

Abstract

The chemical synthesis of cyclic peptides is a well-established area of research. This has been further expanded by development of bio-orthogonal reactions that enable access to peptides of greater structural complexity. One approach utilizes 1,3-dichloroacetone to selectively link free cysteine side-chains with an acetone-like bridge via an SN2 reaction. Here, we have used this reaction to dimerize cyclic peptide monomers to create novel bicyclic dimeric peptides. We investigated a range of reaction parameters to identify the optimal dimerization conditions for our model systems. One of the acetone-linked dimeric peptides was analyzed for proteolytic stability in human serum and was observed to still be fully intact after 48 h. This study provides valuable insights into the application of 1,3-dichloroacetone as a tool in the synthesis of complex, multicyclic peptides.
Introduction
A common phenomenon observed in proteins is their ability to self-associate to form dimers.1 We can mimic this phenomenon using peptide mimetics whereby we replicate specific loops of proteins, cyclize them, then tether them together. Dimerization of peptides has long been considered to be a viable strategy to increase binding affinity, potency, and/or resistance to inactivation for peptide agonists2−4 and antagonists.5 Several ligation strategies have been adopted to dimerize peptides, such as disulfide bond formation, which is a common structural element found in naturally occurring peptides.6−8 However, disulfide bonds are highly sensitive to reducing conditions9,10 and can readily form mixed disulfide isomers via thiolysis.11−13 Amide linkages are more resistant to reduction, but carboxylates and amines are extremely common in peptides, therefore, orthogonal side-chain protection strategies are usually required to achieve site-specific conjugation.
A range of bio-orthogonal ligation chemistries have been developed, including the copper(II)-catalyzed azide–alkyne cycloaddition (“click” chemistry),14 the strain-promoted copper-free click reaction,15,16 Diels–Alder cycloadditions between tetrazines and strained alkenes or alkynes,17,18 and photoactivated 1,3-dipolar cycloaddition reactions.19 However, these reactions require incorporation of often expensive, non-natural functional groups. This is undesirable in drug discovery programs because high production costs of lead compounds can limit their scalability, and the toxicity of metabolites derived from chemically diverse peptides is often unknown. In this environment, efficient, economical, and biocompatible ligation chemistries are essential.
With the prominence of cystine in many naturally occurring peptides of interest (e.g., cyclotides, venoms)20,21 and their application as conformational constraints in medicinal chemistry research,4,22 we have long been interested in utilizing cysteine as the conjugation point for dimer formation. There are numerous reports whereby cysteine has been utilized for macrocyclization or cross-coupling of peptides, with numerous “linker” strategies available.23−25 We initially explored disulfide bonds as an option but always considered the possibility of bond reduction and disulfide shuffling. Given our interest in applying a coupling technique to form bicyclic homodimeric peptides, we scoured the literature for a reaction that was inexpensive, rapid, and would provide symmetry between the two cyclic monomers of the dimer. To that end, we have evaluated the recently reported SN2 reaction26 between thiolates and 1,3-dichloroacetone (DCA) and utilized it to form dimers from several cyclic peptide monomers.
As a bifunctional thiol reactive agent, DCA has long been used as a protein cross-linker27−29 and, more recently, been applied to peptide macrocyclization26 (Figure 1). It can be used to form an acetone-like “bridge” via thioether formation to create a peptide macrocycle, where it was found to stabilize an α-helical structure.26 We hypothesized that DCA would be an ideal reagent to efficiently form a stable “bridge” between two cyclic peptides and enable the rapid preparation of homodimeric cyclic peptides. Furthermore, this reaction does not require non-naturally occurring amino acids, and it can be conveniently performed in aqueous buffer. The carbonyl group in the tether also affords a chemoselective handle via oxime ligation to enable site-specific labeling and the formation of higher order oligomers.26 Herein, we demonstrate the utility of the DCA reaction by dimerizing several backbone-cyclized peptides. To ascertain optimal reaction conditions, we studied its chemoselectivity and several reaction parameters: stoichiometry, concentration, temperature, pH, and buffer.
Figure 1.

Previously reported application of DCA for peptide cyclization.26 aa = any amino acid; C = cysteine—the amino acid side-chain has been explicitly included; TCEP = tris(2-carboxyethyl)phosphine.
Results and Discussion
To examine the utility of DCA as a peptide dimerizing agent, we designed and synthesized cyclic peptides 1a to 6a (Figure 2 and Table 1). These cyclic peptides are loosely based on sequences found in the neurotrophin family of neurotrophic factors, and the ligand-gated purinoceptor ion channel P2X7.4,30,31 As a group, they have different ring sizes (five to eleven residues) and variation in the number of lysine residues (zero to three; Table 1), to determine whether the ε-amino group competes with the β-thiolate of cysteine for DCA.262a was synthesized to specifically evaluate the impact that omitting lysine has on dimerization yields. As a proof-of-concept peptide, 2a was only tested in selected conditions and reaction products were not isolated.
Figure 2.

Structures of cyclic peptide monomers, 1a–6a. Blue circles represent cyclic peptides containing a single cysteine residue (that amino acid side-chain has been explicitly included).
Table 1. Peptide Sequences, Characteristics, and Classifications (ID)a.
| Starting monomer sequence | No. of residues in starting monomer | No. of Lys in starting monomer | Starting monomer ID | Acetone-linked dimer ID | Disulfide-linked dimer ID |
|---|---|---|---|---|---|
| cyclo[pCKKR] | 5 | 2 | 1a | 1b | |
| cyclo[pCRRR] | 5 | 0 | 2a | 2b | |
| cyclo[QLCpAVPVSKG] | 11 | 1 | 3a | 3b | 3c |
| cyclo[QLKCKVPVSKG] | 11 | 3 | 4a | 4b | |
| cyclo[NSPVCpAIKTG] | 11 | 1 | 5a | 5b | |
| cyclo[CKQTLIKVFG] | 10 | 2 | 6a | 6b |
NB: Amino acids are represented in one-letter code; p = D-Pro.
Synthesis of Cyclic Monomer Peptides
1a–5a were synthesized using our previously reported approach30,32 (Scheme 1), which involves assembly of the peptide chain on 2-chlorotrityl resin via solid-phase peptide synthesis (SPPS), cleavage of the protected peptides from the solid support, backbone cyclization, and global deprotection. Importantly, no significant epimerization was observed during cyclization of these peptides. This was to be expected for 3a–5a as the C-terminal amino acid in the linear precursor was a Gly residue. Furthermore, we have previously observed with analogues of 1a and 2a no evidence of significant epimerization of C-terminal arginines (unpublished data). All peptides were isolated in good yield (12–27% overall yield, >95% purity) using reversed-phase high-performance liquid chromatography (RP-HPLC).
Scheme 1. Synthesis of Cyclic Monomer, 1a–5a using 1a as Exemplar.

Amino acids are represented by one-letter codes in a circle; 2CTC resin = 2-chlorotrityl chloride resin; p = D-Pro; SPPS = solid-phase peptide synthesis; DIEA = N,N-diisopropylethylamine; HATU = 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate; DODT = 3,6-dioxa-1,8-octanedithiol. Assembly of the linear peptides and the cyclization step uses dimethylformamide (DMF) as the solvent.
6a is sequentially similar to 3a, 4a, and 5a but lacks proline in its sequence, which can promote cyclization. Therefore, we employed native chemical ligation (NCL), which does not require the presence of bulky side-chain protecting groups that can hinder cyclization. C-terminal peptide hydrazides were prepared and subsequently oxidized to the reactive acyl azide, followed by thioesterification to form the critical thioester moiety that is essential for the reaction to proceed (Scheme 2).336a was assembled on a hydrazine resin, then cleaved with trifluoroacetyl (TFA) to yield the linear peptide hydrazide. Head-to-tail cyclization via NCL was then performed to produce 6a. However, trace amounts of 4-mercaptophenylacetic acid (MPAA) were also present in the crude peptide mixture, which coeluted with the parent peptide during the purification step. This necessitated multiple HPLC purifications and resulted in a lower-than-expected yield (6% yield). It is essential that all traces of MPAA are removed prior to dimerization because its thiol group can compete with the peptide for DCA. For this reason, we recommend that NCL is not the preferred method of peptide cyclization for this application (although other thiol reagents could have been used to avoid the side-product we observed here23,24).
Scheme 2. Synthesis of Peptide 6a via Native Chemical Ligation.

Amino acids are represented by one-letter codes in a circle; SPPS = solid-phase peptide synthesis; DODT = 3,6-dioxa-1,8-octanedithiol; MPAA = 4-mercaptophenylacetic acid.
Synthesis of Dimeric Peptides
Synthesis of the peptide dimers was achieved via adaptation of a previously reported method;26 although, notably the peptide concentration was increased from 0.1 mM in the literature to 4 mM (Scheme 3). All monomeric peptides were incubated with 1.1 equiv tris(2-carboxyethyl)phosphine (TCEP) in 50 mM NH4HCO3 buffer (pH 8.0) for 15 min, before the first aliquot of 0.275 equiv of DCA was added. Each reaction proceeded for 30 min before a second 0.275 equiv of DCA was added. Reactions were monitored by reversed-phase liquid chromatography mass spectrometry (RP-LCMS) at the following time-points: 15 min after the addition of TCEP (t = 0), 30 min after the first DCA addition (t = 30 min), and 30 min after the second DCA addition (t = 1 h). Reaction monitoring RP-LCMS data appears in Figure S1. The conditions described above will from this point be referred to as condition 1 (Table 2).
Scheme 3. Synthetic Scheme of Dimeric Peptides Using Condition 1.

Blue circles represent cyclic peptides bearing a single cysteine residue (side-chain explicitly included).
Table 2. Conditions Investigated for DCA Dimerization Reaction and the Optimal Parameters Determined from the Resultsa.

Original condition (condition 1) is shaded in light grey. Variables altered are shaded in dark grey. All equivalents are relative to the starting monomer.
Optimal parameters are compared to those from condition 1, using LCMS to quantify success of each reaction parameter assessed.
Using condition 1, all monomeric starting material (1a–6a) was consumed at t = 1 h, but notably the acetone-linked dimer yields varied (Figure 3). Two common side products were detected for all six peptides. Side product 1 (Figure 4A) had a mass of +56 Da compared with the monomeric peptides. We initially thought that this was due to the presence of monochloroacetone (MCA; Figure S2) in the DCA starting material, which can be a side product that can form during DCA synthesis. However, NMR analysis of our DCA found that there was no detectable MCA present (Figure S3). We speculate that the MCA is forming in situ, although the precise mechanism currently remains unclear. Side product 2 is a TCEP-related adduct which is +304 Da relative to the starting monomer (Table S1). A side product whereby one end of DCA was hydrolyzed (side product 3; Figure 4B) was observed during formation of 6b. An MPAA adduct of this peptide was also detected (vide supra).
Figure 3.

Yield of each species at t = 1 h of the dimerization reaction using condition 1. Identity of intermediate and side products 1–4 are also described in Table S1.
Figure 4.

Suspected identities of (A) side product 1, (B) side product 3, (C) side product 4, and (D) the dimerization reaction intermediate.
As a negative control, the reaction was carried out in the absence of DCA, whereby 1a–6a were incubated in 50 mM NH4HCO3 buffer (pH 8.0) with 1.1 equiv of TCEP at 4 mM for 1 h. All monomers remained unreacted. This demonstrates that disulfide bond formation was not a competing reaction, which was not surprising due to the presence of the reductant TCEP. Subsequently, the impact of each reaction parameter was investigated systematically as listed in Table 2, based on condition 1.
Impact of Varying the Stoichiometry of TCEP
Peptide precursors to be dimerized must be preincubated with TCEP to suppress formation of disulfide-linked dimers via oxidation. The reaction was performed without TCEP (condition 2) to investigate the impact of a reductant-free reaction (Figure S4). It was found that preincubation of TCEP is important for obtaining good yields for most of the peptides except 3b and 5b. Side product 3 (Figure 4B) was found in condition 2 for all six peptides. Small quantities of disulfide-linked dimers were also observed in condition 2 in most cases. Yields of 3b and 5b were not notably affected by the absence of TCEP. Nevertheless, we recommend utilizing TCEP, as the minor disulfide-linked product can be difficult to separate from the desired dimer.
Impact of Varying the Stoichiometry of DCA
We found that it is preferable for DCA to be added to the reaction slowly and not used in a large excess. In condition 1, 0.55 equiv of DCA was added over two equal additions at 30 min increments. Subsequently, conditions where 0.275, 0.55, and 1.1 equiv of DCA relative to the starting monomer were all added simultaneously (conditions 3–5) were also tested in 1a, 3a–6a (Figure S5). As expected, reactions were not complete at t = 1 h with 0.275 equiv of DCA (condition 3) as there was still starting monomer left unreacted. Interestingly, the dimerization yields of condition 3 do not significantly differ from the standard condition reaction, suggesting that most of the desired product has formed using the first 0.275 equiv of DCA in the standard condition reactions; the second aliquot mostly caused formation of the side products that were observed using condition 1.
The addition of a single quantity of either 0.55 equiv (condition 4) or 1.1 equiv of DCA (condition 5) into the reaction (especially the latter) tended to decrease dimerization yields but the starting monomer had reacted fully. It suggests that high concentrations of unreacted DCA result in an increase in the side product formation, which verifies the previous finding.
Impact of Varying the Starting Monomer Concentration
As expected, peptide concentration played an important role in determining the reaction outcome. The dimerization reaction was tested with peptide monomers at 12 mM (condition 6), 1 mM (condition 7), and the standard 4 mM (condition 1; Figure S6). For all six peptides, reactions progressed slowly when the starting monomer concentration was increased, compared with the standard condition (12 mM; condition 6), with a substantial quantity of starting monomer still present (varied between 48 and 72%), and no more than 25% desired acetone-linked dimer formed at t = 1 h. The monofunctionalized DCA intermediate (intermediate; Figure 4D) was observed in condition 6. Minimal side product was observed when starting monomer concentration is 12 mM, suggesting that higher concentrations slow the reaction kinetics.
The 1 mM starting monomer concentration condition (condition 7) was tested for all peptides except 2a. This low concentration favored the production of side products 1 and 2 (Table S1), and therefore reduced the desired peptide dimer yield of 1b and 3b–5b. However, the yield of 6b increased from 60% (condition 1; 4 mM) to 72% at 1 mM. However, in general, using a 4 mM starting concentration of monomer appears to be preferential for the DCA dimerization reaction.
Impact of Varying the Reaction pH
The pKa value of cysteine thiols is 8–934 and that of lysine amines is typically near 10.5,35 which indicates that chemoselectivity is achievable. In addition to the originally reported pH 8.0 in 50 mM NH4HCO3 buffer, 50 mM NH4HCO3 buffer at a range of pH values (6.0, 7.0, and 9.0; conditions 8–10; Figure S7), and three other commonly used buffers at their original pH (100 mM Na2CO3 at pH 11.3, phosphate-buffered saline (PBS) at pH 7.4 and 0.1% TFA at pH 1.8; conditions 11–13; Figure S8) were tested in the dimerization reactions of 1a and 3a–6a. Based on the results from pH and buffer combinations that we tested, we recommend carrying out the DCA dimerization reaction at pH 7.0 in 50 mM NH4HCO3 buffer.
Performing the reactions at pH 9.0 (condition 10) in 50 mM NH4HCO3 did not have a notable impact on the yield of desired peptide dimers increase in side products. Reactions at pH 6.0 (condition 8) were extremely slow and most starting monomers remained unreacted at t = 1 h. A small proportion of intermediates were observed at t = 1 h across the five peptides tested, whereas none or only trace of the desired dimers was formed.
We observed a reduction in side products when the reaction was carried out at pH 7.0 in 50 mM NH4HCO3 (condition 9), in the formation of 1b, 3b, 5b, and 6b. It is believed that this was due to protonation of lysine amines at this pH, which decreased interference of the desired reaction. However, dimer yields at t = 1 h were not improved for most reactions compared to condition 1, with some monomer peptides left unreacted or partially reacted. This is suspected to be a result of incomplete deprotonation of cysteine thiols at this pH. Nevertheless, although the dimer yield was not increased, reaction at pH 7.0 improved the ratio of products to side products. We concluded that pH 7.0 to 9.0 is a workable pH range for the DCA dimerization reaction; within this range, pH 7.0 provides the highest purity of the desired product, but requires a longer reaction time.
As expected, reactions in 0.1% TFA (condition 13) progressed extremely slowly, as cysteine thiols should be protonated at pH 1.8. However, reactions were also slow in PBS, even though its starting pH (7.4) is within the workable pH range that we had determined for 50 mM of NH4HCO3. This is explained by the relatively low buffer strength of PBS compared to 50 mM NH4HCO3. Indeed, when we measured the pH of PBS post-reaction, we observed it was closer to pH 6.0, which would explain the similarity of the reaction profiles between using PBS at pH 7.4 or 50 mM NH4HCO3 at pH 6.0.
Using 0.1% TFA or PBS both slow the reaction kinetics with little to no dimers observed after 1 h, and no side products were observed in these conditions. Conversely, using 100 mM Na2CO3 at pH 11.3 (condition 11) gave not only drastically lower yields compared to condition 1, but also resulted in a marked increase in side products. In addition to the common side products seen in condition 1, a hydrolyzed side product (side product 3) was also found in the reactions of all but one of the peptides. Importantly, no DCA adduct through lysine was observed even though the lysine amine was expected to be deprotonated at this pH.
Our thorough investigation of pH and buffer combinations served to reinforce the report by Assem and colleagues that 50 mM NH4HCO3 buffer was the optimum choice for the dimerization reaction.26 However, we found that a pH of 7.0 rather than 8.0 provided the highest purity of desired products and is our recommendation when using DCA to dimerize peptides.
Impact of Varying the Reaction Temperature
A reaction temperature of 0 °C slowed down the reaction kinetics. However, the lower temperature also served to improve yields by suppressing the side product formation. In total, we tested three reaction temperatures (0, 40, and 60 °C; conditions 14–16) for the dimerization reaction in addition to room temperature (condition 1). 1a and 3a–6a were tested in all temperature conditions and 2a using conditions 1 and 11 (Figure S9).
An increase in side product 1 (Figure 4A) with increased reaction temperature was observed in all peptides tested. Side product 3 (Figure 4B) also started to appear at the elevated temperature in some cases. For side products 1 and 3, the dimerization yield decreased at high temperature (conditions 15 and 16) for all peptides except peptide 6b, which at 40 °C yielded a comparable dimer (6b) to that obtained at room temperature; at 60 °C increased dimer yield by 11%.
We observed that a reaction temperature of 0 °C suppressed the formation of side products, with improved yields for 1b, 2b, 4b, and 6b (particularly 1b and 6b), which were 8 and 14% higher, respectively, relative to condition 1. Although the dimerization yield decreased by 15% at t = 1 h when reacting 3a in condition 14, 17% of the reaction intermediate was observed; yields should improve with a longer reaction time. Likewise, utilizing condition 14 for 5a resulted in a 3% yield reduction at t = 1 h; 17% of reaction intermediate was also present. These results suggest that reducing the reaction temperature can improve the acetone-linked dimer yield. However, longer reaction times are required.
Impact of Side-Chain Protection on Lysine
Assem and colleagues speculated that the presence of lysine amines as alternative nucleophiles could complicate reactions using DCA.26 We were mindful of this possibility and included peptides in this study with multiple lysines to investigate this possibility. However, we did not observe any lysine adduct in our results, suggesting that the ε-amine does not participate in the reaction with DCA using our conditions, but it may instead interfere with the desired reaction of β-thiolate of cysteine with DCA in an indirect manner. To test whether unprotected lysine is really an important factor affecting the yield of acetone-linked dimers, we synthesized a version of 1a (Scheme 4) whereby an orthogonal side-chain protecting group on cysteine was selectively removed using reducing conditions, leaving the side-chain protecting groups of lysines and arginine intact.
Scheme 4. Synthesis of Cyclic Monomer Peptide 1a with Side-Chain of Lysines Protected.

Amino acids are represented by one-letter codes in a circle; PPh3 = triphenylphosphine.
Semi-protected 1a was dimerized using our standard conditions (condition 1). Notably, we made one alteration and used a 1:1 solution of 50 mM NH4HCO3, pH 8.0 and DMF to dissolve the hydrophobic peptide. We chose DMF as a cosolvent because the DCA “stock” solution was also prepared in DMF. A yield of 77% of acetone-linked dimer was obtained, which was a 10% improvement of the dimerization step compared to using “unprotected” 1a (67%). Semi-protected 1b then underwent side-chain deprotection in TFA to yield 1b. The results verify that although DCA does not directly conjugate to lysine side-chains, unprotected lysines can still appear to affect acetone-linked dimer yield. Protecting the lysine side-chains is a potential way to improve dimerization yields, especially for lysine-rich peptides. Importantly, this result also demonstrated that the reaction was exclusively occurring via the Cys residues, as the product from this synthesis of 1b and the first synthesis using deprotected 1a were characterized and shown to have identical LCMS retention times and m/z.
In Vitro Serum Stability Assays
As we are particularly interested in the possible utility of dimers linked via acetone-like bridges as potential pharmacological tools, their stability in biological fluids is critical. Thus, we chose to examine 3b in an in vitro model of serum stability in a solution of human serum (Figure 5). In our assay, there appeared to be no loss of 3b over a 48 h period. To investigate if the acetone-like linkage itself has any impact on peptide stability in serum, we synthesized and tested an analogous disulfide-linked dimer 3c (Figure S10), which was found to also be intact over this 48 h period. This demonstrates that the acetone-like linkage has no negative impact on peptide stability in serum. In contrast, the linear control peptide (substance P) was hydrolyzed completely within 3 h, with a half-life of 72 min.
Figure 5.
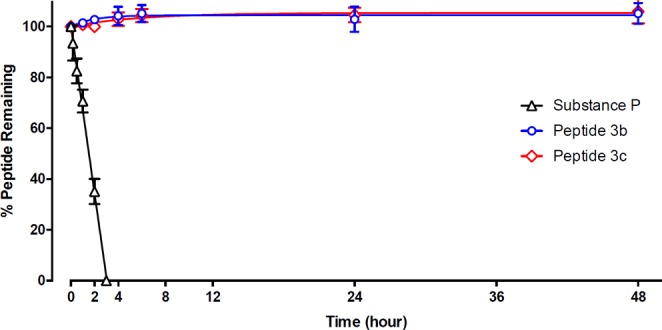
Stability of acetone-linked dimer peptide 3b, disulfide-linked dimer peptide 3c, and a linear control peptide substance P in human serum in vitro. Peptides (0.5 mg/mL) were incubated at 37 °C with human serum. The percentage of peptide remaining in samples taken at various time points was determined by RP-HPLC, using 4-isopropylbenzyl alcohol as an internal standard.
It is well established that head-to-tail cyclized peptides are typically highly resistant to proteolytic cleavage.36 Our stability assays show that combining circular peptides via the enzymatically and redox-stable acetone-like “bridge” results in dimeric bicyclic peptides that are metabolically stable. Metabolic stability is only one contributor—along with renal excretion—to peptide half-life in vivo. Nevertheless, the stable nature of acetone-linked cyclic dimers suggests that they could be suitable for administration in vivo, either as pharmacological tools or as lead compounds for preclinical development.
Conclusions
The utilization of DCA in combination with cysteine-bearing peptides is an effective bio-orthogonal conjugation strategy for the synthesis of dimeric bicyclic peptides. However, there are several aspects of this reaction that need to be carefully considered. The key findings of this study showed that DCA should be added to the reaction mixture slowly to minimize the concentration of unreacted DCA, and thereby minimize side product formation. Furthermore, pH and temperature are also critical, with 50 mM NH4HCO3 buffer at pH 7.0 and 0 °C determined to be the optimal conditions. We note that in peptides with multiple lysines, selective side-chain protection strategies can be applied to shield lysine side chains during the dimerization step.
In summary, DCA has proven to be a useful addition to the peptide synthesis toolkit and anticipate that it will become more commonly utilized in the preparation of complex multicyclic peptides.
Experimental Section
General Procedure A: Preparation of Fmoc-Amino Acid-Substituted Resins
Fmoc protected C-terminal amino acid (2.0 equiv relative to resin) was dissolved in DCM (10 mL/g of resin), followed by addition of N,N-diisopropylethylamine (DIEA) (6.0 equiv relative to resin) and 2-chlorotrityl chloride resin. The mixture was gently shaken for 4 h at room temperature. Unloaded sites were then capped by 1 mL of methanol for 10 min. Afterwards, the loaded resin was washed with DMF (5 × 10 mL), methanol (5 × 10 mL), and diethyl ether (5 × 10 mL) and dried in vacuo.
General Procedure B: Solid-Phase Peptide Synthesis—Linear Peptides
Peptides were synthesized on Fmoc-amino acid substituted 2-chlorotrityl chloride resin (see general method A) using standard Fmoc protocols on a CEM Liberty automated microwave synthesizer. Fmoc-deprotections were performed using 20% piperidine in DMF (2 × 5 mL, 5 min). Couplings were performed using Fmoc-amino acids (0.2 M, dissolved in DMF) and 0.45 M O-(1H-6-chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate, activating with 18% DIEA in DMF. Between successive steps, the peptide resin was washed three times with DMF.
General Procedure C: Peptide Cleavage from 2-Chlorotrityl Chloride Resin
Peptide-resin was treated with 0.8% TFA in DCM (5 mL) for 5 min at room temperature, shaking gently. The mixture was then filtered, and the filtrate was collected into a flask containing 1:2 acetonitrile (ACN)/H2O (50 mL). The treatment with 0.8% TFA solution was repeated an additional seven times. DCM in the filtrate was removed in a rotary evaporator, and the remaining filtrate was then lyophilized.
General Procedure D: Peptide Head-to-Tail Cyclization
Linear side-chain-protected peptide was dissolved in DMF or 500 μL DMF and the remaining volume comprised ACN to give a peptide concentration of 4 mM. Then, 6.0 equiv (relative to peptide) of DIEA and 3.0 equiv (relative to peptide) of HATU were added to the peptide solution. The mixture was gently stirred at room temperature for 4 h. Reaction progression was monitored by analytical RP-LCMS, and when complete, the mixture was diluted in 1:1 ACN/H2O and lyophilized.
General Procedure E: Removal of Acid Sensitive Protecting Groups on Peptide
To the peptide was added 5 mL of deprotection solution (95% TFA, 2% H2O, 2% DODT, 1% TIPS), which was stirred gently for 2 h at room temperature. The solution was concentrated under a stream of N2 gas to ∼10% of the original volume, and 15 mL cold ether was added to precipitate the peptide. The reaction vessel was centrifuged (2500 rpm, 15 min, 4 °C), and the ether was decanted to yield a residual solid. The peptide was subsequently purified by semi-preparative RP-HPLC (buffer A was 0.1% v/v TFA in milli Q water, and buffer B was 0.08% v/v TFA in acetonitrile).
Synthesis of Cyclo[D-Pro-Cys-Lys-Lys-Arg] (1a)
Peptide 1a was prepared following procedures A, B, C, D, and E using an Arg-preloaded 2CTC resin (0.25 mmol) and Fmoc-D-Pro-OH, Fmoc-Cys(Trt)-OH, and Fmoc-Lys(Boc)-OH. Purification by preparative RP-HPLC (0–40% buffer B in buffer A in 60 min) and yielded peptide 1a in high purity (>95%) as a white fluffy powder (35.6 mg, 23%). Analytical RP-LCMS: (0–60% buffer B in buffer A in 9 min), tR = 4.4 min, m/z = 613.3 [M + H]+. HRMS (C26H48N10O5S1): calcd 613.3623 [M + H]+; found, 613.3615 [M + H]+.
Synthesis of Cyclo[D-Pro-Cys-Lys(Boc)-Lys(Boc)-Arg(Pbf)] (Side-Chain-Protected 1a)
Cyclo[d-Pro-Cys(StBu)-Lys(Boc)-Lys(Boc)-Arg(Pbf)] was prepared following procedures A, B, C, and D using an Arg-preloaded 2CTC resin (0.25 mmol) and Fmoc-D-Pro-OH, Fmoc-Cys(StBu)-OH, and Fmoc-Lys(Boc)-OH. The side-chain-protecting group StBu was removed by incubating the peptide with 5.0 equiv triphenylphosphine (PPh3) for 5 days (in 5:1 ACN/H2O, at 40 °C). Purification by preparative RP-HPLC (30–80% buffer B in buffer A in 50 min) yielded cyclo[d-Pro-Cys-Lys(Boc)-Lys(Boc)-Arg(Pbf)] in high purity (>97%) as a white fluffy powder (10.1 mg, 4%). Analytical RP-LCMS: (0–60% buffer B in buffer A in 9 min), tR = 10.8 min, m/z = 1065.4 [M + H]+.
Synthesis of Cyclo[D-Pro-Cys-Arg-Arg-Arg] (2a)
Peptide 2a was prepared following procedures A, B, C, D, and E using an Arg-preloaded 2CTC resin (0.1 mmol) and Fmoc-D-Pro-OH and Fmoc-Cys(Trt)-OH. Purification by preparative RP-HPLC (0–40% buffer B in buffer A in 60 min) and yielded peptide 2a in high purity (>95%) as a white fluffy powder (8.1 mg, 12%). Analytical RP-LCMS: (0–60% buffer B in buffer A in 9 min), tR = 4.4 min, m/z = 669.3 [M + H]+. HRMS (C26H48N14O5S1): calcd 669.3755 [M + H]+, 335.1917 [M + 2H]2+; found, 335.1908 [M + 2H]2+.
Synthesis of Cyclo[Gln-Leu-Cys-D-Pro-Ala-Val-Pro-Val-Ser-Lys-Gly] (3a)
Peptide 3a was prepared following procedures A, B, C, D, and E using a Gly-preloaded 2CTC resin (0.25 mmol) and Fmoc-Gln(Trt)-OH, Fmoc-Leu-OH, Fmoc-Cys(Trt)-OH, Fmoc-D-Pro-OH, Fmoc-Ala-OH, Fmoc-Val-OH, Fmoc-Pro-OH, Fmoc-Ser(tBu)-OH, and Fmoc-Lys(Boc)-OH. Purification by preparative RP-HPLC (20–60% buffer B in buffer A in 60 min) yielded peptide 3a in high purity (>95%) as a white fluffy powder (70.8 mg, 26%). Analytical RP-LCMS: (0–60% buffer B in buffer A in 9 min), tR = 7.1 min, m/z = 1080.6 [M + H]+. HRMS (C48H81N13O13S1): calcd 1080.5904 [M + H]+; found, 1080.5895 [M + H]+.
Synthesis of Cyclo[Gln-Leu-Lys-Cys-Lys-Val-Pro-Val-Ser-Lys-Gly] (4a)
Peptide 4a was prepared following procedures A, B, C, D, and E using a Gly-preloaded 2CTC resin (0.25 mmol) and Fmoc-Gln(Trt)-OH, Fmoc-Leu-OH, Fmoc-Lys(Boc)-OH, Fmoc-Cys(Trt)-OH, Fmoc-Val-OH, Fmoc-Pro-OH, and Fmoc-Ser(tBu)-OH. Purification by preparative RP-HPLC (0–40% buffer B in buffer A in 60 min) yielded peptide 4a in high purity (>95%) as a white fluffy powder (79.9 mg, 27%). Analytical RP-LCMS: (0–60% buffer B in buffer A in 9 min), tR = 6.4 min, m/z = 1168.6 [M + H]+. HRMS (C52H93N15O13S1): calcd 1168.6900 [M + H]+, 584.8489 [M + 2H]2+; found, 584.8492 [M + 2H]2+.
Synthesis of Cyclo[Asn-Ser-Pro-Val-Cys-D-Pro-Ala-Ile-Lys-Thr-Gly] (5a)
Peptide 5a was prepared following procedures A, B, C, D, and E using a Gly-preloaded 2CTC resin (0.25 mmol) and Fmoc-Asn(Trt)-OH, Fmoc-Ser(tBu)-OH, Fmoc-Pro-OH, Fmoc-Val-OH, Fmoc-Cys(Trt)-OH, Fmoc-D-Pro-OH, Fmoc-Ala-OH, Fmoc-Ile-OH, Fmoc-Lys(Boc)-OH, Fmoc-Thr(tBu)-OH, and Fmoc-Gly-OH. Purification by preparative RP-HPLC (20–60% buffer B in buffer A in 40 min) yielded peptide 5a in high purity (>95%) as a white fluffy powder (18.5 mg, 22%). Analytical RP-LCMS: (0–60% buffer B in buffer A in 9 min), tR = 7.2 min, m/z = 1068.4 [M + H]+. HRMS (C46H77N13O14S1): calcd 1068.5527 [M + H]+; found, 1068.5518 [M + H]+.
Synthesis of Cyclo[Cys-Lys-Gln-Thr-Leu-Ile-Lys-Val-Phe-Gly] (6a)
To generate hydrazine-Trt-(2-Cl) resin, 2CTC resin (0.25 mmol) was treated with 200 μL hydrazine in 5 mL DCM; the mixture was shaken at room temperature for 1 h. The linear sequence was synthesized on hydrazine-Trt-(2-Cl) resin according to general procedure B. Peptide hydrazide was cleaved from the resin and acid-sensitive side-chain-protecting groups simultaneously removed according to general procedure E. The peptide hydrazide product was then purified by preparative RP-HPLC (0–40% buffer B in buffer A in 40 min). Linear peptide hydrazide was recovered as a white fluffy powder (124.0 mg; 43%). Analytical RP-LCMS: (0–60% buffer B in buffer A in 9 min), tR = 7.0 min. LCMS (m/z): 1150.6 [M + H]+.
To perform peptide backbone cyclization with NCL, purified peptide hydrazide was dissolved in an aqueous ligation buffer (6.0 M Gn·HCl, 0.2 M Na2HPO4, pH 3.0) at −10 °C for 20 min; the peptide concentration was 2 mM. The reaction mixture was maintained at −10 °C (the flask was immersed in an ice bath) and gently stirred. NaNO2 (7 equiv relative to the peptide) was solubilized in H2O and added dropwise to the peptide solution and stirred for 30 min at −10 °C. MPAA (7 equiv relative to the peptide) was then added to the reaction. The acidity of the solution was adjusted to pH 7.0 and the temperature raised to 22 °C. The reaction was monitored by LCMS, with each analytical aliquot being reduced by TCEP (pH 7.0) prior to analysis. The reaction was complete after 1 h. Purification by preparative RP-HPLC (30–45% buffer B in buffer A in 50 min) yielded peptide 6a in high purity (>95%) as a white fluffy powder (16.5 mg, 6%). Analytical RP-LCMS: (0–60% buffer B in buffer A in 9 min), tR = 8.6 min, m/z = 1118.6 [M + H]+. HRMS (C52H87N13O12S1): calcd 1118.6406 [M + H]+, 559.8242 [M + 2H]2+; found, 559.8235 [M + 2H]2+.
DCA Dimerization Reaction Trial, Condition 1
Peptides 1a–6a (0.32 μmol) were prepared in 40 mM stock in H2O. TCEP (1.1 equiv, 100 mM in H2O) was added to the peptide before the reaction was diluted to 4 mM with 50 mM NH4HCO3 buffer (pH 8.0). The reaction was shaken gently for 15 min before the first 0.275 equiv of DCA (40 mM in DMF) was added. The reaction was allowed to proceed for 30 min, and 5 μL of the reaction was diluted in 15 μL of H2O and analyzed in RP-LCMS (0–60% B in A in 9 min). The second 0.275 equiv of DCA (40 mM in DMF) was then added to the reaction. The reaction was shaken gently for another 30 min before 5 μL of the reaction was diluted in 15 μL of H2O and analyzed in RP-LCMS (0–60% B in A in 9 min). Percentage of each species was determined by area under the peak of the 215 nm chromatograph. Reaction conditions 2–16 were based on this condition.
Synthesis of Acetone-Linked Dimer of Cyclo[d-Pro-Cys-Lys-Lys-Arg] (1b)
Method 1, Using Peptide 1a
Peptide 1a (6.0 mg) was mixed with 1.1 equiv of TCEP before being solubilized in 50 mM NH4HCO3 buffer (pH 8.0) at 4 mM concentration. Reaction was shaken gently in an ice bath for 15 min before the first 0.275 equiv of DCA (40 mM in DMF) was added dropwise. The second 0.275 equiv of DCA (40 mM in DMF) was added dropwise 30 min afterwards. The reaction was shaken in the ice bath gently for another 30 min. Purification by preparative RP-HPLC (0–40% buffer B in buffer A in 60 min) yielded peptide 1b in high purity (>99%) as a white fluffy powder (4.7 mg, 75%).
Method 2, Using Side-Chain-Protected 1a
Side-chain-protected 1a (5.2 mg) was mixed with 1.1 equiv of TCEP before being solubilized in 1:1 50 mM NH4HCO3 buffer (pH 8.0)/DMF at 4 mM concentration. The reaction was shaken gently at room temperature (r.t.) for 15 min before the first 0.275 equiv of DCA (40 mM in DMF) was added dropwise. The second 0.275 equiv of DCA (40 mM in DMF) was added dropwise 30 min afterwards. The reaction was shaken at r.t. gently for another 30 min. Side-chain-protecting groups on the dimer peptide were then removed following procedure E. Purification by preparative RP-HPLC (0–40% buffer B in buffer A in 60 min) yielded peptide 1b in high purity (>97%) as a white fluffy powder (0.8 mg, 27%).
Analytical RP-LCMS: (0–60% B in A in 9 min), tR = 5.0 min, m/z = 1279.5 [M + H]+. HRMS (C55H98N20O11S2): calcd 1279.7274 [M + H]+, 640.3676 [M + 2H]2+; found, 640.3667 [M + 2H]2+.
Synthesis of Acetone-Linked Dimer of Cyclo[Gln-Leu-Cys-d-Pro-Ala-Val-Pro-Val-Ser-Lys-Gly] (3b)
Peptide 3a (9.3 mg) was mixed with 1.1 equiv of TCEP before being solubilized in 50 mM NH4HCO3 buffer (pH 8.0) at 4 mM concentration. The reaction was shaken gently in an ice bath for 15 min before the first 0.275 equiv of DCA (40 mM in DMF) was added dropwise. The second 0.275 equiv of DCA (40 mM in DMF) was added dropwise 30 min afterwards. The reaction was shaken in the ice bath gently for another 30 min. Purification by preparative RP-HPLC (20–60% buffer B in buffer A in 60 min) yielded peptide 3b in high purity (>98%) as a white fluffy powder (6.0 mg, 63%). Analytical RP-LCMS: (0–60% B in A in 9 min), tR = 7.7 min, m/z = 1108.0 [M + 2H]2+. HRMS (C99H164N26O27S2): calcd 2214.1800 [M + H]+, 1107.5939 [M + 2H]2+; found, 1108.0948 [M + 2H]2+.
Synthesis of Acetone-Linked Dimer of Cyclo[Gln-Leu-Lys-Cys-Lys-Val-Pro-Val-Ser-Lys-Gly] (4b)
Peptide 4a (11.5 mg) was mixed with 1.1 equiv of TCEP before being solubilized in 50 mM NH4HCO3 buffer (pH 8.0) at 4 mM concentration. The reaction was shaken gently in an ice bath for 15 min before the first 0.275 equiv of DCA (40 mM in DMF) was added dropwise. The second 0.275 equiv of DCA (40 mM in DMF) was added dropwise 30 min afterwards. The reaction was shaken in the ice bath gently for another 30 min. Purification by preparative RP-HPLC (10–50% buffer B in buffer A in 60 min) yielded peptide 4b in high purity (>98%) as a white fluffy powder (6.9 mg, 58%). Analytical RP-LCMS: (0–60% buffer B in buffer A in 9 min), tR = 6.6 min, m/z = 1195.6 [M + 2H]2+. HRMS (C107H188N30O27S2): calcd 2390.3857 [M + H]+, 598.3523 [M + 4H]4+; found, 598.6021 [M + 4H]4+.
Synthesis of Acetone-Linked Dimer of Cyclo[Asn-Ser-Pro-Val-Cys-d-Pro-Ala-Ile-Lys-Thr-Gly] (5b)
Peptide 5a (37.8 mg) was mixed with 1.1 equiv TCEP before being solubilized in 50 mM NH4HCO3 buffer (pH 8.0) at 4 mM concentration. The reaction was shaken gently in an ice-salt bath for 15 min before the first 0.275 equiv DCA (40 mM in DMF) was added dropwise. The second 0.275 equiv DCA (40 mM in DMF) was added dropwise 30 min afterwards. The reaction was shaken in the ice-salt bath gently for another 30 min. Purification by preparative RP-HPLC (0–60% buffer B in buffer A in 60 min) yielded peptide 5b in high purity (>97%) as a white fluffy powder (21.3 mg, 56%). Analytical RP-LCMS: (0–60% buffer B in buffer A in 9 min), tR = 7.6 min, m/z = 1095.6 [M + 2H]2+. HRMS (C95H156N26O29S2): calcd 2190.1172 [M + H]+, 1095.5625 [M + 2H]2+; found, 1096.0586 [M + 2H]2+.
Synthesis of Disulfide-Linked Dimer of Cyclo[Gln-Leu-Cys-d-Pro-Ala-Val-Pro-Val-Ser-Lys-Gly] (3c)
Peptide 3a (10.0 mg) was solubilized by 100 mM NH4HCO3 buffer (pH 8.0) at 4 mM concentration. The reaction was stirred gently while exposed to air. The reaction was not completed at t = 3 d. Purification by preparative RP-HPLC (20–60% buffer B in buffer A in 60 min) yielded peptide 3c in high purity (>97%) as a white fluffy powder (0.8 mg, 8%); unreacted peptide 3a was also recovered in high purity (>98%) as a white fluffy powder (4.5 mg, 45%). Analytical RP-LCMS: (0–60% buffer B in buffer A in 9 min), tR = 8.0 min, m/z = 1079.9 [M + 2H]2+. HRMS (C96H160N26O26S2): calcd 2158.1541 [M + H]+, 1079.5810 [M + 2H]2+; found, 1080.0808 [M + 2H]2+.
Synthesis of Acetone-Linked Dimers of cyclo[D-Pro-Cys-Arg-Arg-Arg] (2b) and cyclo[Cys-Lys-Gln-Thr-Leu-Ile-Lys-Val-Phe-Gly] (6b)
Peptides 2b and 6b were synthesised as proof of concept only, and were not scaled up for isolation.
In Vitro Serum Stability Assay for Peptide 3b, 3c, and Substance P
Human serum from male AB plasma (purchased from Sigma-Aldrich, H4522) in a capped Eppendorf tube was incubated at 37 °C for 10 min in a water bath, then mixed with the peptide (dissolved in PBS, pH 7.4) to a final concentration of 0.5 mg/mL, along with 4-isopropylbenzyl alcohol (0.05% v/v) as an internal standard. Aliquots (15 μL) were taken at t = 0 (taken immediately after mixing) and specified timepoints (substance P: 0, 10, 30, 60, 120, 180 min; peptide 3b/3c: 0, 1, 2, 4, 6, 24, 48 h) when reaction tube was kept incubated at 37 °C. Each aliquot was immediately transferred into an Eppendorf tube of 45 μL ACN and 60 μL water sitting on ice to quench the reaction, followed by centrifuge (4 °C, 10 min, 12,000 rpm) to pellet any precipitated proteins. Experiments were carried out in triplicate for each peptide. The area under the curve for each species was determined using the 215 nm chromatograph. Percentage of peptide remaining was calculated following the equation below. Data was analyzed using GraphPad Prism, version 7.04.
 |
Instrumentation
LCMS was performed on an Agilent 1260 Infinity II system. The photodiode array detector HS (215 nm or unless otherwise stated) coupled directly to an electrospray ionization source and an Agilent 6120 single quadrupole mass analyzer. Standard RP-HPLC analysis was performed at 40 °C using an Agilent InfinityLab Poroshell 120 EC-C8 3.0 × 50 mm 2.7 μm column, fitted with an InfinityLab Poroshell 120 EC-C8 3.0 × 5 mm 2.7 μm guard column. The column eluted with a gradient of 0–60% ACN in 0.05% aqueous TFA over 9 min at a flow rate of 0.5 mL/min. Mass spectra were obtained in the positive mode with a scan range of 2–2000 m/z. Buffer A was 0.05% v/v TFA in milli Q water, and buffer B was 0.05% v/v TFA in acetonitrile.
Identities of final products were confirmed by high-resolution mass spectrometry (HRMS) spectra, obtained using an Agilent MS Q-TOF (model G6545XT) using the (+)-ion mode with a Dual AJS ESI ion source.
Analytical reverse-phase HPLC was performed using an Agilent 1200 series HPLC system, fitted with an Eclipse XD8-C8 4.6 Å, 5 μm column. Buffer A was 0.1% v/v TFA in milli Q water, and buffer B was 0.08% v/v TFA in acetonitrile. The column eluted with a gradient of 0–60% ACN in 0.08% aqueous TFA over 10 min at a flow rate of 1 mL/min.
Acknowledgments
Project received funding from Multiple Sclerosis Research Australia, SN was supported by a Melbourne Neuroscience Institute fellowship. We thank Dr Dalia Ponce and the HRMS facility at the Department of Pharmacology and Therapeutics at the University of Melbourne for assistance in peptide characterization, Prof Philip Thompson and Dr Simon Mountford for NMR analysis on the DCA sample, and Prof Philip Thompson and A/Prof Akhter Hossain for proofreading.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b03152.
Experimental procedures and spectral and other characterization data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Marianayagam N. J.; Sunde M.; Matthews J. M. The power of two: protein dimerization in biology. Trends Biochem. Sci. 2004, 29, 618–625. 10.1016/j.tibs.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Wrighton N. C.; Balasubramanian P.; Barbone F. P.; Kashyap A. K.; Farrell F. X.; Jolliffe L. K.; Barrett R. W.; Dower W. J. Increased potency of an erythropoietin peptide mimetic through covalent dimerization. Nat. Biotechnol. 1997, 15, 1261–1265. 10.1038/nbt1197-1261. [DOI] [PubMed] [Google Scholar]
- Johnson D. L.; Farrell F. X.; Barbone F. P.; McMahon F. J.; Tullai J.; Kroon D.; Freedy J.; Zivin R. A.; Mulcahy L. S.; Jolliffe L. K. Amino-terminal dimerization of an erythropoietin mimetic peptide results in increased erythropoietic activity. Chem. Biol. 1997, 4, 939–950. 10.1016/s1074-5521(97)90302-1. [DOI] [PubMed] [Google Scholar]
- O’Leary P. D.; Hughes R. A. Design of potent peptide mimetics of brain-derived neurotrophic factor. J. Biol. Chem. 2003, 278, 25738–25744. 10.1074/jbc.m303209200. [DOI] [PubMed] [Google Scholar]
- Cheronis J. C.; Whalley E. T.; Nguyen K. T.; Eubanks S. R.; Allen L. G.; Duggan M. J.; Loy S. D.; Bonham K. A.; Blodgett J. K. A new class of bradykinin antagonists: synthesis and in vitro activity of bissuccinimidoalkane peptide dimers. J. Med. Chem. 1992, 35, 1563–1572. 10.1021/jm00087a010. [DOI] [PubMed] [Google Scholar]
- Góngora-Benítez M.; Tulla-Puche J.; Albericio F. Multifaceted Roles of Disulfide Bonds. Peptides as Therapeutics. Chem. Rev. 2014, 114, 901–926. 10.1021/cr400031z. [DOI] [PubMed] [Google Scholar]
- Vlahov I. R.; Wang Y.; Kleindl P. J.; Leamon C. P. Design and regioselective synthesis of a new generation of targeted chemotherapeutics. Part II: Folic acid conjugates of tubulysins and their hydrazides. Bioorg. Med. Chem. Lett. 2008, 18, 4558–4561. 10.1016/j.bmcl.2008.07.041. [DOI] [PubMed] [Google Scholar]
- Hällbrink M.; Florén A.; Elmquist A.; Pooga M.; Bartfai T.; Langel Ü. Cargo delivery kinetics of cell-penetrating peptides. Biochim. Biophys. Acta 2001, 1515, 101–109. 10.1016/s0005-2736(01)00398-4. [DOI] [PubMed] [Google Scholar]
- Wong C. T. T.; Rowlands D. K.; Wong C.-H.; Lo T. W. C.; Nguyen G. K. T.; Li H.-Y.; Tam J. P. Orally Active Peptidic Bradykinin B1 Receptor Antagonists Engineered from a Cyclotide Scaffold for Inflammatory Pain Treatment. Angew. Chem. Int. Ed. 2012, 51, 5620–5624. 10.1002/anie.201200984. [DOI] [PubMed] [Google Scholar]
- Thell K.; Hellinger R.; Sahin E.; Michenthaler P.; Gold-Binder M.; Haider T.; Kuttke M.; Liutkevičiu̅tė Z.; Göransson U.; Gründemann C.; Schabbauer G.; Gruber C. W. Oral activity of a nature-derived cyclic peptide for the treatment of multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 3960–3965. 10.1073/pnas.1519960113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves R. P. P.; Fernandes P. A.; Varandas A. J. C.; Ramos M. J. Benchmarking of Density Functionals for the Accurate Description of Thiol-Disulfide Exchange. J. Chem. Theory Comput. 2014, 10, 4842–4856. 10.1021/ct500840f. [DOI] [PubMed] [Google Scholar]
- Bach R. D.; Dmitrenko O.; Thorpe C. Mechanism of thiolate-disulfide interchange reactions in biochemistry. J. Org. Chem. 2008, 73, 12–21. 10.1021/jo702051f. [DOI] [PubMed] [Google Scholar]
- Kolšek K.; Aponte-Santamaria C.; Grater F. Accessibility explains preferred thiol-disulfide isomerization in a protein domain. Sci. Rep. 2017, 7, 9858. 10.1038/s41598-017-07501-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong V.; Presolski S. I.; Ma C.; Finn M. â. G. Analysis and optimization of copper-catalyzed azide-alkyne cycloaddition for bioconjugation. Angew. Chem. Int. Ed. 2009, 48, 9879–9883. 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescher J. A.; Bertozzi C. R. Chemistry in living systems. Nat. Chem. Biol. 2005, 1, 13–21. 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem. Int. Ed. 2009, 48, 6974–6998. 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman M. L.; Royzen M.; Fox J. M. Tetrazine ligation: Fast bioconjugation based on inverse-electron-demand Diels-Alder reactivity. J. Am. Chem. Soc. 2008, 130, 13518–13519. 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj N. K.; Weissleder R.; Hilderbrand S. A. Tetrazine-Based Cycloadditions: Application to Pretargeted Live Cell Imaging. Bioconjugate Chem. 2008, 19, 2297–2299. 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W.; Wang Y.; Qu J.; Madden M. M.; Lin Q. A photoinducible 1,3-dipolar cycloaddition reaction for rapid, selective modification of tetrazole-containing proteins. Angew. Chem. Int. Ed. 2008, 47, 2832–2835. 10.1002/anie.200705805. [DOI] [PubMed] [Google Scholar]
- Jin A.-H.; Muttenthaler M.; Dutertre S.; Himaya S. W. A.; Kaas Q.; Craik D. J.; Lewis R. J.; Alewood P. F. Conotoxins: Chemistry and Biology. Chem. Rev. 2019, 119, 11510. 10.1021/acs.chemrev.9b00207. [DOI] [PubMed] [Google Scholar]
- Northfield S. E.; Wang C. K.; Schroeder C. I.; Durek T.; Kan M.-W.; Swedberg J. E.; Craik D. J. Disulfide-rich macrocyclic peptides as templates in drug design. Eur. J. Med. Chem. 2014, 77, 248–257. 10.1016/j.ejmech.2014.03.011. [DOI] [PubMed] [Google Scholar]
- O’Leary P. D.; Hughes R. A. Structure-Activity Relationships of Conformationally Constrained Peptide Analogues of Loop 2 of Brain-Derived Neurotrophic Factor. J. Neurochem. 1998, 70, 1712–1721. 10.1046/j.1471-4159.1998.70041712.x. [DOI] [PubMed] [Google Scholar]
- Thompson R. E.; Liu X.; Alonso-García N.; Pereira P. J. B.; Jolliffe K. A.; Payne R. J. Trifluoroethanethiol: An Additive for Efficient One-Pot Peptide Ligation–Desulfurization Chemistry. J. Am. Chem. Soc. 2014, 136, 8161–8164. 10.1021/ja502806r. [DOI] [PubMed] [Google Scholar]
- Johnson E. C. B.; Kent S. B. H. Insights into the Mechanism and Catalysis of the Native Chemical Ligation Reaction. J. Am. Chem. Soc. 2006, 128, 6640–6646. 10.1021/ja058344i. [DOI] [PubMed] [Google Scholar]
- Spokoyny A. M.; Zou Y.; Ling J. J.; Yu H.; Lin Y.-S.; Pentelute B. L. A Perfluoroaryl-Cysteine SNAr Chemistry Approach to Unprotected Peptide Stapling. J. Am. Chem. Soc. 2013, 135, 5946–5949. 10.1021/ja400119t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assem N.; Ferreira D. J.; Wolan D. W.; Dawson P. E. Acetone-Linked Peptides: A Convergent Approach for Peptide Macrocyclization and Labeling. Angew. Chem., Int. Ed. 2015, 54, 8665–8668. 10.1002/anie.201502607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L.; Krantz B.; Russell N. S.; Deshpande S.; Wilkinson K. D. Nonhydrolyzable Diubiquitin Analogues Are Inhibitors of Ubiquitin Conjugation and Deconjugation. Biochemistry 2000, 39, 10001–10010. 10.1021/bi0007019. [DOI] [PubMed] [Google Scholar]
- Shandiz A. T.; Capraro B. R.; Sosnick T. R. Intramolecular cross-linking evaluated as a structural probe of the protein folding transition state. Biochemistry 2007, 46, 13711–13719. 10.1021/bi701042e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shandiz A. T.; Baxa M. C.; Sosnick T. R. A “Link-Psi” strategy using crosslinking indicates that the folding transition state of ubiquitin is not very malleable. Protein Sci. 2012, 21, 819–827. 10.1002/pro.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher J. M.; Morton C. J.; Zwar R. A.; Murray S. S.; O’Leary P. D.; Hughes R. A. Design of a conformationally defined and proteolytically stable circular mimetic of brain-derived neurotrophic factor. J. Biol. Chem. 2008, 283, 33375–33383. 10.1074/jbc.m802789200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasawa A.; Kawate T. Structural basis for subtype-specific inhibition of the P2X7 receptor. eLife 2016, 5, e22153 10.7554/elife.22153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher J. M.; Hughes R. A. Modified low molecular weight cyclic peptides as mimetics of BDNF with improved potency, proteolytic stability and transmembrane passage in vitro. Bioorg. Med. Chem. 2009, 17, 2695–2702. 10.1016/j.bmc.2009.02.053. [DOI] [PubMed] [Google Scholar]
- Fang G.-M.; Li Y.-M.; Shen F.; Huang Y.-C.; Li J.-B.; Lin Y.; Cui H.-K.; Liu L. Protein Chemical Synthesis by Ligation of Peptide Hydrazides. Angew. Chem., Int. Ed. 2011, 50, 7645–7649. 10.1002/anie.201100996. [DOI] [PubMed] [Google Scholar]
- Madzelan P.; Labunska T.; Wilson M. A. Influence of peptide dipoles and hydrogen bonds on reactive cysteine pKavalues in fission yeast DJ-1. FEBS J. 2012, 279, 4111–4120. 10.1111/febs.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama Y.; Castañeda C. A.; Chimenti M.; García-Moreno B.; Iwahara J. Direct evidence for deprotonation of a lysine side chain buried in the hydrophobic core of a protein. J. Am. Chem. Soc. 2008, 130, 6714–6715. 10.1021/ja801731g. [DOI] [PubMed] [Google Scholar]
- Nielsen D. S.; Shepherd N. E.; Xu W.; Lucke A. J.; Stoermer M. J.; Fairlie D. P. Orally Absorbed Cyclic Peptides. Chem. Rev. 2017, 117, 8094–8128. 10.1021/acs.chemrev.6b00838. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.