

Hepatitis C virus (HCV), like all positive-stranded RNA viruses, remodels intracellular membranes to establish protected sites of viral RNA replication (reviewed in den Boon et al1). These replication compartments, also called membranous webs, are thought to provide a platform for components required for virus replication and to shield the viral RNA from cytosolic nucleases and innate immune sensors. The mechanism for the formation of these structures is poorly characterized. Although initial evidence pointed to a role for HCV nonstructural protein (NS)4B in replication compartment formation,2 recent data suggest that NS5A plays a primary role. Expressing NS5A alone results in the formation of double membrane vesicles that are likely sites of viral replication.3 The integrity of the replication compartment is influenced by the interaction of NS5A with the cellular phosphatidylinositol-4-kinase-α(PI4KA).4–6 In this issue of Gastroenterology, 2 papers focus on NS5A-mediated replication complex formation. Wang et al7 investigate a role for the PI4KA effector oxysterol binding protein (OSBP) in the transport of cholesterol to aid replication complex formation. In the second paper, Madan et al8 present evidence that the mechanism of action for cyclophilin antagonists, which target NS5A and are under evaluation in clinical trials, is the inhibition of replication compartment formation.
The current treatment regimen for HCV includes ribavirin, pegylated interferon (IFN), and a viral protease inhibitor like telaprevir or boceprevir. At least 30 additional direct-acting antivirals (DAAs) are also under several stages of clinical trials in the hope of eliminating IFN from the treatment. Most of these DAAs either target the protease, polymerase, or NS5A.9 The recent approval of sofosbuvir, a nucleotide inhibitor of the polymerase, plus ribavirin as an IFN-free treatment regime is the first step toward this approach. In addition, several host proteins are also being investigated as potential drug targets due to their higher genetic barrier to resistance. Alisporivir, a cyclophilin A (CypA) inhibitor, has high efficacy, pangenotypic activity, and low viral breakthrough rates. CypA inhibitors are non-immunosuppressive derivatives of cyclosporine. Although cases of pancreatitis have halted alisporivir trials in combination with IFN and ribavirin, no such toxicity was observed in IFN-free regimens. Thus, alisporivir is still being evaluated in IFN-free HCV treatment regimens.10 CypA is an ubiquitous protein with peptidyl prolyl isomerase activity and this activity is thought to be important for proper folding of certain proteins. Drug resistance mutations against CypA inhibitors occur in NS5A, suggestive of an interaction between NS5A and CypA. NS5A-CypA interaction has been observed in vitro and the cis-trans isomerization of NS5A by CypA is thought to induce conformational change that might facilitate NS5A interaction with NS5B and RNA.11–15
NS2 was suggested to be an additional drug target for cyclophilin inhibitors. HCV genomes with NS2 were more sensitive to CypA inhibitors than subgenomic replicons that harbor only NS3-5B, suggesting a possible role for NS2 in replication via cyclophilin interaction.12,16 However, Madan et al8 suggest that viral replication fitness, as opposed to NS2, influences sensitivity to CypA inhibitors. Proteolytic processing at NS2/3 junction is a rate-limiting step determining HCV replication fitness, thus leading to an indirect contribution of NS2/3 to CypA inhibitor susceptibility. Interestingly, HCV replication fitness affects some inhibitor classes, but not others. The nucleoside analog 2′-C-methyl-cytidine and the NS5A DAA BMS-553 were also dependent on replication fitness, but not the protease inhibitor telaprevir or the PI4KA inhibitor AL-9. The reasons for this differential influence of HCV replication fitness are unclear.
The authors then show that CypA inhibitors block the establishment of new replication compartments, but have little effect on previously established replication compartments. Indeed, when they isolated crude replication compartments and added the CypA inhibitor in vitro, viral RNA synthesis was unaffected. The authors next studied the effect of CypA inhibitors on double membrane vesicle formation using electron microscopy. Using an HCV protein expression system that forms membranous webs independent of replication, the authors show that CypA inhibitors dramatically reduce the number of double membrane vesicles formed in the context of wild-type NS5A, but not in the context of a CypA inhibitor-resistant mutant of NS5A. These data suggest that CypA-dependent modification of NS5A is required for the biogenesis of HCV replication compartments.
In addition to NS5A-CypA interaction, NS5A-PI4KA interaction also influences HCV replication compartments. HCV infection stimulates the accumulation of the PI4KA product, phosphatidylinositol-4-phosphate (PI4P).4–6,17,18 Inhibition of PI4KA with small interfering RNAs or inhibitors, or mutating NS5A to preclude its interaction with PI4KA, results in altered replication compartment morphology.4,19 One possibility is that PI4P or a further modified PI(4,5)P2 may be involved in binding cellular and/or viral proteins required for replication.20,21 Alternatively, NS5A-PI4KA interaction may affect HCV replication in a PI4P-independent fashion, such as regulation of NS5A phosphorylation.19
Wang et al7 investigated the role of the PI4P-binding protein, OSBP, in HCV replication. OSBP has been previously shown to be important for HCV virion secretion.22,23 In their study,7 Wang et al observed an additional role for OSBP in HCV replication: The recruitment of cholesterol to replication compartments. Nonvesicular transport of lipids within the cell occurs through lipid transfer proteins. Several of these proteins contain a pleckstrin homology (PH) domain that interacts with PI4P and FFAT (di-phenylalanine in an acidic tract) motif that interacts with vesicle-associated membrane protein-associated protein (VAP) and a lipid transfer domain that interacts with the lipid that needs to be transferred between membranes.24 Both PI4KA and VAP are required for optimal HCV replication, suggesting that nonvesicular lipid transport might occur in these compartments (reviewed in Bishe et al21). OSBP is a PH domain-containing lipid transfer protein that transports cholesterol across membranes. Wang et al7 show that OSBP is important for HCV replication using short hairpin RNA-mediated knockdown and pharmacologic degradation of OSBP. Co-localization of OSBP and NS5A is lost when PI4KA is silenced, suggesting that OSBP may be recruited to replication compartment via PI4P. Inhibition of OSBP caused clustering and reduction in the diameter of the membranous vesicles, suggesting that OSBP is also required for the integrity of replication compartments. Using fluorescent cholesterol analogs, the authors confirm that cholesterol is enriched in NS5A-positive structures and that this cholesterol trafficking requires both PI4KA and OSBP. Using rescue experiments with different mutants of OSBP, the authors also show that PH domain is important for co-localization with NS5A; however, all 3 domains—PH, FFAT, and the lipid transfer domain—are important for cholesterol transport, as expected. Drugs that effectively perturbed the OSBP-dependent delivery of cholesterol to sites of replication and HCV replication complex formation include the PI4KA inhibitor AL9 and the OSBP inhibitor OSW-1.
PI4P is also induced during replication of another genus of positive strand RNA viruses, the enteroviruses, although they use a different isoform of PI4K, PI4K-beta (PI4KB) to generate PI4P.18 Wang et al7 demonstrated that OSBP is also required for replication of the prototype enterovirus, poliovirus, but not for dengue virus replication, which does not induce PI4P. It was also recently published that OSBP is recruited by PI4KB and that it is required for unesterified cholesterol transport to poliovirus replication compartments.25 Minor enviroxime compounds that inhibit HCV and enterovirus replication have been recently shown to be acting via OSBP inhibition.26 Thus, OSBP could be a potential cellular drug target that can block both HCV and enterovirus replication. In addition to OSBP-mediated delivery, there may also be vesicle-mediated delivery of cholesterol to replication compartments. Indeed, enteroviruses coopt host endocytic machinery to traffic cholesterol to replication compartments.27 The same endocytic process is also required for efficient HCV replication, suggesting that it may play a secondary role in cholesterol transport to sites of HCV replication.28
It is likely that PI4KA and PI4P have other functions in addition to the recruitment of OSBP in HCV infection. Other cellular and viral proteins that bind PI4P or PI(4,5)P2 have yet to be investigated for a role in HCV replication complex formation. Furthermore, the NS5A-PI4KA interaction modulates the NS5A phosphorylation status, which is critical for viral replication.19 Indeed, a stronger correlation exists for PI4KA modulated NS5A hyperphosphorylation, as opposed to PI4P levels, with HCV replication efficiency.19 Thus, PI4KA may perform PI4P-independent functions in HCV replication. It is likely that a basal level of PI4P production at replication compartments is required for their fidelity, whereas PI4KA either directly or indirectly also regulates NS5A hyperphosphorylation. Further studies will help to delineate various functions of PI4KA in HCV replication.
Overall, both papers in this issue highlight the point that plus strand RNA replication complex formation is a fertile ground for identifying cellular drug targets (Figure 1). Strengths of host-targeted antivirals include their high resistance barrier and pangenotypic activity. Indeed, CypA inhibitors show very low viral breakthrough during treatment.10 However, high barrier to resistance is not a universal attribute of host-based therapies. Enterovirus resistance to enviroximes, which target PI4KB for enterovirus replication, develops very easily, such that a single point mutation eliminates the need for the kinase and its product PI4P altogether in the viral replication.29,30 Certain classes of DAAs might also impact viral replication compartment formation. NS5A DAAs have 2 antiviral properties. They inhibit an NS5A-dependent step in virion assembly and also inhibit RNA replication with kinetics similar to the CypA inhibitor.31 Further studies will determine whether these DAAs directly or indirectly manipulate NS5A interaction with cellular factors like PI4KA or OSBP.
Figure 1.
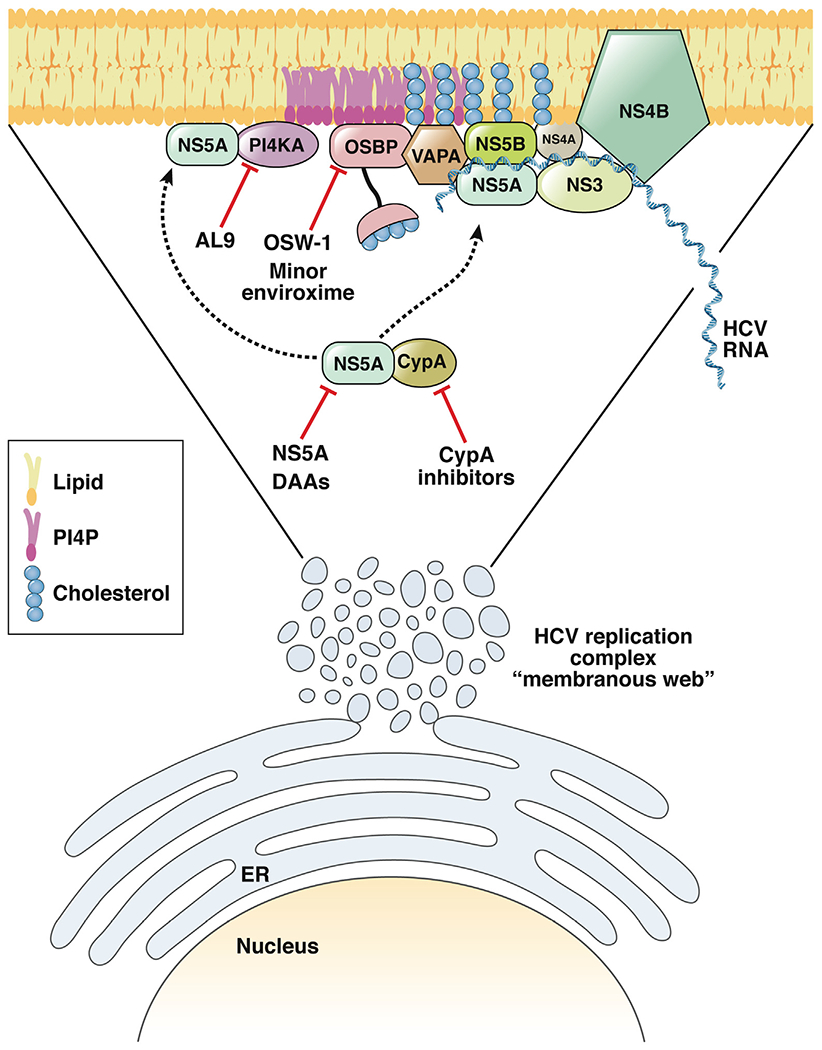
Hepatitis C virus (HCV) infection remodels the endoplasmic reticulum to create its replication compartment, termed the membranous web. Cyclophilin A (CypA) interaction with nonstructural protein (NS)5A is important for the biogenesis of these membranous webs.8 In addition, NS5A interacts with and activates phosphatidylinositol-4-kinase-alpha (PI4KA) to induce its product phosphatidylinositol-4-phosphate (PI4P), which recruits oxysterol binding protein (OSBP) to transfer cholesterol to these membranes.7 OSBP and vesicle-associated membrane protein-associated protein A (VAPA) likely recruit replication complex proteins, including NS5A and NS5B. CypA inhibitors perturb replication compartment formation and NS5A RNA binding. NS5A DAAs may also inhibit this process, either directly or indirectly. The PI4KA inhibitor AL-9 and OSBP inhibitors OSW1 and minor enviroxime-like compounds, could inhibit HCV replication by blocking cholesterol recruitment to the membranous web.
See “Oxysterol-binding protein is a phosphatidylinositol 4-kinase effector required for HCV replication membrane integrity and cholesterol trafficking,” by Wang H, Perry JW, Lauring AS, et al, on page 1373; and “Inhibition of HCV replication by cyclophilin antagonists is linked to replication fitness and occurs by inhibition of membranous web formation,” by Madan V, Paul D, Lohmann V, et al on page 1361.
Acknowledgments
The authors thank Ana Shulla and Tristan Jordan for critical reading of the manuscript.
Funding
G.R. is supported by NIAID (AI080703 and AI102236), the American Cancer Society (118676-RSG-10-059-01-MPC), and Susan and David Sherman. V.C. is supported by the American Heart Association.
Footnotes
Conflicts of interest
The authors disclose no conflicts.
References
- 1.den Boon JA, Ahlquist P. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu Rev Microbiol 2010;64:241–256. [DOI] [PubMed] [Google Scholar]
- 2.Egger D, Wolk B, Gosert R, et al. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol 2002;76:5974–5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Romero-Brey I, Merz A, Chiramel A, et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog 2012;8:e1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reiss S, Rebhan I, Backes P, et al. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 2011;9:32–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berger KL, Kelly SM, Jordan TX, et al. Hepatitis C virus stimulates the phosphatidylinositol 4-kinase III alpha-dependent phosphatidylinositol 4-phosphate production that is essential for its replication. J Virol 2011; 85:8870–8883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tai AW, Salloum S. The role of the phosphatidylinositol 4-kinase PI4KA in hepatitis C virus-induced host membrane rearrangement. PLoS One 2011;6:e26300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H, Perry JW, Lauring AS, et al. Oxysterol-binding protein is a phosphatidylinositol 4-kinase effector required for HCV replication membrane integrity and cholesterol trafficking. Gastroenterology 2014;146:1373–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Madan V, Paul D, Lohmann V, et al. Inhibition of HCV replication by cyclophilin antagonists is linked to replication fitness and occurs by inhibition of membranous web formation. Gastroenterology 2014;146:1361–1372. [DOI] [PubMed] [Google Scholar]
- 9.Manns MP, von Hahn T. Novel therapies for hepatitis C - one pill fits all? Nat Rev Drug Discov 2013;12:595–610. [DOI] [PubMed] [Google Scholar]
- 10.Gallay PA, Lin K. Profile of alisporivir and its potential in the treatment of hepatitis C. Drug Des Dev Ther 2013; 7:105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chatterji U, Bobardt M, Selvarajah S, et al. The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. J Biol Chem 2009;284:16998–17005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaul A, Stauffer S, Berger C, et al. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog 2009;5:e1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Z, Yang F, Robotham JM, et al. Critical role of cyclophilin A and its prolyl-peptidyl isomerase activity in the structure and function of the hepatitis C virus replication complex. J Virol 2009;83:6554–6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coelmont L, Hanoulle X, Chatterji U, et al. DEB025 (Alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS One 2010;5:e13687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foster TL, Gallay P, Stonehouse NJ, et al. Cyclophilin A interacts with domain II of hepatitis C virus NS5A and stimulates RNA binding in an isomerase-dependent manner. J Virol 2011;85:7460–7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ciesek S, Steinmann E, Wedemeyer H, et al. Cyclosporine A inhibits hepatitis C virus nonstructural protein 2 through cyclophilin A. Hepatology 2009;50: 1638–1645. [DOI] [PubMed] [Google Scholar]
- 17.Bianco A, Reghellin V, Donnici L, et al. Metabolism of phosphatidylinositol 4-kinase III alpha-dependent PI4P Is subverted by HCV and is targeted by a 4-anilino quinazoline with antiviral activity. PLoS Pathog 2012;8: e1002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu NY, Ilnytska O, Belov G, et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010;141:799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reiss S, Harak C, Romero-Brey I, et al. The lipid kinase phosphatidylinositol-4 kinase III alpha regulates the phosphorylation status of hepatitis C virus NS5A. PLoS Pathog 2013;9:e1003359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berger KL, Randall G. Potential roles for cellular cofactors in hepatitis C virus replication complex formation. Commun Integr Biol 2009;2:471–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bishe B, Syed G, Siddiqui A. Phosphoinositides in the hepatitis C virus life cycle. Viruses 2012;4:2340–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amako Y, Sarkeshik A, Hotta H, et al. Role of oxysterol binding protein in hepatitis C virus infection. J Virol 2009; 83:9237–9246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amako Y, Syed GH, Siddiqui A. Protein kinase D negatively regulates hepatitis C virus secretion through phosphorylation of oxysterol-binding protein and ceramide transfer protein. J Biol Chem 2011;286:11265–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lev S Non-vesicular lipid transport by lipid-transfer proteins and beyond. Nat Rev Mol Cell Biol 2010; 11:739–750. [DOI] [PubMed] [Google Scholar]
- 25.Arita M Phosphatidylinositol-4 kinase III beta and oxysterol-binding protein accumulate unesterified cholesterol on poliovirus-induced membrane structure. Microbiol Immunol 2014. February 15 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 26.Arita M, Kojima H, Nagano T, et al. Oxysterol-binding protein family I is the target of minor enviroxime-like compounds. J Virol 2013;87:4252–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ilnytska O, Santiana M, Hsu NY, et al. Enteroviruses harness the cellular endocytic machinery to remodel the host cell cholesterol landscape for effective viral replication. Cell Host Microbe 2013;14:281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berger KL, Cooper JD, Heaton NS, et al. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc Natl Acad Sci U S A 2009;106:7577–7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van der Schaar HM, van der Linden L, Lanke KH, et al. Coxsackievirus mutants that can bypass host factor PI4KIIIbeta and the need for high levels of PI4P lipids for replication. Cell Res 2012;22:1576–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arita M, Kojima H, Nagano T, et al. Phosphatidylinositol 4-kinase III beta is a target of enviroxime-like compounds for antipoliovirus activity. J Virol 2011; 85:2364–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guedj J, Dahari H, Rong L, et al. Modeling shows that the NS5A inhibitor daclatasvir has two modes of action and yields a shorter estimate of the hepatitis C virus half-life. Proc Natl Acad Sci U S A 2013;110:3991–3996. [DOI] [PMC free article] [PubMed] [Google Scholar]