

Abstract
Post-translational modification of substrate proteins plays crucial roles in the regulation of their activity, cellular localization, and ability to be recognized by other proteins. One of those modifications involves the installment of adenosine-diphosphate-ribose (ADPr) onto nucleophilic side-chain groups of amino acid residues. This highly dynamic process is regulated by ADP-ribosyl transferases (ARTs) that install the ADPr-molecules on selected proteins and poly(ADP-ribosyl) glycohydrolases (PARGs) and (ADP-ribosyl)hydrolases (ARHs) that trim down and remove ADPr-chains. In this mini-review, the most recent advances in the chemical synthesis of ADPr-conjugates, poly-ADP-ribose, ADPr-peptides, and -proteins, and other tools to investigate ADPr-biology are discussed.
1. Introduction
Among the spectrum of post-translational modifications (PTMs), the attachment of adenosine-diphosphate-ribose (ADPr) to protein substrates in either its monomeric form, mono-ADP-ribose (MAR), or polymeric form, poly-ADP-ribose (PAR), has gained relatively little attention when compared to, e.g., phosphorylation or glycosylation events. ADP-ribosylation plays crucial roles in DNA-repair pathways and maintenance of genomic stability but is also implicated in cell differentiation, immunity, transcription regulation, and stress responses.1
The field of ADPr-research has grown over the past two decades due to, among others, the evolvement of highly sensitive mass spectrometric techniques and sophisticated chemical methodologies. The identification of ADP-ribosylation sites on protein substrates and enzymes involved in installing and removing this PTM has led to new insights into biochemical pathways that ADP-ribosylation is involved in. The FDA approval of olaparib, the first poly-ADPr-polymerase inhibitor in the treatment of BRCA1/2-mutated ovarian cancers, highlights the importance of gaining an understanding of this dynamic modification and the enzymes involved.
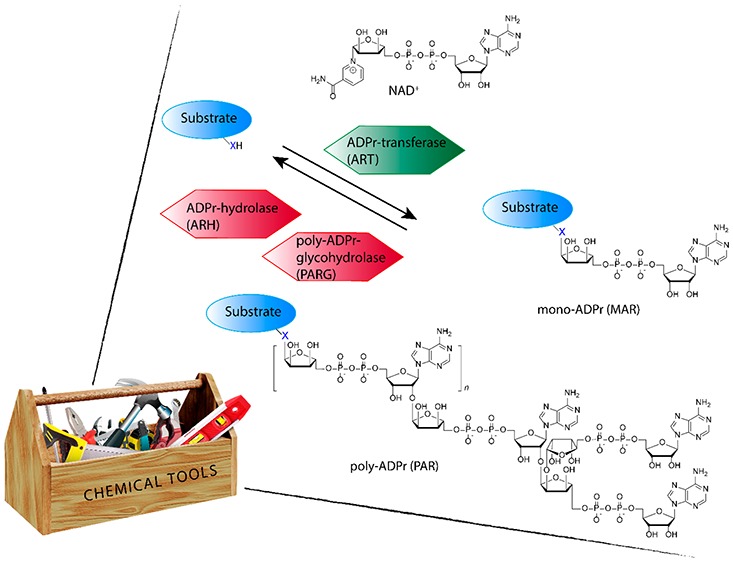
The enzymes that catalyze the transfer of ADPr from nicotinamide adenine dinucleotide (NAD+) to a nucleophilic amino acid side-chain functionality, while expelling nicotinamide, are collectively classified as ADP-ribosyl transferases (ARTs) (see Figure 1).2,3 ARTs can be clustered into two distinct groups based on their active site architecture. The first group is known as ADP-ribosyltransferase diphteria-toxin like (ARTD), sharing a similar active site histidine-tyrosine-glutamate (HYE) motif as their bacterial ancestor diphteria toxin. The second group is known as ADP-ribosyltransferase cholera-toxin like (ARTC) that share the same arginine-serine-glutamate (RSE) motif as the cholera toxin founding enzyme. The 17 known human enzymes catalyzing ADP-ribosylation collectively are called poly-ADP-ribosyl polymerases (PARPs). Although the name implies this family is able to form PAR, only 4 of the family members are validated to indeed produce PAR, while most others produce MAR.2 As in most post-translational modifications, ADP-ribosylation is a dynamic process in which the polymeric chains can be trimmed down by poly(ADP-ribosyl)glycohydrolases (PARGs) that cleave the glycoside bonds in PAR, and monomeric ADPr is removed by (ADP-ribosyl)hydrolases (ARHs), terminal ADP-ribose protein glycohydrolases (TARGs), or MacroDomain enzymes that cleave the amino acid ribose bond.4,5 Targeted substrate proteins have been found to be ADP-ribosylated at arginine, lysine, aspartic acid, glutamic acid, asparagine, glutamine, serine, threonine, and cysteine, leading to O-glycosidic, N-glycosidic, or S-glycosidic linkages, all with subtle differences in intrinsic stabilities and dedicated transferases and hydrolases responsible for their attachment and removal, respectively.3 One example is the recent identification of serine-ADP-ribosylation sites on histone proteins during DNA damage,6,7 in which histone PARylation Factor 1 (HPF1) switches PARylation by PARP1 and PARP2 from aspartate/glutamate residues to serine residues specifically.8 Counteracting HPF1/PARP action is hydrolase ARH3 that is found to be the main enzyme responsible for reversing serine-ADP-ribosylation.9 Another example is the opposing actions of PARP10 that install MAR on acidic amino acid residues (aspartate/glutamate) in kinase substrates and MacroD1 and MacroD2 that are able to remove MAR from these PARP10 substrates.10 In addition to the variety of transferases and hydrolases, over 800 “reader” proteins have been identified to have an ADPr-binding domain, showing the broad impact of ADP-ribosylation and ADPr-dependent signaling.3
Figure 1.

Schematic representation of enzymatic activities involved in regulating ADP-ribosylation.
2. Chemical Synthesis
Several methodologies facilitating the chemical synthesis of model peptides carrying MAR and PAR have been developed in order to create well-defined model substrates that can be used to measure the affinity between ADPr “reader”-enzymes and ADPr-substrates or study the cleavage preference of hydrolases. The main challenges in chemically preparing such ADPr-materials are the construction of the α-glycosidic bond between the amino acid and ribose parts, the efficient formation of the pyrophosphate linkage, and finding protecting group schemes compatible with maintaining the integrity of the delicate modification.11
2.1. α-Ribosyl Amino Acids
ART-mediated displacement of nicotinamide from β-oriented NAD+ by a nucleophilic heteroatom in the side chain of the incoming amino acid leads to an α-oriented linkage between the amino acid and ribose moiety (see Figure 2A). A variety of orthogonally α-ribosylated amino acids have been prepared chemically in order to allow incorporation into peptides, followed by the installment of the adenosine-diphosphate moiety afterward. Orthogonally protected ribosyl-asparagine and -glutamine were the first to be prepared, making use of an anomeric ribosyl azide that was reduced using a heterogeneous catalyst to yield the anomeric amine in situ, that was subsequently coupled to the side-chain carboxylic acid functionality of aspartic or glutamic acid (see Figure 2B).12,13 The reduction of anomeric azides leads to a mixture of α- and β- configured amines due to the occurrence of epimerization in the hemiaminal stage, which on one hand is essential for obtaining the desired α-configuration but on the other hand leads to a drop in yield as the nondesired β-product will also be formed. When using traceless Staudinger-ligation methodology on similar ribosyl azides, the reaction can be directed to result in the α-configuration selectively, giving an improved yield.14 An alternative methodology relying on glycosylation chemistry using ribosyl N-phenyl trifluoroacetimidates allowed the preparation of not only ribosylated asparagine and glutamine but also serine, aspartic acid, glutamic acid, and citrulline, as an arginine isoster (see Figure 2C).15,16
Figure 2.

(A) ADP-ribosylated glutamine residue highlighting the anomeric configuration. (B) Retrosynthesis of α-ribosyl glutamine from β-azido riboside and protected glutamic acid. (C) Overview and retrosynthesis of all ribosyl amino acids obtained using imidate glycosylations.
2.2. (Pyro)phosphate Formation
The chemical formation of the pyrophosphate bond in synthetic PAR- or MAR-peptides relies on the coupling of a phospho-monoester (phosphate) to an activated phosphorus species. The Filippov group employs the coupling of adenosine monophosphate to the in situ prepared phosphorimidazolidate of the ribosylated peptide on solid support, as the first chemical synthesis of ADPr-peptides (see Figure 3A).13 Although ADPr-peptide was formed, the installment of the reactive phosphorimidazolidate on the immobilized peptide was prone to side reactions, reducing overall efficiency. Reversing the chemical reactions in this approach by coupling of the phosphorimidazolidate of adenosine to a ribosyl peptide that was in situ phosphorylated proved more efficient (see Figure 3A). This method was further optimized by introducing a ribosylated amino acid in the peptide sequence that already carries a protected phosphate group, which in turn could be easily deprotected and subsequently reacted with the phosphoramidite of adenosine followed by oxidation to yield ADPr-peptides (see Figure 3B).15 The Hergenrother group shows a slightly deviating approach in constructing the pyrophosphate linkage in a solution-based approach toward ADPr-dimers. In this method, phosphitylation of suitably protected ribose is followed by hydrolysis toward the H-phosphonate, which in turn can be activated using N-chloro-succinimide to give a phosphochloridate in situ, that can be condensed with adenosine phosphate to yield the desired pyrophosphate linkage (see Figure 3C).17 Although not compared side by side on the same substrates, the authors mention that in constructing the pyrophosphate linkage in dimeric ADPr the H-phosphonate methodology is faster and better yielding then the more conventional phosphomorpholidate or phosphorimidazolide methods used to form generic pyrophosphates.
Figure 3.

Construction of ADPr using (A) the phosphate–phosphoramidate coupling protocol on solid support linked peptide, (B) phosphate–phosphoramidite coupling protocol on solid support linked peptide, and (C) phosphate–H-phosphonate coupling protocol on ribosyl adenosine in a solution-based method.
3. ADP-Ribosylated Peptides and Analogues Thereof
In classical Fmoc/Boc-based peptide synthesis, strong acidic conditions are used to liberate peptides from the resin and remove protecting groups at the same time. Such harsh conditions are incompatible with maintaining ADPr integrity and hence protecting groups, and solid support linkers that can be cleaved under alkaline conditions need to be used when constructing ADPr-peptides. Alternatively, the ADPr-moiety can be introduced post peptide synthesis, in order to conduct the strong acidic treatment of the peptide prior to installment of the ADPr-moiety.
The Filippov lab relies on constructing ADP-ribosylated peptides on resin equipped with a base-labile linker and base-labile protecting groups, preventing exposure of the ADPr-peptides to acidic conditions. Using this methodology, ADPr-peptides carrying the relevant serine-ADPr linkage and glutamine-, asparagine-, or citrulline-ADPr linkage, as stable isosters of glutamic acid-, aspartic acid-, or arginine-ADPr, respectively, can be prepared. Although only relatively short ADPr-peptides can be constructed using this approach, such peptides were applied to investigate the binding affinity to macroD2 or TARG1.15 Furthermore, they prepared a biotinylated histone H2B peptide carrying ADPr on glutamine, as a stabile isostere of the native glutamate site, that was used to pulldown recombinant ARH3 and PARG in an in vitro experiment under conditions limiting hydrolytic activity of these enzymes. This pulldown showed only an interaction between the ADPr-peptide and ARH3 but not PARG, revealing that of the two hydrolases only ARH3 was able to recognize MARylated peptides.18 The mutagenesis of several residues in ARH3 followed by comparing the interaction with this H2B-ADPr peptide disclosed six residues in ARH3 to be crucial for MAR binding. A mass spectrometry approach, used to identify the true ADPr-acceptor sites that could be demodified by ARH3, identified mostly serine-ADP-ribosylation on the lysine-serine consensus sequence6,7 to be recognized by ARH3.18 Chemical preparation using the Filippov protocol of the reported physiologically relevant substrate, Histone H2B ADP-ribosylated at serine-10, gave acces to the native α-glycosidic ADPr linkage and its non-native β-isomer.19 Both peptides were incubated with ARH3 in a study to reveal that ARH3 hydrolase activity is selective for the α-ADPr-linkage.
The Muir lab uses a post peptide synthesis approach to install ADPr-groups on short peptides by introducing amino-oxy-containing amino acids into the peptide and subsequently performing oxime ligation on ADP-ribose (see Figure 4A).20 This strategy leads to ring-opened furanose conjugates, but also ring-closed isomers can be generated using N-methyl amino-oxy-containing peptides, although this last reaction is slow and low yielding. Such ADPr-peptide conjugates interacted with macrodomains on histone proteins, and these interactions, being in the micromolar affinity range, were strong enough to facilitate pulldown of the histone macrodomain. When introducing a benzophenone photo-cross-linking group to these ADPr–peptide conjugates, covalent capture of interacting proteins with the ADPr-peptide and pulldown from cell lysates was shown to be feasible. Using such an oxime-linked ADPr-peptide in a high-throughput screen on human macrodomain proteins, a highly selective allosteric inhibitor able to modulate macrodomain 2 activity of PARP14 was identified.21 This inhibitor prevents PARP14 to localize on sites of DNA damage and hence might be an interesting drug lead, as PARP14 is associated with inflammatory diseases and different types of cancer.
Figure 4.

Conjugation chemistries toward ADPr-peptides using (A) oxime ligation and (B) CuAAC ligation.
Another approach uses a copper-catalyzed azide alkyne cycloaddition reaction (CuAAC or commonly called “click chemistry”), where an alkyne-modified ADPr-synthon is conjugated to azide-modified peptides (see Figure 4B). Similar to oxime ligation, this allows for the post-peptide synthesis introduction of ADPr.22 Not only can peptides be ADP-ribosylated using this method but also small proteins can be conjugated, as was shown by the introduction of ADPr on azides substituting Arg42- or Gly76-sites in the 8.5 kDa ubiquitin protein. Although this conjugation chemistry leads to an artificial triazole linkage between ADPr and the protein, autoubiquitination assays using the Legionella effector SdeA enzyme show significant autoubiquitinating occurring, although at a reduced rate compared to the wild-type native ADPr-Ub. These results however show the adaptability of this click approach to ADPribosylate, not only peptides but also full-length proteins, and use them in assaying enzymatic reactions in vitro.
4. Poly-ADP-Ribose Chains
The synthesis of poly-ADPr brings a distinct challenge, as an α-oriented ribosyl linkage to adenosine needs to be constructed rather than an α-oriented ribosyl linkage to an amino acid. The crucial repeating unit in poly-ADPr is ribosyl-adenosine (see Figure 5B) which has been synthesized using different routes.23,24 Analogues of this repeating unit have been shown to have an inhibitory effect on PARP-1.25 The pioneering attempts were followed by syntheses introducing orthogonally protected phosphorus species at both ends of the ribosyl-adenosine, allowing the preparation of pyrophosphate linkages and the formation of ADPr-dimers.17 The authors showed that these synthetically prepared ADPr-dimers were hydrolyzed by active PARG and not by the catalytically inactive glycohydrolase. The authors also report the crystal structure of inactive human PARG with the dimeric ADPr-substrate at a resolution of 1.9 Å, representing the first structure of the human PARG-enzyme and its substrate. By introducing an alkyne moiety on the anomeric position of the ADPr-dimer, a fluorescent label (Cy3) or affinity tag (biotin) could be introduced using copper-catalyzed click chemistry. The fluorescently labeled ADPr-dimer was applied in a general fluorescence polarization-based PAR–protein binding assay to determine the dissociation constants between an inactive (E756N or E755N) human PARG mutant with Kd values of 83 nM and 208 nM, respectively.17
Figure 5.

(A) Solid-phase-based poly-ADPr synthesis, (B) repeating unit for linear poly-ADPr, and (C) repeating unit for branched poly-ADPr.
The embracement of solid-support-based synthesis in an automated fashion has led to the construction of larger ADPr-oligomers in a straightforward manner (see Figure 5A).26 In this approach, a phosphoramidite-equipped ribosyl-adenosine building block, carrying a protected phosphate, is coupled to a phosphate riboside attached to a resin. After coupling and oxidation, a pyrophosphate bond is formed giving rise to mono-ADPr. Subsequent deprotection of the terminal phosphate is the start of the next cycle of elongation to yield a dimeric ADPr, which in turn can be elongated to yield trimeric ADPr. The thus-prepared PAR fragments were applied in studying the chromatin remodeler ALC1 in vitro.(27) The affinity for the ADP-ribose monomer, dimer, and trimer with the macrodomain of ALC1 was determined using isothermal titration calorimetry (ITC). The intramolecular ALC1 ATPase-macrodomain shows an increase in affinity from micromolar (dimer) to nanomolar (trimer), whereas the monomer hardly shows interaction at all. A thermal shift assay also reveals significant stabilization of the macrodomain upon binding to the apparent minimal signal, tri-ADPr. The interaction of the trimer with the macrodomain was found to be an allosteric trigger to the conformational change of ALC1 that releases the autoinhibitory effect and switches on its enzymatic chromatin relaxation activity. The access to such synthetically prepared ADPr-oligomers was indispensable in the mechanistic studies of the oncogenic ALC1-protein.
In addition, the synthesis of the core motif found in branched ADPr-chains, ribosyl-ribosyl-adenosine28 and its triple monophosphate29 will without doubt lead to more complex branched poly-ADPr-chains in the near future, when applied in a similar automated solid-phase procedure (see Figure 5C).
5. Fluorescence-Based Assays
ADPr-based substrates carrying fluorogenic 4-(trifluoromethyl)umbelliferone on the anomeric position of ribose allows for determination of kinetic parameters of PARGs and ARHs.30 This substrate is not fluorescent until enzymatic actions hydrolyze the glycosidic bond upon which an increase in fluorescence can be observed as a direct measure of enzyme activity (see Figure 6A). Neither reactions with β-oriented substrates nor catalytic inactive mutants of the enzymes gave an increase of fluorescent signal, validating the mode of action of these enzymes. Another assay relies on the chemical reaction of NAD+ with benzamidine to create a fluorophore in situ. The fluorescence, therefore, is a direct measure of nonconsumed NAD+, which can be directly correlated to PARP activity and inhibition thereof (see Figure 6B).31 Although this last assay does not monitor the formation of ADPr, but rather focuses on the presence (and consumption) of NAD+, its utility in a high-throughput format to identify PARP inhibitors makes it a very useful technique in the ADPr-toolbox.
Figure 6.

(A) Fluorogenic ADPr substrate and (B) chemical conversion of nonconsumed NAD+ to a fluorescent NAD analogue.
6. Tools to Target ADP-Ribosylation in a Cellular Context
Most of the tools described above are applied in vitro, on recombinantly expressed and purified proteins, without the broader context of protein–protein interactions or the dynamic environment of the cell. Approaches to study ADPr-dynamics and localization as well as an unbiased identification of substrate proteins modified by this PTM are of added value to the in vitro tools. Severals methods have been devised to use small-molecule probes that utilize the native ADP-ribosylation machinery inside cells. One such an example is the development of the amino-oxy probes by Cohen and co-workers that allow trapping of ADPr-proteins with a glutamate or aspartate site of conjugation. ADPr-peptide ester linkages can undergo ADPr transfer from the 1′- to 2′-hydroxyl moiety, resulting in an equilibrium state of the ribose between a closed 1′-hydroxyl and opened 1′-aldehyde stage (see Figure 7A). This aldehyde can react with the amino-oxy probe at acidic pH (4.0–4.5) to generate a stable oxime linkage. The alkyne on the amino-oxy reagent can subsequently be used in click conjugation of a fluorophore. This approach was used to visualize cellular ADPr-proteins effected by PARPs during oxidative stress conditions.32
Figure 7.

(A) Amino-oxy probes for use in oxime labeling of ester-linked ADPr-proteins. (B) Two alkyne-modified NAD analogues to be incorporated in ADPr-chains in cellular applications.
Another approach relies on chemically prepared NAD+ analogues carrying alkynes, cyclopropene, or cyclooctyne as a bio-orthogonal handle that can also be applied in cellular systems, allowing the formation of labeled ADPr-chains, that can be visualized after conjugation of fluorescent dyes onto the bio-orthogonal functionality in real time (see Figure 7B).33−36 Conjugation of a biotin tag to such alkyne-modified ADPr-proteins followed by MS–MS proteomics gives insight into the ADPribosylome.36 This method resembles the earlier reported approach where biotin-NAD is used to mark ADPr-chains with the affinity tag, with the difference that in the two-step labeling approach modification of the NAD structure by the handle is minimal and hence reduces interference of the handle when incorporated into ADPr-chains.37 A chemo-enzymatic approach or fully chemical synthesis of 3′-azido-modified NAD+ is recently reported.38 This NAD+ analogue has high activity and specificity for PARylation mediated by PARP1. The modified PAR polymers are found to be less efficiently degraded by PARG than the native polymers, allowing these tools to be used to visualize PARylation in cells and label PARylated proteins in the cell lysate.
A recent report by the Leung group introduces the enzymatic labeling of a terminal ADP-ribose (ELTA) technique in which the enzyme 2′-5′-oligoadenylate synthetase 1 (OAS1) uses a modified dATP analogue to label a protein conjugated to ADPr.39 Using dATP-analogues carrying a radioactive, fluorescent, or affinity label, this technique can be applied to investigate a MAR- or PARylated proteome.
The group of Cohen reports a different approach using alkyne carrying NAD+ analogues by finding combinations of genetically engineered mARTs that utilize chemically modified NAD+-alkyne analogues. In this approach, the modified NAD+ can only be used by the genetically engineered mART to ADPribosylate its targets.40,41 Subsequent click chemistry on the alkyne and MS–MS analysis reveals the substrates modified by the engineered mART. Using this methodology, ADPr-substrates can be identified and simultaneously linked to the transferase that performed the action.42
7. Conclusion and Outlook
Due to its delicate nature and instability during acidic chemical treatments, ADP-ribosylation gained relatively little attention when compared to other post-translational modifications such as, for instance, glycosylation. ADP-ribosylation, however, is a prevalent PTM, and ADPr-chains are even considered to be the third type of nucleic acid, after DNA and RNA. To study the broad impact this modification has on a variety of cellular (signaling) processes, tools to visualize and modulate the activity of the conjugating and deconjugating enzymes have been under development. The development of chemical methodology to create ADPr-based material avoiding strong acidic conditions by making use of protecting groups that can be removed using alkaline treatment allowed the construction of native ADPr-peptides and ADPr-chains. Such peptides and chains were shown to be very useful in studying the details and preferences of ARH and PARG enzymes. Translating these chemistries to solid-phase approaches led to more advanced constructs and will expand the scope and complexity of chemically prepared ADPr-material even further in the near future. The adaptation of commonly known ligation methodology, such as oxime ligation and copper-mediated azide alkyne cycloaddition reactions, has also proven to be of value in terms of making ADPr-protein conjugates. This method can be further applied in the near future by using expression-based systems to include azides into native proteins that can be ADPribosylated using CuAAC chemistry. The preparation of both ADPr-peptides carrying the native linkage and those joined together via bio-orthogonal reactions resulted in the controlled synthesis of well-defined ADPr-materials. Equipping such molecules with fluorescent dyes, affinity tags such as biotin or photo-cross-linking groups makes them useful tools in chemical biology approaches, casting a light on enzymatic properties such as substrate preference, affinity, and structural details that could not have been obtained through enzymatic preparation of (nonmodified) ADPr-chains. The development of modified NAD+ analogues and their incorporation into ADPr-chains using the ADPr-machinery in cellular settings, combined with the development of more sensitive mass spectrometry methods, opens up a new field of research, in which the ADPribosylome can be studied. In addition, the development of assays facilitating high-throughput screening of modulators of ADPr-metabolism is essential for the future development of, e.g., inhibitors of ART-activity. Furthermore, new substrates are being identified showing ADP-ribosylation on: (1) the phosphorylated ends of RNA by PARP10,43 (2) thymidine residues in single-stranded DNA by Mycobacterium tuberculosis ADPr-transferase DarT,44 and (3) the phosphate ends of DNA duplexes or single-stranded oligonucleotides by PARP1/2.45 The biological role of such hybrid oligonucleotides remains largely speculative, but their identification shows that there is still a lot to be unveiled in the realm of ADPr-biology, a quest that will benefit from the preparation of specific assay reagents and probes.
Acknowledgments
This work is funded by a ZonMw Off-Road grant and a NWO-VIDI grant.
Biography
Gerbrand J. van der Heden van Noort obtained his Ph.D. degree from Leiden University (2012) under the guidance of Dr. Dmitri Filippov and Prof. Gijs van der Marel developing organic chemistry driven methodology to construct hybrid biopolymers consisting of peptide, carbohydrate, and nucleic acid parts. A part of this work led to the first chemical synthesis of ADP-ribosylated peptide isosteres and ribosyl-adenosine, the repeating unit of ADP-ribose polymers. After postdoctoral research at the Humboldt University (Berlin, Germany) and The Netherlands Cancer Institute (Amsterdam, Netherlands), he started his own line of research focusing on the interplay between the post-translational modifications ADP-ribosylation and ubiquitination, using organic chemistry and chemical biology approaches at the Leiden University Medical Centre.
The author declares no competing financial interest.
References
- Bock F. J.; Chang P. New directions in poly(ADP-ribose) polymerase biology. FEBS J. 2016, 283, 4017–4031. 10.1111/febs.13737. [DOI] [PubMed] [Google Scholar]
- Vyas S.; Matic I.; Uchima L.; Rood J.; Zaja R.; Hay R. T.; Ahel I.; Chang P. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat. Commun. 2014, 5, 4426–4426. 10.1038/ncomms5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M. S.; Chang P. Insights into the biogenesis, function, and regulation of ADP-ribosylation. Nat. Chem. Biol. 2018, 14, 236–243. 10.1038/nchembio.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slade D.; Dunstan M. S.; Barkauskaite E.; Weston R.; Lafite P.; Dixon N.; Ahel M.; Leys D.; Ahel I. The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature 2011, 477, 616–620. 10.1038/nature10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rack J. G. M.; Ariza A.; Drown B. S.; Henfrey C.; Bartlett E.; Shirai T.; Hergenrother P. J.; Ahel I. (ADP-ribosyl)hydrolases: Structural Basis for Differential Substrate Recognition and Inhibition. Cell Chem. Biol. 2018, 25, 1533–1546. 10.1016/j.chembiol.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidecker O.; Bonfiglio J. J.; Colby T.; Zhang Q.; Atanassov I.; Zaja R.; Palazzo L.; Stockum A.; Ahel I.; Matic I. Serine is a new target residue for endogenous ADP-ribosylation on histones. Nat. Chem. Biol. 2016, 12, 998–1000. 10.1038/nchembio.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo L.; Leidecker O.; Prokhorova E.; Dauben H.; Matic I.; Ahel I. Serine is the major residue for ADP-ribosylation upon DNA damage. eLife 2018, 7, e34334 10.7554/eLife.34334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfiglio J. J.; Fontana P.; Zhang Q.; Colby T.; Gibbs-Seymour I.; Atanassov I.; Bartlett E.; Zaja R.; Ahel I.; Matic I. Serine ADP-Ribosylation Depends on HPF1. Mol. Cell 2017, 65, 932–940. 10.1016/j.molcel.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana P.; Bonfiglio J. J.; Palazzo L.; Bartlett E.; Matic I.; Ahel I. Serine ADP-ribosylation reversal by the hydrolase ARH3. eLife 2017, 6, e28533 10.7554/eLife.28533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal F.; Feijs K. L. H.; Frugier E.; Bonalli M.; Forst A. H.; Imhof R.; Winkler H. C.; Fischer D.; Caflisch A.; Hassa P. O.; Lüscher B.; Hottiger M. O. Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat. Struct. Mol. Biol. 2013, 20, 502–507. 10.1038/nsmb.2521. [DOI] [PubMed] [Google Scholar]
- Liu Q.; van der Marel G. A.; Filippov D. V. Chemical ADP-ribosylation: mono-ADPr-peptides and oligo-ADP-ribose. Org. Biomol. Chem. 2019, 17, 5460–5474. 10.1039/C9OB00501C. [DOI] [PubMed] [Google Scholar]
- Bonache M. A.; Nuti F.; Le Chevalier Isaad A.; Real-Fernández F.; Chelli M.; Rovero P.; Papini A. M. Synthesis of new ribosylated Asn building blocks as useful tools for glycopeptide and glycoprotein synthesis. Tetrahedron Lett. 2009, 50, 4151–4153. 10.1016/j.tetlet.2009.04.124. [DOI] [Google Scholar]
- van der Heden van Noort G. J.; van der Horst M. G.; Overkleeft H. S.; van der Marel G. A.; Filippov D. V. Synthesis of Mono-ADP-Ribosylated Oligopeptides Using Ribosylated Amino Acid Building Blocks. J. Am. Chem. Soc. 2010, 132, 5236–5240. 10.1021/ja910940q. [DOI] [PubMed] [Google Scholar]
- Speciale G.; Bernardi A.; Nisic F. A Facile Synthesis of α-N-Ribosyl-Asparagine and α-N-Ribosyl-Glutamine Building Blocks. Molecules 2013, 18, 8779–8785. 10.3390/molecules18088779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kistemaker H. A. V.; Nardozza A. P.; Overkleeft H. S.; van der Marel G. A.; Ladurner A. G.; Filippov D. V. Synthesis and Macrodomain Binding of Mono-ADP-Ribosylated Peptides. Angew. Chem., Int. Ed. 2016, 55, 10634–10638. 10.1002/anie.201604058. [DOI] [PubMed] [Google Scholar]
- Kistemaker H. A. V.; van der Heden van Noort G. J.; Overkleeft H. S.; van der Marel G. A.; Filippov D. V. Stereoselective Ribosylation of Amino Acids. Org. Lett. 2013, 15, 2306–2309. 10.1021/ol400929c. [DOI] [PubMed] [Google Scholar]
- Lambrecht M. J.; Brichacek M.; Barkauskaite E.; Ariza A.; Ahel I.; Hergenrother P. J. Synthesis of Dimeric ADP-Ribose and Its Structure with Human Poly(ADP-ribose) Glycohydrolase. J. Am. Chem. Soc. 2015, 137, 3558–3564. 10.1021/ja512528p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abplanalp J.; Leutert M.; Frugier E.; Nowak K.; Feurer R.; Kato J.; Kistemaker H. V. A.; Filippov D. V.; Moss J.; Caflisch A.; Hottiger M. O. Proteomic analyses identify ARH3 as a serine mono-ADP-ribosylhydrolase. Nat. Commun. 2017, 8, 2055. 10.1038/s41467-017-02253-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorneveld J.; Rack J. G. M.; Ahel I.; Overkleeft H. S.; van der Marel G. A.; Filippov D. V. Synthetic α- and β-Ser-ADP-ribosylated Peptides Reveal α-Ser-ADPr as the Native Epimer. Org. Lett. 2018, 20, 4140–4143. 10.1021/acs.orglett.8b01742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyle P. M.; Muir T. W. Method for the Synthesis of Mono-ADP-ribose Conjugated Peptides. J. Am. Chem. Soc. 2010, 132, 15878–15880. 10.1021/ja1064312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller M.; Riedel K.; Gibbs-Seymour I.; Uth K.; Sieg C.; Gehring A. P.; Ahel I.; Bracher F.; Kessler B. M.; Elkins J. M.; Knapp S. Discovery of a Selective Allosteric Inhibitor Targeting Macrodomain 2 of Polyadenosine-Diphosphate-Ribose Polymerase 14. ACS Chem. Biol. 2017, 12, 2866–2874. 10.1021/acschembio.7b00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Kistemaker H. A. V.; Bhogaraju S.; Dikic I.; Overkleeft H. S.; van der Marel G. A.; Ovaa H.; van der Heden van Noort G. J.; Filippov D. V. A General Approach Towards Triazole-Linked Adenosine Diphosphate Ribosylated Peptides and Proteins. Angew. Chem., Int. Ed. 2018, 57, 1659–1662. 10.1002/anie.201710527. [DOI] [PubMed] [Google Scholar]
- van der Heden van Noort G. J.; Overkleeft H. S.; van der Marel G. A.; Filippov D. V. Ribosylation of Adenosine: An Orthogonally Protected Building Block for the Synthesis of ADP-Ribosyl Oligomers. Org. Lett. 2011, 13, 2920–2923. 10.1021/ol200971z. [DOI] [PubMed] [Google Scholar]
- Mikhailov S. N.; Kulikova I. V.; Nauwelaerts K.; Herdewijn P. Synthesis of 2′-O-α-d-ribofuranosyladenosine, monomeric unit of poly(ADP–ribose). Tetrahedron 2008, 64, 2871–2876. 10.1016/j.tet.2008.01.028. [DOI] [Google Scholar]
- Zheng M.; Mex M.; Götz K. H.; Marx A. Synthesis of disaccharide nucleoside analogues as potential poly(ADP-ribose) polymerase-1 inhibitors. Org. Biomol. Chem. 2018, 16, 8904–8907. 10.1039/C8OB01894D. [DOI] [PubMed] [Google Scholar]
- Kistemaker H. A. V.; Lameijer L. N.; Meeuwenoord N. J.; Overkleeft H. S.; van der Marel G. A.; Filippov D. V. Synthesis of Well-Defined Adenosine Diphosphate Ribose Oligomers. Angew. Chem., Int. Ed. 2015, 54, 4915–4918. 10.1002/anie.201412283. [DOI] [PubMed] [Google Scholar]
- Singh H. R.; Nardozza A. P.; Möller I. R.; Knobloch G.; Kistemaker H. A. V.; Hassler M.; Harrer N.; Blessing C.; Eustermann S.; Kotthoff C.; Huet S.; Mueller-Planitz F.; Filippov D. V.; Timinszky G.; Rand K. D.; Ladurner A. G. A Poly-ADP-Ribose Trigger Releases the Auto-Inhibition of a Chromatin Remodeling Oncogene. Mol. Cell 2017, 68, 860–871. 10.1016/j.molcel.2017.11.019. [DOI] [PubMed] [Google Scholar]
- Kistemaker H. A. V.; Overkleeft H. S.; van der Marel G. A.; Filippov D. V. Branching of poly(ADP-ribose): Synthesis of the Core Motif. Org. Lett. 2015, 17, 4328–4331. 10.1021/acs.orglett.5b02143. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Kistemaker H. A. V.; Overkleeft H. S.; van der Marel G. A.; Filippov D. V. Synthesis of ribosyl-ribosyl-adenosine-5′,5″,5‴(triphosphate)—the naturally occurring branched fragment of poly(ADP ribose). Chem. Commun. 2017, 53, 10255–10258. 10.1039/C7CC05755E. [DOI] [PubMed] [Google Scholar]
- Drown B. S.; Shirai T.; Rack J. G. M.; Ahel I.; Hergenrother P. J. Monitoring Poly(ADP-ribosyl)glycohydrolase Activity with a Continuous Fluorescent Substrate. Cell Chem. Biol. 2018, 25, 1562–1570. 10.1016/j.chembiol.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putt K. S.; Hergenrother P. J. A nonradiometric, high-throughput assay for poly(ADP-ribose) glycohydrolase (PARG): application to inhibitor identification and evaluation. Anal. Biochem. 2004, 333, 256–264. 10.1016/j.ab.2004.04.032. [DOI] [PubMed] [Google Scholar]
- Morgan R. K.; Cohen M. S. A Clickable Aminooxy Probe for Monitoring Cellular ADP-Ribosylation. ACS Chem. Biol. 2015, 10, 1778–1784. 10.1021/acschembio.5b00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallrodt S.; Buntz A.; Wang Y.; Zumbusch A.; Marx A. Bioorthogonally Functionalized NAD+ Analogues for In-Cell Visualization of Poly(ADP-Ribose) Formation. Angew. Chem., Int. Ed. 2016, 55, 7660–7664. 10.1002/anie.201600464. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Rösner D.; Grzywa M.; Marx A. Chain-Terminating and Clickable NAD+ Analogues for Labeling the Target Proteins of ADP-Ribosyltransferases. Angew. Chem., Int. Ed. 2014, 53, 8159–8162. 10.1002/anie.201404431. [DOI] [PubMed] [Google Scholar]
- Buntz A.; Wallrodt S.; Gwosch E.; Schmalz M.; Beneke S.; Ferrando-May E.; Marx A.; Zumbusch A. Real-Time Cellular Imaging of Protein Poly(ADP-ribos)ylation. Chain-Terminating and Clickable NAD+ Analogues for Labeling the Target Proteins of ADP-Ribosyltransferases. Angew. Chem., Int. Ed. 2016, 55, 11256–11260. 10.1002/anie.201605282. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Kim J. H.; Frizzell K. M.; Kraus W. L.; Lin H. Clickable NAD Analogues for Labeling Substrate Proteins of Poly(ADP-ribose) Polymerases. J. Am. Chem. Soc. 2010, 132, 9363–9372. 10.1021/ja101588r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.Use of biotinylated NAD to label and purify ADP-ribosylated proteins. In Methods in Enzymology; Academic Press: 1997; Vol. 280, pp 255–265. [DOI] [PubMed] [Google Scholar]
- Zhang X.-N.; Cheng Q.; Chen J.; Lam A. T.; Lu Y.; Dai Z.; Pei H.; Evdokimov N. M.; Louie S. G.; Zhang Y. A ribose-functionalized NAD+ with unexpected high activity and selectivity for protein poly-ADP-ribosylation. Nat. Commun. 2019, 10, 4196. 10.1038/s41467-019-12215-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando Y.; Elkayam E.; McPherson R. L.; Dasovich M.; Cheng S.-J.; Voorneveld J.; Filippov D. V.; Ong S.-E.; Joshua-Tor L.; Leung A. K. L. ELTA: Enzymatic Labeling of Terminal ADP-Ribose. Mol. Cell 2019, 73, 845–856. 10.1016/j.molcel.2018.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter-O’Connell I.; Jin H.; Morgan R. K.; David L. L.; Cohen M. S. Engineering the Substrate Specificity of ADP-Ribosyltransferases for Identifying Direct Protein Targets. J. Am. Chem. Soc. 2014, 136, 5201–5204. 10.1021/ja412897a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter-O’Connell I.; Jin H.; Morgan R. K.; Zaja R.; David L. L.; Ahel I.; Cohen M. S. Identifying Family-Member-Specific Targets of Mono-ARTDs by Using a Chemical Genetics Approach. Cell Rep. 2016, 14, 621–631. 10.1016/j.celrep.2015.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter-O’Connell I.; Vermehren-Schmaedick A.; Jin H.; Morgan R. K.; David L. L.; Cohen M. S. Combining Chemical Genetics with Proximity-Dependent Labeling Reveals Cellular Targets of Poly(ADP-ribose) Polymerase 14 (PARP14). ACS Chem. Biol. 2018, 13, 2841–2848. 10.1021/acschembio.8b00567. [DOI] [PubMed] [Google Scholar]
- Munnur D.; Bartlett E.; Mikolčević P.; Kirby I. T.; Matthias Rack J. G.; Mikoč A.; Cohen M. S.; Ahel I. Reversible ADP-ribosylation of RNA. Nucleic Acids Res. 2019, 47, 5658–5669. 10.1093/nar/gkz305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankevicius G.; Ariza A.; Ahel M.; Ahel I. The Toxin-Antitoxin System DarTG Catalyzes Reversible ADP-Ribosylation of DNA. Mol. Cell 2016, 64, 1109–1116. 10.1016/j.molcel.2016.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talhaoui I.; Lebedeva N. A.; Zarkovic G.; Saint-Pierre C.; Kutuzov M. M.; Sukhanova M. V.; Matkarimov B. T.; Gasparutto D.; Saparbaev M. K.; Lavrik O. I.; Ishchenko A. A. Poly(ADP-ribose) polymerases covalently modify strand break termini in DNA fragments in vitro. Nucleic Acids Res. 2016, 44, 9279–9295. 10.1093/nar/gkw675. [DOI] [PMC free article] [PubMed] [Google Scholar]