

Abstract
Background
Astrocytes are the most abundant glial cells in a brain that mediate inflammatory responses and provide trophic support for neurons. We have previously disclosed that paroxetine, a common selective serotonin reuptake inhibitor, ameliorates LPS-induced microglia activation. However, it remains elusive for the role of paroxetine in astrocytic responses.
Methods
Isolated primary astrocytes were pretreated with paroxetine and stimulated with different stimuli, lipopolysaccharide (LPS) or microglia conditioned medium pre-activated with LPS (M/Lps). Inflammatory and neurotrophic responses, underlying mechanisms and the impact on neuronal survival were assessed.
Results
Paroxetine had no impact on LPS-stimulated iNOS, TNF-α, and IL-1β expression, but inhibited M/Lps-induced TNF-α and IL-1β expression in primary astrocytes. Paroxetine suppressed M/Lps- but not LPS-induced activation of NF-κB and had no impact on the activation of MAPKs and STAT3. Incubation with the resulted astrocyte conditioned media caused no change in the viability of SH-SY5Y cells. BDNF and MANF mRNA expressions were upregulated by M/Lps and paroxetine, respectively. However, M/Lps- or LPS-induced extracellular releases of NO, TNF-α, and/or BDNF in astrocytes were in minor amount compared to those by microglia.
Conclusions
Paroxetine ameliorates the reactive microglia-mediated inflammatory responses in astrocytes partially via inhibition of the NF-κB pathway but has no impact on LPS-stimulated astrocyte activation. While the effects of paroxetine on secondary astrocytic responses are not robust compared to its effect on the innate immune responses of microglia, the results together may implicate a therapeutic potential of paroxetine against neuroinflammation-associated neurological disorders such as Parkinson’s disease.
Keywords: Paroxetine, Astrocytes, Microglia, Neuroinflammation, Parkinson’s disease
Background
Parkinson’s disease (PD) is a common neurodegenerative disease characterized by the selective death of dopaminergic neurons in the substantia nigra. Although its exact etiology remains elusive, accumulating evidence has suggested that neuroinflammation plays a key role in the pathogenesis of PD [1–3]. In the central nervous system, innate immune responses are collectively mediated by microglia and astrocytes while the former dominates the way to neuroinflammation [4, 5]. Indeed, reactive microglia and astrocytes are both found in the nigrostriatal bundle of PD patients or PD animal models [6–9]. A number of PD-associated gene products such as α-synuclein, PINK1, and DJ-1 have been implicated in astrocyte dysfunction. Exposure to mutant α-synuclein and deficiency in PINK1 lead to activation of both microglia and astrocytes and generation of a large amount of neuroinflammatory factors [10–12]. PINK1 deficiency also inhibits the differentiation of neural stem cells into astrocytes [13] and causes proliferation defect in astrocytes which may result in a delay in the wound healing process [14]. DJ-1 is abundantly expressed in reactive astrocytes of PD patients [15]. Its deficiency impairs glutamate uptake into astrocytes [16] and leads to increased susceptibility to inflammatory signaling [17]. Different from microglia, reactive astrocytes depending on the context may release pro-inflammatory factors such as tumor necrosis factor α (TNF-α) and interleukin-1β (IL-1β), or provide trophic support for neurons by releasing neurotrophic factors such as brain-derived neurotrophic factor (BDNF) and mesencephalic astrocyte-derived neurotrophic factor (MANF) [4, 5, 18]. The interaction between microglia and astrocytes leads to collective outcomes for neurons. For instance, astrocytes were reported to enhance the inflammatory responses of activated microglia, resulting in more dopaminergic neuron death [19].
Studies have suggested the negative immunoregulatory effects of antidepressant drugs [20–23]. Amongst, paroxetine is a common selective serotonin reuptake inhibitor and is used for disorders such as major depressive disorder, generalized anxiety disorder, and obsessive-compulsive disorder, and also with fewer side effects than the first generation antidepressants tricyclic antidepressants [24, 25]. Depression is the most common psychiatric disturbance reported in PD patients. Paroxetine is thus also used for relieving depressive symptomatology and regulating behavioral control in PD patients and is generally well tolerated [26, 27]. Paroxetine is reported to protect against dopaminergic neuronal loss in an MPTP-induced mouse model of PD, potentially through its inflammatory alleviation in the substantia nigra [28]. Lipopolysaccharide (LPS) is an endotoxin often used for modeling PD in the context of neuroinflammation both in vivo [29, 30] and in vitro [31–35], particularly when the neuroinflammation aspect of mechanisms is focused. By applying LPS, we have previously disclosed that paroxetine ameliorates the microglia activation through regulation of MAPK signaling [36]. However, it remains elusive for the role of paroxetine in astrocytic responses. Thus, in this study, we aimed to understand the impact of paroxetine on astrocyte activation induced by LPS and reactive microglia using a conditioned medium culture system.
Methods
Cell culture and primary astrocyte isolation
Dulbecco’s modified eagle medium (DMEM; C11995500BT) and DMEM/F-12 (C11330500BT) were purchased from Gibco (Grand Island, NY, USA). Fetal bovine serum (FBS; VS500T, Australia origin) was from Ausbian (Shanghai, China). BV2 microglia cells (provided by Dr. Zhu CQ, Fudan University) and SH-SY5Y cells (Cell Bank of Chinese Academy of Sciences, Shanghai, China) were grown in DMEM supplemented with 10% FBS and penicillin/streptomycin (100 U/mL and 100 μg/mL, respectively; P1400, Solarbio, Beijing, China). Cells were cultured at 37 °C in a humidified atmosphere of 5% CO2.
Primary astrocytes were prepared as previously described with slight modification [37, 38]. In brief, cerebral cortices were dissected from brains of new-born pups of the Institute of Cancer Research (ICR) mice (postnatal 1–2 days; purchased from the Experimental Animal Center of Wenzhou Medical University) in cold Hank’s buffered saline. The animals used in this study were treated in accordance with protocols approved by the Institutional Animal Care and Use Committee of Wenzhou Medical University. Cortices were gently triturated and digested with 0.25% Trypsin-EDTA solution (25,200,056, Gibco, Grand Island, NY, USA) for 15 min at 37 °C. Thereafter, an equal volume of culture medium, that is, DMEM/F-12 supplemented with 10% FBS and penicillin/streptomycin, was added and centrifuged at 200 g for 5 min. The pellets were gently resuspended in culture medium and filtered through a 100-μm-pore mesh. The cells were then seeded in 75 cm2 flasks coated with poly-L-lysine (1 mg/mL) and cultured at 37 °C with medium changed every 4 days. When cells reached a confluency of 90–95%, the flasks were placed on an orbital shaker at 250 rpm and 37 °C for 16 h. The remaining attached cells were then collected and re-cultured. The purity of astrocytes was evaluated by immunostaining of glial fibrillary acidic protein (GFAP). GFAP positive cells were counted in 3-random fields each time. Cultures were used for subsequent experiments when GFAP positivity was over 95% (Fig. 1a).
Fig. 1.

Isolation of primary astrocytes and experimental design. a Representative images of isolated primary astrocytes. Green, GFAP; blue, nuclei; bar size, 100 μm. b Illustration of experimental design and conditioned media. A/Lps, conditioned medium of astrocytes cultured with LPS; A/MC, conditioned medium of astrocytes cultured with M/C; A/MLps, conditioned medium of astrocytes cultured with M/Lps; GFAP, glial fibrillary acidic protein; LPS, lipopolysaccharide; M/C, microglia conditioned medium without LPS stimulation; M/Lps, microglia conditioned medium pre-activated with LPS
Immunofluorescence
Cells were fixed in 4% paraformaldehyde for 30 min, followed by permeabilization in 0.2% Triton X-100 for 15 min and then blocked with 5% bovine serum albumin (Beyotime, Shanghai, China) for 1 h at room temperature. The washing buffer is phosphate-buffered saline. Primary antibodies against GFAP and p65 were from Millipore (MAB360, Billerica, MA, USA) and Cell Signaling (8242S, Boston, MA, USA), respectively. Alexa Fluor 488-conjugated anti-mouse (A11001) and 555-conjugated anti-rabbit (A21428) IgG antibodies and Hoechst 33258 (H3569) were from Thermo Fisher (Rockford, IL, USA).
Treatments and preparation of conditioned media
Microglia conditioned media were prepared as previously [19]. BV2 cells were seeded at a density of 2–3 × 106 in a 10-cm culture plate and cultured for 24 h followed by serum starvation overnight. Cells were then treated with or without LPS (100 ng/mL; L4391, Sigma, St. Louis, MO, USA) for 2 h, and then refreshed with serum-free medium with a continued incubation for 24 h. Following centrifugation at 20,000 g for 5 min, the media were collected as microglia conditioned medium without LPS stimulation (M/C) or microglia conditioned medium pre-activated with LPS (M/Lps), and stored at − 80 °C till use (Fig. 1b).
Primary astrocytes of passage 3–5 were seeded at a density of 2–3 × 106 per well in a 6-well plate and cultured for 24 h. Following starvation overnight, cells were pretreated with paroxetine (P9623, Sigma, St. Louis, MO, USA) at indicated concentrations for 30 min, followed by treatments of LPS (100 ng/mL) or the above M/C and M/Lps for the indicated time. The resulted media of astrocytes treated with M/C, M/Lps or LPS for 24 h were centrifuged at 20,000 g for 5 min and designated as A/MC (conditioned medium of astrocytes cultured with M/C), A/MLps (conditioned medium of astrocytes cultured with M/Lps), and A/Lps (conditioned medium of astrocytes cultured with LPS), respectively (Fig. 1b). Media were stored at − 80 °C till use.
Cell viability measurement
Cell viability was determined by the tetrazolium salt 3-[4,5-dimethylthiazol-2-yl] -2,5-diphenyltetrazolium bromide (MTT; C0009, Beyotime, Shanghai, China) assay [39]. Primary astrocytes or SH-SY5Y cells were seeded in triplicates in 96-well plates at 5 × 104 and 1 × 104 per well, respectively, and cultured for 24 h. Astrocytes were treated with paroxetine at different concentrations for 24 h following overnight starvation. SH-SY5Y cells were incubated with A/MC, A/MLps, A/Lps, or LPS directly (100 ng/mL) for 24 h. Thereafter, MTT (5 mg/mL prepared in phosphate-buffered saline) was added into each well and incubated at 37 °C for 4 h. The resulting formazan crystals were dissolved in dimethyl sulfoxide. Optical density was measured at 490 nm.
RNA isolation and quantitative PCR
Total RNA was isolated using TRIzol Reagent (15596018, Invitrogen, Carlsbad, CA, USA), and reverse-transcribed to cDNA using the PrimeScript RT kit (RR037A, TaKaRa, Dalian, China). Quantitative PCR was performed using SYBR Green Supermix (1725260, Bio-Rad, Hercules, CA, USA) following the manufacturer’s instruction. Primers were as follows: TNF-α, 5′-CGT CAG CCG ATT TGC TAT CT-3′ and 5′-CGG ACT CCG CAA AGT CTA AG-3′; IL-1β, 5′-GAA ATG CCA CCT TTT GAC AGT G-3′ and 5′-TGG ATG CTC TCA TCA GGA CAG-3′; BDNF, 5′-TCA TAC TTC GGT TGC ATG AAG G-3′ and 5′-AGA CCT CTC GAA CCT GCC C-3′; MANF, 5′-TCT GGG ACG ATT TTA CCA GGA-3′ and 5′-CTT GCT TCA CGG CAA AAC TTT-3′; β-actin, 5′-AGC CAT GTA CGT AGC CAT CC-3′ and 5′-CTC TCA GCT GTG GTG GTG AA-3′.
Determination of TNF-α and BDNF production
Medium TNF-α and BDNF were measured using ELISA kits respectively from R&D Systems (VAL609, Minneapolis, MN, USA) and Westang (F10200, Shanghai, China) according to the manufacturers’ instructions. Absorbance was read at 450 nm. The concentration of each sample was calculated from the standard curve prepared using the included standards.
NO production assay
Medium nitrite was measured as an indicator of NO production [40]. In brief, 50 μl of supernatant was mixed with an equal volume of Griess reagent I, followed by the addition of 50 μl of Griess reagent II (S0021, Beyotime, Shanghai, China) at room temperature. Absorbance was immediately measured at 540 nm. The concentration of each sample was calculated from a standard curve generated using sodium nitrite.
Western blot analysis
Cells were lysed in sample buffer containing 60 mM Tri-HCl, pH 6.8, 2% SDS and 5% glycerol, and boiled for 5 min. Total protein concentration was measured using a BCA kit (P0010, Beyotime, Shanghai, China). An equal amount of protein from each sample was loaded and analyzed by western blot as previously described [41]. Pyrrolidine dithiocarbamic acid (PDTC; P8765) was purchased from Sigma (St. Louis, MO, USA). Primary antibodies against p-p65 (3031), p65 (3034), p-JNK1/2 (9251), JNK1/2 (9252), p-p38 (9211), p38 (9212), p-ERK1/2 (9101), ERK1/2 (9102), p-STAT3 (9145), STAT3 (12640), iNOS (2977) and β-actin (4970), anti-rabbit (7074) and anti-mouse (7076) secondary antibodies, and LumiGLO® Reagent and Peroxide chemiluminescence detection kit (7003) were all purchased from Cell Signaling (Boston, MA, USA).
Statistical analysis
Data were analyzed by one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test for multiple comparisons and two-way ANOVA for factorial design experiments using the SPSS 23.0 statistics software. Values were expressed as mean ± SE from at least three independent experiments. The p < 0.05 was considered statistically significant.
Results
Effect of paroxetine on LPS-induced inflammatory responses in primary astrocytes
To understand the potential toxicity of paroxetine on primary astrocytes, we treated cells with paroxetine at 0, 1, 3, 5, 10, and 20 μM for 24 h. Results showed that paroxetine at 20 μM, but not at the other doses, induced significant cell death [Fig. 2; F(5,12) = 15.38, p < 0.001; post hoc, p = 0.001 (0 μM vs 20 μM)].
Fig. 2.
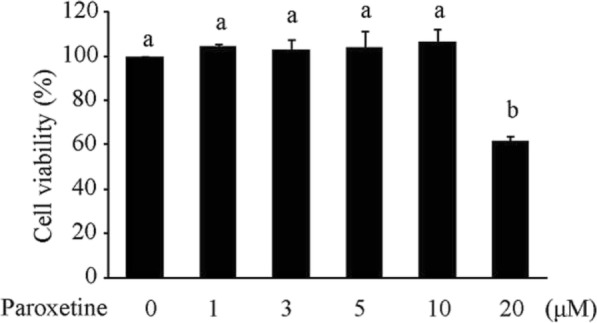
Impact of paroxetine on the viability of primary astrocytes. Cells were treated with paroxetine at different concentrations for 24 h. Cell viability was expressed as percentage of the control (0 μM), which was set as 100%. Values are means ± SE, n = 3. Statistical comparisons were performed using one-way ANOVA, followed by Tukey’s post hoc test. Different letters indicate p < 0.05
LPS stimulated light iNOS expression in isolated primary astrocytes [Fig. 3a; F(6,14) = 31.82, p < 0.001; full blots listed in Additional file 1: Figure S1], as the induction is much weaker than that in microglia following the same treatment [36]. As a consequence, no significant elevation in NO levels was detected in astrocytes upon LPS treatment (Fig. 3b). LPS induced a significant increase in TNF-α and IL-1β mRNA expression [Fig. 3c; TNF-α: F(6,21) = 56.12, p < 0.001; IL-1β: F(6,21) = 67.68, p < 0.001] and extracellular release of TNF-α [Fig. 3d; F(6,14) = 225.56, p < 0.001]. However, paroxetine pretreatments at different concentrations did not lead to apparent changes in iNOS expression and NO production (Fig. 3a, b), neither in TNF-α and IL-1β mRNA expression and TNF-α production (Fig. 3c, d).
Fig. 3.

Effect of paroxetine on LPS-induced inflammatory responses in primary astrocytes. Cells were pretreated with paroxetine at different concentrations for 30 min followed by stimulation with LPS at 100 ng/mL for 24 h. a Western blot analysis of iNOS expression. Protein levels were quantified and normalized to their respective β-actin levels. Values were expressed relative to the one treated with LPS alone, which was set as 100. b NO production indicated by nitrite levels in culture media. c Quantitative PCR analyses of TNF-α and IL-1β expression. The mRNA levels were expressed relative to the one treated with LPS alone, which was set as 100. d Concentrations of TNF-α in culture media. Values are means ± SE, n = 3 for a, b, and d, n = 4 for c. Statistical comparisons were performed using one-way ANOVA, followed by Tukey’s post hoc test. Different letters indicate p < 0.05. IL-1β, interleukin 1β; iNOS, inducible nitric oxide synthase; LPS, lipopolysaccharide; NO, nitric oxide; TNF-α, tumor necrosis factor α
Effect of paroxetine on M/Lps-induced inflammatory responses in primary astrocytes
The iNOS expression upon M/Lps incubation was barely detectable (data not shown). To determine whether there was an increase in the production of cytokines derived from astrocytes, the NO, and TNF-α carried from M/C or M/Lps per se (without treating astrocytes) were measured being their baseline levels (Fig. 4a, c; the right last two columns). There was no significant production of NO by astrocytes treated with M/Lps compared to its baseline level (Fig. 4a). M/Lps induced a significant increase in TNF-α and IL-1β mRNA expression in primary astrocytes. The induction was suppressed by pretreatments of paroxetine, in particular at doses of 5 and 10 μM [Fig. 4b; TNF-α: F(6,21) = 14.77, p < 0.001; post hoc, p = 0.037 (M/Lps vs M/Lps + paroxetine 10 μM; down by 42.2%); IL-1β: F(6,21) = 15.88, p < 0.001; post hoc, p = 0.027 (M/Lps vs M/Lps + paroxetine 5 μM; down by 46.7%), p = 0.002 (M/Lps vs M/Lps + paroxetine 10 μM; down by 61.8%)]. Although not significant, a slight increase appeared in the production of TNF-α in media of astrocytes incubated with M/Lps compared to its baseline level (the third vs the ninth). This increase was blunted by pretreatments of paroxetine at doses of 5 and 10 μM [Fig. 4c; F(8,27) = 99.06, p < 0.001; post hoc, p = 0.013 (M/Lps vs M/Lps + paroxetine 5 μM; down by 21.9%), p = 0.011 (M/Lps vs M/Lps + paroxetine 10 μM; down by 22.3%)].
Fig. 4.

Paroxetine suppresses M/Lps-induced inflammatory responses in primary astrocytes. Cells were pretreated with paroxetine at different concentrations for 30 min followed by incubation with microglia conditioned media for 24 h. a NO production indicated by nitrite levels in culture media. b Quantitative PCR analyses of TNF-α and IL-1β expression. The mRNA levels were expressed relative to the one incubated with M/Lps alone, which was set as 100. c Concentrations of TNF-α in culture media. Values are means ± SE, n = 3 for a, n = 4 for b, c. Statistical comparisons were performed using one-way ANOVA, followed by Tukey’s post hoc test. Different letters indicate p < 0.05. IL-1β, interleukin 1β; M/C, microglia conditioned medium without LPS stimulation; M/Lps, microglia conditioned medium pre-activated with LPS; NO, nitric oxide; TNF-α, tumor necrosis factor α
Paroxetine inhibits M/Lps-induced inflammatory responses in primary astrocytes partially through NF-κB pathway
To understand the underlying mechanism of paroxetine inhibition on M/Lps-induced cytokine expression, we analyzed the mitogen-activated protein kinase (MAPK), NF-κB and STAT3 signaling pathways. Cells were pretreated with paroxetine for 30 min followed by treatment of M/Lps for the indicated time. As shown in the comparisons of 0 min (Fig. 5), paroxetine alone had little impact on the activation of these proteins. The p38, JNK1/2, ERK1/2, p65/NF-κB, and STAT3 were all markedly activated in primary astrocytes upon M/Lps treatments (Fig. 5a). However, the phosphorylation of p38, JNK1/2, ERK1/2, and STAT3 was not suppressed by paroxetine pretreatment except that of p65/NF-κB (Fig. 5b–f). The inhibition rate for p-p65 was 33.7, 40.2, 49.4, and 53.6%, respectively, at 15, 30, 60, and 120 min [Fig. 5e; M/Lps*time interaction: F(4,30) = 3.08, p = 0.031; M/Lps + paroxetine vs M/Lps: p = 0.001 (15 min), p = 0.004 (30 min), p < 0.001 (60 min), p = 0.001 (120 min)]. Immunofluorescence further showed that M/Lps stimulated nuclear translocation of p65 and this translocation was blocked by paroxetine pretreatment [Fig. 6; time*paroxetine interaction: F(1,8) = 12.30, p = 0.008; M/Lps + paroxetine vs M/Lps: p = 0.012 (30 min)]. Pretreatment of PDTC (a specific inhibitor of NF-κB pathway; Fig. 7a; M/Lps*PDTC interaction: F(1,8) = 27.24, p = 0.001; M/Lps + PDTC vs M/Lps: p < 0.001) reduced the M/Lps-induced elevation of TNF-α and IL-1β mRNA expression by 18.0% and 39.0%, respectively, in the astrocytes (Fig. 7b; TNF-α: F(1,8) = 22.14, p = 0.002; M/Lps + PDTC vs M/Lps: p < 0.001; IL-1β: F(1,8) = 18.73, p = 0.003; M/Lps + PDTC vs M/Lps: p < 0.001).
Fig. 5.

Effect of paroxetine on M/Lps-induced signaling activation in primary astrocytes. Cells were pretreated with or without 10 μM of paroxetine for 30 min followed by stimulation with M/Lps for 0–120 min. a Western blot analyses of p38, JNK1/2, ERK1/2, p65/NF-κB, and STAT3 activation. b–f Levels of phosphorylation forms were quantified and normalized to their respective total levels. Values were expressed relative to the one stimulated with M/Lps alone for 15 min, which was set as 100. Data are means ± SE, n = 4. Statistical comparisons were performed using two-way ANOVA. Different letters and * indicate p < 0.05. M/Lps, microglia conditioned medium pre-activated with LPS
Fig. 6.

Effect of paroxetine on M/Lps-induced p65/NF-κB nuclear translocation in astrocytes. Cells were pretreated with or without 10 μM of paroxetine for 30 min followed by stimulation with M/Lps for 30 min. a Representative images of p65 immunostaining. Green, GFAP; red, p65; blue, nuclei; bar size, 200 μm. b Fluorescence intensity of nuclear p65 was determined by quantifying 15 cells in multiple random fields, followed by normalization to their respective nuclei staining. Data are expressed as means ± SE from three independent experiments. Statistical comparisons were performed using two-way ANOVA. Different letters and * indicate p < 0.05. GFAP, glial fibrillary acidic protein; M/Lps, microglia conditioned medium pre-activated with LPS
Fig. 7.
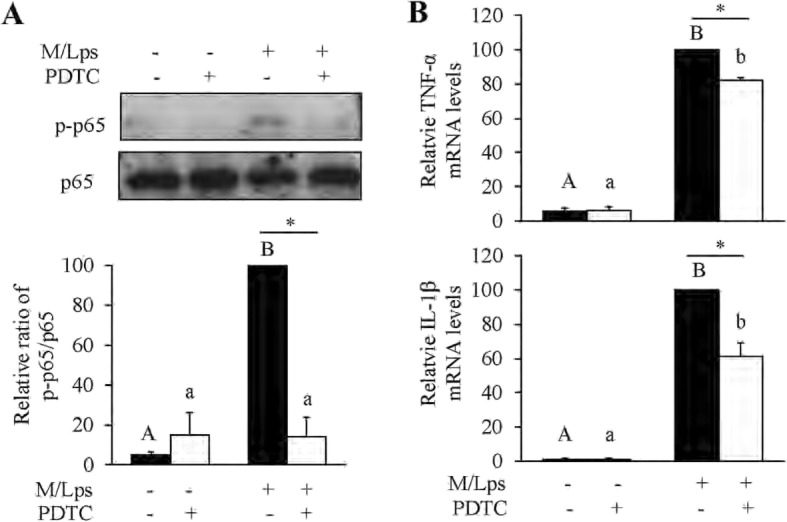
Inhibition of p65/NF-κB signaling on M/Lps-stimulated inflammatory responses in astrocytes. a Effect of PDTC on p65 activation. Cells were pretreated with 20 μM of PDTC for 2 h prior to stimulation with M/Lps for 30 min. Levels of p-p65 were quantified and normalized to total p65 levels. b Effect of PDTC on M/Lps-induced mRNA expression of TNF-α and IL-1β. Cells were pretreated with 20 μM of PDTC for 2 h prior to stimulation with M/Lps for 24 h. Values were expressed relative to the one stimulated with M/Lps alone, which was set as 100. Data are means ± SE, n = 4. Statistical comparisons were performed using two-way ANOVA. Different letters and * indicate p < 0.05. IL-1β, interleukin 1β; M/C, microglia conditioned medium without LPS stimulation; M/Lps, microglia conditioned medium pre-activated with LPS; PDTC, pyrrolidine dithiocarbamic acid; TNF-α, tumor necrosis factor α
We additionally examined the effect of paroxetine on p65/NF-κB activation in the context of LPS stimulation. Results showed that LPS induced little p65 phosphorylation (data not shown) but stimulated apparent p65 nuclear translocation (p < 0.05). However, in concord with its effect on cytokine production, paroxetine did not blunt this translocation (Fig. 8).
Fig. 8.

Effect of paroxetine on LPS-induced p65/NF-κB nuclear translocation in primary astrocytes. Cells were pretreated with or without 10 μM of paroxetine for 30 min followed by stimulation with LPS for 30 min. a Representative images of p65 nuclear translocation. Green, GFAP; red, p65; blue, nuclei; bar size, 200 μm. b Fluorescence intensity of nuclear p65 was determined by quantifying 15 cells in multiple random fields, followed by normalization to their respective nuclei staining. Data are expressed as means ± SE from three independent experiments. Statistical comparisons were performed using two-way ANOVA. Different letters indicate p < 0.05. GFAP, glial fibrillary acidic protein; LPS, lipopolysaccharide
A/MLps with paroxetine has little impact on SH-SY5Y cell viability
To investigate whether neuronal survival was affected by the reactive-microglia-mediated activation of astrocytes, LPS-activated astrocytes, and LPS per se, we cultured SH-SY5Y cells, a neuroblastoma cell line commonly used for PD study, with A/MLps, A/Lps, and LPS. Results showed that A/MLps, A/Lps, and LPS regardless of paroxetine pretreatment resulted in little impact on the survival of SH-SY5Y cells (Fig. 9).
Fig. 9.
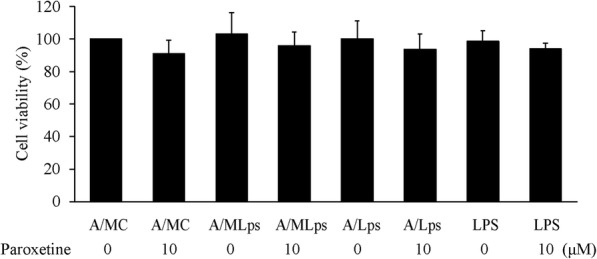
Effect of astrocyte conditioned media on SH-SY5Y cell survival. SH-SY5Y cells were incubated with A/MC, A/MLps, A/Lps, or LPS for 24 h. Cell viability was expressed as percentage of the A/MC alone-treated group, which was set as 100%. Values are means ± SE, n = 3. Statistical comparisons were performed using two-way ANOVA. A/Lps, conditioned medium of astrocytes cultured with LPS; A/MC, conditioned medium of astrocytes cultured with M/C; A/MLps, conditioned medium of astrocytes cultured with M/Lps
We further investigated whether potential neurotrophic support of the astrocytes played a role amidst. The mRNA expression of neurotrophic factors, in particular BDNF, was increased in astrocytes following the incubation with M/Lps [Fig. 10a; BDNF: F(6,14) = 16.41, p < 0.001; post hoc, p = 0.012 (M/C vs M/Lps; up by 89%); MANF: F(6,14) = 5.06, p = 0.006]. In striking contrast to the results in cytokines, paroxetine did not suppress the M/Lps-induced expression of these neurotrophic factors. Instead, paroxetine at 10 μM led to higher mRNA expression of BDNF and MANF in astrocytes [BDNF: post hoc, p < 0.001 (M/C vs M/C + paroxetine 10 μM; up by 152%), p = 0.003 (M/Lps vs M/Lps + paroxetine 10 μM; up by 56%); MANF: p = 0.037 (M/C vs M/C + paroxetine 10 μM; up by 88%)]. Similarly, paroxetine at 10 μM resulted in a trend of elevation in medium BDNF levels compared with their respective controls (Fig. 10b). BDNF concentration was significantly increased in media of the astrocytes incubated with M/Lps compared to the M/C incubation (F(5,18) = 15.61, p < 0.001; post hoc, p = 0.003). Interestingly, compared to the baseline levels in the conditioned media, medium BNDF levels appeared to be reduced upon incubation with the astrocytes (Fig. 10b; the first vs fifth column, the third vs sixth column).
Fig. 10.
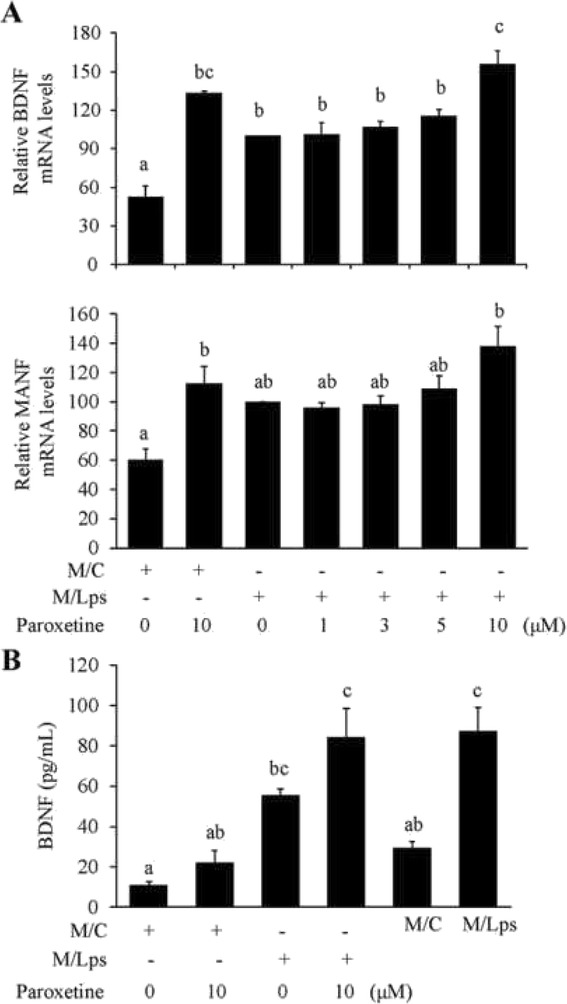
Effect of paroxetine on neurotrophic responses of astrocytes. Cells were pretreated with paroxetine at different concentrations for 30 min followed by incubation with microglia conditioned media for 24 h. a Quantitative PCR analyses of BDNF and MANF expression. The mRNA levels were expressed relative to the one incubated with M/Lps alone, which was set as 100. b Concentrations of BDNF in culture media. Values are means ± SE, n = 3 for a, n = 4 for b. Statistical comparisons were performed using one-way ANOVA, followed by Tukey’s post hoc test. Different letters indicate p < 0.05. BDNF, brain-derived neurotrophic factor; MANF, mesencephalic astrocyte-derived neurotrophic factor; M/C, microglia conditioned medium without LPS stimulation; M/Lps, microglia conditioned medium pre-activated with LPS
Discussion
Neuroinflammation is involved in the pathophysiology of neurodegenerative diseases such as PD and Alzheimer’s disease [1–3]. Targeting microglia- and astrocyte-mediated immune responses is therapeutically promising against these disorders [42]. Besides being a drug against depression, paroxetine is shown to inhibit MPTP-induced loss of nigrostriatal dopaminergic neurons and neuroinflammatory responses, and to suppress LPS-induced microglia activation [28, 36, 43]. Results of the current study in primary astrocytes suggest that (1) paroxetine inhibits reactive microglia-mediated, but not LPS-induced, astrocytic inflammatory responses; (2) LPS-induced microglia activation dominates over its subsequent activation of astrocytes; (3) paroxetine-mediated astrocytic responses during LPS insult has a little actual impact on neuronal survival.
Astrocytes are often activated following activation of microglia, which may stimulate astrocytes in a paracrine pathway [44]. Incubation of astrocytes with microglia conditioned medium indicates a paracrine regulation of microglia on astrocytic responses. Indeed, reactive microglia stimulate mRNA expression of both cytokines such as TNF-α and IL-1β and neurotrophic factors such as BDNF and MANF in astrocytes. However, compared to the reactive microglia per se, this paracrine activation of astrocytes may have a rather small effect on the surroundings as suggested by the light extracellular release of effectors in media. It is not surprising given that microglia are presumably more potent than astrocytes leading to neuroinflammation [4, 5]. In consistency, the induction of iNOS by LPS in astrocytes is considerably less than that in microglia [36], which may explain why the NO production is not significantly elevated in astrocytes. Sheng et al. also showed that NO production was not detectable in either immortalized rat astrocytes or primary astrocytes upon treatment of LPS at 100 ng/mL [45].
However, while reactive microglia indeed release neurotrophic factors [46, 47], we initially thought that astrocytes should have been more potent secreting neurotrophic factors upon being activated. Unexpectedly, there is no drastic increase of BDNF in the astrocyte media compared to its baseline levels of microglia conditioned media. Instead, astrocytes seem to absorb microglia-released neurotrophic factors as observed. On the other hand, paroxetine indeed upregulates mRNA expression and extracellular release of neurotrophic factors such as BDNF as being previously noted [48].
Saijo et al. reported that microglia-mediated inflammatory responses were magnified by astrocytes leading to more neuronal death [19]. However, as above discussed, this magnification is not displayed in the current study. Both studies used primary astrocytes and BV2 cells for preparing conditioned media. In an effort to interpret the above discrepancy, we observed that their BV2 cells used to make conditioned media were all infected with lentivirus [19]. As manifested in one of their results, LPS-activated conditioned media from this type of cells exhibited significantly higher toxicity to tyrosine hydroxylase positive neurons compared to those prepared from uninfected cells, suggesting that microglia upon lentivirus infection are more reactive and potent generating pro-inflammatory cytokines. Consequently, paracrine factors in their microglia conditioned media are presumably of much higher concentrations, then leading to more drastic astrocytic responses than in the current study. BV2 cell is considered as a valid substitute for primary microglia in complex cell-cell interaction studies [49]. Nonetheless, the obtained results should be confirmed using conditioned medium derived from primary microglia cultures.
Paroxetine prevents LPS-induced inflammatory responses in microglia [36], but not in astrocytes. Toll-like receptor distribution varies considerably between microglia and astrocytes [50]. For instance, TLR4, the major LPS receptor, is expressed extensively in microglia, but this receptor and its downstream signaling component MYD88 are deficient in rodent astrocytes [51, 52]. Thus, the signaling pathways in response to LPS much differ in these two types of immune cells. Paroxetine markedly suppresses the reactive microglia-induced activation of p65/NF-κB in astrocytes including both phosphorylation and nuclear translocation. Interestingly, the nuclear translocation of p65 induced by a direct LPS treatment was not blunted by paroxetine. These results suggest that the two kinds of stimuli, LPS and cytomix from conditioned media, have different intermediate effectors in astrocytes. Completely blocking NF-κB pathway by PDTC results in less inhibition in cytokine expression compared to those by paroxetine (18.0% vs 42.2%, and 39.0% vs. 61.8%, respectively, for the reductions in TNF-α and IL-1β expression). Thus, additional pathways should be involved in the paroxetine suppression of astrocytic inflammatory responses. STAT3 and MAPKs including p38, JNK, and ERK are considered to be key regulators in astrocytes in response to neuroinflammation besides NF-κB [53]. However, it turns out that both MAPKs and STAT3 are not the answer. Paroxetine is also reported to prevent the downregulation of L-Glu transporters in astrocytes by inhibiting L-Glu release from reactive microglia, which otherwise causes excitotoxicity [54, 55]. In addition, paroxetine antagonizes P2X4 receptor, resulting in inhibition of ATP-induced calcium influx in 1321 N1 astrocytic cell line [56]. Thus, further investigation is needed to fully understand signaling participated in this regulation by paroxetine.
We have previously reported that incubation with LPS-stimulated microglia conditioned media leads to a significant death of SH-SY5Y cells by about 15%, where for example the medium TNF-α level is at approximately 8500 pg/mL [36]. No significant impact on SH-SY5Y cell viability is observed when incubated with the reactive microglia- or LPS-stimulated astrocyte conditioned media, where the level of TNF-α is roughly only at 1100 and 100 pg/mL, respectively, as a comparison. Minor amounts of cytokines and neurotrophic factors are derived from astrocytes as above mentioned. Paroxetine thus renders little impact on the neuronal survival under this condition.
Conclusions
The current study demonstrates that paroxetine ameliorates the reactive microglia-mediated inflammatory responses in astrocytes partially via inhibition of NF-κB pathway, but has no impact on LPS-stimulated astrocyte activation. Paroxetine also stimulates neurotrophic support of astrocytes. While the effect of paroxetine on this secondary astrocytic response appears minor compared to its effect on the innate immune responses of microglia, paroxetine can indeed alleviate neuroinflammation by suppressing microglia activation [36] and its paracrine inflammatory activation of astrocytes. The microglia and astrocyte results combined may implicate a therapeutic potential of paroxetine against neuroinflammation-associated neurological disorders such as PD.
Supplementary information
Additional file 1:. Figure S1. Full blots for the Figs. 3a, 6a and 7a.
Acknowledgements
Not applicable.
Abbreviations
- A/Lps
Conditioned medium of astrocytes cultured with LPS
- A/MC
Conditioned medium of astrocytes cultured with M/C
- A/MLps
Conditioned medium of astrocytes cultured with M/Lps
- BDNF
Brain-derived neurotrophic factor
- ERK
Extracellular signal-regulated kinase
- GFAP
Glial fibrillary acidic protein
- IL-1β
Interleukin 1β
- iNOS
Inducible nitric oxide synthase
- JNK
c-jun N-terminal kinase
- LPS
Lipopolysaccharide
- M/C
Microglia conditioned medium without LPS stimulation
- M/Lps
Microglia conditioned medium pre-activated with LPS
- MANF
Mesencephalic astrocyte-derived neurotrophic factor
- MAPK
Mitogen-activated protein kinase
- NF-κB
Nuclear factor κB
- NO
Nitric oxide
- PD
Parkinson’s disease
- PDTC
Pyrrolidine dithiocarbamic acid
- STAT3
Signal transducer and activator of transcription 3
- TLR
Toll-like receptor
- TNF-α
Tumor necrosis factor α
Authors’ contributions
\XZ, JHZ, and CPH designed the study. LBZ, JHH, HQZ, SYJ, CNZ, and NNH conducted the experiments. XZ, JHH, LBZ, and JHZ analyzed the data. JHZ, CPH, and JHH wrote the manuscript. All authors read and approved the final manuscript.
Funding
The study was supported by funding from the Zhejiang Provincial Natural Science Foundation (LD19H090001, and LZ19H090002), National Natural Science Foundation of China (81571087, 81771380, and 81771510), Wenzhou Municipal Science and Technology Bureau (Y20170071 and C20170003).
Availability of data and materials
All data supporting the conclusions of this study are included in the article.
Ethics approval and consent to participate
The animals used in this study were treated in accordance with protocols approved by the Institutional Animal Care and Use Committee of Wenzhou Medical University, China.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Xiong Zhang, Lan-Bing Zhu and Jia-Hui He contributed equally to this work.
Contributor Information
Xiong Zhang, Email: zhangxiong98@gmail.com.
Lan-Bing Zhu, Email: 1024547355@qq.com.
Jia-Hui He, Email: 121509197@qq.com.
Hong-Qiu Zhang, Email: 1572400504@qq.com.
Shu-Ya Ji, Email: 1605675878@qq.com.
Chao-Nan Zhang, Email: 2210491069@qq.com.
Na-Na Hou, Email: 1241269563@qq.com.
Chen-Ping Huang, Email: hcp@wmu.edu.cn.
Jian-Hong Zhu, Email: jhzhu@wmu.edu.cn.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12974-020-1712-0.
References
- 1.Poewe W, Seppi K, Tanner C, Halliday G, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Primers. 2017;3:17013. doi: 10.1038/nrdp.2017.13. [DOI] [PubMed] [Google Scholar]
- 2.Ransohoff R. How neuroinflammation contributes to neurodegeneration. Science. 2016;353:777–783. doi: 10.1126/science.aag2590. [DOI] [PubMed] [Google Scholar]
- 3.Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561:258–262. doi: 10.1038/s41586-018-0448-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Halliday G, Stevens C. Glia: initiators and progressors of pathology in Parkinson's disease. Mov Disord. 2011;26:6–17. doi: 10.1002/mds.23455. [DOI] [PubMed] [Google Scholar]
- 5.Shastri A, Bonifati DM, Kishore U. Innate immunity and neuroinflammation. Mediat Inflamm. 2013;2013:342931. doi: 10.1155/2013/342931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Croisier E, Moran LB, Dexter DT, Pearce RK, Graeber MB. Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. J Neuroinflammation. 2005;2:14. doi: 10.1186/1742-2094-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duffy M, Collier T, Patterson J, Kemp C, Luk K, Tansey M, et al. Lewy body-like alpha-synuclein inclusions trigger reactive microgliosis prior to nigral degeneration. J Neuroinflammation. 2018;15:129. doi: 10.1186/s12974-018-1171-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Booth H, Hirst W, Wade-Martins R. The role of astrocyte dysfunction in Parkinson’s disease pathogenesis. Trends Neurosci. 2017;40:358–370. doi: 10.1016/j.tins.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miklossy J, Doudet D, Schwab C, Yu S, McGeer E, McGeer P. Role of ICAM-1 in persisting inflammation in Parkinson disease and MPTP monkeys. Exp Neurol. 2006;197:275–283. doi: 10.1016/j.expneurol.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 10.Yang WL, Wang GH, Wang CE, Guo XY, Yin P, Gao JQ, et al. Mutant alpha-synuclein causes age-dependent neuropathology in monkey brain. J Neurosci. 2015;35:8345–8358. doi: 10.1523/JNEUROSCI.0772-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun LK, Shen RF, Agnihotri SK, Yun C, Huang ZW, Büeler H. Lack of PINK1 alters glia innate immune responses and enhances inflammation-induced, nitric oxide-mediated neuron death. Sci Rep. 2018;8:383. doi: 10.1038/s41598-017-18786-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Torres-Odio S, Key J, Hans-Hermann H, Canet-Pons J, Valek L, Roller B, et al. Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. J Neuroinflammation. 2017;14:154. doi: 10.1186/s12974-017-0928-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi I, Choi DJ, Yang H, Woo JH, Chang MY, Kim JY, et al. PINK1 expression increases during brain development and stem cell differentiation, and affects the development of GFAP-positive astrocytes. Mol Brain. 2016;9:5. doi: 10.1186/s13041-016-0186-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi I, Kim J, Jeong HK, Kim B, Jou I, Park SM, et al. PINK1 deficiency attenuates astrocyte proliferation through mitochondrial dysfunction, reduced AKT and increased p38 MAPK activation, and downregulation of EGFR. Glia. 2013;61:800–812. doi: 10.1002/glia.22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manuela N, Veronika M, Karin G, Hans AK, Christian H, Philipp JK. Pathological properties of the Parkinson’s disease-associated protein DJ-1 in alpha-synucleinopathies and tauopathies: relevance for multiple system atrophy and Pick's disease. Acta Neuropathol. 2004;107:489–496. doi: 10.1007/s00401-004-0834-2. [DOI] [PubMed] [Google Scholar]
- 16.Kim J, Cha S, Choi YR, Jou I, Joe E, Park SM. DJ-1 deficiency impairs glutamate uptake into astrocytes via the regulation of flotillin-1 and caveolin-1 expression. Sci Rep. 2016;6:28823. doi: 10.1038/srep28823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim KS, Kim JS, Park J-Y, Suh YH, Jou I, Joe E-H, et al. DJ-1 associates with lipid rafts by palmitoylation and regulates lipid rafts-dependent endocytosis in astrocytes. Hum Mol Genet. 2013;22:4805–4817. doi: 10.1093/hmg/ddt332. [DOI] [PubMed] [Google Scholar]
- 18.Sofroniew M, Vinters H. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137:47–59. doi: 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwang J, Zheng L, Ock J, Lee M, Kim S, Lee H, et al. Inhibition of glial inflammatory activation and neurotoxicity by tricyclic antidepressants. Neuropharmacology. 2008;55:826–834. doi: 10.1016/j.neuropharm.2008.06.045. [DOI] [PubMed] [Google Scholar]
- 21.Mansouri M, Naghizadeh B, Ghorbanzadeh B, Alboghobeish S, Amirgholami N, Houshmand G, et al. Venlafaxine prevents morphine antinociceptive tolerance: the role of neuroinflammation and the l-arginine-nitric oxide pathway. Exp Neurol. 2018;303:134–141. doi: 10.1016/j.expneurol.2018.02.009. [DOI] [PubMed] [Google Scholar]
- 22.Vollmar P, Haghikia A, Dermietzel R, Faustmann PM. Venlafaxine exhibits an anti-inflammatory effect in an inflammatory co-culture model. Int J Neuropsychopharmacol. 2008;11:111–117. doi: 10.1017/S1461145707007729. [DOI] [PubMed] [Google Scholar]
- 23.Zhao Y, Pan Y, Tang M, Lin W. Blocking p38 signaling reduces the activation of pro-inflammatory cytokines and the phosphorylation of p38 in the Habenula and reverses depressive-like behaviors induced by Neuroinflammation. Front Pharmacol. 2018;9:511. doi: 10.3389/fphar.2018.00511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gunasekara NS, Noble S, Benfield P. Paroxetine. An update of its pharmacology and therapeutic use in depression and a review of its use in other disorders. Drugs. 1998;55:85–120. doi: 10.2165/00003495-199855010-00007. [DOI] [PubMed] [Google Scholar]
- 25.Dechant K, Clissold S. Paroxetine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in depressive illness. Drugs. 1991;41:225–253. doi: 10.2165/00003495-199141020-00007. [DOI] [PubMed] [Google Scholar]
- 26.Ceravolo R, Nuti A, Piccinni A, Agnello GD, Bellini G, Gambaccini G, et al. Paroxetine in Parkinson’s disease: effects on motor and depressive symptoms. Neurology. 2000;55:1216–1218. doi: 10.1212/WNL.55.8.1216. [DOI] [PubMed] [Google Scholar]
- 27.Richard IH, McDermott MP, Kurlan R, Lyness JM, Como PG, Pearson N, et al. A randomized, double-blind, placebo-controlled trial of antidepressants in Parkinson disease. Neurology. 2012;78:1229–1236. doi: 10.1212/WNL.0b013e3182516244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chung Y, Kim S, Jin B. Paroxetine prevents loss of nigrostriatal dopaminergic neurons by inhibiting brain inflammation and oxidative stress in an experimental model of Parkinson's disease. J Immunol. 2010;185:1230–1237. doi: 10.4049/jimmunol.1000208. [DOI] [PubMed] [Google Scholar]
- 29.Meredith GE, Sonsalla PK, Chesselet MF. Animal models of Parkinson's disease progression. Acta Neuropathol. 2008;115:385–398. doi: 10.1007/s00401-008-0350-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao H, Zhang F, Zhou H, Kam W, Wilson B, Hong J. Neuroinflammation and α-synuclein dysfunction potentiate each other, driving chronic progression of neurodegeneration in a mouse model of Parkinson's disease. Environ Health Perspect. 2011;119:807–814. doi: 10.1289/ehp.1003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du R, Zhou Y, Xia M, Lu M, Ding J, Hu G. α-Synuclein disrupts the anti-inflammatory role of Drd2 via interfering β-arrestin2-TAB1 interaction in astrocytes. J. Neuroinflammation. 2018;15:258. doi: 10.1186/s12974-018-1302-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen P, Peng G, Li G, Yang S, Wu S, Wang C, et al. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol Psychiatry. 2006;11:1116–1125. doi: 10.1038/sj.mp.4001893. [DOI] [PubMed] [Google Scholar]
- 33.Kim MJ, Park M, Kim DW, Shin MJ, Son O, Jo HS, et al. Transduced PEP-1-PON1 proteins regulate microglial activation and dopaminergic neuronal death in a Parkinson's disease model. Biomaterials. 2015;64:45–56. doi: 10.1016/j.biomaterials.2015.06.015. [DOI] [PubMed] [Google Scholar]
- 34.Lee Y, Park J, Leem Y, Park J, Kim D, Choi Y, et al. The phosphodiesterase 10 inhibitor papaverine exerts anti-inflammatory and neuroprotective effects via the PKA signaling pathway in neuroinflammation and Parkinson's disease mouse models. J Neuroinflammation. 2019;16:246. doi: 10.1186/s12974-019-1649-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang W, Nguyen LTT, Burlak C, Chegini F, Guo F, Chataway T, et al. Caspase-1 causes truncation and aggregation of the Parkinson's disease-associated protein α-synuclein. Proc Natl Acad Sci U S A. 2016;113:9587–9592. doi: 10.1073/pnas.1610099113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu R, Zou M, Wang J, Zhu J, Lai J, Zhou L, et al. Paroxetine ameliorates lipopolysaccharide-induced microglia activation via differential regulation of MAPK signaling. J Neuroinflammation. 2014;11:47. doi: 10.1186/1742-2094-11-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCarthy KD. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shen Y, Qin H, Chen J, Mou L, He Y, Yan Y, et al. Postnatal activation of TLR4 in astrocytes promotes excitatory synaptogenesis in hippocampal neurons. J Cell Biol. 2016;215:719–734. doi: 10.1083/jcb.201605046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu JH, Lei XG. Double null of selenium-glutathione peroxidase-1 and copper, zinc-superoxide dismutase enhances resistance of mouse primary hepatocytes to acetaminophen toxicity. Exp Biol Med (Maywood) 2006;231:545–552. doi: 10.1177/153537020623100508. [DOI] [PubMed] [Google Scholar]
- 40.Wilms H, Sievers J, Rickert U, Rostami-Yazdi M, Mrowietz U, Lucius R. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1beta, TNF-alpha and IL-6 in an in-vitro model of brain inflammation. J Neuroinflammation. 2010;7:30. doi: 10.1186/1742-2094-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu JH, Chen CL, Flavahan S, Harr J, Su B, Flavahan NA. Cyclic stretch stimulates vascular smooth muscle cell alignment by redox-dependent activation of Notch3. Am J Physiol Heart Circ Physiol. 2011;300:H1770–H1780. doi: 10.1152/ajpheart.00535.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Q, Liu Y, Zhou J. Neuroinflammation in Parkinson's disease and its potential as therapeutic target. Transl Neurodegener. 2015;4:19. doi: 10.1186/s40035-015-0042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tynan RJ, Weidenhofer J, Hinwood M, Cairns MJ, Day TA, Walker FR. A comparative examination of the anti-inflammatory effects of SSRI and SNRI antidepressants on LPS stimulated microglia. Brain Behav Immun. 2012;26:469–479. doi: 10.1016/j.bbi.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 44.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sheng W, Zong Y, Mohammad A, Ajit D, Cui J, Han D, et al. Pro-inflammatory cytokines and lipopolysaccharide induce changes in cell morphology, and upregulation of ERK1/2, iNOS and sPLA2-IIA expression in astrocytes and microglia. J Neuroinflammation. 2011;8:121. doi: 10.1186/1742-2094-8-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trang T, Beggs S, Wan X, Salter M. P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J Neurosci. 2009;29:3518–3528. doi: 10.1523/JNEUROSCI.5714-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang X, Zeng L, Yu T, Xu Y, Pu S, Du D, et al. Positive feedback loop of autocrine BDNF from microglia causes prolonged microglia activation. Cell Physiol Biochem. 2014;34:715–723. doi: 10.1159/000363036. [DOI] [PubMed] [Google Scholar]
- 48.Allaman I, Fiumelli H, Magistretti P, Martin J. Fluoxetine regulates the expression of neurotrophic/growth factors and glucose metabolism in astrocytes. Psychopharmacology. 2011;216:75–84. doi: 10.1007/s00213-011-2190-y. [DOI] [PubMed] [Google Scholar]
- 49.Henn A, Lund S, Hedtjarn M, Schrattenholz A, Porzgen P, Leist M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. Altex. 2009;26:83–94. doi: 10.14573/altex.2009.2.83. [DOI] [PubMed] [Google Scholar]
- 50.Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. 2016;113:E1738–E1746. doi: 10.1073/pnas.1525528113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. 2016;89:37–53. doi: 10.1016/j.neuron.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Colombo E, Farina C. Astrocytes: Key regulators of Neuroinflammation. Trends Immunol. 2016;37:608–620. doi: 10.1016/j.it.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 54.Fujimori K, Takaki J, Shigemoto-Mogami Y, Sekino Y, Suzuki T, Sato K. Paroxetine prevented the down-regulation of astrocytic L-Glu transporters in neuroinflammation. J Pharmacol Sci. 2015;127:145–149. doi: 10.1016/j.jphs.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 55.Takaki J, Fujimori K, Miura M, Suzuki T, Sekino Y, Sato K. L-glutamate released from activated microglia downregulates astrocytic L-glutamate transporter expression in neuroinflammation: the ‘collusion’ hypothesis for increased extracellular L-glutamate concentration in neuroinflammation. J Neuroinflammation. 2012;9:275. doi: 10.1186/1742-2094-9-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nagata K, Imai T, Yamashita T, Tsuda M, Tozaki-Saitoh H, Inoue K. Antidepressants inhibit P2X4 receptor function: a possible involvement in neuropathic pain relief. Mol Pain. 2009;5:20. doi: 10.1186/1744-8069-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1:. Figure S1. Full blots for the Figs. 3a, 6a and 7a.
Data Availability Statement
All data supporting the conclusions of this study are included in the article.