

Significance Statement
Although sickle cell trait and sickle cell disease are associated with CKD among black people in the United States, longitudinal data on GFR decline in such individuals is sparse. In a cohort of black patients having sickle cell trait and sickle cell disease was associated with a significantly faster annual eGFR decline, and eGFR declined significantly faster in patients with sickle cell disease than in those with sickle cell trait. Male sex, diabetes mellitus, and high baseline GFRs were associated with faster eGFR decline in both phenotypes. In sickle cell trait, high hemoglobin S and elevated hemoglobins F and A2 were associated with a slower eGFR decline. Physicians caring for black patients need to consider sickle cell trait and sickle cell disease status and interactions with comorbidities when evaluating CKD risk.
Keywords: Sickle cell trait, Sickle cell disease, chronic kidney disease, African American, kidney function decline, estimated glomerular filtration rat
Visual Abstract

Abstract
Background
Sickle cell trait and sickle cell disease are thought to be independent risk factors for CKD, but the trajectory and predictors of kidney function decline in patients with these phenotypes are not well understood.
Methods
Our multicenter, observational study used registry data (collected January 2005 through June 2018) and included adult black patients with sickle cell trait or disease (exposures) or normal hemoglobin phenotype (reference) status (ascertained by electrophoresis) and at least 1 year of follow-up and three eGFR values. We used linear mixed models to evaluate the difference in the mean change in eGFR per year.
Results
We identified 1251 patients with sickle cell trait, 230 with sickle cell disease, and 8729 reference patients, with a median follow-up of 8 years. After adjustment, eGFR declined significantly faster in patients with sickle cell trait or sickle cell disease compared with reference patients; it also declined significantly faster in patients with sickle cell disease than in patients with sickle cell trait. Male sex, diabetes mellitus, and baseline eGFR ≥90 ml/min per 1.73 m2 were associated with faster eGFR decline for both phenotypes. In sickle cell trait, low hemoglobin S and elevated hemoglobin A were associated with faster eGFR decline, but elevated hemoglobins F and A2 were renoprotective.
Conclusions
Sickle cell trait and disease are associated with faster eGFR decline in black patients, with faster decline in sickle cell disease. Low hemoglobin S was associated with faster eGFR decline in sickle cell trait but may be confounded by concurrent hemoglobinopathies. Prospective and mechanistic studies are needed to develop best practices to attenuate eGFR decline in such patients.
Sickle cell trait (SCT), a carrier state characterized by the inheritance of one copy of the sickle hemoglobin gene, was previously thought to be benign.1 Recently, SCT has emerged as a significant contributor to the burden of CKD in black patients.2–5 Approximately 8%–9% of all black Americans have SCT,6,7 therefore, there is a need to improve our understanding of how SCT affects kidney function over time.
It is important to note that SCT is not a disease and is not an intermediate phenotype of sickle cell disease (SCD). However, SCT and SCD share one similarity that contributes to renal injury, i.e., renal medullary damage over time due to recurrent hemoglobin sickling.8
SCD is caused by the inheritance of two abnormal hemoglobin genes (at least one of which is the sickle hemoglobin gene).9 SCD is associated with severe morbidity and early mortality.9,10 Because of improved care over the past few decades, patients with SCD now live longer.11–13 Consequently, the chronic end-organ complications of hemoglobin sickling, such as CKD, have increased in SCD.11–13 CKD is associated with nearly one in five deaths in SCD12; however, there is a lack of longitudinal data on renal outcomes in SCD.
Despite studies describing the association between sickle hemoglobin and CKD,2,4,8,11,14–17 there is limited understanding of the effect of sickle hemoglobin on eGFR decline over time compared with a population with no sickle hemoglobin. Furthermore, no studies have described which factors significantly affect eGFR decline in SCT over time. Importantly, eGFR decline in SCT and SCD have not been investigated simultaneously to assess a dose-response relationship between sickle hemoglobin and eGFR. Also, there is sparse data on the association between hemoglobin phenotypes (hemoglobin S [HbS], hemoglobin F [HbF], hemoglobin A [HbA], and hemoglobin A2 [HbA2]) and eGFR decline among patients with SCT, which could provide insights into a dose-response relationship.
The aim of this observational study was to compare the annual change in mean eGFR among black Americans with SCT and SCD to black Americans with a normal hemoglobin phenotype. We also investigated the risk for incident stage 3 CKD and factors associated with eGFR decline in SCT and SCD.
Methods
Study Population
This study utilized data from the Research Patient Data Registry (RPDR) collected between January 1, 2005 and June 30, 2018. RPDR is a centralized clinical data registry that gathers detailed inpatient and outpatient clinical data from the member hospitals and institutions of Partners Healthcare, Boston, Massachusetts. RPDR has been described in detail in previously published studies.18–21
Inclusion and Exclusion Criteria
We applied the following inclusion criteria: age ≥18 years at first available serum creatinine after January 1, 2005, at least three serum creatinine values, at least 1 year between first and last serum creatinine dates, self-identified as black and a hemoglobin electrophoresis at any time. The following exclusion criteria were applied: an eGFR <30 ml/min per 1.73 m2 at first serum creatinine after January 1, 2005 and an eGFR >180 ml/min per 1.73 m2 (Figure 1). This cutoff was selected on the basis of hyperfiltration studies in patients with pre-diabetes showing eGFR values up to 180 ml/min per 1.73 m2, and because SCD, black patients and young patients tend to have higher baseline eGFRs.22–24
Figure 1.

Flow chart of inclusion in study.
This study was approved by the Institutional Review Board at Partners Healthcare, Boston, Massachusetts and the need for informed consent was waived.
Exposures
We defined our exposure as patients with SCT or SCD confirmed by a hemoglobin electrophoresis test performed at any time in the electronic medical record and interpreted by a pathologist. The reference group was defined as patients with a normal hemoglobin phenotype confirmed by a hemoglobin electrophoresis test interpreted by a pathologist. These hemoglobin electrophoresis tests were ordered by physicians for clinical indications and not for research purposes.
Outcomes
Our primary outcome of interest was the difference between exposure and reference patients in the mean annual change in eGFR (Figure 2). We also examined the risk for incident CKD defined as time to first eGFR<60 ml/min per 1.73 m2 in patients with a baseline eGFR ≥65 ml/min per 1.73 m2 (Figure 3). eGFR was calculated using the CKD Epidemiology Collaboration [CKD-EPI] creatinine equation.25,26
Figure 2.

Predicted slopes of mean change in eGFR over study period. Both SCT and SCD were associated with significantly faster GFR decline compared to the reference. GFR decline in the reference was -1.22 (95% CI, -1.23 to -1.20) ml/min per 1.73 m2 per year. GFR decline in SCT was -1.67 (95% CI, -1.72 to -1.62) ml/min per 1.73 m2 per year. GFR decline in SCD was -2.50 (95% CI, -2.58 to -2.42) ml/min per 1.73 m2 per year. Adjusted for baseline age, sex, hypertension, diabetes mellitus, history of cardiovascular disease, smoking status, acute kidney injury, ACEi/ARB use, urine albumin: creatinine ratio categories and baseline eGFR. SCT models were also adjusted for hemoglobin electrophoresis indication. SCT, sickle cell trait; SCD, sickle cell disease. GFR, glomerular filtration rate.
Figure 3.

Association of sickle cell trait and sickle cell disease with incident stage 3 chronic kidney disease. Both SCT and SCD were associated with a significantly increased risk for incident CKD after adjustment. Within each phenotype, this risk was higher in males on sub-analysis. Adjusted for baseline age, sex, hypertension, diabetes mellitus, history of cardiovascular disease, smoking status, acute kidney injury, ACEi/ARB use, urine albumin: creatinine ratio categories and baseline eGFR. SCT models were also adjusted for hemoglobin electrophoresis indications. SCT, sickle cell trait; SCD, sickle cell disease.
Covariates
Baseline characteristics were determined at the time of first serum creatinine between January 1, 2005 and June 30, 2018 (Table 1). Demographics and smoking status were obtained by chart review. Comorbidities (hypertension; diabetes mellitus [DM]; cardiovascular disease [CVD], defined as stroke or coronary artery disease; and AKI) were obtained using algorithms on the basis of International Classification of Disease, Tenth Revision diagnosis codes and their International Classification of Disease, Ninth Revision equivalents (see Supplemental Material).21 Laboratory values and medication use at any time during follow-up were obtained by chart review.
Table 1.
Baseline characteristics of cohort
| Variable | Reference, n=8,729 | SCT, n=1,251 | P Value | SCD, n=230 | P Value |
|---|---|---|---|---|---|
| Demographics | |||||
| Mean age (SD), yr | 36 (±12) | 40 (±14) | <0.01 | 33 (±12) | <0.01 |
| Age ≥65 yr | 3% | 7% | <0.01 | 1% | 0.01 |
| Women | 88% | 78% | <0.01 | 50% | <0.01 |
| Median follow-up (IQR), yr | 8.1 (4.6–11.3) | 8.5 (4.7–11.6) | 0.04 | 7.3 (4.2–11.4) | 0.21 |
| Comorbidities | |||||
| Hypertension | 20% | 30% | <0.01 | 11% | <0.01 |
| DM | 16% | 25% | <0.01 | 10% | <0.01 |
| CVD | 10% | 14% | <0.01 | 28% | <0.01 |
| Smoking | <0.01 | <0.01 | |||
| Never | 50% | 45% | 39% | ||
| Ever | 16% | 14% | 9% | ||
| Missing | 34% | 41% | 52% | ||
| AKI | 7% | 13% | <0.01 | 25% | <0.01 |
| Medications | |||||
| ACEis/ARBs | 20% | 29% | <0.01 | 21% | 0.75 |
| Aspirin | 28% | 22% | <0.01 | 35% | <0.01 |
| Statins | 15% | 23% | <0.01 | 12% | 0.15 |
| Hydroxyurea | 0.2% | 0.3% | 0.21 | 48% | <0.01 |
| Laboratory | |||||
| Mean eGFR (SD), ml/min per 1.73 m2 | 114 (±26) | 103 (±27) | <0.01 | 128 (±32) | <0.01 |
| eGFR categories | <0.01 | <0.01 | |||
| ≥120 ml/min per 1.73 m2 | 45% | 30% | 66% | ||
| 90–119 ml/min per 1.73 m2 | 36% | 40% | 22% | ||
| 60–89 ml/min per 1.73 m2 | 16% | 25% | 8% | ||
| 30–59 ml/min per 1.73 m2 | 3% | 5% | 4% | ||
| Urine ACR | <0.01 | 0.32 | |||
| <30 mg/g | 2% | 3% | 1% | ||
| 30–299 mg/g | 1% | 2% | 0% | ||
| >300 mg/g | 0% | 0% | 0% | ||
| Missing | 97% | 95% | 99% | ||
| Mean hemoglobin (SD), g/dla | 12.3 (±1.6) | 12.6 (±1.7) | <0.01 | 9.9 (±2.1) | <0.01 |
| Mean leukocyte count (SD), ×105 cells/mm3a | 7.6 (±3.4) | 7.2 (±3.0) | <0.01 | 12.3 (±6.4) | <0.01 |
| Hemoglobin electrophoresis indications | <0.01 | — | — | ||
| Anemia | 39.4% | 24.5% | — | — | |
| Perinatal testing | 51.8% | 37.9% | — | — | |
| Other | 3.4% | 16.8% | — | — | |
| Unknown | 5.4% | 20.8% | — | — | |
| Mean HbS percentage (SD) | — | 37.0 (±4.3) | — | — | — |
| HbF>0.4% | — | 24.6% | — | — | — |
| Mean HbA percentage levels (SD) | — | 59.4 (±4.4) | — | — | — |
| Mean HbA2 percentage (SD) | — | 3.2 (±0.7) | — | — | — |
AKI was during follow-up; eGFR was obtained using CKD-EPI creatinine equation.
Missing data: hemoglobin (13 reference, 8 SCT, and 0 SCD), leukocyte count (1422 reference, 227 SCT, and 18 SCD), HbS (21), HbF (77), HbA (23), and HbA2 (74).
Statistical Analyses
All analyses were conducted using STATA 14.2 (StataCorp., College Station, TX). Two-sided P values ≤0.05 were considered statistically significant. We compared baseline categorical variables using chi-squared test and continuous variables using t test (with reporting of the mean and SDs) or Wilcoxon rank-sum test (with reporting of the median and interquartile ranges [IQR]) depending on the distribution of data (parametric and nonparametric, respectively).
SCT versus the reference group and SCD versus the reference group were each analyzed independently. To evaluate the association between our exposure groups and the mean rate of change in eGFR per year, we used linear mixed models with random intercepts and slopes to estimate the linear trend in eGFR over time in the exposure group and compared it with that of the reference group, as well as accounting for correlations of observations within each patient.27 All available eGFR values were used for this longitudinal analysis. As illustrated by the equation below, the jth eGFR of the ith patient was estimated using the following equation:
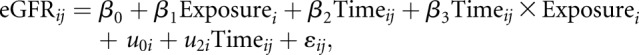 |
where 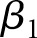 represents the estimated difference between exposure and reference groups,
represents the estimated difference between exposure and reference groups, 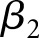 represents the estimated rate of decline in eGFR in the reference group, and the interaction term (
represents the estimated rate of decline in eGFR in the reference group, and the interaction term (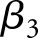 ) represents the difference in the linear trend between the exposure group and reference group. The terms
) represents the difference in the linear trend between the exposure group and reference group. The terms 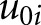 and
and 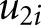 represent a random intercept and a random slope, respectively. The estimated difference in slopes (
represent a random intercept and a random slope, respectively. The estimated difference in slopes (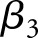 ) and their 95% confidence intervals (95% CIs) for each exposure group were reported (Table 2).
) and their 95% confidence intervals (95% CIs) for each exposure group were reported (Table 2).
Table 2.
Difference in mean eGFR change per year in SCT and SCD compared with the reference
| Exposure | Median Number of eGFR Values (IQR) | Unadjusted β (95% CI), ml/min per 1.73 m2 per yr | Adjusted β (95% CI), ml/min per 1.73 m2 per yra |
|---|---|---|---|
| Reference | 17 (10–29) | 0 | 0 |
| SCT | 19 (10–34) | −0.45 (−0.50 to −0.40) | −0.45 (−0.50 to −0.39) |
| SCD | 48 (22–125) | −1.28 (−1.35 to −1.21) | −1.29 (−1.37 to −1.22) |
| SCD versus SCT | — | −0.83 (−0.92 to −0.74) | −0.86 (−0.95 to −0.76) |
Adjusted for baseline age, sex, hypertension, DM, history of CVD, smoking status, AKI, ACEi/ARB use, urine ACR categories, and baseline eGFR. SCT models were also adjusted for hemoglobin electrophoresis indication.
The association between SCT/SCD and risk for incident CKD was evaluated using Cox proportional hazard models with results summarized using hazard ratios (HRs) and Wald asymptotic 95% CIs (Figure 3). To allow for time to the event in our incident CKD analysis, we excluded patients with a baseline eGFR <65 ml/min per 1.73 m2. This threshold was chosen on the basis of expected age-related eGFR decline of 0.75 ml/min per 1.73 m2 per year in the general population28,29 and the cohort median follow-up.
Multivariable models for both outcomes (difference in mean annual change in eGFR and incident CKD) were adjusted for baseline age, sex, hypertension, DM, CVD, angiotensin-converting enzyme inhibitors or angiotensin receptor blockers (ACEis/ARBs), smoking status (never, former/current, and missing), AKI, baseline eGFR, and categorical urine albumin-to-creatinine ratio (urine ACR; <30 mg/g, 30–299 mg/g, ≥300 mg/g, and missing). SCT models were also adjusted for hemoglobin electrophoresis indication. The covariates used for model adjustment were selected on the basis of prior studies on kidney function decline30,31 and suspected clinical relevance to the analyses.
To evaluate risk factors for eGFR decline within each exposure group (Table 3), we generated slope estimates by including an interaction term with time (in years) and the following variables: sex, hypertension, DM, CVD, ACEis/ARBs, aspirin, statins, time-varying hemoglobin, and time-varying leukocyte counts. We also investigated the association between HbS, HbF, HbA, and HbA2 and the mean annual change in eGFR among patients with SCT.
Table 3.
Association of covariates with mean eGFR change per year among patients with SCT and patients with SCD
| Covariates | SCT β (95% CI), ml/min per 1.73 m2 per yr | SCD β (95% CI), ml/min per 1.73 m2 per yr |
|---|---|---|
| Men | −1.47 (−1.58 to −1.37) | −0.24 (−0.41 to −0.08) |
| Hypertension | −1.21 (−1.31 to −1.11) | −0.13 (−0.36 to 0.09) |
| DM | −1.62 (−1.72 to −1.52) | −2.76 (−3.07 to −2.45) |
| CVD | −0.75 (−0.85 to −0.64) | −0.38 (−0.55 to −0.22) |
| ACEi/ARB use | −1.39 (−1.49 to −1.29) | −2.34 (−2.52 to −2.16) |
| Aspirin use | −1.19 (−1.29 to −1.09) | −1.11 (−1.29 to −0.93) |
| Statin use | −1.45 (−1.54 to −1.35) | −1.13 (−1.31 to −0.95) |
| Hemoglobin | 0.36 (0.34 to 0.39) | 0.34 (0.30 to 0.38) |
| Leukocyte count | −0.07 (−0.09 to −0.06) | 0.04 (0.03 to 0.06) |
| Low HbS (<35%) | −0.57 (−0.68 to −0.46) | — |
| Low HbS (<35%) adjusting for MCV | −0.51 (−0.64 to −0.37) | |
| HbS (versus 19%–34.8%) | ||
| 34.9%–38.9% | 0.67 (0.55 to 0.80) | — |
| 39%–47.2% | 0.49 (0.36 to 0.61) | — |
| Elevated HbF (>0.4%) | 1.05 (0.92 to 1.18) | — |
| Elevated HbA (>70%) | −1.92 (−2.11 to −1.74) | — |
| Elevated HbA2 (>3.5%) | 0.84 (0.73 to 0.95) | — |
| Hydroxyurea | — | −1.17 (−1.38 to −0.95) |
| Hospital visits/year for crises (versus <1/yr) | ||
| 1–2 per yr | — | −2.06 (−2.42 to −1.71) |
| >2 per yr | — | −2.92 (−3.17 to −2.66) |
eGFR obtained using CKD-EPI creatinine equation. All coefficients were adjusted for baseline age, sex, hypertension, DM, history of CVD, AKI during follow-up, smoking status, ACEi/ARB use, urine ACR categories, and baseline eGFR. Hemoglobin electrophoresis indication was included in SCT models. Hemoglobin phenotype models were adjusted for all phenotypes. Hydroxyurea was included in SCD models. MCV, mean corpuscular volume.
Among patients with SCD, we investigated the effect of prevalent hydroxyurea use and frequency of sickle cell crises hospital visits per year as risk factors for eGFR decline. SCD crisis was determined using International Classification of Disease codes (see Supplemental Material).
We explored three-way interaction terms between each exposure, follow-up time, and covariates of interest using the main multivariable models. Subsequently, we compared the differences in the mean change in eGFR over time between exposures and the reference within each covariate subgroup (Table 4).
Table 4.
Differences in mean change in eGFR per year in SCT versus reference and SCD versus reference, within each covariate subgroup
| Covariates | SCT versus Referencea | P Value: SCT Interaction with Covariate and Time | SCD versus Referencea | P Value: SCD Interaction with Covariate and Time | |
|---|---|---|---|---|---|
| Sex | Male | −1.22 (−1.33 to −1.10) | <0.01 | −1.16 (−1.28 to −1.03) | 0.34 |
| Female | −0.07 (−0.13 to −0.01) | −1.24 (−1.33 to −1.14) | |||
| HTN | Yes | −0.58 (−0.66 to −0.51) | <0.01 | −1.08 (−1.26 to −0.90) | <0.01 |
| No | 0.04 (−0.04 to 0.12) | −1.54 (−1.62 to −1.46) | |||
| DM | Yes | −0.80 (−0.88 to −0.72) | <0.01 | −3.40 (−3.68 to −3.13) | <0.01 |
| No | 0.13 (0.06 to 0.20) | −1.34 (−1.41 to −1.26) | |||
| CVD | Yes | −0.26 (−0.35 to −0.16) | 0.01 | −0.80 (−0.91 to −0.68) | <0.01 |
| No | −0.44 (−0.50 to −0.38) | −1.36 (−1.46 to −1.26) | |||
| ACEi/ARB | Yes | −0.58 (−0.65 to −0.50) | <0.01 | −2.50 (−2.64 to −2.35) | <0.01 |
| No | 0.02 (−0.05 to 0.10) | −0.95 (−1.03 to −0.87) | |||
| Aspirin | Yes | −0.69 (−0.76 to −0.61) | <0.01 | −1.38 (−1.48 to −1.29) | <0.01 |
| No | −0.001 (−0.07 to 0.08) | −0.78 (−0.90 to −0.66) | |||
| Statins | Yes | −0.74 (−0.82 to −0.66) | <0.01 | −1.69 (−1.83 to −1.55) | <0.01 |
| No | 0.07 (−0.001 to 0.14) | −1.22 (−1.31 to −1.14) | |||
eGFR obtained using CKD-EPI creatinine equation. HTN, hypertension.
All models adjusted for baseline age, sex, hypertension, DM, history of CVD, smoking status, AKI, ACEi/ARB use, urine ACR categories, and baseline eGFR. SCT models were also adjusted for hemoglobin electrophoresis indications.
Finally, subgroup analyses were performed to estimate the adjusted mean annual change in eGFR within each baseline eGFR category. eGFR categories used were ≥120 ml/min per 1.73 m2, 90–119 ml/min per 1.73 m2, 60–89 ml/min per 1.73 m2, and <60 ml/min per 1.73 m2.
For sensitivity analysis, 1:2 matching was performed between SCT and reference, and SCD and reference patients, using the following criteria: age±3 years, sex, hypertension, DM, and categorical urine ACR.
Results
Baseline Characteristics
We identified 10,210 black patients (1251 SCT, 230 SCD, and 8729 reference) (Figure 1). Baseline characteristics for all cohort participants are displayed in Table 1. The mean age of all participants was 36±13 years. Median follow-up for the entire cohort was 8.2 (IQR, 4.6–11.3) years. The mean eGFR at baseline for all participants was 113±27 ml/min per 1.73 m2. Hemoglobin electrophoresis indications were primarily routine perinatal testing (49%) and anemia workup (37%).
Compared with the reference population, patients with SCT were significantly older (40±14 versus 36±12 years; P<0.01), with fewer women (78% versus 88%; P<0.01), longer follow-up (8.4 years; IQR, 4.7–11.6 years versus 8.1 years; IQR, 4.6–11.3 years; P=0.04), and had a higher prevalence of comorbidities. Patients with SCT also had a lower mean eGFR (103±27 versus 114±26 ml/min per 1.73 m2; P<0.01). Perinatal testing was the most common hemoglobin electrophoresis indication in SCT and the reference; however, a higher proportion of patients with SCT had unknown reasons for testing.
Patients with SCD were significantly younger than the reference patients (33±12 versus 36±12 years; P<0.01), with fewer women (50% versus 88%; P<0.01), shorter follow-up (7.3 years; IQR, 4.2–11.4 years versus 8.1 years; IQR, 4.6–11.3 years; P=0.21), and a lower prevalence of comorbidities, except for CVD where the prevalence was higher in SCD (32% versus 11%; P<0.01). Patients with SCD had a higher mean eGFR (128±32 versus 114±26 ml/min per 1.73 m2; P<0.01) and a higher mean leukocyte count (12.3±6.4 versus 7.6±3.4 ×105 cells/mm3; P<0.01).
In the sensitivity analysis cohorts, the differences in demographics, comorbidities, and baseline eGFR were attenuated (Supplemental Table 1, A and B).
SCT/SCD and Differences in Mean Change in eGFR per Year
Reference patients had a median of 17 (IQR, 10–29) eGFR values over 8 (IQR, 4–11) years. The unadjusted mean change in eGFR in the reference patients was −1.22 (95% CI, −1.23 to −1.20) ml/min per 1.73 m2 per year.
The median number of eGFR values for patients with SCT was 19 (IQR, 10–34) over 8 (IQR, 5–12) years. The unadjusted mean change in eGFR in all patients with SCT was −1.67 (95% CI, −1.72 to −1.62) ml/min per 1.73 m2 per year. eGFR declined significantly faster in SCT compared with reference patients (0.45 ml/min per 1.73 m2 per year faster; P<0.01) and did not change after multivariable adjustment (Figure 2, Table 2).
The median number of eGFR values for patients with SCD was 48 (IQR, 22–125) over 7 (IQR, 4–11) years. The unadjusted mean change in eGFR in patients with SCD was −2.50 (95% CI, −2.58 to −2.42) ml/min per 1.73 m2 per year. The unadjusted decline in eGFR in patients with SCD was significantly faster than in reference patients (1.29 ml/min per 1.73 m2 per year faster; P<0.01) (Figure 2, Table 2). This difference persisted after multivariable adjustment (Figure 2, Table 2).
Comparison of SCD to SCT confirmed a significantly faster eGFR decline in patients with SCD (0.83 ml/min per 1.73 m2 per year faster; P<0.01), which persisted after adjustment (Figure 2, Table 2).
Results of sensitivity analysis did not change the direction or significance of these results (Supplemental Table 2).
SCT/SCD and Incident Stage 3 CKD
Patients with a baseline eGFR <65 ml/min per 1.73 m2 were excluded from this analysis, leaving 9733 patients (1141 SCT, 216 SCD, and 8376 reference). Baseline characteristics of this subcohort are reported in Supplemental Table 3. Across all patients, there were 753 events (155 SCT, 16 SCD, and 582 reference patients) over a median follow-up of 8 (IQR, 5–11) years. Findings are displayed in Figure 3.
Compared with reference patients, the risk for incident CKD in SCT was increased both in unadjusted (HR, 2.07; 95% CI, 1.74 to 2.47) and adjusted (HR, 1.25; 95% CI, 1.05 to 1.51) analysis.
The risk of incident CKD in the patients with SCD was not significant in univariate analysis (HR, 1.27; 95% CI, 0.77 to 2.09), but was significant after adjustment (HR, 2.37; 95% CI, 1.43 to 3.93).
The sensitivity cohort produced similar findings in SCT and SCD both in direction and magnitude (Supplemental Table 4).
Factors associated with eGFR Decline in SCT/SCD
Risk factors associated with eGFR decline in SCT/SCD are summarized in Table 3. Among patients with SCT, male sex, hypertension, DM, CVD, ACEis/ARBs, aspirin, statins, and higher leukocyte counts were significantly associated with a faster decline in eGFR. Higher hemoglobin levels were associated with a significantly slower decline in eGFR among patients with SCT. Low HbS (defined as <35%32) was associated with a faster decline in eGFR. Subsequent evaluation of HbS tertiles (on the basis of the distribution of the data) confirmed lower HbS was associated with faster eGFR decline. Elevated HbF (defined as >0.4%33) was associated with a slower eGFR decline. Elevated HbA (defined as ≥70%) was associated with faster eGFR decline. Elevated HbA2 (defined as >3.5%) was associated with slower eGFR decline. Stratified analysis by covariates comparing eGFR decline in SCT with reference patients showed that the fastest decline occurred in men (Table 4).
Among patients with SCD, male sex, DM, CVD, ACEis/ARBs, aspirin, and statins, but not hypertension, were associated with faster rates of eGFR decline (Table 3). Higher hemoglobin and leukocyte counts were associated with slower decline in eGFR in patients with SCD. Hydroxyurea use and more frequent SCD crisis hospital visits were associated with faster eGFR decline. Stratified analysis by covariates comparing eGFR decline in SCD with reference patients showed the fastest decline was seen among patients with DM (Table 4). Further analysis of SCD by severity phenotype (see Appendix) showed SS and S-beta thalassemia zero were associated with faster GFR decline (Supplemental Table 5).
Mean Change in eGFR by Baseline Kidney Function
Results are reported in Supplemental Table 6. Among reference patients, the adjusted mean change in eGFR per year was −1.21 (95% CI, −1.23 to −1.19) ml/min per 1.73 m2. Rates of eGFR decline were fastest in patients with an eGFR ≥120 ml/min per 1.73 m2 and <60 ml/min per 1.73 m2.
The adjusted mean change in eGFR among all patients with SCT in our cohort was −1.64 (95% CI, −1.69 to −1.59) ml/min per 1.73 m2 per year. The fastest rates of eGFR decline in patients with SCT was noted in the eGFR≥90 ml/min per 1.73 m2 categories.
In the SCD cohort, the adjusted mean change in eGFR was −2.48 (95% CI, −2.56 to −2.40) ml/min per 1.73 m2 per year. Similar to the SCT group, the fastest rates of adjusted eGFR decline in patients with SCD occurred in the eGFR≥90 ml/min per 1.73 m2 categories.
Discussion
In this longitudinal study of black patients with a baseline eGFR ≥30 ml/min per 1.73 m2, we found that SCT is associated with a faster rate of eGFR decline compared with patients with a normal hemoglobin phenotype. This decline is significantly faster in male patients with SCT who have traditional risk factors for CKD, lower hemoglobin levels, and a baseline eGFR ≥90 ml/min per 1.73 m2. In addition, we confirmed an accelerated rate of eGFR decline in patients with SCD. To our knowledge, this is the first study to investigate factors associated with eGFR decline in SCT and simultaneously compare the mean change in eGFR in patients with SCT, patients with SCD, and patients with a normal hemoglobin phenotype.
Our findings suggest that black Americans with SCT lose nearly half an eGFR unit (ml/min per 1.73 m2) more of kidney function every year than black Americans with a normal hemoglobin phenotype. Only one prior study reported eGFR decline in SCT compared with a reference population (0.214 ml/min per 1.73 m2 per year faster in SCT).2 However, unlike this prior study, our cohort was derived from a multihospital system concentrated in the New England region of the United States. Thus, the differences in our findings may be influenced by a higher burden of comorbidities. Severe environmental stressors such as extremes of temperature are known to promote red blood cell sickling34; however, this has not been demonstrated in SCT. Nevertheless, both studies confirm that the effect of SCT on kidney function decline is insidious but significant.
The risk factors for eGFR decline in SCT have not been elucidated in prior studies. Our findings suggest a synergistic effect between male sex and SCT on eGFR decline that is more pronounced than the effect of male sex on eGFR decline described in the general population.35–39 Insights into possible reasons may be gained from previously described sex differences in SCD. One such study suggested that estrogen may ameliorate hemoglobin sickling and attenuate some of its downstream effects (endothelial dysfunction and inflammation).40 Other studies have shown that HbF (which, in significant amounts, inhibits sickling) is inherently higher in adult women compared with men.41–43 Another mechanism that has been described is greater responsiveness and bioavailability of nitric oxide in women with SCD compared with men, thus resulting in improved endothelium dependent blood flow in women, which, in turn, could protect the renal medulla.44 However, it is important to note that women in our study may have been healthier because their hemoglobin electrophoresis indication was more often perinatal testing.
The data from this study also showed that several typical risk factors for CKD increase the risk for eGFR decline in SCT. Our findings comparing patients with SCT with diabetes to reference patients with diabetes (Table 4) are consistent with one study that demonstrated significantly higher levels of markers of vascular dysfunction in individuals with SCT and DM compared with individuals with normal hemoglobin phenotype and DM.45 The effect of HbS percentage on eGFR decline in SCT have not been evaluated. Our study shows that, counterintuitively, lower HbS is associated with faster eGFR decline in SCT. Prior SCT studies found lower HbS improved urine concentrating ability.32 However, no research has demonstrated whether hyposthenuria is a precursor to CKD in SCT. Lower HbS percentage in SCT suggests, but is not diagnostic of, a concurrent α-thalassemia.46 α-Thalassemia in SCT is renoprotective, presumably by reducing the amount of HbS.47 Microcytosis is a prominent feature of α-thalassemia48; however, adding mean corpuscular volumes to the multivariable model (Table 3) only mildly attenuated the effect of lower HbS, presumably because microcytosis may also be due to iron deficiency anemia. However, it is worth noting that variations in other hemoglobin levels can affect HbS percentages. Of note, in this cohort, HbA strongly correlated inversely with HbS (r=−0.92; P<0.001), and HbA≥70% was strongly associated with faster eGFR decline (Table 3). This may be evidence of another underlying hemoglobinopathy or could suggest a complex relationship between HbA and HbS in the pathophysiology of kidney disease in SCT. No prior studies investigating renal outcomes in SCT evaluated the effect of HbA.32,47 Similarly, HbA2 in our cohort weakly correlated inversely with HbS (r=−0.10; P<0.001). Studies have demonstrated that HbA2, like HbF, inhibits red blood cell sickling.49,50 Our results suggest that elevated HbA2 and HbF may be renoprotective in SCT.
The mechanisms for kidney disease in SCT are not well described but evidence exists for renal medullary disruption and endothelial dysfunction in SCT, similar to, but less severe than SCD.45,51–55 Recurrent hemoglobin sickling results in ischemia-reperfusion injury in the renal medulla and, over time, these insults induce local inflammation and fibrosis. Unlike SCD, glomerular hyperfiltration has not been described in SCT. Despite the lower baseline eGFR in this SCT cohort, the fastest rates of eGFR decline in SCT occurred in the eGFR≥90 ml/min per 1.73 m2 categories. Faster rates of eGFR decline have been described in diabetic patients with a baseline eGFR ≥120 ml/min per 1.73 m2 due to increased intraglomerular pressures and oxidative stress.56–58 Prospective studies are needed to better understand the natural history of eGFR decline and glomerular hyperfiltration in SCT.
In contrast to SCT, SCD is a well defined cause of CKD.8,12,59,60 Few studies, however, have looked at longitudinal eGFR decline in SCD. One report described a mean annual change in eGFR of −1.82 ml/min per 1.73 m2,16 but a Jamaican investigation found SCD was associated with a mean annual eGFR decline of 3.2 ml/min per 1.73 m2.61 The differences in our studies suggest environmental or clinical factors that were not accounted for. Nevertheless, eGFR decline in SCD can progress toward stage 3 CKD, as demonstrated by this study. The univariate Cox analysis did not reach significance because of few events and a higher baseline eGFR in SCD. Consequently, adjustment for baseline eGFR changed the estimates significantly (Figure 3).
Notably, our findings did not show an association between hypertension and eGFR decline in SCD. Hypertension in SCD may be underdiagnosed62 and therefore not captured by International Classification of Disease codes. In contrast, DM had the strongest association with eGFR decline in our SCD cohort. As patients with SCD live longer, DM has become increasingly common and this has serious implications for renal outcomes.13 From birth, patients with SCD develop glomerular hyperfiltration and renal medullary disruption.17,59,63 Second hits by comorbidities such as DM could exacerbate this pathology. Frequent hospital visits for pain crises also play a significant role in eGFR decline in SCD as shown by our data (Table 3). Significant tubular injury occurs during SCD pain crises even in the absence of clinical AKI, leading to progressive damage.64 Interestingly, a higher leukocyte count, which has been associated with increased morbidity and mortality in SCD,65 was associated with slower eGFR decline in our SCD cohort. This may be because neutrophilic leukocytosis is a marker of inflammation induced by the intravascular release of free heme molecules in SCD.8,10 This same process of heme-induced inflammation in SCD has been suggested as a possible mechanism for glomerular hyperfiltration.8 Hydroxyurea was associated with faster SCD eGFR decline in our study. There is a lack of literature on the long- term effects of hydroxyurea on eGFR decline.16,66 Given the retrospective nature of this study, medication results must be interpreted with caution due to the potential for confounding by indication. Glomerular hyperfiltration is a major contributor to sickle cell nephropathy.8 In our cohort, patients with SCD with a baseline eGFR ≥90 ml/min per 1.73 m2 experienced faster eGFR decline.
Our results substantially add to the otherwise sparse data on kidney function decline in SCT and SCD. This study has several strengths including large sample size, numerous data points per patient and a robust sensitivity analysis and subanalyses. However, this was a retrospective study dependent on hemoglobin electrophoresis testing to identify patients, which may limit external validity. The indications identified for the use of hemoglobin electrophoresis suggest this cohort is sicker than the average black population and therefore prone to faster eGFR decline. Perinatal testing for hemoglobinopathies led to a preponderance of female patients; however, adjusting for sex did not markedly alter results. The other major indication for hemoglobin electrophoresis was anemia. However, the non-SCD study cohort had normal hemoglobin levels (Table 1). Furthermore, the overall unadjusted mean eGFR change in our entire cohort (−1.37 ml/min per 1.73 m2 per year) was similar to the eGFR change described in the black subpopulation of the Multi-Ethnic Study of Atherosclerosis cohort (−1.39 ml/min per 1.73 m2 per year).30 Other limitations include the absence of APOL1 status (which has no known association with sickle cell inheritance and therefore may be randomly distributed in our cohort), the use of International Classification of Disease codes to verify comorbidities, lack of socioeconomic status data, lack of data on renal tubular acidosis or rhabdomyolysis, missing urine ACR data (owing to most patients not having a clinical indication to check urine ACR), and an inability to account for individual changes in therapy, which could alter eGFR decline over this long follow-up period. Despite the long follow-up time in this cohort, we do not expect that there were significant variations in practice because of changes in guidelines that could affect the outcomes reported here. Concerns regarding the accuracy of the CKD-EPI equation67 in patients with eGFR>60 ml/min per 1.73 m2 are valid; however, using rate of eGFR decline as an outcome should address this concern. SCD physiologically renders creatinine-based eGFRs inaccurate.63,68,69 However, using rate of eGFR decline as an outcome should also nullify this concern. Furthermore, CKD-EPI creatinine estimations are the most reliable creatinine-based GFR estimation in SCD.68,69
In conclusion, our study observed a significantly faster rate of decline in eGFR among black Americans with SCT/SCD compared with black Americans with a normal hemoglobin phenotype. This rate of decline worsened from SCT to SCD, confirming that carrying one copy of the sickle cell gene confers less harm to the kidneys compared with SCD. However, among patients with SCT, counterintuitively, lower HbS was associated with faster eGFR decline. But, as expected, elevated HbF and HbA2 were highly protective among patients with SCT. Given our lack of understanding of whether there are variations in HbS over time in SCT, how factors other than α-thalassemia affect HbS levels in SCT, and the interplay between HbA and HbS in SCT, our findings need to be evaluated further. We identified several risk factors that need to be investigated in prospective studies to define best practices and interventions to attenuate eGFR decline and prevent incident CKD in black patients with SCT/SCD.
Disclosures
Dr. Achebe reports personal fees from Bluebird Bio, Fulcrum Therapeutics, and Global Blood Therapeutics, outside the submitted work. Dr. Thadhani is a consultant to Fresenius Medical Care North America. All remaining authors have nothing to disclose.
Funding
Dr. Olaniran is supported by the Ben J. Lipps Research Fellowship Award of the American Society of Nephrology. Dr. Allegretti is supported by the American Heart Association Career Development Award 18CDA34110131. Dr. Eneanya is supported by National Institutes of Health (NIH) grant K23DK114526-01. Dr. Thadhani is supported by NIH grant R01DK094486-06. Dr. Nigwekar is supported by the American Heart Association National Center for Research Program Winter 2015 Fellow-to-Faculty Transition Award 15FTF25980003, and by the Harvard Catalyst, The Harvard Clinical and Translational Science Center (National Center for Research Resources and the National Center for Advancing Translational Sciences, NIH), KL2/Catalyst Medical Research Investigator Training Award TR001100 (an appointed KL2 award). Dr. Kalim is supported by NIH award K23DK106479.
Supplementary Material
Acknowledgments
Dr. Olaniran designed the study with input from all authors. Dr. Olaniran, Dr. Zhao, Dr. Allegretti, Dr. Nigwekar, and Dr. Kalim performed data analysis. Dr. Olaniran created the figures and drafted the manuscript. All authors revised the manuscript and approved the final version of the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
See related editorial, “Renal Functional Decline in Sickle Cell Disease and Trait,” on pages 236–238.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2019050502/-/DCSupplemental.
Supplemental Table 1. (A) Baseline characteristics of SCT sensitivity analysis cohort. (B) Baseline characteristics of SCD sensitivity analysis cohort.
Supplemental Table 2. Difference in mean eGFR change per year in SCT and SCD using sensitivity analysis cohort.
Supplemental Table 3. Baseline characteristics of cohort with baseline eGFR ≥65 ml/min per 1.73 m2.
Supplemental Table 4. Association of SCT and SCD with incident stage 3 CKD using sensitivity analysis cohort.
Supplemental Table 5. Unadjusted and adjusted coefficients of the difference in mean eGFR change per year in SCD by severity.
Supplemental Table 6. Adjusted coefficients describing mean annual change in eGFR by baseline eGFR category in reference patients, patients with SCT, and patients with SCD.
References
- 1.Gibson JS, Rees DC: How benign is sickle cell trait? EBioMedicine 11: 21–22, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naik RP, Derebail VK, Grams ME, Franceschini N, Auer PL, Peloso GM, et al.: Association of sickle cell trait with chronic kidney disease and albuminuria in African Americans. JAMA 312: 2115–2125, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Naik RP, Haywood C Jr.: Sickle cell trait diagnosis: Clinical and social implications. Hematology Am Soc Hematol Educ Program 2015: 160–167, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bucknor MD, Goo JS, Coppolino ML: The risk of potential thromboembolic, renal and cardiac complications of sickle cell trait. Hemoglobin 38: 28–32, 2014 [DOI] [PubMed] [Google Scholar]
- 5.Naik RP, Irvin MR, Judd S, Gutiérrez OM, Zakai NA, Derebail VK,et al. : Sickle cell trait and the risk of ESRD in blacks. J Am Soc Nephrol 28: 2180–2187, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ojodu J, Hulihan MM, Pope SN, Grant AM; Centers for Disease Control and Prevention (CDC) : Incidence of sickle cell trait — United States, 2010. MMWR Morb Mortal Wkly Rep 63: 1155–1158, 2014 [PMC free article] [PubMed] [Google Scholar]
- 7.Centers for Disease Control and Prevention: Data and Statistics on Sickle Cell Disease, 2016. Available at: https://www.cdc.gov/ncbddd/sicklecell/data.html. Accessed June 8, 2017
- 8.Nath KA, Hebbel RP: Sickle cell disease: Renal manifestations and mechanisms. Nat Rev Nephrol 11: 161–171, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rees DC, Williams TN, Gladwin MT: Sickle-cell disease. Lancet 376: 2018–2031, 2010 [DOI] [PubMed] [Google Scholar]
- 10.Williams TN, Thein SL: Sickle cell anemia and its phenotypes. Annu Rev Genomics Hum Genet 19: 113–147, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Powars DR, Chan LS, Hiti A, Ramicone E, Johnson C: Outcome of sickle cell anemia: A 4-decade observational study of 1056 patients. Medicine (Baltimore) 84: 363–376, 2005 [DOI] [PubMed] [Google Scholar]
- 12.Powars DR, Elliott-Mills DD, Chan L, Niland J, Hiti AL, Opas LM, et al.: Chronic renal failure in sickle cell disease: Risk factors, clinical course, and mortality. Ann Intern Med 115: 614–620, 1991 [DOI] [PubMed] [Google Scholar]
- 13.Sandhu MK, Cohen A: Aging in sickle cell disease: Co-morbidities and new issues in management. Hemoglobin 39: 221–224, 2015 [DOI] [PubMed] [Google Scholar]
- 14.Derebail VK, Nachman PH, Key NS, Ansede H, Falk RJ, Kshirsagar AV: High prevalence of sickle cell trait in African Americans with ESRD. J Am Soc Nephrol 21: 413–417, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dueker ND, Della-Morte D, Rundek T, Sacco RL, Blanton SH: Sickle cell trait and renal function in hispanics in the United States: The Northern Manhattan Study. Ethn Dis 27: 11–14, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Derebail VK, Ciccone EJ, Zhou Q, Kilgore RR, Cai J, Ataga KI: Progressive decline in estimated GFR in patients with sickle cell disease: An observational cohort study. Am J Kidney Dis 74: 47–55, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nath KA, Katusic ZS: Vasculature and kidney complications in sickle cell disease. J Am Soc Nephrol 23: 781–784, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nalichowski R, Keogh D, Chueh HC, Murphy SN: Calculating the benefits of a research patient data repository. AMIA Annu Symp Proc 1044, 2006 [PMC free article] [PubMed]
- 19.Liao KP, Cai T, Gainer V, Goryachev S, Zeng-treitler Q, Raychaudhuri S, et al.: Electronic medical records for discovery research in rheumatoid arthritis. Arthritis Care Res (Hoboken) 62: 1120–1127, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nigwekar SU, Solid CA, Ankers E, Malhotra R, Eggert W, Turchin A, et al.: Quantifying a rare disease in administrative data: The example of calciphylaxis. J Gen Intern Med 29[Suppl 3]: S724–S731, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olaniran KO, Eneanya ND, Allegretti AS, Zhao SH, Achebe MM, Thadhani RI: Cardiovascular outcomes in African Americans with sickle cell trait and chronic kidney disease. Am J Nephrol 49: 93–102, 2019 [DOI] [PubMed] [Google Scholar]
- 22.Hirschberg R: Glomerular hyperfiltration in sickle cell disease. Clin J Am Soc Nephrol 5: 748–749, 2010 [DOI] [PubMed] [Google Scholar]
- 23.Tonneijck L, Muskiet MH, Smits MM, van Bommel EJ, Heerspink HJ, van Raalte DH, et al.: Glomerular hyperfiltration in diabetes: Mechanisms, clinical significance, and treatment. J Am Soc Nephrol 28: 1023–1039, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palatini P: Glomerular hyperfiltration: A marker of early renal damage in pre-diabetes and pre-hypertension. Nephrol Dial Transplant 27: 1708–1714, 2012 [DOI] [PubMed] [Google Scholar]
- 25.Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D: A more accurate method to estimate glomerular filtration rate from serum creatinine: A new prediction equation. Modification of diet in renal disease study group. Ann Intern Med 130: 461–470, 1999 [DOI] [PubMed] [Google Scholar]
- 26.Inker LA, Schmid CH, Tighiouart H, Eckfeldt JH, Feldman HI, Greene T, et al.: CKD-EPI Investigators : Estimating glomerular filtration rate from serum creatinine and cystatin C. N Engl J Med 367: 20–29, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laird NM, Ware JH: Random-effects models for longitudinal data. Biometrics 38: 963–974, 1982 [PubMed] [Google Scholar]
- 28.Lindeman RD, Tobin J, Shock NW: Longitudinal studies on the rate of decline in renal function with age. J Am Geriatr Soc 33: 278–285, 1985 [DOI] [PubMed] [Google Scholar]
- 29.Glassock RJ, Winearls C: Ageing and the glomerular filtration rate: Truths and consequences. Trans Am Clin Climatol Assoc 120: 419–428, 2009 [PMC free article] [PubMed] [Google Scholar]
- 30.Peralta CA, Katz R, DeBoer I, Ix J, Sarnak M, Kramer H, et al.: Racial and ethnic differences in kidney function decline among persons without chronic kidney disease. J Am Soc Nephrol 22: 1327–1334, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsai CW, Ting IW, Yeh HC, Kuo CC: Longitudinal change in estimated GFR among CKD patients: A 10-year follow-up study of an integrated kidney disease care program in Taiwan. PLoS One 12: e0173843, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta AK, Kirchner KA, Nicholson R, Adams JG 3rd, Schechter AN, Noguchi CT, et al.: Effects of alpha-thalassemia and sickle polymerization tendency on the urine-concentrating defect of individuals with sickle cell trait. J Clin Invest 88: 1963–1968, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leonova JYe, Kazanetz EG, Smetanina NS, Adekile AD, Efremov GD, Huisman TH: Variability in the fetal hemoglobin level of the normal adult. Am J Hematol 53: 59–65, 1996 [DOI] [PubMed] [Google Scholar]
- 34.Tewari S, Brousse V, Piel FB, Menzel S, Rees DC: Environmental determinants of severity in sickle cell disease. Haematologica 100: 1108–1116, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ricardo AC, Yang W, Sha D, Appel LJ, Chen J, Krousel-Wood M, et al.: CRIC Investigators : Sex-related disparities in CKD progression. J Am Soc Nephrol 30: 137–146, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carrero JJ: Gender differences in chronic kidney disease: Underpinnings and therapeutic implications. Kidney Blood Press Res 33: 383–392, 2010 [DOI] [PubMed] [Google Scholar]
- 37.Silbiger S, Neugarten J: Gender and human chronic renal disease. Gend Med 5[Suppl A]: S3–S10, 2008 [DOI] [PubMed] [Google Scholar]
- 38.Yium J, Gabow P, Johnson A, Kimberling W, Martinez-Maldonado M: Autosomal dominant polycystic kidney disease in blacks: Clinical course and effects of sickle-cell hemoglobin. J Am Soc Nephrol 4: 1670–1674, 1994 [DOI] [PubMed] [Google Scholar]
- 39.Eriksen BO, Ingebretsen OC: The progression of chronic kidney disease: A 10-year population-based study of the effects of gender and age. Kidney Int 69: 375–382, 2006 [DOI] [PubMed] [Google Scholar]
- 40.Gladwin MT, Schechter AN, Ognibene FP, Coles WA, Reiter CD, Schenke WH, et al.: Divergent nitric oxide bioavailability in men and women with sickle cell disease. Circulation 107: 271–278, 2003 [DOI] [PubMed] [Google Scholar]
- 41.Chang YC, Smith KD, Moore RD, Serjeant GR, Dover GJ: An analysis of fetal hemoglobin variation in sickle cell disease: The relative contributions of the X-linked factor, beta-globin haplotypes, alpha-globin gene number, gender, and age. Blood 85: 1111–1117, 1995 [PubMed] [Google Scholar]
- 42.Chang YP, Maier-Redelsperger M, Smith KD, Contu L, Ducroco R, de Montalembert M, et al.: The relative importance of the X-linked FCP locus and beta-globin haplotypes in determining haemoglobin F levels: A study of SS patients homozygous for beta S haplotypes. Br J Haematol 96: 806–814, 1997 [DOI] [PubMed] [Google Scholar]
- 43.Akinsheye I, Alsultan A, Solovieff N, Ngo D, Baldwin CT, Sebastiani P, et al.: Fetal hemoglobin in sickle cell anemia. Blood 118: 19–27, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gladwin MT, Schechter AN, Ognibene FP, Coles WA, Reiter CD, Schenke WH,et al. : Divergent nitric oxide bioavailability in men and women with sickle cell disease. Circulation 107: 271–278, 2003 [DOI] [PubMed] [Google Scholar]
- 45.Diaw M, Pialoux V, Martin C, Samb A, Diop S, Faes C, et al.: Sickle cell trait worsens oxidative stress, abnormal blood rheology, and vascular dysfunction in type 2 diabetes. Diabetes Care 38: 2120–2127, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steinberg MH, Hsu H, Nagel RL, Milner PF, Adams JG, Benjamin L, et al.: Gender and haplotype effects upon hematological manifestations of adult sickle cell anemia. Am J Hematol 48: 175–181, 1995 [DOI] [PubMed] [Google Scholar]
- 47.Raffield LM, Ulirsch JC, Naik RP, Lessard S, Handsaker RE, Jain D, et al.: NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium, Hematology & Hemostasis, Diabetes, and Structural Variation TOPMed Working Groups : Common α-globin variants modify hematologic and other clinical phenotypes in sickle cell trait and disease. PLoS Genet 14: e1007293, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Piel FB, Weatherall DJ: The α-Thalassemias. N Engl J Med 371: 1908–1916, 2014 [DOI] [PubMed] [Google Scholar]
- 49.Steinberg MH, Rodgers GP: HbA2: Biology, clinical relevance and a possible target for ameliorating sickle cell disease. Br J Haematol 170: 781–787, 2015 [DOI] [PubMed] [Google Scholar]
- 50.Griffin PJ, Sebastiani P, Edward H, Baldwin CT, Gladwin MT, Gordeuk VR, et al.: The genetics of hemoglobin A2 regulation in sickle cell anemia. Am J Hematol 89: 1019–1023, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Statius van Eps LW, Pinedo-Veels C, de Vries GH, de Koning J: Nature of concentrating defect in sickle-cell nephropathy. Microradioangiographic studies. Lancet 1: 450–452, 1970 [DOI] [PubMed] [Google Scholar]
- 52.Podduturi V, Guileyardo JM: Sickle cell trait as a contributory cause of death in natural disease. J Forensic Sci 60: 807–811, 2015 [DOI] [PubMed] [Google Scholar]
- 53.Monchanin G, Serpero LD, Connes P, Tripette J, Wouassi D, Francina A, et al.: Plasma levels of adhesion molecules ICAM-1 and VCAM-1 in athletes with sickle cell trait with or without alpha-thalassemia during endurance exercise and recovery. Clin Hemorheol Microcirc 40: 89–97, 2008 [PubMed] [Google Scholar]
- 54.Folsom AR, Tang W, Roetker NS, Kshirsagar AV, Derebail VK, Lutsey PL, et al.: Prospective study of sickle cell trait and venous thromboembolism incidence. J Thromb Haemost 13: 2–9, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Naik RP, Wilson JG, Ekunwe L, Mwasongwe S, Duan Q, Li Y, et al.: Elevated D-dimer levels in African Americans with sickle cell trait. Blood 127: 2261–2263, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ruggenenti P, Porrini EL, Gaspari F, Motterlini N, Cannata A, Carrara F, et al.: GFR Study Investigators : Glomerular hyperfiltration and renal disease progression in type 2 diabetes. Diabetes Care 35: 2061–2068, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bjornstad P, Cherney DZ, Snell-Bergeon JK, Pyle L, Rewers M, Johnson RJ, et al.: Rapid GFR decline is associated with renal hyperfiltration and impaired GFR in adults with Type 1 diabetes. Nephrol Dial Transplant 30: 1706–1711, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Premaratne E, Verma S, Ekinci EI, Theverkalam G, Jerums G, MacIsaac RJ: The impact of hyperfiltration on the diabetic kidney. Diabetes Metab 41: 5–17, 2015 [DOI] [PubMed] [Google Scholar]
- 59.Olaniran KO, Eneanya ND, Nigwekar SU, Vela-Parada XF, Achebe MM, Sharma A, et al.: Sickle cell nephropathy in the pediatric population. Blood Purif 47: 205–213, 2019 [DOI] [PubMed] [Google Scholar]
- 60.Ranque B, Menet A, Diop IB, Thiam MM, Diallo D, Diop S, et al.: Early renal damage in patients with sickle cell disease in sub-Saharan Africa: A multinational, prospective, cross-sectional study. Lancet Haematol 1: e64–e73, 2014 [DOI] [PubMed] [Google Scholar]
- 61.Asnani M, Serjeant G, Royal-Thomas T, Reid M: Predictors of renal function progression in adults with homozygous sickle cell disease. Br J Haematol 173: 461–468, 2016 [DOI] [PubMed] [Google Scholar]
- 62.Moodalbail DG, Falkner B, Keith SW, Mathias RS, Araya CE, Zaritsky JJ, et al.: Ambulatory hypertension in a pediatric cohort of sickle cell disease. J Am Soc Hypertens 12: 542–550, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ware RE, Rees RC, Sarnaik SA, Iyer RV, Alvarez OA, Casella JF, et al.: BABY HUG Investigators : Renal function in infants with sickle cell anemia: Baseline data from the BABY HUG trial. J Pediatr 156: 66–70.e1, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Audard V, Moutereau S, Vandemelebrouck G, Habibi A, Khellaf M, Grimbert P, et al.: First evidence of subclinical renal tubular injury during sickle-cell crisis. Orphanet J Rare Dis 9: 67, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang D, Xu C, Manwani D, Frenette PS: Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood 127: 801–809, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bartolucci P, Habibi A, Stehlé T, Di Liberto G, Rakotoson MG, Gellen-Dautremer J, et al.: Six months of hydroxyurea reduces albuminuria in patients with sickle cell disease. J Am Soc Nephrol 27: 1847–1853, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, et al.: CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) : A new equation to estimate glomerular filtration rate. Ann Intern Med 150: 604–612, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arlet JB, Ribeil JA, Chatellier G, Eladari D, De Seigneux S, Souberbielle JC, et al.: Determination of the best method to estimate glomerular filtration rate from serum creatinine in adult patients with sickle cell disease: A prospective observational cohort study. BMC Nephrol 13: 83, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yee MEM, Lane PA, Archer DR, Joiner CH, Eckman JR, Guasch A: Estimation of glomerular filtration rate using serum cystatin C and creatinine in adults with sickle cell anemia. Am J Hematol 92: E598–E599, 2017 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.