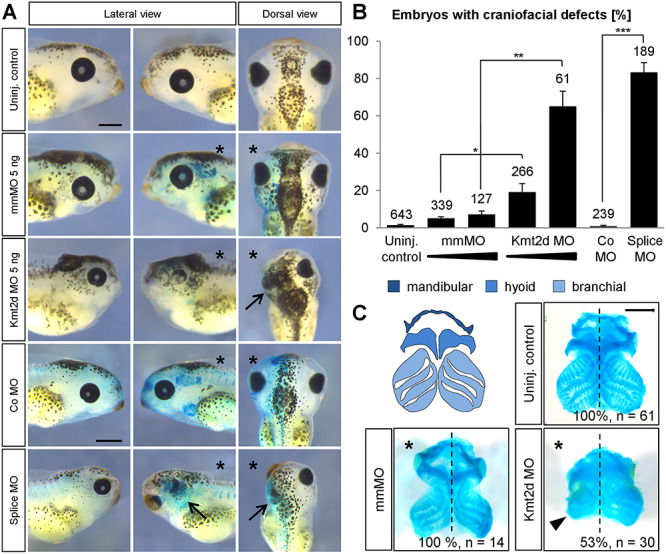
Kmt2d loss-of-function causes craniofacial malformations. (A) Phenotypic analysis of craniofacial defects at stage 42. Embryos were injected with 2.5–5 ng of mmMO or Kmt2d MO or 5 ng control (Co) MO or splice MO into one blastomere at the two-cell stage. 150 pg lacZ mRNA was co-injected as a lineage tracer. The injected side of the embryo is marked (asterisk). Both the Kmt2d MO and the splice MO cause severe craniofacial malformations (arrows), compared to mmMO- and Co MO-treated controls. Scale bars = 500 μm. (B) Graph summarizing the results from at least four independent experiments; ±SEM and the number of analyzed embryos are given for each condition. A two-tailed unpaired Student’s t-test was applied. (C) Cartilage staining. Schematic representation of the cranial cartilage structures of a Xenopus laevis embryo (upper left panel). Embryos were injected with 2.5 ng mmMO or Kmt2d MO in combination with 50 pg mGFP mRNA as a lineage tracer into one blastomere at the two-cell stage, and the cranial cartilage was visualized by Alcian Blue staining at stage 44. Kmt2d knockdown leads to reduced cranial cartilage (arrowhead) on the injected side (asterisk). Scale bar = 500 μm.
Kmt2d is expressed in NC cells and required for their migration
As Kmt2d loss-of-function leads to severe craniofacial defects, we further dissected its function in NC development. Previously we have shown that kmt2d RNA is already present in the anterior neural plate at early neurula stages and continues to be expressed in pre- and migratory NC cells (37). Here we also analyzed the temporal expression pattern by RT-PCR and detected high levels of maternal RNA with a slight decrease in expression during gastrulation and a peak of kmt2d expression at late neurula stages (Supplementary Material, Fig. S1A). Using transverse sections through the head region of a stage 29 embryo subjected to kmt2d whole mount in situ hybridization, we further confirmed kmt2d expression in the eyes, the branchial arches, the developing brain as well as in the otic placode and ear vesicles (Supplementary Material, Fig. S1B–D). Collectively, these data confirm the expression of kmt2d in the NC throughout pre-migratory as well as migratory stages, indicating a role for Kmt2d during several steps of NC development.
Based on our hypothesis that loss-of-function of Kmt2d affects NC development, we investigated whether Kmt2d is required for normal NC migration. Embryos were injected with Kmt2d MOs and NC migration was analyzed by whole mount in situ hybridization against the NC marker twist. Injection of the translation-blocking as well as the splice-blocking MO led to a significant inhibition of NC migration in Xenopus embryos (Fig. 2A and B and Supplementary Material, Fig. S2A and B). We also observed a dose-dependent effect on the NC phenotype. While the NC cells primarily exhibited a reduced migration distance in embryos injected with 2.5 ng Kmt2d MO, we additionally noticed a reduction of twist-positive cells upon injection of 5 ng Kmt2d MO (Fig. 2A). The specificity of the Kmt2d knockdown NC phenotype was further confirmed through co-injection of the Kmt2d MO and mRNA of a human KMT2D fragment, containing several interaction domains as well as the catalytic SET domain (Fig. 2C–E). Overexpression of the human KMT2D fragment (SET) led to a partial rescue of the NC migration defects caused by the Kmt2d MO (Fig. 2D and E). In addition, the binding specificity of the Kmt2d MO was verified by Western Blot analysis of an HA-tagged Kmt2d fragment comprising the 5′UTR, translation start site and complete MO binding site (Fig. 2F and G). Injection of the Kmt2d MO inhibited the expression of the construct, while its expression was unaffected by the 5 base pair mismatch MO. Taken together, these data provide evidence that Kmt2d is required for in vivo NC migration.
Figure 2.
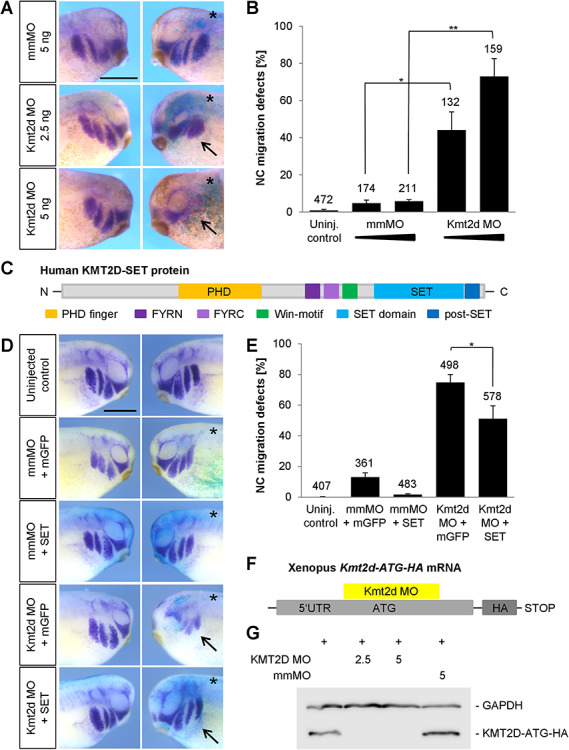
Kmt2d is required for cranial NC cell migration. (A) Embryos were injected with 2.5 or 5 ng mmMO or Kmt2d MO and 100 pg lacZ mRNA into one blastomere at the two-cell stage. At stage 27 NC migration was analyzed by twist whole mount in situ hybridization. Kmt2d loss-of-function leads to an inhibition of NC migration (arrows), compared to mismatch controls. The injected side of the embryo is marked (asterisk). Scale bar = 500 μm. (B) Graph summarizing the data from three independent experiments; ±SEM and the number of analyzed embryos are indicated for each condition. A two-tailed unpaired Student’s t-test was applied. (C–E) Rescue experiments. (C) Schematic view of motifs and functional domains of the human KMT2D-SET construct used for rescue experiments. (D) Embryos were injected into one blastomere at the two-cell stage with 3 ng mmMO or Kmt2d MO in combination with 400–500 pg mGFP or KMT2D-SET mRNA and 100 pg lacZ as a lineage tracer. NC migration was analyzed by twist in situ hybridization at stage 25. Overexpression of the human KMT2D-SET construct partially restores NC migration in Kmt2d morphants, compared to embryos injected with Kmt2d MO and mGFP mRNA (arrows). The injected side of the embryos is marked (asterisk). Error bar = 500 μm. (E) Graph summarizing the percentage of embryos with twist defects from six independent experiments; ±SEM and number of analyzed embryos are given for each condition. A two-tailed unpaired Student’s t-test was applied. (F) Schematic view of the Xenopus laevis Kmt2d-ATG-HA mRNA which contains the Kmt2d MO binding site. (G) To analyze the binding efficiency of the Kmt2d MO, embryos were injected at the one-cell stage with 1 ng of Xenopus Kmt2d-ATG-HA mRNA either alone or in combination with 2.5–5 ng mmMO or the translation-blocking Kmt2d MO. Protein extracts were prepared at stage 20, and the expression of Kmt2d-ATG-HA was assessed by Western Blot analysis. Injection of 2.5 and 5 ng Kmt2d MO led to a depletion of the Kmt2d-ATG-HA protein, while the expression was unaffected upon co-injection of 5 ng mmMO. GAPDH expression was used as a loading control.
NC cells require Kmt2d for cell dispersion but not for cell protrusion formation
As the loss-of-function data suggest a critical role for Kmt2d during NC migration, we analyzed if Kmt2d has a cell-autonomous function in NC cells. Therefore, we performed transplantation assays and grafted GFP-labeled pre-migratory NC tissue from mmMO- or Kmt2d MO-injected embryos into wild-type donor embryos, whose NC cells had been removed. At tailbud stages, the migration behavior of the morphant NC cells was phenotypically evaluated. Indeed, migration of the Kmt2d-depleted NC cells was significantly inhibited, while the majority of mmMO-injected transplants showed normal migration (Fig. 3A–C), suggesting that Kmt2d function is cell-autonomously required in NC cells.
Figure 3.
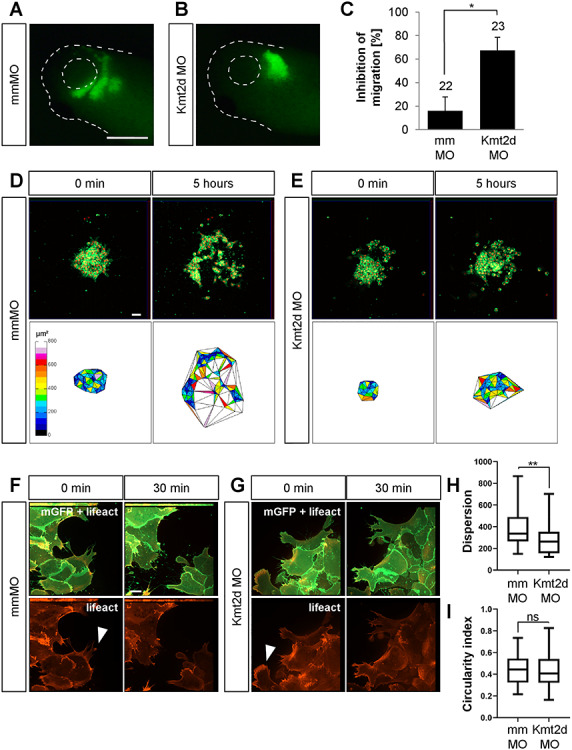
Kmt2d is required for cell dispersion in explanted NC cells. (A and B) Transplantation assay. Embryos were injected with 2.5 ng mmMO or Kmt2d MO in combination with 50 pg mGFP mRNA into one blastomere at the two-cell stage. Fluorescently labeled NC cells from injected embryos were transplanted into a wild-type host embryo, and their migration behavior was analyzed at stage 27. Scale bar = 500 μm. (A) While transplanted mismatch control NC cells migrate normally, (B) the migration of Kmt2d-depleted NC cells was inhibited. (C) Graph summarizing three independent experiments with ±SEM and the number of analyzed embryos given for each condition. A two-tailed unpaired Student’s t-test was applied. (D–G) Explant assay. (D and E) Embryos were injected with 2.5 ng mmMO or Kmt2d MO in combination with 50 pg mGFP and 250 pg H2B-mCherry mRNA to label the cell membrane and the nucleus, respectively. NC cells were explanted, cultured on fibronectin, and their migration behavior was imaged for 8 h. Cell dispersion was assessed using ImageJ. Scale bar = 50 μm. (D) mmMO NC explants show normal cell dispersion after 5 h of cultivation, while (E) Kmt2d MO-treated cells display inhibited dispersion. (F and G) Embryos were injected with 2.5 ng mmMO or Kmt2d MO in combination with 50 pg mGFP and 300 pg lifeact-RFP mRNA to label the cell membrane and f-actin, respectively. Higher magnification of explanted NC cells reveals normal protrusion formation (arrowheads) in both (F) mmMO- and (G) Kmt2d MO-treated NC cells. Scale bar = 10 μm. (H) Dispersion of mmMO- and Kmt2d MO-injected NC explants defined as the mean triangle size per explant (μm2) calculated by Delaunay triangulation after 5 h of cultivation in three independent experiments. Box plots show median and 25th to 75th percentiles, and Whiskers display min to max values. Mann-Whitney U test was applied (n = 33 mmMO explants and n = 28 Kmt2d MO explants analyzed). (I) Analysis of circularity showed no difference in cell shape between mmMO- and Kmt2d MO-treated NC cells (complete circle = 1). Box plots show median and 25th to 75th percentiles. Whiskers display min to max values. Mann-Whitney U test was applied (n = 53 cells analyzed from 16 mmMO NC explants; n = 105 cells analyzed from 27 Kmt2d MO NC explants; data from three independent experiments).
To further dissect Kmt2d function on a cellular level, we analyzed NC migration in vitro. Xenopus embryos were injected with 2.5 ng Kmt2d MO in combination with mGFP and H2B-mCherry mRNA to mark the membrane and nucleus, respectively. NC cells were explanted at stage 17 followed by cultivation on a fibronectin matrix for 8 h. Live-cell imaging of Kmt2d-deprived NC cells revealed impaired cell dispersion. After 5 h the area of dispersion, determined by Delaunay triangulation followed by calculation of the mean triangle size, was strongly reduced in Kmt2d morphant explants compared to mmMO-injected controls (Fig. 3D, E and H and Supplementary Material, Movies S1 and S2). Interestingly, like control cells, Kmt2d-depleted NC cells were able to form cell protrusions. However, they were not able to leave the explant or seemed to die rapidly upon detaching from the cell cluster, as inferred from their round cell shape. To examine the cell shape and the actin cytoskeleton in further detail, we used expression of lifeact-RFP (Fig. 3F and G). In line with the formation of morphologically normal protrusions, the actin cytoskeleton of NC cells was not affected by Kmt2d loss-of-function (Fig. 3G) compared to mismatch controls (Fig. 3F). Analysis of the cell shape also showed no change in the circularity of NC cells upon Kmt2d knockdown (Fig. 3I). In addition, manual tracking of single cells indicated that there is no change in the velocity and persistence of migrating NC cells upon Kmt2d knockdown (Supplementary Material, Fig. S3 and Movies S3 and S4). These results strongly suggest that Kmt2d morphant NC cells are generally motile, and the migration defect is likely caused by disturbed dispersion rather than cell motility.
Loss of Kmt2d causes defects in neural plate border establishment and NC specification
To characterize the role of Kmt2d in NC development, we also analyzed its function at earlier stages of NC development. First, we analyzed the effect of Kmt2d loss-of-function on the expression of the NC specification markers foxd3, slug/snai2 and twist at stage 17–18. Marker analysis via whole mount in situ hybridization revealed a dose-dependent reduction of foxd3, slug/snai2 and twist expression at pre-migratory stages (Fig. 4B–D and F–H), which could be reproduced using the splice-blocking MO (Supplementary Material, Figs S2D, F and S4). Notably, injection of 5 ng translation-blocking Kmt2d MO led to a severe depletion of NC specifiers and an increase in apoptotic cells (Supplementary Material, Fig. S6), while 2.5 ng Kmt2d MO had only minor effects on marker gene expression. These observations indicate that Kmt2d may also have earlier functions in NC development. Thus, we analyzed if the neural plate border (NPB), which corresponds to the territory between the neural plate and the epidermis and defines the prospective NC (38), is correctly induced. Expression of the NPB marker genes pax3 and zic1 was analyzed via whole mount in situ hybridization and revealed a dose-dependent impairment of NPB patterning in Kmt2d MO-injected embryos. Injection of 5 ng translation-blocking Kmt2d MO led to a significant expansion of pax3 and zic1 expression (Fig. 4A and Supplementary Material, Fig. S5), which could also be reproduced using the splice-blocking MO (Supplementary Material, Fig. S2C and E). Notably, the majority of embryos injected with 2.5 ng MO showed normal NPB patterning, suggesting a dose-dependent effect. Transversal sectioning of embryos stained for zic1 confirmed that the NPB territory is broader in Kmt2d morphants compared to mismatch control animals (Supplementary Material, Fig. S5E and F). The broadening of NPB marker expression was accompanied by a reduced epidermal keratin signal in the dorsal most part of the epidermis (Fig. 5A–E) and an expansion of sox2 positive cells in the neural plate (Fig. 5F–J), further indicating an early defect in ectodermal patterning at high MO dosage. In summary, these data support a role for Kmt2d not only during NC migration but also in the establishment of the NPB as well as NC specification.
Figure 4.
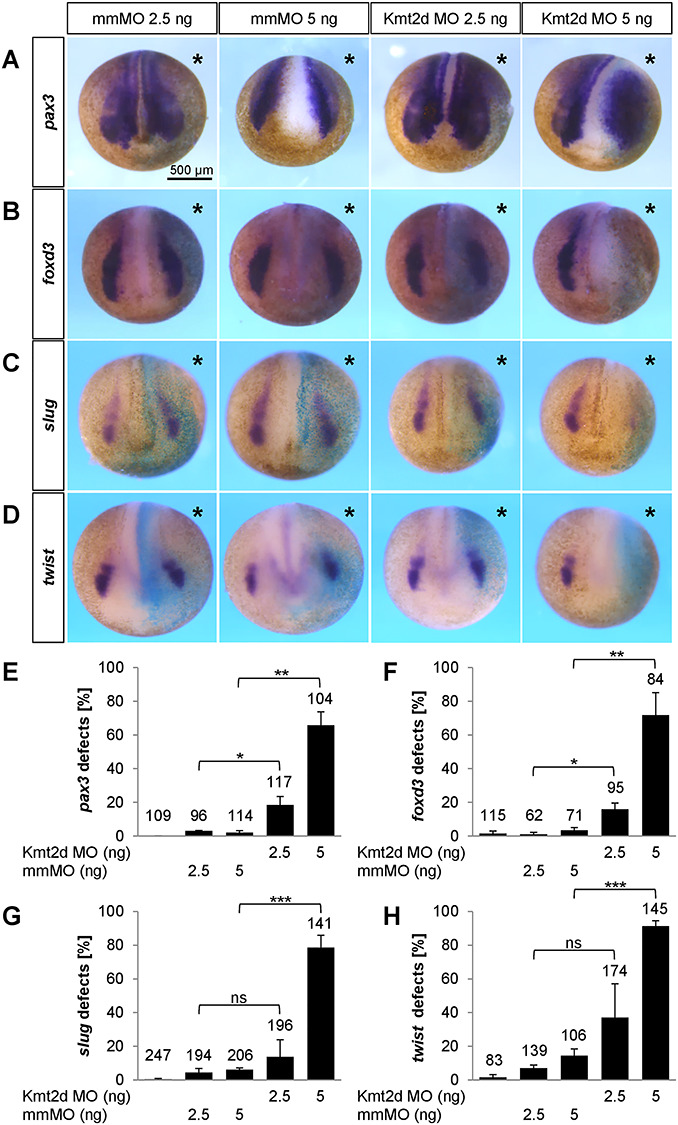
Kmt2d knockdown affects NPB formation and NC specification in a dose-dependent manner. (A–D) Embryos were injected with 2.5 or 5 ng mmMO or Kmt2d MO into one blastomere at the two-cell stage. 100 pg lacZ mRNA were used for lineage tracing. At neurula stages, the expression of several NC marker genes was analyzed by whole mount in situ hybridization. Injection of 5 ng Kmt2d MO leads to (A) a broader pax3 expression in the NPB region and (B) a severe reduction of foxd3, (C) slug/snai2 and (D) twist expression in the pre-migratory NC. (E–H) Graphs summarizing the results from three independent experiments for each NC marker gene analyzed in (A–D). Kmt2d loss-of-function affects the expression of (E) pax3, (F) foxd3, (G) slug/snai2 and (H) twist in a dose-dependent manner; ±SEM and the number of analyzed embryos are indicated for each condition. A two-tailed unpaired Student’s t-test was applied.
Figure 5.
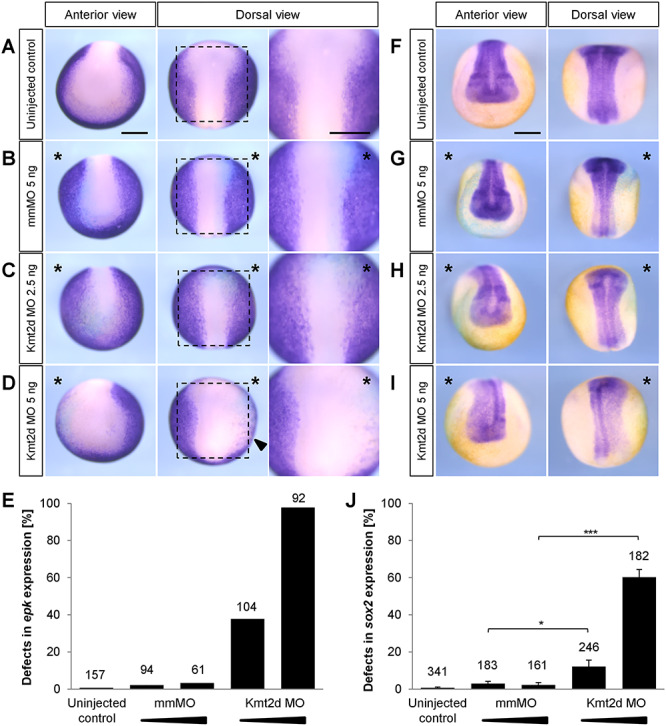
Kmt2d loss-of-function leads to an expansion of the neural plate as indicated by altered epidermal keratin and sox2 expression. Two-cell stage embryos were injected with 2.5 or 5 ng of mismatch (mm) MO or Kmt2d MO in combination with 100 pg lacZ mRNA as a lineage tracer. The injected side of the embryo is marked (asterisk). Scale bars = 500 μm. (A–D) In situ hybridization for the non-neural ectoderm marker epidermal keratin (epk) was performed at stage 14. (A) Uninjected control embryos and (B) mmMO treated controls show normal epk expression in the epidermal ectoderm. (C) While 2.5 ng Kmt2d MO has no effect in the majority of embryos, (D) 5 ng Kmt2d MO leads to a dorsolaterally reduced epk expression. (E) The graph summarizes the mean percentage of embryos with epk expression defect from two independent experiments. The total number of analyzed embryos is indicated for each condition. (F–I) In situ hybridization for the neural plate marker sox2 was performed at stage 19. (F) Uninjected control embryos and (G) mmMO treated controls show normal sox2 expression in the neural plate. (H) While 2.5 ng Kmt2d MO has no effect in the majority of embryos, (I) 5 ng Kmt2d MO leads to an expansion of the sox2-positive territory. (J) Data from four independent experiments. Graph summarizes the mean percentage of embryos with sox2 expression defect. The total number of analyzed embryos and ±SEM are indicated for each condition. A two-tailed unpaired Student’s t-test was applied.
Kmt2d loss-of-function correlates with a decrease in H3K4me1 and H3K27ac
As a SET1 family histone methyltransferase (HMT), the primary function of KMT2D is to catalyze the transfer of up to three methyl groups to lysine 4 on histone H3 (H3K4). Thus, we investigated if Kmt2d loss-of-function may affect H3K4 methylation levels in Xenopus embryos. To analyze the effect on the H3K4 methylation status, embryos were injected with mmMO or the translation-blocking Kmt2d MO, and protein extracts were prepared at stage 20. We performed Western Blot analysis using an antibody against the H3K4me1 mark and detected a significant decrease in bulk H3K4me1 levels in Kmt2d MO-injected embryos, compared to controls (Fig. 6A and B). In contrast, the bulk H3K4me3 level was not significantly affected (data not shown), suggesting that in Xenopus Kmt2d functions primarily as a monomethyltransferase. As H3K4 monomethylation in combination with H3K27 acetylation is associated with active enhancers, we further analyzed if H3K27 acetylation is also affected by Kmt2d loss-of-function. Western Blot analysis using an antibody against H3K27ac revealed a decrease in the H3K27ac level upon injection of the Kmt2d MO (Fig. 6C and D). Taken together, our results link the Kmt2d knockdown-mediated NC defects to the regulation of epigenetic marks associated with transcriptional activation and confirm a role for Kmt2d in the establishment of active enhancer marks.
Figure 6.
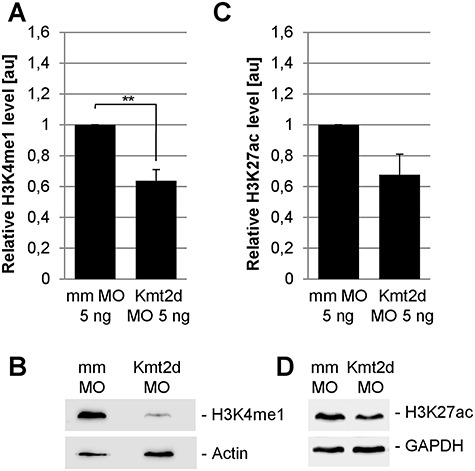
Loss of Kmt2d leads to reduced H3K4me1 and H3K27ac levels in Xenopus embryos. One-cell stage Xenopus embryos were injected with 5 ng mmMO or Kmt2d MO or 7.5 ng control MO or splice MO in combination with 50 pg mGFP mRNA as a lineage tracer. Protein extracts were prepared at neurula stage 20. (A and B) Western Blot analysis of the H3K4me1 mark reveals a reduction of the H3K4me1 level in Kmt2d MO treated embryos compared to controls. Graph summarizes three independent experiments and ±SEM is indicated. A two-tailed unpaired Student’s t-test was applied. (C and D) H3K27ac Western Blot analysis shows that injection of the Kmt2d MO leads to a decrease in the H3K27ac level, compared to mismatch controls. Graph summarizes the results from four independent experiments and ±SEM is indicated; P-value = 0.0519 in a two-tailed unpaired Student’s t-test.
Sema3F expression is affected by Kmt2d loss-of-function
In search of genes regulated by Kmt2d, we used a candidate approach. Class 3 semaphorins have been shown to play a role in the pathogenesis of CHARGE syndrome (9,39,40), a disease with phenotypic and molecular links to Kabuki syndrome (41). Furthermore, Sema3F was recently identified as an UTX/KDM6A target gene, a histone H3 lysine 27 demethylase that is mutated in a small percentage of Kabuki syndrome patients (42). In Xenopus Sema3F is expressed in the branchial arches and required for NC migration (43,44). Thus, we tested if the expression of Sema3F is affected by Kmt2d loss-of-function. To this end we injected mmMO or Kmt2d MO at the two-cell stage and performed in situ hybridization for sema3f. Indeed, we observed that Kmt2d loss-of-function inhibited Sema3F expression on the injected side, while it was unaffected in mismatch control embryos (Fig. 7A and B). To analyze if Semaphorin overexpression is able to rescue the Kmt2d morphant phenotype, two-cell stage embryos were co-injected with either mmMO or Kmt2d MO in combination with Sema3F mRNA or mGFP mRNA as a negative control. NC migration was analyzed by in situ hybridization for the NC marker twist. Interestingly, we observed that Sema3F overexpression partially rescued the Kmt2d loss-of-function NC migration defects (Fig. 7C and D) indicating that Sema3F is likely a Kmt2d target gene.
Figure 7.
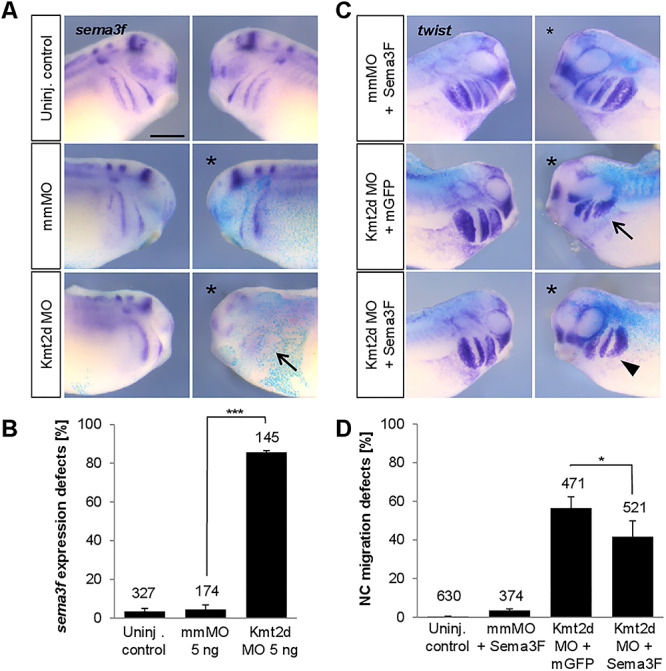
Kmt2d loss-of-function inhibits Sema3F expression and ectopic Sema3F can partially substitute for Kmt2d. (A) Sema3F expression is reduced in Kmt2d morphants. Embryos were co-injected with 5 ng mmMO or Kmt2d MO and lacZ mRNA as a lineage tracer. In situ hybridization for sema3f confirms that Kmt2d knockdown inhibits Sema3F expression in Xenopus embryos (arrow). (B) Graph summarizing the data from three independent experiments. Total number of analyzed embryos and ±SEM are indicated for each condition. A two-tailed unpaired Student’s t-test was applied. (C) Two-cell stage embryos were co-injected with 2.5–3 ng Kmt2d MO and 250 pg sema3F mRNA or mGFP mRNA and subjected to twist in situ hybridization. Overexpression of Sema3F results in a partial rescue of NC migration in Kmt2d-depleted embryos. (D) Graph summarizing the data from six independent experiments. Total number of analyzed embryos and ±SEM are indicated for each condition. A Fisher’s exact test was applied.
Discussion
The developmental mechanisms leading to the distinct features of Kabuki syndrome are largely unknown, although the complex clinical symptoms as well as the KMT2D mutational landscape in patients have been well described (45–50). Therefore, functional studies addressing the role of KMT2D during embryonic development are required to shed light on the complexity of the Kabuki syndrome pathogenesis. Here we used the Xenopus system to analyze the effect of Kmt2d knockdown on NC development. Our findings confirm that Kabuki syndrome shows central features of a neurocrestopathy, since multiple steps of NC development, including the development of NC derivatives such as the cranial cartilage, are severely disrupted upon Kmt2d knockdown.
Although non-NC tissues likely also contribute to the etiology of Kabuki symptoms, we suggest that the most defining features are caused by defects in NC development. This is further supported by a recent study addressing the function of UTX/KDM6A, an H3K27 demethylase that removes repressive chromatin modifications and is mutated in a low percentage of Kabuki syndrome patients (46,51–53). Shpargel et al. showed that the majority of clinical features can be modeled in mice carrying a NC-specific deletion of UTX/KDM6A. They demonstrate that KDM6A plays a role in cranial NC viability at post-migratory stages and is required for the formation of craniofacial structures (42). Here we show that Kmt2d loss-of-function affects both NC specification and migration in a dose-dependent manner. Low doses of the Kmt2d MO affect later processes like NC migration and cartilage differentiation, while high doses inhibit NPB patterning. Likely the low doses are a better representation of the heterozygous and sometimes also mosaic KMT2D mutations (54) found in Kabuki syndrome patients.
Using transplantation as well as explantation techniques, we demonstrate that Kmt2d has NC-specific functions. Interestingly, although in vivo as well as in vitro NC migration is severely inhibited by Kmt2d loss-of-function, morphant NC cells are able to form cell protrusions. Thus, the migration defects are likely caused by an inhibition of cell dispersion. Since we find no evidence for an impaired cell motility, the underlying cause for this phenotype may be an altered cell adhesion profile in Kmt2d knockdown NC cells. Consistent with this, it was shown in HeLa cells that a KMT2D-containing complex regulates genes involved in cell adhesion and cytoskeletal organization. Furthermore, loss-of-function was accompanied by changes in the spreading behavior of cells (16) supporting our findings in Xenopus NC cells.
The direct or indirect involvement of chromatin effectors like KMT2D in the regulation of diverse signaling pathways makes it a major challenge to dissect the pathomechanism underlying the Kabuki syndrome phenotype. In line with a number of recent reports (25,27), our Xenopus data indicate that Kmt2d primarily acts as a H3K4 monomethyltransferase in vivo. The combination of the histone modifications H3K4me1 and H3K27ac represents a characteristic enhancer signature (23,55,56). Interestingly, the Kmt2d knockdown phenotype in Xenopus embryos was correlated with a decrease in the H3K4me1 level as well as a reduction of the H3K27ac mark. The concomitant effect of Kmt2d knockdown on both of these histone modifications is consistent with other recent reports stating that KMT2D and its homologue KMT2C cooperatively activate enhancers together with the H3K27 acetyltransferase p300 (15,26,57,58). Moreover, a crucial role for KMT2D in the activation of developmental enhancers during cellular differentiation has been reported in various cell types (15,28,59,60). Taken together, these data suggest that the KMT2D loss-of-function phenotype may be caused by an altered epigenetic regulation at distal regulatory elements of genes required for NC development (Fig. 8).
Figure 8.
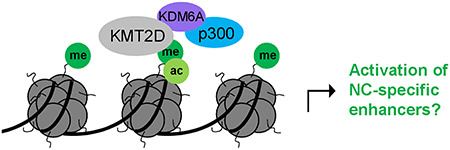
Working model of how KMT2D may function to activate NC-specific enhancers. KMT2D deposits H3K4me1 marks at genomic targets. p300 interaction is mediated via the KMT2D/KDM6A complex, finally leading to H3K27 acetylation and a fully active enhancer state.
Recently, a growing body of evidence indicates the importance of chromatin-regulating factors for NC development (reviewed in 61). During NC formation, a certain level of histone deacetylase (HDAC) activity is required in order to retain pluripotency characteristics, which is a prerequisite for the acquisition of a NC cell fate. Accordingly, HDAC1/2 have been shown to repress the expression of lineage-specifying genes by maintaining low levels of H3K9ac and H3K27ac at promoters and enhancers, respectively (62). On the other hand, NC-related enhancers have to be specifically activated via the deposition of H3K4me1 and H3K27ac marks in order to promote the expression of NC specification and migration genes (55). For the chromatin remodeling factor CHD7, the gene mutated in most CHARGE patients, Bajpai and colleagues demonstrated an important role in the direct binding and activation of slug/snai2, sox9 and twist enhancers or other regulatory elements (4). Furthermore, the NC specifier and transcription factor Slug/Snai2 interacts with the Sin3A/HDAC complex, thereby mediating the repression of E-cadherin and facilitating epithelial-to-mesenchymal transition (63,64). At later stages, HDAC8 activity was required to promote the differentiation of NC cells into craniofacial cartilage and bone (65). Collectively, these studies demonstrate that epigenetic regulation is involved at all steps of NC development, which is in line with our findings that Kmt2d is crucial for the formation and proper migration of NC cells in vivo.
Previously, a number of reports demonstrated a phenotypic and molecular link between Kabuki and CHARGE syndrome (9–11,41,66), which represents another neurocrestopathy (67). Indeed, our data shows that the craniofacial dysmorphism observed in Kmt2d morphants is highly reminiscent of the Xenopus CHARGE phenotype observed in embryos injected with Chd7 MO (4,39). Previously, we showed that Sema3A is regulated by CHD7 and overexpression of Sema3A in Xenopus embryos partially rescued the Chd7 loss-of-function phenotype (39). In analogy to these findings, we demonstrate here that Kmt2d regulates Sema3F expression and Sema3F overexpression partially rescues Kmt2d loss-of-function. Thus, CHD7 and KMT2D positively regulate the expression of members of the class 3 semaphorins, Sema3A and Sema3F, which both are required for the directional migration of Xenopus cranial NC cells (43). These data support the hypothesis that the phenotypic overlap of CHARGE and Kabuki syndrome is based on a common molecular link.
Taken together, our data provides new evidence that Kabuki syndrome belongs to the class of neurocrestopathies. This should highlight the importance of future studies focusing on the mechanism of action of KMT2D in NC cells.
Materials and Methods
Xenopus microinjection
Staging of Xenopus embryos was performed following the normal table by Nieuwkoop and Faber (68). All procedures were performed according to the German animal use and care law (Tierschutzgesetz) and approved by the German state administration Hesse (Regierungspräsidium Giessen).
Female adult frogs were injected with 500 units of hCG (Ovogest, MSD animal health) on the day before egg laying. Embryos were obtained by in vitro fertilization and cultivated in 0.1× MBS. All microinjections were performed into one blastomere at the two-cell stage except for one-cell stage injection of embryos used for Western Blot analysis. Two previously described MOs against Xenopus kmt2d were used in this study, one translation-blocking MO targeting the 5′UTR of the kmt2d transcript (5′-GCCTTTTGTTCATCCATTCCTGTTC-3′) and a splice-blocking MO (5′-TATCGCCAAACATACCTCCTCTCCT-3′) resulting in aberrant splicing of the precursor mRNA of kmt2d (37). A 5 bp mismatch MO (5′-GaCTTTTcTTCATaCATTaCTcTTC-3′) served as a control for the translation-blocking Kmt2d MO, and a standard control MO (5′-CCTCTTACCTCAGTTACAATTTATA-3′) was used as a control for the splice-blocking MO. All MOs were obtained from Gene Tools (Philomath, USA). Concentrations of 2.5–5 ng of the translation-blocking Kmt2d MO or mismatch MO and 5–7.5 ng of the splice-blocking or control MO were injected, as indicated in the figure legends. lacZ mRNA was co-injected as a lineage tracer in experiments requiring phenotypic analysis of craniofacial defects or in situ hybridization. These embryos were fixed in MEMFA and subjected to XGal staining, followed by re-fixation overnight. For Alcian Blue staining of embryos, NC explantation, Western Blot analysis and TUNEL assays, 50 pg mGFP mRNA was used as a lineage tracer, and the embryos were processed as described below. Capped sense mRNA of the following constructs was synthesized using the mMESSAGE mMACHINE™ SP6 or T7 Transcription Kit (Invitrogen): mGFP (69), H2B-mCherry (70), Lifeact-RFP (71), Semaphorin 3F (44), hKMT2D-SET (this study) and Kmt2d-ATG-HA (this study). The Kmt2d-ATG-HA construct used to test for morpholino binding was amplified from Xenopus laevis cDNA using the following primers (5′ → 3′): fw_CAGGGAATTCGGACGGGAGGATCACAGATT and rv_GCCTCTCGAGTGCTGACCAAGATGCACACC. The fragment was digested with EcoRI and XhoI and ligated into the digested pCS2 + -HA vector using T4 DNA ligase (Thermo Fisher Scientific), followed by transformation of competent E. coli cells. The human KMT2D-SET rescue construct consists of 2067 bp of the KMT2D 3′ coding region containing a PHD finger domain, the FYRN and FYRC regions, the WDR5-interacting (Win) motif as well as the catalytic SET domain and post-SET domain. The construct was cloned into the pcDNA3.1 vector.
Whole mount in situ hybridization and Alcian Blue cartilage staining
Whole mount in situ hybridization of Xenopus embryos was performed as previously described (72). To visualize kmt2d expression in the head region of Xenopus tailbud stage embryos, wild-type albino embryos were analyzed by whole mount in situ hybridization using a previously described construct (37). Subsequently, 50 μm sections were prepared using a Leica VT1000S vibratome and mounted in Mowiol.
For cartilage staining Xenopus embryos were cultivated until stage 42–44 and fixed in Dent’s fixative for 1 h. They were rinsed in EtOH/acetic acid (80:20) and transferred into the Alcian Blue staining solution containing 0.05% Alcian Blue in EtOH/acetic acid (80:20) for 12–24 h at RT. Xenopus tadpoles were then cleared in EtOH/acetic acid (80:20) for 24 h to remove background staining. Afterwards, the embryos were rehydrated in 1× PTw containing 1% KOH (w/v) and bleached in 1% KOH/3% (v/v) H2O2/1× PTw solution. After rinsing in 1% KOH/1× PTw, the embryos were transferred into glycerol/1× PTw (1:1) and immediately used for dissection and documentation of the cartilage, using a Leica M165 FC stereomicroscope.
RT-PCR of Xenopus Kmt2d
For time course analysis of the Kmt2d expression level throughout embryonic development, total RNA was isolated from wild-type Xenopus embryos of different stages. Therefore, embryos were frozen in liquid nitrogen and lysed using a 30G syringe. RNA isolation was performed with the illustra RNAspin Mini RNA Isolation Kit (GE Healthcare) according to the manufacturer’s protocol, including DNase I treatment. cDNA was synthesized with the RevertAid Reverse Transcriptase (Thermo Fisher Scientific). cDNA of embryos from three different batches was pooled for each stage condition, and PCRs were conducted with 28 cycles using the DreamTaq DNA Polymerase (Thermo Fisher Scientific). The following primer sequences were used to amplify a 287 bp Kmt2d fragment starting 7 bp downstream of the TSS: KMT2D_fw 5′CAAAAGGCATCCAGCGAGGA3′ and KMT2D_rv 5′GTTTCTTGCACCTGAATGGGGC3′. Amplification of histone H4 served as a control (H4_fw 5′CGGGATAACATTCAGGGTATCACT3′, H4_rv 5′ATCCATGGCGGTAACTGTCTTCCT3′). PCR products were loaded on an agarose gel and documented via the Odyssey® FC Imaging System (LI-COR Biosciences).
NC explants, transplants, and live-cell imaging
To analyze the migration behavior of NC cells by live-cell imaging, embryos were injected with 50 pg mGFP and 250 pg H2B-mCherry or 300 pg lifeact-RFP mRNA in combination with 2.5 ng mmMO or Kmt2d MO into one blastomere at the two-cell stage. Xenopus NC explants were dissected at stage 17 and cultivated on fibronectin (1:100 in sterile 1× PBS, Sigma-Aldrich) in 0.8× MBS. The NC cells were left to migrate for 4–5 h at room temperature for the detailed analysis of protrusion formation (63× oil objective). The circularity of NC cells was measured by manually selecting border cells in ImageJ. To document the migration behavior of NC cells in overview movies, they were cultivated overnight at 18°C (10× objective). The dispersion of NC cells was analyzed by Delaunay triangulation using the dispersion tool in ImageJ (plugin kindly provided by Roberto Mayor). Therefore, the nuclei of living NC cells were marked at the starting point of migration and after 5 h of cultivation. Dispersion was quantified by calculating the mean triangle size between individual nuclei of NC cells for each explant after 5 h of cultivation (73). To measure the velocity of migrating single cells, NC cells were explanted in 0.8× MBS at neurula stage 17. Explants were immediately transferred into calcium-free 0.8× MBS and incubated for 2 min to dissociate the cells and obtain small clusters. After dissociation, NC cells were transferred into a fibronectin-coated chamber and cultivated at room temperature in 0.8× MBS. Migration of individual NC cells was documented for 45 min by live-cell imaging (25× oil objective), and single cells were tracked using the manual tracking plugin in ImageJ, followed by calculation of cell velocity and persistence (directionality) via the chemotaxis tool plugin (ImageJ). All live-cell imaging was performed using spinning disk confocal microscopy (Axio Observer Z1, Zeiss).
For NC transplantation, Xenopus embryos were injected with 2.5 ng Kmt2d MO or 5 bp mmMO in combination with mGFP mRNA in one blastomere at the two-cell stage. At stage 17 the fluorescently labeled premigratory NC cells of MO-injected embryos were transplanted into a wild-type host embryo as previously described (74). The migration behavior of transplanted NC cells was documented and assessed at stage 25 using a Leica M165 FC stereomicroscope.
Preparation of protein extracts and Western Blot
Protein extracts of 20 stage 20 embryos per condition were prepared in Co-IP buffer (50 mm Tris pH 7.5, 150 mm NaCl, 0.5% (v/v) NP-40) containing 1× Complete Protease Inhibitor (Roche). The respective volume of Laemmli loading buffer was added and the samples were incubated at 95°C for 3 min to promote denaturation. Protein extracts were loaded onto a discontinuous 15% SDS-PAGE gel, followed by semidry blotting (Trans-Blot® Turbo™ Transfer System, Bio-Rad) onto a nitrocellulose membrane and blocking in 5% milk/TBST for 1 h at RT. The following antibodies were used for detection: anti-actin (1:5000, Millipore MAB1501), anti-GAPDH (1:4000, Thermo Fisher Scientific #AM4300), anti-H3K4me1 (1:1000, Abcam ab8895), anti-H3K4me3 (1:1000, Cell Signaling Technology #9751), anti-H3K27ac (1:750, Abcam ab4729), anti-HA (1:1000, BioLegend 901 503), anti-mouse-HRP (1:4000, Santa Cruz sc-516 102) and anti-rabbit-HRP (1:2000, Cell Signaling Technology #7074). Chemiluminescence was detected and analyzed using the SuperSignal™ West Dura Extended Duration Substrate (Thermo Fisher Scientific) and the LI-COR FC Odyssey® Imaging System.
TUNEL assay
To detect and label apoptotic cells, terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick end labeling (TUNEL) was performed on neurula embryos. Xenopus embryos were co-injected with 5 ng Kmt2d or 5 bp mismatch MO and mGFP RNA as a lineage tracer in one blastomere at the two-cell stage. Stage 18–20 embryos were fixed for 3 h in MEMFA and bleached in 4% H2O2 solution, followed by storage in 100% MeOH overnight. The embryos were rehydrated, washed in 1× PTw and incubated in 1× TdT buffer (in 1× PBS) for 1.5 h before treatment with the terminal deoxynucleotidyl transferase (Invitrogen, 1:100 in 1× TdT buffer/1× PBS) together with Dig-11-UTP (1:500, Roche) for 2 days at RT. To stop the reaction, the samples were incubated in 1 mm EDTA/1× PBS for 2 h at 65°C, followed by several washing steps in 1× PTw. After blocking in 2% blocking reagent (Roche) and 20% horse serum (life technologies) in 1× MAB for at least 60 min, the antibody reaction (anti-digoxigenin 1:5000 in blocking solution, Roche) was performed for 4 h at RT. NBT/BCIP (Roche) was diluted in alkaline phosphatase buffer and used for the staining reaction. Embryos were considered to have an apoptotic phenotype if the number of TUNEL-positive cells was at least three times higher on the injected side compared to the uninjected half.
Statistical analysis
The number of biological replicates (N) and the total number of analyzed embryos, explants or cells (n) are indicated for each experiment and condition, respectively. The P-values were calculated by applying either a two-tailed unpaired Student’s t-test (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001), a Fisher’s exact test or a Mann-Whitney U test as indicated in the figure legends. Standard errors of the mean (±SEM) are given in the graphs.
Supplementary Material
Acknowledgements
We thank Silke Pauli for critical reading and valuable discussion of the manuscript, Roser Ufartes for providing the KMT2D rescue plasmid and Roberto Mayor for the ImageJ plugin used for analyzing NC cell dispersion. Furthermore, we thank Christiane Rohrbach and Ingrid Bohl-Maser for excellent technical assistance.
Funding
German Research Foundation (DFG) (BO1978/5-1).
Conflict of interest statement.
The authors declare no conflict of interest.
References
- 1. Bolande R.P. (1974) Neurocristopathies—unifying concept of disease arising in neural crest maldevelopment. Hum. Pathol., 5, 409–429. [DOI] [PubMed] [Google Scholar]
- 2. Pagon R.A., Graham J.M. Jr., Zonana J. and Yong S.L. (1981) Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J. Pediatr., 99, 223–227. [DOI] [PubMed] [Google Scholar]
- 3. Sperry E.D., Hurd E.A., Durham M.A., Reamer E.N., Stein A.B. and Martin D.M. (2014) The chromatin remodeling protein CHD7, mutated in CHARGE syndrome, is necessary for proper craniofacial and tracheal development. Dev. Dyn., 243, 1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bajpai R., Chen D.A., Rada-Iglesias A., Zhang J., Xiong Y., Helms J., Chang C.P., Zhao Y., Swigut T. and Wysocka J. (2010) CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature, 463, 958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Patten S.A., Jacobs-McDaniels N.L., Zaouter C., Drapeau P., Albertson R.C. and Moldovan F. (2012) Role of Chd7 in zebrafish: a model for CHARGE syndrome. PLoS One, 7, e31650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Asad Z., Pandey A., Babu A., Sun Y., Shevade K., Kapoor S., Ullah I., Ranjan S., Scaria V., Bajpai R. et al. (2016) Rescue of neural crest-derived phenotypes in a zebrafish CHARGE model by Sox10 downregulation. Hum. Mol. Genet., 25, 3539–3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kuroki Y., Suzuki Y., Chyo H., Hata A. and Matsui I. (1981) A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J. Pediatr., 99, 570–573. [DOI] [PubMed] [Google Scholar]
- 8. Niikawa N., Matsuura N., Fukushima Y., Ohsawa T. and Kajii T. (1981) Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J. Pediatr., 99, 565–569. [DOI] [PubMed] [Google Scholar]
- 9. Schulz Y., Wehner P., Opitz L., Salinas-Riester G., Bongers E.M., van Ravenswaaij-Arts C.M., Wincent J., Schoumans J., Kohlhase J., Borchers A. et al. (2014) CHD7, the gene mutated in CHARGE syndrome, regulates genes involved in neural crest cell guidance. Hum. Genet., 133, 997–1009. [DOI] [PubMed] [Google Scholar]
- 10. Verhagen J.M., Oostdijk W., Terwisscha van Scheltinga C.E., Schalij-Delfos N.E. and van Bever Y. (2014) An unusual presentation of kabuki syndrome: clinical overlap with CHARGE syndrome. Eur. J. Med. Genet., 57, 510–512. [DOI] [PubMed] [Google Scholar]
- 11. Sakata S., Okada S., Aoyama K., Hara K., Tani C., Kagawa R., Utsunomiya-Nakamura A., Miyagawa S., Ogata T., Mizuno H. et al. (2017) Individual clinically diagnosed with CHARGE syndrome but with a mutation in KMT2D, a gene associated with kabuki syndrome: a case report. Front. Genet., 8, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Badalato L., Farhan S.M., Dilliott A.A., Care4Rare Canada C., Bulman D.E., Hegele R.A. and Goobie S.L. (2017) KMT2D p.Gln3575His segregating in a family with autosomal dominant choanal atresia strengthens the kabuki/CHARGE connection. Am. J. Med. Genet. A, 173, 183–189. [DOI] [PubMed] [Google Scholar]
- 13. Ng S.B., Bigham A.W., Buckingham K.J., Hannibal M.C., McMillin M.J., Gildersleeve H.I., Beck A.E., Tabor H.K., Cooper G.M., Mefford H.C. et al. (2010) Exome sequencing identifies MLL2 mutations as a cause of kabuki syndrome. Nat. Genet., 42, 790–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Prasad R., Zhadanov A.B., Sedkov Y., Bullrich F., Druck T., Rallapalli R., Yano T., Alder H., Croce C.M., Huebner K. et al. (1997) Structure and expression pattern of human ALR, a novel gene with strong homology to ALL-1 involved in acute leukemia and to drosophila trithorax. Oncogene, 15, 549–560. [DOI] [PubMed] [Google Scholar]
- 15. Lee J.E., Wang C., Xu S., Cho Y.W., Wang L., Feng X., Baldridge A., Sartorelli V., Zhuang L., Peng W. et al. (2013) H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. elife, 2, e01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Issaeva I., Zonis Y., Rozovskaia T., Orlovsky K., Croce C.M., Nakamura T., Mazo A., Eisenbach L. and Canaani E. (2007) Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol. Cell Biol., 27, 1889–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shilatifard A. (2008) Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr. Opin. Cell Biol., 20, 341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Y., Mittal A., Reid J., Reich S., Gamblin S.J. and Wilson J.R. (2015) Evolving catalytic properties of the MLL family SET domain. Structure, 23, 1921–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dillon S.C., Zhang X., Trievel R.C. and Cheng X. (2005) The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol., 6, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heintzman N.D., Stuart R.K., Hon G., Fu Y., Ching C.W., Hawkins R.D., Barrera L.O., Van Calcar S., Qu C., Ching K.A. et al. (2007) Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet., 39, 311–318. [DOI] [PubMed] [Google Scholar]
- 21. Ruthenburg A.J., Allis C.D. and Wysocka J. (2007) Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol. Cell, 25, 15–30. [DOI] [PubMed] [Google Scholar]
- 22. Santos-Rosa H., Schneider R., Bannister A.J., Sherriff J., Bernstein B.E., Emre N.C., Schreiber S.L., Mellor J. and Kouzarides T. (2002) Active genes are tri-methylated at K4 of histone H3. Nature, 419, 407–411. [DOI] [PubMed] [Google Scholar]
- 23. Calo E. and Wysocka J. (2013) Modification of enhancer chromatin: what, how, and why? Mol. Cell, 49, 825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ardehali M.B., Mei A., Zobeck K.L., Caron M., Lis J.T. and Kusch T. (2011) Drosophila Set1 is the major histone H3 lysine 4 trimethyltransferase with role in transcription. EMBO J., 30, 2817–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Herz H.M., Mohan M., Garruss A.S., Liang K., Takahashi Y.H., Mickey K., Voets O., Verrijzer C.P. and Shilatifard A. (2012) Enhancer-associated H3K4 monomethylation by Trithorax-related, the drosophila homolog of mammalian Mll3/Mll4. Genes Dev., 26, 2604–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dorighi K.M., Swigut T., Henriques T., Bhanu N.V., Scruggs B.S., Nady N., Still C.D. 2nd, Garcia B.A., Adelman K. and Wysocka J. (2017) Mll3 and Mll4 facilitate enhancer RNA synthesis and transcription from promoters independently of H3K4 Monomethylation. Mol. Cell, 66, 568, e564–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hu D., Gao X., Morgan M.A., Herz H.M., Smith E.R. and Shilatifard A. (2013) The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol. Cell Biol., 33, 4745–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang S.P., Tang Z., Chen C.W., Shimada M., Koche R.P., Wang L.H., Nakadai T., Chramiec A., Krivtsov A.V., Armstrong S.A. et al. (2017) A UTX-MLL4-p300 transcriptional regulatory network Coordinately shapes active enhancer landscapes for eliciting transcription. Mol. Cell, 67, 308, e306–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tsai I.C., McKnight K., McKinstry S.U., Maynard A.T., Tan P.L., Golzio C., White C.T., Price D.J., Davis E.E., Amrine-Madsen H. et al. (2018) Small molecule inhibition of RAS/MAPK signaling ameliorates developmental pathologies of kabuki syndrome. Sci. Rep., 8, 10779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ansari K.I., Hussain I., Shrestha B., Kasiri S. and Mandal S.S. (2011) HOXC6 is transcriptionally regulated via coordination of MLL histone methylase and estrogen receptor in an estrogen environment. J. Mol. Biol., 411, 334–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oswald F., Rodriguez P., Giaimo B.D., Antonello Z.A., Mira L., Mittler G., Thiel V.N., Collins K.J., Tabaja N., Cizelsky W. et al. (2016) A phospho-dependent mechanism involving NCoR and KMT2D controls a permissive chromatin state at notch target genes. Nucleic Acids Res., 44, 4703–4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guo C., Chang C.C., Wortham M., Chen L.H., Kernagis D.N., Qin X., Cho Y.W., Chi J.T., Grant G.A., McLendon R.E. et al. (2012) Global identification of MLL2-targeted loci reveals MLL2's role in diverse signaling pathways. Proc. Natl. Acad. Sci. USA, 109, 17603–17608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goo Y.H., Sohn Y.C., Kim D.H., Kim S.W., Kang M.J., Jung D.J., Kwak E., Barlev N.A., Berger S.L., Chow V.T. et al. (2003) Activating signal cointegrator 2 belongs to a novel steady-state complex that contains a subset of trithorax group proteins. Mol. Cell Biol., 23, 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bjornsson H.T., Benjamin J.S., Zhang L., Weissman J., Gerber E.E., Chen Y.C., Vaurio R.G., Potter M.C., Hansen K.D. and Dietz H.C. (2014) Histone deacetylase inhibition rescues structural and functional brain deficits in a mouse model of kabuki syndrome. Sci. Transl. Med., 6, 256ra135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Van Laarhoven P.M., Neitzel L.R., Quintana A.M., Geiger E.A., Zackai E.H., Clouthier D.E., Artinger K.B., Ming J.E. and Shaikh T.H. (2015) Kabuki syndrome genes KMT2D and KDM6A: functional analyses demonstrate critical roles in craniofacial, heart and brain development. Hum. Mol. Genet., 24, 4443–4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ang S.Y., Uebersohn A., Spencer C.I., Huang Y., Lee J.E., Ge K. and Bruneau B.G. (2016) KMT2D regulates specific programs in heart development via histone H3 lysine 4 di-methylation. Development, 143, 810–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schwenty-Lara J., Nurnberger A. and Borchers A. (2019) Loss of function of Kmt2d, a gene mutated in kabuki syndrome, affects heart development in Xenopus laevis. Dev. Dyn., in press, 248, 465–476. [DOI] [PubMed] [Google Scholar]
- 38. Groves A.K. and LaBonne C. (2014) Setting appropriate boundaries: fate, patterning and competence at the neural plate border. Dev. Biol., 389, 2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ufartes R., Schwenty-Lara J., Freese L., Neuhofer C., Moller J., Wehner P., Ravenswaaij-Arts C.M.A., Wong M.T.Y., Schanze I., Tzschach A. et al. (2018) Sema3a plays a role in the pathogenesis of CHARGE syndrome. Hum. Mol. Genet., 27, 1343–1352. [DOI] [PubMed] [Google Scholar]
- 40. Lalani S.R., Safiullah A.M., Molinari L.M., Fernbach S.D., Martin D.M. and Belmont J.W. (2004) SEMA3E mutation in a patient with CHARGE syndrome. J. Med. Genet., 41, e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schulz Y., Freese L., Manz J., Zoll B., Volter C., Brockmann K., Bogershausen N., Becker J., Wollnik B. and Pauli S. (2014) CHARGE and kabuki syndromes: a phenotypic and molecular link. Hum. Mol. Genet., 23, 4396–4405. [DOI] [PubMed] [Google Scholar]
- 42. Shpargel K.B., Starmer J., Wang C., Ge K. and Magnuson T. (2017) UTX-guided neural crest function underlies craniofacial features of kabuki syndrome. Proc. Natl. Acad. Sci. USA, 114, E9046–E9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bajanca F., Gouignard N., Colle C., Parsons M., Mayor R. and Theveneau E. (2019) In vivo topology converts competition for cell-matrix adhesion into directional migration. Nat. Commun., 10, 1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Koestner U., Shnitsar I., Linnemannstons K., Hufton A.L. and Borchers A. (2008) Semaphorin and neuropilin expression during early morphogenesis of Xenopus laevis. Dev. Dyn., 237, 3853–3863. [DOI] [PubMed] [Google Scholar]
- 45. Bogershausen N. and Wollnik B. (2013) Unmasking kabuki syndrome. Clin. Genet., 83, 201–211. [DOI] [PubMed] [Google Scholar]
- 46. Bogershausen N., Gatinois V., Riehmer V., Kayserili H., Becker J., Thoenes M., Simsek-Kiper P.O., Barat-Houari M., Elcioglu N.H., Wieczorek D. et al. (2016) Mutation update for kabuki syndrome genes KMT2D and KDM6A and further delineation of X-linked kabuki syndrome subtype 2. Hum. Mutat., 37, 847–864. [DOI] [PubMed] [Google Scholar]
- 47. Micale L., Augello B., Maffeo C., Selicorni A., Zucchetti F., Fusco C., De Nittis P., Pellico M.T., Mandriani B., Fischetto R. et al. (2014) Molecular analysis, pathogenic mechanisms, and readthrough therapy on a large cohort of kabuki syndrome patients. Hum. Mutat., 35, 841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Makrythanasis P., van Bon B.W., Steehouwer M., Rodriguez-Santiago B., Simpson M., Dias P., Anderlid B.M., Arts P., Bhat M., Augello B. et al. (2013) MLL2 mutation detection in 86 patients with kabuki syndrome: a genotype-phenotype study. Clin. Genet., 84, 539–545. [DOI] [PubMed] [Google Scholar]
- 49. Banka S., Veeramachaneni R., Reardon W., Howard E., Bunstone S., Ragge N., Parker M.J., Crow Y.J., Kerr B., Kingston H. et al. (2012) How genetically heterogeneous is kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur. J. Hum. Genet., 20, 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Adam M.P. and Hudgins L. (2005) Kabuki syndrome: a review. Clin. Genet., 67, 209–219. [DOI] [PubMed] [Google Scholar]
- 51. Lederer D., Grisart B., Digilio M.C., Benoit V., Crespin M., Ghariani S.C., Maystadt I., Dallapiccola B. and Verellen-Dumoulin C. (2012) Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with kabuki syndrome. Am. J. Hum. Genet., 90, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Miyake N., Koshimizu E., Okamoto N., Mizuno S., Ogata T., Nagai T., Kosho T., Ohashi H., Kato M., Sasaki G. et al. (2013) MLL2 and KDM6A mutations in patients with kabuki syndrome. Am. J. Med. Genet. A, 161A, 2234–2243. [DOI] [PubMed] [Google Scholar]
- 53. Miyake N., Mizuno S., Okamoto N., Ohashi H., Shiina M., Ogata K., Tsurusaki Y., Nakashima M., Saitsu H., Niikawa N. et al. (2013) KDM6A point mutations cause kabuki syndrome. Hum. Mutat., 34, 108–110. [DOI] [PubMed] [Google Scholar]
- 54. Banka S., Howard E., Bunstone S., Chandler K.E., Kerr B., Lachlan K., McKee S., Mehta S.G., Tavares A.L., Tolmie J. et al. (2013) MLL2 mosaic mutations and intragenic deletion-duplications in patients with kabuki syndrome. Clin. Genet., 83, 467–471. [DOI] [PubMed] [Google Scholar]
- 55. Rada-Iglesias A., Bajpai R., Prescott S., Brugmann S.A., Swigut T. and Wysocka J. (2012) Epigenomic annotation of enhancers predicts transcriptional regulators of human neural crest. Cell Stem Cell, 11, 633–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A. et al. (2010) Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA, 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lai B., Lee J.E., Jang Y., Wang L., Peng W. and Ge K. (2017) MLL3/MLL4 are required for CBP/p300 binding on enhancers and super-enhancer formation in brown adipogenesis. Nucleic Acids Res., 45, 6388–6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lin-Shiao E., Lan Y., Coradin M., Anderson A., Donahue G., Simpson C.L., Sen P., Saffie R., Busino L., Garcia B.A. et al. (2018) KMT2D regulates p63 target enhancers to coordinate epithelial homeostasis. Genes Dev., 32, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang J., Dominguez-Sola D., Hussein S., Lee J.E., Holmes A.B., Bansal M., Vlasevska S., Mo T., Tang H., Basso K. et al. (2015) Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat. Med., 21, 1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lindsley A.W., Saal H.M., Burrow T.A., Hopkin R.J., Shchelochkov O., Khandelwal P., Xie C., Bleesing J., Filipovich L., Risma K. et al. (2016) Defects of B-cell terminal differentiation in patients with type-1 kabuki syndrome. J. Allergy Clin. Immunol., 137, 179, e110–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hu N., Strobl-Mazzulla P.H. and Bronner M.E. (2014) Epigenetic regulation in neural crest development. Dev. Biol., 396, 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rao A. and LaBonne C. (2018) Histone deacetylase activity has an essential role in establishing and maintaining the vertebrate neural crest. Development, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Strobl-Mazzulla P.H. and Bronner M.E. (2012) A PHD12-Snail2 repressive complex epigenetically mediates neural crest epithelial-to-mesenchymal transition. J. Cell Biol., 198, 999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Taneyhill L.A., Coles E.G. and Bronner-Fraser M. (2007) Snail2 directly represses cadherin6B during epithelial-to-mesenchymal transitions of the neural crest. Development, 134, 1481–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Haberland M., Mokalled M.H., Montgomery R.L. and Olson E.N. (2009) Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes Dev., 23, 1625–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Moccia A., Srivastava A., Skidmore J.M., Bernat J.A., Wheeler M., Chong J.X., Nickerson D., Bamshad M., Hefner M.A., Martin D.M. et al. (2018) Genetic analysis of CHARGE syndrome identifies overlapping molecular biology. Genet. Med., 20, 1022–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pauli S., Bajpai R. and Borchers A. (2017) CHARGEd with neural crest defects. Am. J. Med. Genet. C Semin. Med. Genet., 175, 478–486. [DOI] [PubMed] [Google Scholar]
- 68. Nieuwkoop P.D., Faber J. and Utrecht H.L. (1956) Normal Table of Xenopus laevis (Daudin): A Systematical and Chronological Survey of the Development from the Fertilized Egg Till the End of Metamorphosis. North-Holland Pub. Co., Amsterdam. [Google Scholar]
- 69. Moriyoshi K., Richards L.J., Akazawa C., O'Leary D.D. and Nakanishi S. (1996) Labeling neural cells using adenoviral gene transfer of membrane-targeted GFP. Neuron, 16, 255–260. [DOI] [PubMed] [Google Scholar]
- 70. Kashef J., Kohler A., Kuriyama S., Alfandari D., Mayor R. and Wedlich D. (2009) Cadherin-11 regulates protrusive activity in Xenopus cranial neural crest cells upstream of trio and the small GTPases. Genes Dev., 23, 1393–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Riedl J., Crevenna A.H., Kessenbrock K., Yu J.H., Neukirchen D., Bista M., Bradke F., Jenne D., Holak T.A., Werb Z. et al. (2008) Lifeact: a versatile marker to visualize F-actin. Nat. Methods, 5, 605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Harland R.M. (1991) In situ hybridization: an improved whole-mount method for Xenopus embryos. Methods Cell Biol., 36, 685–695. [DOI] [PubMed] [Google Scholar]
- 73. Barriga E.H., Shellard A. and Mayor R. (1976) (2019) in vivo and in vitro quantitative analysis of neural crest cell migration. Methods Mol. Biol., 135–152. [DOI] [PubMed] [Google Scholar]
- 74. Borchers A., Epperlein H.H. and Wedlich D. (2000) An assay system to study migratory behavior of cranial neural crest cells in Xenopus. Dev. Genes Evol., 210, 217–222. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.