

Abstract
Lack of specific markers for innate lymphoid cells (ILCs) limit our knowledge on their spatial organization in situ. We compared two quadruple-color staining protocols for detection of the three principal human ILC subsets in formalin-fixed paraffin-embedded specimens. ILC subset–associated archetypical transcription factors (TFs) T-bet, GATA3, and RORγt were used as positive identifiers in combination with lymphoid lineage markers to exclude non-ILCs. One method (“virtual quadruple staining”) comprised of iterative single stainings on the same section performing digital scanning and subsequent immunoglobulin and chromogen stripping after each staining round. The second technique (“true-color quadruple staining”) comprised sequential double stainings with permanent colors. Both protocols appeared suitable for accurate detection of each ILC subset, and as added result, concomitant visualization of their T cell subset counterpart. Only true-color quadruple staining enabled simultaneous detection of all three ILC subsets within one section. Furthermore, we found that type 3 and type 1 ILCs (ILC1s) represent the major subsets in colon and that part of the ILC1s typically colocalizes with blood vessels. Our data highlight the utility of TFs combined with lineage markers for the identification of ILC subsets and proposed workflow opens the way to gain deeper insight of their anatomical distribution.
Keywords: immunohistochemistry, ILC1, ILC2, ILC3, multiple immunostaining, transcription factor
Introduction
The family of innate lymphoid cells (ILCs) comprises a distinct group of hematopoietic cells with a lymphoid cell morphology, and which are functionally involved in protective responses against microorganisms, in lymphoid tissue formation, in tissue remodeling after damage, and in the homeostasis of tissue stromal cells.1 In contrast to conventional T and B lymphocytes, ILCs lack the expression of rearranged antigen receptors and as a consequence, they do not show any degree of antigen-specific responsiveness. Instead, they respond to cytokines and other soluble factors in their microenvironment, which are produced by surrounding tissue-resident cells in response to pathogens or other danger signals. ILCs are defined as cell lineage marker-negative (Lin−) lymphocytes, as they lack the expression of markers that identify other known families of immune cells, such as T and B lymphocytes, dendritic cells, monocytes, macrophages, and granulocytes. The ILC family has been classified into three principal groups on the basis of hallmark transcription factor (TF) expression and specific cytokine production profile—a categorization that mirrors the classification of CD4+ T helper (Th) cell subsets Th1, Th2, and Th17, respectively.2 Group 1 ILCs comprise natural killer (NK) cells and ILC1s, group 2 ILCs include a single subset, that is, ILC2s, whereas group 3 ILCs contain lymphoid tissue-inducer cells and natural cytotoxicity receptor-positive and receptor-negative ILC3s. Group 1 ILCs are defined by the expression of TF T-box expressed in T cells (T-bet) and their production of IFN-γ; group 2 ILCs are characterized by the expression of GATA-binding protein 3 (GATA3) and production of type 2 cytokines interleukin (IL)-4, IL-5, IL-9, and IL-13; all subsets within group 3 are dependent on the TF retinoic acid–related orphan receptor (ROR)γt and can secrete IL-17A and/or IL-22. Of note, no specific markers have as yet been described for ILCs. The vast majority of ILC1s, ILC2s, and ILC3s express CD161 and the α-chain of the IL-7 receptor (CD127). Accumulation and/or dysfunction of ILCs has been reported to occur in several pathological conditions, such as Crohn’s disease (ILC1)3 and inflammatory skin disorders atopic dermatitis (ILC2) and psoriasis (ILC3), respectively.4,5
There is a cascade of studies on phenotype and function of tissue-derived purified ILCs or in vitro cultured ILCs, but at present, the literature on detection of tissue ILCs in situ by immunohistochemistry is limited. The investigation on ILCs in situ is hampered by the lack of exclusive markers for these cells. Multiple markers are needed to identify ILCs and ILC subsets, which is feasible when performing flow-cytometry analysis on single cell suspensions, but this method provides no information on the topographic distribution of these cells in (diseased) tissues. However, identification of ILCs in tissues is hard to perform for several reasons. First, at least four markers are needed to identify them and exclude other cell types, when applying the flow-cytometry strategy. Second, it is technically difficult to visualize the many different epitopes in the same section, and even after successful immunostaining it may be hard for the human eye to recognize all the colors and color combinations in just one specimen. Third, many antibodies that are commonly used for the detection of ILCs in flow-cytometry experiments cannot be used in immunohistochemical (IHC) staining approaches. Finally, ILCs are believed to represent only a very minor population of resident cells or infiltrating inflammatory cells, which further restricts their detection in situ.
This study was aimed to set up an IHC quadruple-staining workflow to detect human ILCs and ILC subsets in formalin-fixed, paraffin-embedded (FFPE) tissue sections, using a panel of markers commonly applied to identify ILCs by flow-cytometry and using colon as an example of human tissue. We compared two different staining approaches, each having its own typical advantages and shortcomings. The first method is a classical multiple staining technique, in which we performed two sequential double-stainings combining monoclonal antibodies from different species, and directly visualized the reaction pattern of each antibody in its own permanent color (red, blue, green, and black), having the ability to observe the result by a conventional light microscope. The second technique comprised four sequential single-stainings on the same tissue section, while performing digital scanning of the stained section and subsequent stripping of the immunoglobulins and dye after each staining round. With computer-assisted analysis, these four individual stainings were combined into one composite color image. Our IHC quadruple-staining methods enable to investigate the presence and anatomical distribution of ILC subsets in tissues and, in addition, offer the concomitant identification of Th cell/CD8+ cytotoxic T (Tc) cell subsets, as CD3 is applied as an exclusion marker and the categorization of these cell types is based on similar TFs. Moreover, by visualizing ILCs in situ it is possible to discover co-localization of these cells with other cell types or structures (e.g., blood vessels, epithelial cells) which might provide insight into the function of ILCs.
Materials and Methods
Tissue Samples and Ethical Statement
Normal colon tissues (n=5) were obtained from the archives of the Department of Pathology at the Amsterdam University Medical Centers, location AMC. The colon tissues were FFPE tissue blocks, originally sampled for diagnostic purposes from the resection borders of surgical specimens derived from patients with colon carcinoma. FFPE tonsil tissues served as control for all antibodies. The institutional Medical Ethics Review Committee granted a waiver for the anonymous use of human leftover material of diagnostic procedures. The research was conducted in accordance with the Declaration of Helsinki.
Reagents for Immunohistochemistry
The following primary mouse anti-human protein antibodies (clone; manufacturer) were used in this study: RORγt (6F3.1; Merck Millipore, Darmstadt, Germany), CD3 (A-1; Santa Cruz Biotechnology, CA), CD20 (L26; Immunologic, Duiven, Netherlands), CD79α (JCB117; Dako, Glostrup, Denmark), and Podoplanin (D2-40; Dako). The following rabbit anti-human protein antibodies were used: T-bet/Tbx21 (EPR9302; Abcam, Cambridge, UK), GATA3 (EPR16651; Abcam), CD3 (SP7; Immunologic), CD56 (MRQ-42; Cell Marque, Rocklin, CA), and von Willebrand Factor (polyclonal; Dako).
As secondary reagents we used alkaline phosphatase (AP)- or horseradish peroxidase (HRP)-conjugated anti-mouse and anti-rabbit BrightVision detection kits (Immunologic). Chromogens PermaBlue/AP, PermaRed/AP, and PermaGreen/HRP were from Diagnostic Biosystems (Sanbio, Netherlands), Deep Space Black was purchased from Biocare Medical (Concord, CA) and Nova-RED was from Vector Laboratories (Burlingame, CA). Antibodies and polymers were always diluted in Normal Antibody Diluent from Scytek Laboratories (Logan, UT). We used Tris-buffered saline solution with 0.05% Tween (TBST) as washing buffer between incubation steps. The stained sections were mounted in Glycerol Gelatin aqueous slide mounting medium (Sigma-Aldrich, Zwijndrecht, Netherlands) or in PertexTM (VWR international, Amsterdam, Netherlands).
Virtual Quadruple Staining Protocol
This protocol comprised a series of repeatedly performed immunostainings, with a digitization and elution step after each staining round. Tissue sections (4 µm thickness) were first subjected to deparaffinization in xylol, rehydration in graded alcohols, and blocking of endogenous peroxidase using 0.3% H2O2 in methanol for 10 min. Next, heat-induced epitope retrieval (HIER) was performed for 10 min at 98C in Tris-ethylenediaminetetraacetic acid (Tris-EDTA) buffer (pH 9), using PT Module equipment and PT Module Buffer 4 from Thermo Fisher Scientific (Waltham, MA). Subsequently, sections were treated with protein-free serum block (Super Block; Scytek Laboratories, Logan, UT) followed by incubations with predetermined, appropriate dilutions of primary antibodies. After washing, sections were incubated with relevant HRP-conjugated polymer (either anti-mouse or anti-rabbit, depending on the source of the primary antibodies). HRP activity was visualized in red using NovaRED as substrate (Table 1). After counterstaining with hematoxylin and mounting with glycerol/gelatin, sections were scanned using Philips Intellisite Ultrafast Scanner (Philips Digital Pathology Solutions, Best, Netherlands). Subsequently, the sections were prepared for the following staining round by first removing the coverslips in a water bath at 50C. Next, sections were placed in a stripping buffer (2% SDS/Tris-HCl, 0.7% β-mercaptoethanol) for 30 min at 50C in order to remove the immune complexes and the dye from the sections. After washing the sections in tap water and TBST, a new staining round (without HIER treatment) with a new primary antibody was performed. Negative controls (omitting the primary antibody) were included in all staining rounds. Human tonsil tissue was used as positive control for all antibodies in our panel, and in addition, to set up the experimental IHC workflow of the quadruple stainings to detect ILCs, because this tissue is known to contain all ILC subsets.
Table 1.
Virtual Quadruple Staining Sequence for the Detection of ILC1, ILC2, or ILC3.
| Staining 1 |
Staining 2 |
Staining 3 |
Staining 4 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell Types to Be Detected | Primary Antibody (Species) | Secondary Antibody–Conjugate >Chromogen | Primary Antibody (Species) | Secondary Antibody– Conjugate >Chromogen |
Primary Antibody (Species) | Secondary Antibody–Conjugate >Chromogen |
Primary Antibody (Species) | Secondary
Antibody–Conjugate >Chromogen |
||||
| ILC1 Th1/Tc1 |
T-bet (rabbit) |
anti-rabbit–HRP >NovaRED |
Digital scanning/stripping immune complexes and dye | CD56 (rabbit) |
anti-rabbit–HRP >NovaRED |
Digital scanning/stripping immune complexes and dye | CD3 (rabbit) |
anti-rabbit–HRP >NovaRED |
Digital scanning/stripping immune complexes and dye | CD20/CD79α (mouse) |
anti-mouse–HRP >NovaRED |
Digital scanning/composition of virtual 4-color image |
| ILC2 Th2/Tc2 |
GATA3 (rabbit) |
anti-rabbit–HRP >NovaRED |
CD56 (rabbit) |
anti-rabbit–HRP >NovaRED |
CD3 (rabbit) |
anti-rabbit–HRP >NovaRED |
CD20/CD79α (mouse) |
anti-mouse–HRP >NovaRED |
||||
| ILC3 Th17/Tc17 |
RORγt (mouse) |
anti-mouse–HRP >NovaRED |
CD56 (rabbit) |
anti-rabbit–HRP >NovaRED |
CD3 (rabbit) |
anti-rabbit–HRP >NovaRED |
CD20/CD79α (mouse) |
anti-mouse–HRP >NovaRED |
||||
Abbreviations: ILC, innate lymphoid cell; Th, helper T cell; Tc, cytotoxic T cell; HRP, horseradish peroxidase.
After completion of a series of four single stainings, digital images were combined into one virtual quadruple staining image. To create these, digital image files were downloaded from the Philips Image Management System. Areas of interest (5000 * 5000 pixels, ~ 1.6 µm2) were selected, and registered, and individual images saved in the PNG raster-graphics file-format as previously described,6 and further processed in Fiji.7 In short, registered images were converted to an imagestack, followed by color deconvolution to create a stack of 8-bit images containing only the NovaRed signal. The resulting stack was inverted, and subsequently converted to a composite, false color image. Finally, color channels were assigned to each of the different immunostainings: magenta for the TFs (T-bet, GATA-3, RORγt), yellow for CD3, green for CD56, and white for CD20/CD79α.
True-color Quadruple Staining Protocol
This quadruple staining technique was performed on 4-µm-thick sections to obtain sections stained in red, blue, black, and green, based on the method originally described by van der Loos and Teeling.8 In brief, we sequentially performed two IHC double-stainings with a HIER treatment (10 min) in between, which strips antibodies from the section without affecting the color deposition obtained in the first double-staining step. Each of these separate double stainings were combinations of a mouse and a rabbit antibody, followed by species-specific HRP- and AP-conjugated polymers. In short, after deparaffinization, rehydration, and blocking endogenous HRP activity, HIER was performed as described above. After a serum-free protein block for 10 min, a mixture of a mouse and rabbit primary antibodies was applied to the sections for 1 hr at room temperature (Table 2). Next, after washing, a mixture of HRP-conjugated anti-mouse and AP-conjugated anti-rabbit polymers was applied to the sections. Then, HRP activity was detected in black and AP activity was visualized in red using Deep Space Black and PermaRed, respectively. Subsequently, HIER was performed for 10 min, sections were washed, and a second double-staining step was performed, essentially as described above, but in this case, HRP activity was visualized in green and AP activity in blue, using PermaGreen and PermaBlue, respectively. Without counterstaining sections were dried on a hotplate at 50C and mounted with Pertex.
Table 2.
True-color Quadruple Staining Sequence for the Detection of ILC1, ILC2, or ILC3.
| Cell Types to Be Detected | Double Staining 1 |
Stripping Immune Complexes | Double Staining 2 |
||||
|---|---|---|---|---|---|---|---|
| Primary Antibody (Species) |
Secondary
Antibody Enzyme-Conjugate |
Chromogen | Primary Antibody (Species) |
Secondary
Antibody Enzyme-Conjugate |
Chromogen | ||
| ILC1 Th1/Tc1 |
T-bet (rabbit) CD3 (mouse) |
anti-rabbit–AP anti-mouse–HRP |
PermaRed Deep Space Black |
HIER 10’ | CD56(rabbit) CD20/CD79α (mouse) |
anti-rabbit–AP anti-mouse–HRP |
PermaBlue PermaGreen |
| ILC2 Th2/Tc2 |
GATA3 (rabbit) CD3 (mouse) |
anti-rabbit–AP anti-mouse–HRP |
PermaRed Deep Space Black |
HIER 10’ | CD56(rabbit) CD20/CD79α (mouse) |
anti-rabbit–AP anti-mouse–HRP |
PermaBlue PermaGreen |
| ILC3 Th17/Tc17 |
RORγt (mouse) CD3 (rabbit) |
anti-mouse–AP anti-rabbit–HRP |
PermaRed Deep Space Black |
HIER 10’ | CD56(rabbit) CD20/CD79α (mouse) |
anti-rabbit–AP anti-mouse–HRP |
PermaBlue PermaGreen |
Abbreviations: ILC, innate lymphoid cell; Th, helper T cell; Tc, cytotoxic T cell; AP, alkaline phosphatase; HRP, horseradish peroxidase; HIER, heat-induced epitope retrieval.
Results
ILC Staining Strategy
With a panel of multiple monoclonal antibodies directed against a defined set of cell surface molecules human ILC subsets can be identified by flow cytometry in complex tissue-derived cell suspensions3,5: ILCs are generally defined as CD45+Lin−CD127+CD161+ cells, which can be subdivided into CRTH2+CD117–/+ ILC2s, CRTH2−CD117+ ILC3s, and CRTH2−CD117− ILC1s. As a first step to develop an IHC protocol to identify ILC in situ in FFPE tissue sections, we tested the usability of the antibody panel used for flow cytometry. Our initial series of IHC experiments revealed that all commercially available anti-human CD127, CD161, and CRTH2 antibodies were inapplicable (data not shown), implying that this approach is not suitable to visualize ILCs on FFPE specimens in situ.
As the three different ILC subsets are defined by their archetypical TF (ILC1s express T-bet, ILC2s display GATA3, while ILC3s show RORγt expression), this offered an alternative opportunity to identify ILCs in situ. However, because these TFs are not only expressed by ILCs, additional stainings are necessary to exclude other cell types which also express these TFs, such as (subtypes of) T-, B-, plasma-, and NK cells. To this end, we included lineage markers CD3 (T cells), CD20/CD79α (B cells/plasma cells), and CD56 (NK cells) for all of which well-documented FFPE-suitable antibodies are available. We did not include lineage markers for granulocytes, since pilot experiments revealed T-bet, GATA3, and RORγt were not expressed in this cell type. Accordingly, ILCs can be defined as TF+ cells (nuclear staining) that are negative for all lineage markers. As the TFs and CD3 are determined by their own color, a further advantage of this multiple staining approach is the possibility for concomitant detection of the three different ILC-subsets (expressing either T-bet, GATA3, or RORγt) and their T cell counterparts Th1/Tc1, Th2/Tc2, and Th17/Tc17 (TF+CD3+ CD56−CD20−CD79α−). For the simultaneous IHC detection of four different epitopes in the same FFPE tissue section we next evaluated two different multiple staining approaches. On the one hand, we developed a “virtual” quadruple immunostaining, in which the actual quadruple staining image is created by a computer, and a “conventional” quadruple staining, where each epitope was visualized in situ, each with its own permanent-color label.
Virtual Quadruple Immunostaining
The virtual quadruple staining technique consists of four sequentially performed single IHC stainings in one tissue specimen, with a digitization (scanning) step in between the staining cycles, followed by a stripping step of the primary and secondary antibodies. An obvious point of concern is that these sequential rounds of antibody and dye removal steps may be detrimental to any target epitope to be detected in later staining cycles and consequently generate a false negative IHC staining. To check for this, we performed all iterative staining steps with the same primary antibody and scanned the section after each staining round. The resulting image series offered the possibility to observe any loss in staining intensity of each individual antibody after each staining round. In addition, comparison of these serially created images also enables to observe for any morphological tissue damage in later staining cycles.
We found that RORγt could no longer be detected after stripping once and that the staining intensity of T-bet and GATA3 was considerably reduced. The intensity of CD56 staining showed a gradual decline after every stripping step, while the epitopes of CD3, CD20, and CD79α were not affected, and could easily be stained without obvious loss of staining intensity even after 3 elution rounds. The tissue integrity of the colon specimen remained intact, and we did not observe any morphological tissue damage during our experiments. After having addressed the question of potential epitope loss by dye/antibody stripping, we were able to designate the optimal order of labeling the target molecules for detection of ILCs in situ. The TFs should be stained first, then CD56 followed by CD3 and a mix of CD20/CD79α in the third and fourth round, respectively (Table 1).
A representative example for the detection of ILC1s by virtual quadruple immunostaining is illustrated in Fig. 1, showing a colon specimen successively stained with positive marker T-bet (Fig. 1A) and exclusion markers CD56 (Fig. 1B), CD3 (Fig. 1C), and a mix of CD20/CD79α (Fig. 1D). Although it is relatively easy to study the phenotype of individual cells, by iterating through the different layers of registered scans in the user interface of the digital pathology image management system, it is also possible to create a single, false-color composite image in which coexpression of any of these different markers can be appreciated (Fig. 1E and F). In these figures, ILC1s can clearly be recognized as T-bet+ Lin− cells and type-1 T cells as T-bet+ CD3+ CD56‒ CD20‒ CD79α‒ cells. Representative examples for the visualization of ILC2s and ILC3s in colon specimens are shown in Supplemental Figs. 1 and 2, respectively.
Figure 1.

Identification of ILC1s in normal human colon by virtual quadruple staining. A single section of colon tissue is successively stained with T-bet (A), CD56 (B), CD3 (C), and a mix of CD20/CD79α (D), using NovaRED as substrate and hematoxylin as counterstaining. Between each staining round, sections were digitized, decolorized, and stripped from antibodies. The red color in the digital images A–D, representing the immunopositive signal, is converted to and combined in one false color composite image (E): T-bet (magenta), CD56 (green), CD3 (yellow), and a mix of CD20/CD79α (white). Figure F shows a higher magnification of the framed area in (E). Scale bar in A (=B, C, D, E): 100 μm; F: 50 μm. Abbreviations: ILC, innate lymphoid cell.
True-color Quadruple Immunostaining
As an alternative for the virtual quadruple immunostaining, we performed a more conventional multiple staining technique, in which each antibody has its own associated color. This protocol comprises two separate double-stainings with one step in between to remove immunoreagents, which can be accomplished by 10’ HIER. In contrast to the false color method described above, the chromogens we used for true-color quadruple immunostaining are permanent, and are not removed during the HIER step between the two staining rounds. Both double stainings were based on differences in the species type of the primary antibodies used, in our experiments which are always a combination of a mouse and a rabbit antibody, followed by HRP- or AP-conjugated anti-mouse and anti-rabbit polymers, respectively. For the true-color quadruple immunostaining technique, similar to the virtual quadruple staining, it was necessary to evaluate if the staining pattern of the antibodies was affected by the second HIER procedure. This test revealed that none of the antibodies in our panel showed a decrease in staining quality when used in the second staining round after the second HIER (data not shown). As anti-TF antibodies had to be used in the first staining round in the virtual quadruple staining technique, we decided to use the anti-TFs antibodies first in the true-color quadruple immunostaining as well. An optimal staining protocol to detect ILCs in tissue is illustrated in Table 2. The first pair of applied antibodies included an antibody against one of the ILC-associated TF (either T-bet, GATA-3 or RORγt) in combination with an antibody against CD3. After the second HIER treatment in the second staining round, we applied the CD20/CD79α (as mix) and CD56 combination. We found that with this quadruple immunostaining technique ILC1s, ILC2s, and ILC3s can clearly be visualized as TF+ Lin− cells, as illustrated in Fig. 2A to C, respectively.
Figure 2.
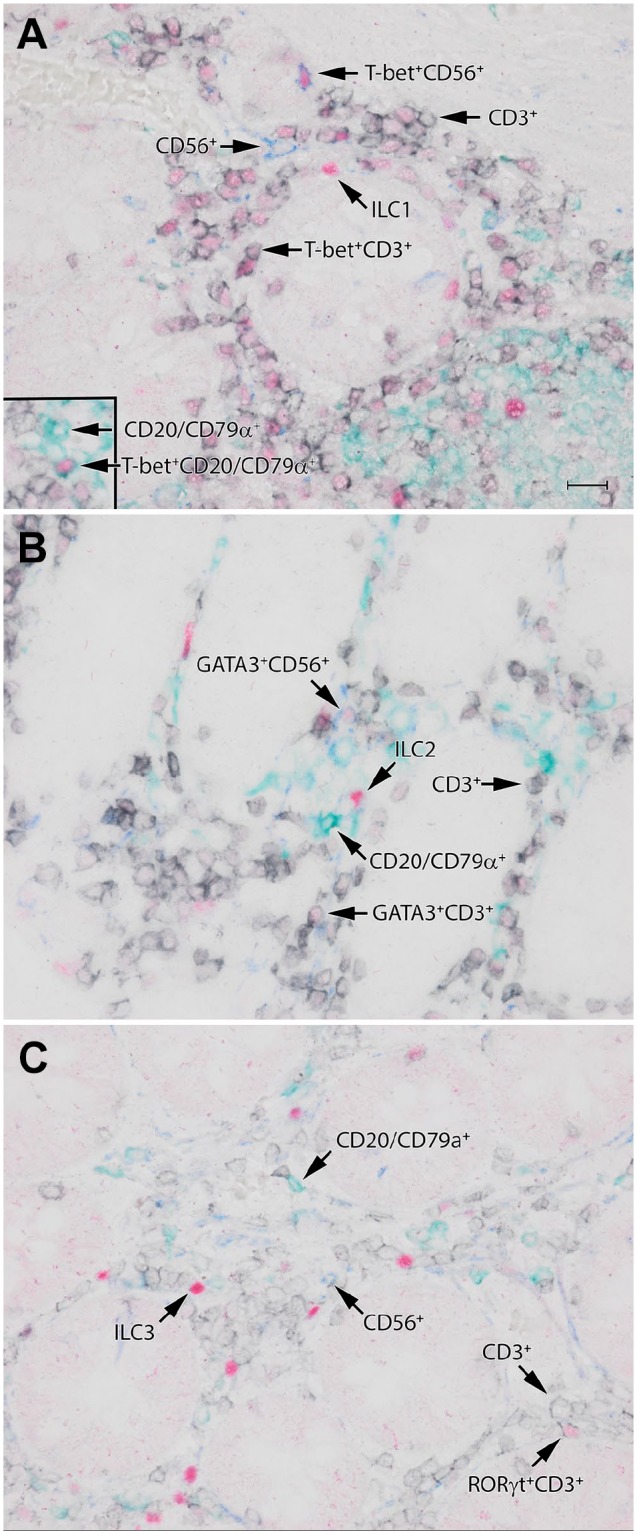
Identification of ILC1s (A), ILC2s (B), and ILC3s (C) in normal human colon by the true color quadruple staining technique. These ILC subsets can be appreciated as TF-positive cells (red), which are negative for lineage markers CD3 (black), CD56 (blue), and CD20/CD79α (green). The TF used to identify ILC1s (A), ILC2s (B), and ILC3s (C) are T-bet, GATA3, and RORγt, respectively. Scale bar: 50 μm, (A, B, C) similar magnification. Abbreviations: ILC, innate lymphoid cell; TF, transcription factor.
Distribution of ILC Subsets in Human Colon
Both quadruple staining techniques described above enable the detection of ILCs in human FFPE specimens, and are suitable to study the distribution of these cells in situ. We found that the ILC subtypes differed in number and localization in the human colon. The majority of the ILC1s showed a scattered distribution in the lamina propria (Figs. 1 and 2A), and occasionally, they were encountered in the submucosa and MALT follicles. Interestingly, ILC1s (T-bet+Lin−) were frequently present in the lumen of vessels in the submucosa (Fig. 3). Often, but not always, these cells were in close proximity to the endothelium. Additional staining revealed that these vessels were vWF+, and always podoplanin−, indicating that these structures are blood vessels and not lymphatic vessels. ILC2s (GATA3+Lin−) were found less frequently when compared to ILC1s and ILC3. They were present in the lamina propria and occasionally in the MALT. ILC2s were not encountered in the submucosa (Fig. 2B and Supplemental Fig. 1). ILC3s (RORγt+Lin−) were found in similar amounts as ILC1s and were mostly observed in the lamina propria (Fig. 2C, Supplemental Fig. 2), but occasionally also in the submucosa and MALT.
Figure 3.

ILC1s are frequently encountered in the lumen of blood vessels, but not in lymph vessels. ILC1s are identified by T-bet (red) and are Lin− (CD3−CD20/79α−, in black). Blood vessels are stained with vWF (green), lymph vessels with podoplanin (blue). Scale bar: 50 μm. Abbreviations: ILC, innate lymphoid cell.
Expression of T-bet was not restricted to ILC1s and T cells (T-bet+CD3+), as we also occasionally observed T-bet co-expression with CD56+ cells and CD20/CD79α+ cells (Fig. 2). Expression of GATA3 and RORγt was confined to ILCs and CD3+ cells (Th17), as no coexpression was found with CD56 or CD20/CD79α. Remarkably, the expression of T-bet and RORγt was much more intense in ILC1s and ILC3s, respectively, in comparison to the expression of these TFs in Th1/Tc1 (CD3+T-bet+) or Th17/Tc17 (CD3+RORγt+) cells (Fig. 2 and Supplemental Fig. 2).
Simultaneous Detection of ILC1s, ILC2s, and ILC3s in a Single Section
Application of the true-color quadruple staining technique allowed simultaneous visualization of all ILC subsets in a single section, detecting GATA3 and a mixture for all lineage markers except CD56 in the first staining round, and T-bet and RORγt in the second round (Table 3). The anti-CD56 had to be left out from the antibody mixture as this rabbit-derived antibody could not be applied together with rabbit anti-GATA3 in the same staining step. The consequence of leaving out anti-CD56 is fairly small, as no CD56+ cell expressed GATA3 and RORγt and only an occasional CD56+ cell showed co-expression with T-bet (see above).
Table 3.
Staining Sequence for the Simultaneous Detection of ILC1s, ILC2s, and ILC3s, and Presence of ILC1s in Blood Vessels.
| Double Staining 1 |
Double Staining 2 |
||||||
|---|---|---|---|---|---|---|---|
| Cell Types to Be Detected | Primary Antibody (Species) |
Secondary Antibody Enzyme–Conjugate | Chromogen | Stripping Immune Complexes | Primary Antibody (Species) | Secondary Antibody Enzyme–Conjugate | Chromogen |
| ILC1 ILC2 ILC3 |
GATA3 (rabbit) CD3/CD20/CD79α (mouse) |
anti-rabbit–AP anti-mouse–HRP |
PermaBlue Deep Space Black |
HIER 10’ | T-bet (rabbit) RORγt (mouse) |
anti-rabbit–AP anti-mouse–HRP |
PermaGreen PermaRed |
| ILC1 blood vessels lymph vessels |
T-bet (rabbit) CD3/CD20/CD79α (mouse) |
anti-rabbit–AP anti-mouse–HRP |
PermaRed Deep Space Black |
HIER 10’ | vWF (rabbit) Podoplanin (mouse) |
anti-rabbit–HRP anti-mouse-AP |
PermaGreen PermaBlue |
Abbreviations: ILC, innate lymphoid cell; AP, alkaline phosphatase; HRP, Horseradish peroxidase; HIER, heat-induced epitope retrieval.
The experiments to visualize simultaneously all ILC subsets in a single section (Fig. 4) revealed similar ILC numbers and distribution as found in our earlier experiments, in which the quadruple staining was just focused on one ILC subset (Figs. 1 and 2). In most cases, T-bet, GATA3, and RORγt were mutually exclusive (Fig. 4), but occasionally, indecisive double staining of TFs was observed, but it was difficult to determine if red (RORγt+) cells double stained with either green (T-bet) or blue (GATA3). Unfortunately, the virtual quadruple staining technique is not suitable for the simultaneous detection of ILC1s, ILC2s, and ILC3s in a single section, as the epitopes of T-bet, GATA3, and RORγt are already reduced/lost after the first immunoglobulin and chromogen stripping round (see above), implying that one of the two cannot be detected in the subsequent staining step.
Figure 4.

Simultaneous detection of ILC1s, ILC2s, and ILC3s in a single section by the sequential quadruple staining technique. Identification is based on the expression of archetypical transcription factors T-bet (green), GATA3 (blue), and RORγt (red) to identify ILC1s, ILC2s, and ILC3s, respectively, and lack of expression of lineage markers CD3/CD20/CD79α (black). Scale bar: 10 μm. Abbreviations: ILC, innate lymphoid cell.
Discussion
As specific markers for ILCs still need to be discovered, it is challenging to unambiguously identify ILCs in human tissues in relation to their microenvironment (in situ) and differentiate them from other leukocytes, in particular Th/Tc cell subsets. In the present study, we developed and evaluated two different IHC quadruple staining approaches for the identification of ILC subtypes in FFPE human tissue. In both staining approaches, identification of ILC1s, ILC2s, and ILC3s is essentially based on detection of their archetypal TF (T-bet, GATA3, and RORγt, respectively), and concomitant detection of lymphocyte lineage markers CD3, CD20, CD56, and CD79α in the same section to exclude T cells, B cells, and NK cells sharing the same TF. The first method was a virtual quadruple staining method, in which serial IHC single stainings of a single section were digitized and subsequently used to generate a composite image visualizing all individual stains in different false colors (in any optional combination) in one image. In the second method, we performed a more conventional procedure, where a one-color label was coupled to one IHC label. Both techniques required diligent optimization of the antibody dilution and color combinations of the epitopes of interest, and in addition, both were suitable for the accurate detection of ILC subsets in FFPE human tissue sections and concomitant identification of Th/Tc subtypes in a single tissue section.
The two quadruple IHC methods have their own typical benefits and drawbacks. An important advantage of the virtual quadruple staining over the “conventional” true-color quadruple staining is that, in principle, any combination of antibodies is possible, independent of the antibodies’ species origin or isotype. A second important advantage of this technique is that it is much easier to distinguish the multiple different signals in the stained sections. In true-color images, it may be difficult to appreciate colocalization of two or more different colors, especially when the intensity of one color is weak compared to the other(s). In the present study, we also experienced such problems, that is, on a subset of RORγt+ cells: in some instances, it was very difficult to determine if the red chromogen colocalized with blue or green. Using the virtual multiple stains, there are more options to analyze the results. First, it is possible to compare, cell by cell, the individual “single” stainings. This can easily be done using a virtual stack of images which can be generated by the computer image management system. A second option is to create composite false color images, which can combine all or a selection of stained images from a multi-stained sample. In the latter option, a combination of colors can be selected that optimally reveals the most important tissue characteristics. The virtual multiple staining is also a good immunostaining tool for color-blind researchers. Besides easy direct recognition of single or multiple stained cells in the computer user interface (by iterating through the image stack), one can also select a combination of colors which is more easily recognized by color blind researchers, for example, using magenta instead of red.
One of the limitations of the virtual quadruple staining method is that not every specific epitope/antibody combination can “survive” the obligatory stripping steps. Elaborate optimization experiments are often necessary to determine the optimal staining sequence. In our study, we observed that staining of the TFs was strongly reduced (T-bet, GATA3) or lost (RORγt) already after one stripping treatment. Consequently, simultaneous detection of ILC1s, ILC2s, and ILC3s within one section is not possible with this method.
The advantage of the true-color over the virtual quadruple IHC method is that the former just requires a conventional light microscope instead of expensive equipment, that is, a slide scanner. In addition, the true-color quadruple IHC method avoids “aggressive” stripping steps, which cause potential loss of target epitopes and/or tissue integrity and limit the number of cycles that a tissue specimen can be stained. Indeed, this method allowed to stain for the sensitive TFs T-bet and RORγt in the second staining step, and consequently, enabled simultaneous detection of ILC1s, ILC2s, and ILC3s within one section. Compared with the virtual quadruple IHC method, the true-color quadruple IHC method is more dependent on the availability of high-quality antibodies of the right species origin and appropriate isotype. Furthermore, the limited availability of chromogens for IHC restrict the number of colors in this multiplex IHC method, whereas the virtual quadruple IHC technique allows unlimited expansion of the number of antibodies/markers, provided that specific staining and tissue integrity remains optimal after each elution round.
An obvious question that remains, is which of the two methods is most suitable to identify ILCs in FFPE tissue. In our opinion, the virtual method is preferable, as with this method it is easier to objectively distinguish multiple colored cells, consequently resulting in a more accurate identification of ILC subsets. In addition, the procedure is easier to perform, and moreover, fewer (expensive) reagents are required, compared to the true-color method. The virtual method only needs HRP-conjugated anti-mouse- and anti-rabbit polymers as secondary antibodies, and NovaRed kits to visualize enzyme activity, whereas the true-color quadruple staining requires additional AP-conjugated anti-mouse- and anti-rabbit polymers, as well as multiple different chromogens. However, an additional limitation of the virtual staining technique is the necessity of a slide scanner. Depending on the research question, it may not always be necessary to stain for T, B and NK cells separately, like we did, but it is also possible to combine the exclusion antibodies (markers) in a cocktail, and visualize them in the same color, resulting in a simple double staining instead of a quadruple staining.
At present, the vast majority of studies on the composition of ILC subsets in various normal or pathological tissues have been performed by multi-parameter flow cytometry on tissue-derived single cell suspensions. Concerning the human intestinal tract, recent flow-cytometry observations demonstrated that ILC3s and ILC1s are the most abundant populations and ILC2s represent a minor population in the colon,9,10 which is corroborated by our IHC data. In addition, the ILC1s, ILC2s, and ILC3s in the intestinal single-cell suspensions were shown to express T-bet, GATA3, and RORγt, respectively, in line with acknowledged definition of the main three ILC subsets,2 and consistent with our IHC set-up. Flow-cytometry analysis further revealed differential composition of the ILC subsets along the healthy gastrointestinal tract.9,10 In addition, shared and distinct changes in ILC composition have been reported for the intestinal compartments of patients with either established or newly diagnosed inflammatory bowel diseases.9 Unfortunately, these studies lack information about the anatomical localization of these cells in situ. In this case, application of our multiplex IHC approach can mean added value.
Highly sophisticated techniques like single-cell RNA sequencing and mass cytometry revealed heterogeneity in phenotype and frequency of the three human ILC subsets, indicating that each of the ILC subsets can be further subdivided into distinct subpopulations, defined by differentially expressed molecules.11,12 Our virtual multiplex IHC method could be of use to visualize and distinguish these newly discovered ILC subpopulations in situ and determine if they may have a particular spatial distribution.
Literature on the localization of human ILCs in situ is limited and only focused on a single ILC subset. In two studies on skin ILC2s, intravital multiphoton microscopy13 and immunofluorescence microscopy14 was employed for visualization. The ILC subset of interest was typically defined by non-specific identifying markers in combination with excluding marker CD3 only, hence not excluding possible presence of CD3− non-ILCs. Nevertheless, these studies demonstrated that ILC2s accumulate in the papillary dermis of patients with atopic dermatitis, and moreover, reside in close proximity with mast cells13 and basophils,14 suggesting a functional relationship in vivo. In another study, multi-color IHC (comparable with our true-color quadruple IHC method) was applied to show a spatial relationship between GATA-3+CD25+Lin−ILC2s and eosinophils in human nasal polyps with eosinophilia.15 Results of these studies on ILC2s in situ are of keen interest since they revealed intimate anatomical relationship of ILC2s with several other key mediators of type-2 immune responses, and at the same time also underline the relevance of multiplex IHC to investigate ILCs. By applying this in situ technique we noticed the previously unknown localization of ILC1s inside the lumina of blood vessels. This observation may be explained, at least in part, by the finding that T-bet is a master regulator for lymphocyte binding to P-selectin on endothelium.16
In summary, we described two quadruple IHC protocols to detect ILC subsets in FFPE human tissue and showed for the first-time the simultaneous presence and distribution of the three main ILC subsets within a single tissue section. Our universal multiplex IHC protocols should pave the way for a more detailed exploration of tissue-specific distribution of ILC subsets, as well as determination of anatomical association with other cell types. Consequently, this approach may improve our understanding of the functional interaction of distinct ILC subsets with their local environment during homeostasis and under particular pathologic conditions.
Supplemental Material
Supplemental material, 2019-00148R1_Production_Supplemental_Data_online_supp for Comparison of Two Different Immunohistochemical Quadruple Staining Approaches to Identify Innate Lymphoid Cells in Formalin-fixed Paraffin-embedded Human Tissue by Onno J. de Boer, Gabrielle Krebbers, Claire Mackaaij, Sandrine Florquin, Menno A. de Rie, Allard C. van der Wal and Marcel B.M. Teunissen in Journal of Histochemistry & Cytochemistry
Acknowledgments
The authors thank Daniek Kapteyn and Cindy Poelen (Amsterdam University Medical Centers, location AMC) for technical support.
Footnotes
Competing Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: MBMT and OJDB designed the study and drafted the manuscript; CM, GK, MBMT, and OJDB designed the experiments; GK performed the immunohistochemistry; ACVDW, GK, MADR, MBMT, OJDB, and SF analyzed and interpreted the data; GK, MADR, SF, and ACVDW participated in manuscript revision. All authors have read and approved the final manuscript.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Contributor Information
Onno J. de Boer, Department of Pathology, Amsterdam University Medical Centers, University of Amsterdam, Amsterdam, The Netherlands.
Gabrielle Krebbers, Department of Dermatology, Amsterdam University Medical Centers, University of Amsterdam, Amsterdam, The Netherlands.
Claire Mackaaij, Department of Pathology, Amsterdam University Medical Centers, University of Amsterdam, Amsterdam, The Netherlands.
Sandrine Florquin, Department of Pathology, Amsterdam University Medical Centers, University of Amsterdam, Amsterdam, The Netherlands.
Menno A. de Rie, Department of Dermatology, Amsterdam University Medical Centers, University of Amsterdam, Amsterdam, The Netherlands
Allard C. van der Wal, Department of Pathology, Amsterdam University Medical Centers, University of Amsterdam, Amsterdam, The Netherlands
Marcel B.M. Teunissen, Department of Dermatology, Amsterdam University Medical Centers, University of Amsterdam, Amsterdam, The Netherlands
Literature Cited
- 1. Artis D, Spits H. The biology of innate lymphoid cells. Nature. 2015;517(7534):293–301. [DOI] [PubMed] [Google Scholar]
- 2. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, Powrie F, Vivier E. Innate lymphoid cells: a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13(2):145–9. [DOI] [PubMed] [Google Scholar]
- 3. Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, Hreggvidsdottir HS, Heinsbroek SE, Legrand N, Buskens CJ, Bemelman WA, Mjösberg JM, Spits H. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol. 2013;14(3):221–9. [DOI] [PubMed] [Google Scholar]
- 4. Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, Hepworth MR, Van Voorhees AS, Comeau MR, Artis D. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med. 2013;5(170):170ra16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Teunissen MBM, Munneke JM, Bernink JH, Spuls PI, Res PCM, te Velde A, Cheuk S, Brouwer MWD, Menting SP, Eidsmo L, Spits H, Hazenberg MD, Mjösberg J. Composition of innate lymphoid cell subsets in the human skin: enrichment of NCR(+) ILC3 in lesional skin and blood of psoriasis patients. J Invest Dermatol. 2014;134(9):2351–60. [DOI] [PubMed] [Google Scholar]
- 6. Pertiwi KR, van der Wal AC, Pabittei DR, Mackaaij C, van Leeuwen MB, Li X, de Boer OJ. Neutrophil extracellular traps participate in all different types of thrombotic and haemorrhagic complications of coronary atherosclerosis. Thromb Haemost. 2018;118(6):1078–87. [DOI] [PubMed] [Google Scholar]
- 7. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van der Loos CM, Teeling P. A generally applicable sequential alkaline phosphatase immunohistochemical double staining. J Histotechnol. 2008;31(3):119–27. [Google Scholar]
- 9. Forkel M, van Tol S, Höög C, Michaëlsson J, Almer S, Mjösberg J. Distinct alterations in the composition of mucosal innate lymphoid cells in newly diagnosed and established Crohn’s disease and ulcerative colitis. J Crohns Colitis. 2019;13(1):67–78. [DOI] [PubMed] [Google Scholar]
- 10. Krämer B, Goeser F, Lutz P, Glässner A, Boesecke C, Schwarze-Zander C, Kaczmarek D, Nischalke HD, Branchi V, Manekeller S, Hüneburg R, van Bremen T, Weismüller T, Strassburg CP, Rockstroh JK, Spengler U, Nattermann J. Compartment-specific distribution of human intestinal innate lymphoid cells is altered in HIV patients under effective therapy. PLoS Pathog. 2017;13(5):e1006373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Björklund ÅK, Forkel M, Picelli S, Konya V, Theorell J, Friberg D, Sandberg R, Mjösberg J. The heterogeneity of human CD127+ innate lymphoid cells revealed by single-cell RNA sequencing. Nat Immunol. 2016;17(4):451–60. [DOI] [PubMed] [Google Scholar]
- 12. Simoni Y, Fehlings M, Kløverpris HN, McGovern N, Koo SL, Loh CY, Lim S, Kurioka A, Fergusson JR, Tang CL, Kam MH, Dennis K, Lim TKH, Fui ACY, Hoong CW, Chan JKY, Curotto de Lafaille M, Narayanan S, Baig S, Shabeer M, Toh SES, Tan HKK, Anicete R, Tan EH, Takano A, Klenerman P, Leslie A, Tan DSW, Tan IB, Ginhoux F, Newell EW. Human innate lymphoid cell subsets possess tissue-type based heterogeneity in phenotype and frequency. Immunity. 2017;46(1):148–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roediger B, Kyle R, Yip KH, Sumaria N, Guy TV, Kim BS, Mitchell AJ, Tay SS, Jain R, Forbes-Blom E, Chen X, Tong PL, Bolton HA, Artis D, Paul WE, Fazekas de, St Groth B, Grimbaldeston MA, Le Gros G, Weninger W. Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nat Immunol. 2013;14(6):564–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim BS, Wang K, Siracusa MC, Saenz SA, Brestoff JR, Monticelli LA, Noti M, Tait Wojno ED, Fung TC, Kubo M, Artis D. Basophils promote innate lymphoid cell responses in inflamed skin. J Immunol. 2014;193(7):3717–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, van Drunen CM, Lutter R, Jonkers RE, Hombrink P, Bruchard M, Villaudy J, Munneke JM, Fokkens W, Erjefält JS, Spits H, Ros XR. IL-1β, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat Immunol. 2016;17(6):636–45. [DOI] [PubMed] [Google Scholar]
- 16. Lord GM, Rao RM, Choe H, Sullivan BM, Lichtman AH, Luscinskas FW, Glimcher LH. T-bet is required for optimal proinflammatory CD4+ T-cell trafficking. Blood. 2005;106(10):3432–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, 2019-00148R1_Production_Supplemental_Data_online_supp for Comparison of Two Different Immunohistochemical Quadruple Staining Approaches to Identify Innate Lymphoid Cells in Formalin-fixed Paraffin-embedded Human Tissue by Onno J. de Boer, Gabrielle Krebbers, Claire Mackaaij, Sandrine Florquin, Menno A. de Rie, Allard C. van der Wal and Marcel B.M. Teunissen in Journal of Histochemistry & Cytochemistry