

Abstract
The Chinese hamster ovary (CHO) cells used to produce biopharmaceutical proteins are known to contain type‐C endogenous retrovirus (ERV) sequences in their genome and to release retroviral‐like particles. Although evidence for their infectivity is missing, this has raised safety concerns. As the genomic origin of these particles remained unclear, we characterized type‐C ERV elements at the genome, transcriptome, and viral particle RNA levels. We identified 173 type‐C ERV sequences clustering into three functionally conserved groups. Transcripts from one type‐C ERV group were full‐length, with intact open reading frames, and cognate viral genome RNA was loaded into retroviral‐like particles, suggesting that this ERV group may produce functional viruses. CRISPR‐Cas9 genome editing was used to disrupt the gag gene of the expressed type‐C ERV group. Comparison of CRISPR‐derived mutations at the DNA and RNA level led to the identification of a single ERV as the main source of the release of RNA‐loaded viral particles. Clones bearing a Gag loss‐of‐function mutation in this ERV showed a reduction of RNA‐containing viral particle release down to detection limits, without compromising cell growth or therapeutic protein production. Overall, our study provides a strategy to mitigate potential viral particle contaminations resulting from ERVs during biopharmaceutical manufacturing.
Keywords: adventitious agents, Chinese hamster ovary cells, endogenous retroviral elements, genome editing
Endogenous retroviruses in Chinese hamster ovary cells were characterized at the genome, transcriptome and viral particle level. Following CRISPR‐mediated mutagenesis, the authors identified a single endogenous retrovirus locus responsible for the budding of viral particles into the cell supernatant. Loss‐of‐function mutations in this particular locus reduced viral genome‐containing viral particles to detection limits, thus augmenting the safety profile of Chinese hamster ovary cells for biopharmaceutical production.
1. INTRODUCTION
Contamination of biopharmaceutical products by adventitious agents such as viruses can interrupt drug supply and thereby imperil patient safety. Although viral contaminations of biopharmaceuticals are rare, they still occur repeatedly (Berting, Farcet, & Kreil, 2010), and mitigating the risk of viral contaminations in therapeutic protein production remains a top priority.
Chinese hamster ovary (CHO) cells are the most widely used mammalian expression system for biopharmaceutical products. Among others, CHO cells became a preferred production host due to their high safety profile compared to other cell lines used for recombinant protein expression. For instance, it was shown that CHO cells possess reduced susceptibility to certain viral infections (Berting et al., 2010). This includes resistance to infections elicited by many humans as well as murine retroviruses, with some of the latter being known to infect other mammalian cells (Hartley & Rowe, 1976; Shepherd, Wilson, & Smith, 2003). In addition, CHO cells, unlike other rodent cells, appeared to be unable to produce infective retroviruses that could replicate in mammalian cells, notably in human cells (Dinowitz et al., 1992; Emanoil‐Ravier et al., 1991; Hojman, Emanoil‐Raivier, Lesser, & Périès, 1989; Shepherd et al., 2003). However, viral‐like particles (VLPs) have been detected both within CHO cells as well as budding off in the culture medium from which the secreted therapeutic protein is collected (KP Anderson, Low, Lie, Keller, & Dinowitz, 1991; Gould & Borisy, 1977; Heine, Kramarsky, Wendel, & Suskind, 1979; Lieber, Benveniste, Livingston, & Todaro, 1973; Manly, Givens, Taber, & Zeigel, 1978; Reuss, 1992). The presence of such VLPs thus raised safety and regulatory concerns. For instance, there is a risk that these VLPs could lead to a possible hamster to human virus transmission. Moreover, these VLPs can also interfere with the detection of other adventitious agents, as commonly encountered with next‐generation sequencing‐based detection methods.
These VLPs were detected independently by several laboratories, suggesting that they result from endogenous retroviruses (ERVs) that stably integrated into the Chinese hamster and/or CHO cell genome, rather than from an exogenous infection (Lie et al., 1994). CHO cells possess two classes of ERVs. The first one consists of intracisternal type‐A ERVs (IAP) related to retrotransposons or to the genus of the Alpharetrovirus, depending on the env gene presence, and they are considered to be a defective ERV class forming immature particles in the cisternae of the endoplasmic reticulum (Anderson et al., 1990). The budding type‐C ERVs mediating the release of VLPs by CHO cells are another class of ERV that is not fully characterized, but that mostly corresponds to the Gammaretrovirus genus (Dinowitz et al., 1992; Lie et al., 1994). Although type‐C ERV sequences remain incompletely characterized, previous studies estimated that approximately 100–300 type‐C ERV sequences may be present in the CHO genome (Dinowitz et al., 1992; S. Li et al., 2019). Some of them seemed to be full‐length and actively transcribed proviruses, such as the ML2G retrovirus that shows nearly 64% sequence identity to the Murine leukemia virus (MLV) family (Anderson et al., 1991; Lie et al., 1994). However, the previously described ML2G ERV sequences contain frameshift mutations in each of its gag, pol, and env genes, indicating that the ERV sequence at this locus cannot produce VLPs (Lie et al., 1994). In addition, CHO cell VLP was reported to contain viral genomic RNA sequences related to type‐C retroviruses, as would be expected of viral particles (VP; De Wit, Fautz, & Xu, 2000). Nevertheless, the ERV sequences responsible for the release of the VLPs and/or VPs by CHO cells have remained uncharacterized.
As of today, CHO cells are commonly believed to produce noninfective retroviral particles, as their infectivity could not be demonstrated. Furthermore, many ERVs do not bear the full‐length LTR‐gag‐pol‐env‐LTR sequences of proviruses, as they contain many crippling point mutations and/or deletions. Nevertheless, the risk that one or several of the numerous type‐C ERV proviruses in the CHO genome is or may become capable of producing infectious particles cannot be excluded. This may happen if epigenetically silenced ERVs would become expressed, as observed upon some chemical treatments (Tihon & Green, 1973), if dysfunctional ERVs may acquire gain‐of‐function mutations, or if ERVs may recombine or trans complement each other. Such genetic changes are more likely to occur in immortalized cell lines, such as CHO cells, which may have an overall increased genetic instability (Wurm, 2013). Notably, the close similarity of CHO type‐C ERVs to the MLV family, a retrovirus family known to cross the species barrier and to infect even primate cells (Donahue et al., 1992), further indicates that CHO VP may have the potential to become human pathogens, as seen for other retroviruses (Urnovitz & Murphy, 1996). Hence strategies to avoid potential viral contaminations originating from CHO cell endogenous sources are highly desirable.
A promising strategy to efficiently prevent CHO VP release would be to inactivate functional ERVs using CRISPR‐Cas9‐mediated mutagenesis. The programmable RNA‐guided CRISPR‐Cas9 nuclease system has already been employed to introduce DNA double‐strand breaks (DSBs) into proviral sequences in human and porcine cells (Kaminski et al., 2016; Yang et al., 2015). Imprecise DSB repair may lead to inactivating insertions and deletions (indels) within the viral sequences. In a seminal paper, it was demonstrated that the CRISPR‐Cas9 technology could be used to knock‐out all 62 genomic porcine ERV sequences upon the prolonged expression of the nuclease, resulting in a more than 1000‐fold reduction of ERV infectivity (Yang et al., 2015). Although successful, viral inactivation remains technically challenging, as the sheer number of ERV‐like sequences may lead to low editing efficiency, high cytotoxicity, and frequent genomic rearrangements (Niu et al., 2017; Semaan, Ivanusic, & Denner, 2015; Yang et al., 2015). Furthermore, the incomplete characterization of type‐C ERV sequences, as well as the absence of a clear link between known genomic type‐C ERV sequences and VPs, have hampered the establishment of a similar ERV inactivation strategy in CHO cells.
Here we sought to characterize in‐depth the budding type‐C ERV sequences of CHO‐K1 cells at the genome, transcriptome, and viral particle levels. We identified a group of transcribed type‐C ERV sequences yielding full‐length transcripts with open reading frames encoding the three viral proteins, suggesting that this ERV group encodes potentially functional retroviruses. Using transient CRISPR‐Cas9 genome editing, we mutated members of the expressed group 1 type‐C ERV sequences and showed that specific loss‐of‐function mutations within the gag gene of a single ERV suffice to decrease the release of functional viral RNA‐loaded particles to near detection limits. This indicated that a single ERV, termed ETC109F, is responsible for most type‐C viral particle release from CHO cells. Altogether, our study provides a novel strategy to further improve the safety profile of CHO cells, paving the way for the complete eradication of endogenous viral contaminations in CHO cell cultures.
2. MATERIALS AND METHODS
2.1. Cell culture
Suspension‐adapted Chinese hamster ovary (CHO‐K1) derived cells were maintained in serum‐free HyClone SFM4CHO medium supplemented with HyClone Cell boost 5 supplement (GE Healthcare), l‐glutamine (Gibco), HT supplement (Gibco) and antibiotic–antimycotic solution (Gibco). CHO cell viability was assessed by Erythrosin B dye staining (Sigma‐Aldrich), and viable cell density and cell size were quantified using the LUNA‐FL Dual Fluorescence Cell Counter (Logos Biosystems). The cells were cultivated in 50 ml TubeSpin bioreactor tubes (TPP, Switzerland) at 37°C, 5% CO2 in a humidified incubator with 180 rpm agitation rate and passaged every 3–4 days.
2.2. Plasmid construction
The mammalian codon‐optimized Streptococcus pyogenes Cas9 (SpCas9) nuclease expression plasmid JDS246 (Addgene plasmid #43861) was used to introduce site‐specific DSBs (Fu et al., 2013). The CRISPRseek R package (Zhu, Holmes, Aronin, & Brodsky, 2014, p. 20) was applied to design single guide RNA (sgRNA) sequences that target the myristoylation (Myr) or the CHO cell ERV‐specific PPYP budding motifs in the gag consensus sequence of group 1 type‐C ERVs. One Myr (Myr2)‐ and one PPYP (PPYP6)‐specific sgRNA sequences were selected as they mediate DSB cleavages no more than 25 bp apart from the target motif, and as they were predicted to have high sgRNA efficiency using the CRISPRseek (Zhu et al., 2014), sequence scan for CRISPR (H. Xu et al., 2015), and sgRNA scorer 1.0 (Chari, Mali, Moosburner, & Church, 2015) scoring tools (Table S1). Genome‐wide off‐target cleavage analysis for these sgRNA sequences was performed using the CRISPRseek R package using the CHO‐K1 cell genome sequence as reference. SgRNA oligonucleotides were designed using the Zinc Finger Targeter software support tool (Sander et al., 2010; Sander, Zaback, Joung, Voytas, & Dobbs, 2007), and annealed sgRNA oligonucleotides were subsequently cloned into the mammalian sgRNA expression vector MLM3636 (Addgene plasmid #43860), as previously described (Fu et al., 2013). As the Myr2 sgRNA sequence lacks a guanine (G) nucleotide at the 5′‐end, an additional, nonpairing G was appended to improve transcription from the sgRNA expression plasmid (Ran et al., 2013). All primers used in this study were purchased from Microsynth AG (Balgach, Switzerland) and are listed in Table S2.
2.3. Characterization of the CHO genome ERVs
The genomic Chinese hamster DNA, obtained from descendants of the colony originally used to isolate the CHO cells (Puck, 1957), was kindly provided by the Nathan Lewis lab (University of California, San Diego). Genomic DNA was similarly obtained from the CHO‐DG44 cell line (Urlaub, Käs, Carothers, & Chasin, 1983).
The CHO‐K1 genome assemblies were from published databases of the Chinese hamster genome assembly (NCBI CHO‐K1 reference genome; X. Xu et al., 2011) and from sequencing data performed on an RSII (Pacific Biosciences) using a total of 197 SMRT cells. PacBio raw subreads were extracted in FASTA format using DEXTRACTOR (Gene Myers; https://github.com/thegenemyers/DEXTRACTOR) and assembled in a diploid‐aware fashion with FALCON (Chin et al., 2016). Residual errors in the assembly were corrected using quiver and pbalign (smrtpipe v. 2.3). ERVs in the assembled genome was annotated using a combination of alignment to known ERV sequences (Altschul, Gish, Miller, Myers, & Lipman, 1990) and by the discovery of unknown more distant family members using a profile‐based approach (Schuepbach et al., 2013).
ERV sequence alignments were realized with MAFFT aligner version 7 (Nakamura, Yamada, Tomii, & Katoh, 2018), with standard parameters, a scoring matrix of 200PAM/k = 2, a gap open penalty of 2.55 and offer value of 0.123. From these alignments, ERV phylogenetic trees were made using Geneious Tree Builder version 11.1.5, using the genetic distance model of Tamurra and Nei (Tamura & Nei, 1993) and the method of neighbor‐joining based on sequence similarity.
The expressed ERVs integration loci (ETC109F, ETC506F, and ETC386F) were characterized by Sanger sequencing following targeted PCR amplification using primers listed in Table S2.
2.4. RNA sequence analysis of CHO cell RNA
The total cellular RNA of CHO‐K1 cells treated or not with the CRISPR‐Cas9 system targeting the group1 ERV was extracted using a NucleoSpin RNA kit (Macherey Nagel) and reverse‐transcribed into cDNA using oligo(dT)15 primers and the GoScript Reverse Transcription System (Promega), as described by the manufacturer. A total of 6 ng of cDNA was used for quantitative polymerase chain reaction (qPCR) experiments using the SYBR Green I Master kit for the Light Cycler 480 machine (Roche) using the qPCR ref GAPDH primer as a normalization control and the qPCR Type1 ERV specific LTR primer targeting expressed group 1 ERVs (Table S2).
The sequencing libraries were prepared following a polyA purification and the Illumina TruSeq stranded mRNA kit. The complementary DNA was sequenced using Illumina paired‐end technology, resulting in libraries of 36–50 millions of 2 × 150 bp paired reads. These libraries were mapped with BWA (H. Li & Durbin, 2009) to the entire CHO‐K1 transcriptome, as available from the chogenome.org website (version 2014), complemented with six prototypical ERV sequences representing the various ERV groups identified in the CHO‐K1 genome (Figure 1b). The expression level per transcript was computed as reads per kilobase values, normalizing the number of reads mapping to the specific transcript by the transcript length. Expression analysis of 11 housekeeping genes was also performed to ensure the reproducibility of the extraction and the library preparation for the Illumina sequencing.
Figure 1.
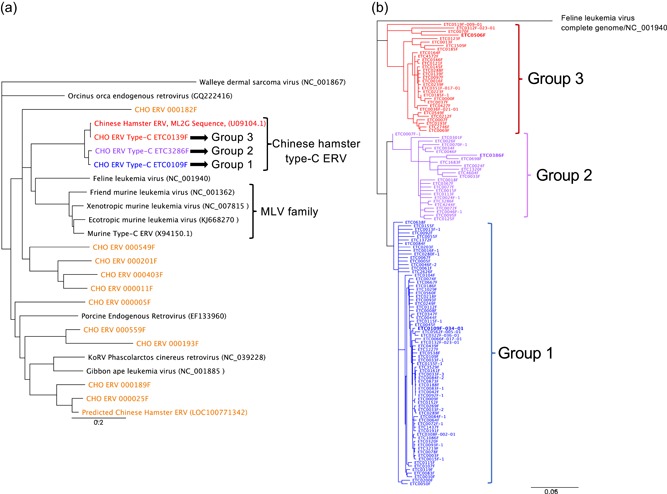
Phylogenetic analyses of Gammaretrovirus‐like endogenous retroviruses (ERV) DNA sequences within the Chinese hamster ovary genome. The ERV phylogenetic trees were constructed from sequence alignments using the neighbor‐joining method and corrected with the DNA evolution model of Tamura and Nei (1993). A total of 10,000 bootstraps were calculated for each tree, and the illustrations represent the consensus of these analyses. (a) The ERV phylogenic tree was based on the alignments of the pol sequences of ERVs and episomal Gammaretroviruses, and the Walleye dermal sarcoma virus was used as outgroup. The ERV sequence families identified in the CHO‐K1 genome are depicted by colors, and only one representative of each group is indicated in the phylogenetic tree. ERVs or Gammaretroviruses described in other species are shown in black letters. (b) The phylogenetic tree of the 131 full‐length type‐C ERV sequences detected in the CHO‐K1 genome was generated based on alignments of the LTR‐gag‐pol‐env–LTR sequences, and FeLV was used as outgroup. Colors represent the different type‐C ERV sequence groups, as in panel A. The ERVs shown in this study to be transcribed in CHO‐K1 cells are indicated by bold letters [Color figure can be viewed at wileyonlinelibrary.com]
2.5. VP RNA extraction and analysis
The viral genomic sequences within the VPs released in CHO cell culture supernatants were characterized as follows. Cell culture supernatants obtained following cell centrifugation were used freshly prepared or after only one freezing and thawing cycle. 450 μl of supernatant was loaded on a Corning Costar Spin‐X column centrifuge tube with a 0.22 μm membrane filter and centrifuged at 16,000g for 1 min. RNase‐free (12.5 units) DNase (Macherey‐Nagel) were added, and the extracts were incubated for 15 min at 37 °C to digest the possibly remaining residual DNA. The total VP RNA was extracted from the resulting extracts using the PureLink Viral RNA/DNA mini kit protocol (Thermo Fisher Scientific) according to the manufacturer's protocol, except that a second DNase treatment was applied to the viral RNA. The extracts were then processed differently depending on the use of the viral RNA for sequencing or for PCR analysis. In the case of sequencing, we used the Turbo DNA free kit from Thermo‐Fisher Scientific, following the manufacturer's instructions. In the case of reverse transcription and PCR or qPCR analysis, the QuantiTect reverse transcription kit from Qiagen was used according to the manufacturer's instructions. The qPCR reactions were performed as for the cellular mRNA but without normalization.
The cDNAs obtained from bulk VP RNAs extracted from cultures of parental CHO cells, from polyclonal cell populations treated with controls empty sgRNA vector or with the Myr and PPYP sgRNA vectors, and from ERV‐edited cell clones were sequenced using Illumina paired‐end technology, yielding libraries of 30–43 millions of 2 × 150 bp paired reads. Reads were first checked for sequence quality using publicly available algorithms (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) before mapping them to the 173 ERV sequences identified in the CHO genome using BWA (H. Li & Durbin, 2009). Expression levels per ERV were computed as read per kilobase values as described in the previous section.
2.6. Fluorescent in‐situ hybridization
To prepare metaphase chromosome spreads, cells were cultured in medium containing 0.3 μg/ml of KaryoMAX Colcemid solution (Life Technologies, REF15212‐012) for 4 hr, and then collected by centrifugation and resuspended in 75 mM hypotonic potassium chloride hypotonic solution (Sigma–Aldrich). Cells were fixed by washing gently in 10 ml of a 3:1 cold methanol/acetic acid mix (Fisher Scientific). The fixation was repeated four times and the cells were collected by centrifugation after each fixation step. Metaphase cells were spotted onto‐polylysine‐coated slides (Thermo Scientific, Menzel‐Gläser) and stored at 4°C in methanol until use.
DNA‐FISH was performed as previously described (Bantignies, Grimaud, Lavrov, Gabut, & Cavalli, 2003) and following these instructions: after RNaseA treatment (Qiagen, 200 μg/ml; 1 hr at 37°C), chromosomal DNA was denatured by incubating the slides in 70% formamide/2 × SSC at 75°C for 2 min, and dehydrated by washing in increasing ethanol concentrations (70%, 90%, and 100% vol:vol in water). The probe was denatured at 95°C for 10 min in hybridization buffer (2 × SSC, 10% dextran sulfate, 50% deionized‐formamide in DEPC‐H2O) and then applied to the slides. Hybridization was conducted overnight at 37°C for 16 hr. The slides were washed two times at 37°C in 2 × SSC and two times at 37°C in 0.5 × SSC and then mounted in an antifade solution containing 4′,6‐diamidino‐2‐phenylindole (DAPI; SouthernBiotech REF.0100–01).
For RNA‐FISH, exponentially growing cells were spotted onto polylysine‐coated coverslip placed in a 24‐well plate, fixed in 3,7% paraformaldehyde at room temperature for 10 min and permeabilized in 0,1% TritonX‐100/DEPC/PBS for 15 min. After rinsing with PBS, probes were applied to cells in hybridization buffer (2XSSC, 10% dextran sulfate, 10% deionized‐formamide in DEPC‐H2O). Hybridization was performed at 37°C overnight. After hybridization, the slides were washed in 2 × SSC, 2 × 20 min and in 0.5SSC, 2 × 20 min at room temperature, and they were mounted in an antifade solution containing DAPI (SouthernBiotech REF.0100–01).
2.7. Image acquisition and FISH signal analysis
Confocal microscopy was performed by using a ZEISS LSM800 microscope (40×; 1.3 N.A. oil Plan‐Apochromat objective). 200–300 metaphase chromosomes spread and interphase cells were scanned and analyzed. Karyotypes were generated automatically using “karyotypeAnalyzer”, an ImageJ based plugins developed by Selexis SA. Interphase cells automatic focus detection and scoring was performed using Find Maxima specific function in ImageJ (a Java‐based image processing program provided by the National Institutes of Health). Statistical analyses were performed using GraphPad Prism8. A Mann–Whitney U test was used to compare ERV‐ETC109F DNA‐FISH signal group and a control signal group.
Pictures of RNA FISH‐hybridized cells were acquired in three dimensions with a confocal microscope ZEISS LSM800 (40×; 1.3 N.A. oil Plan‐Apochromat objective). Image stacks were acquired with z‐steps of 0.5 μm and analyzed using the ImageJ software (NIH) and then Z‐projected using ImageJ.
2.8. Inactivation of ERV sequences in the CHO cell genome
CHO‐K1 cells were seeded at 300,000 cells/ml 1 day before transfection. On the day of transfection, 700,000 cells were electroporated with 3,700 ng of CRISPR‐Cas9 and 1,110 ng of Myr‐ or PPYP‐specific sgRNA expression plasmids using the Neon transfection system (Thermo Fisher Scientific), according to the manufacturer's instructions. CRISPR‐Cas9 and sgRNA expression plasmids were used at the equimolar ratio. A total of 200 ng of pCMV‐DsRed‐Express plasmid (Clonetech) was added to each transfection condition as transfection control. For CRISPR negative control experiments, the Myr‐ or PPYP‐specific sgRNA plasmids were substituted with the empty sgRNA expression vector (empty vector control).
To enrich for transfected and ERV‐mutated CHO cells, at least 70,000 cells were bulk‐sorted for the 30–40% of the cells displaying the highest dsRed fluorescence 48–72 hr after transfection using the MoFlo Astrios EQ or FACSAria II cell sorters (Beckman Colter). Cells were then briefly centrifuged to exchange medium and expanded. To isolate single‐cell clones, CRISPR‐treated cells were incubated at room temperature with DAPI viability dye (BD Biosciences) for 15 min, and single viable cells were sorted into 96‐well plates using the FACSAria Fusion cell sorter (Beckman Coulter). Cell clones were recovered in HyClone SFM4CHO medium supplemented with l‐glutamine, HT supplement, antibiotic–antimycotic solution and ClonaCell‐CHO ACF Supplement (Stemcell Technologies) to increase post‐sorting cell survival. Flow cytometry data were analyzed using FlowJo software v10.4.2. Cells were first gated using side scatter (SSC) versus forward scatter (FSC) to separate the intact cell population from debris, and then selected for single cells in the SSC‐H/SSC‐W and FSC‐H/FSC‐W plots. This single cell population was then sorted for dsRed + cells, using the maximal fluorescence of non‐transfected cells as a gating limit.
To identify ERV mutations among the transcribed ERV sequences, total RNA from CRISPR‐treated single cell‐sorted clones was isolated using the SV 96 Total RNA Isolation System (Promega) and reverse‐transcribed using GoScript Reverse Transcription Mix using the Oligo(dT) primer (Promega). PCR amplification of the CRISPR target regions was carried out using oneTaq DNA polymerase (New England BioLabs) with group 1 type‐C ERV‐specific primers (Table S2) and PCR products were analyzed by Sanger sequencing.
2.9. Deep amplicon sequencing of CRISPR‐targeted genomic regions
To assess the number of CRISPR‐induced ERV mutations at the genome level, DNA was extracted from clones bearing a mutation in the expressed group 1 type‐C ERV RNA sequence, as well as from control cells treated with the empty sgRNA vector and untreated cells, using the DNeasy Blood & Tissue Kit (Qiagen). The extracted genomic DNA was then used to prepare sequencing libraries in a two‐step PCR approach, as described in the Illumina “16S Metagenomic Sequencing Library Preparation” protocol except for the following modifications. Briefly, degenerate primers were designed using the Primer Design‐M tool (Yoon & Leitner, 2015) to amplify approximately 300 bp of the genomic region flanking the Myr2 and PPYP6 sgRNA target sites of all predicted type‐C ERV sequences (290 bp amplicon for Myr; 314 bp amplicon for PPYP; Table S2). Degenerate primers contained various 0–3 bp heterogeneity spacers to increase template complexity (Fadrosh et al., 2014), and Myr or PPYP primers were mixed at the predicted genomic frequency. In the first PCR round, 100 ng of isolated genomic DNA was used to PCR amplify the Myr and PPYP target loci using KAPA HiFi HotStart ReadyMix (2×; Kapa Biosystems) for 23 and 20 cycles, respectively. PCR amplicons were purified with AMPure XP beads (Beckman Coulter) using a 1:1 bead ratio. Amplicon quality and size were verified on an Agilent 2100 Bioanalyzer, and the DNA was quantified using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific). In the second PCR round, Illumina Nextera XT Index sequencing adapters were added to 15 ng of purified amplicons using eight PCR cycles. The final libraries were purified with AMPure XP beads (Beckman Coulter) using a 1:1.12 bead ratio. Library quality and size were verified using Fragment Analyzer (Advanced Analytical) and quantified using Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific). Libraries were pooled at the equimolar ratio, spiked with 25% PhiX and sequenced using 2 × 250 bp paired‐end sequencing on an Illumina Miseq System at the Genomic Technologies Facility of the University of Lausanne (Switzerland).
2.10. Analysis of deep amplicon sequencing reads
All sequenced Illumina paired‐end reads were trimmed using trimmomatic (v0.36; ILLUMINACLIP:config/daf.adapt.fa:2:30:10 LEADING:20 TRAILING:20 MINLEN:50), merged with FLASH2 (v2.2.00;‐‐max‐overlap 350) and converted to FASTA format. Spacer and primer sequences were translated into sequence profiles, flagged on all reads using pfsearch (pftools v3.0) and subsequently excised.
Weighted profiles for both Myr and PPYP CRISPR‐targeted regions were created as following: merged paired‐end reads from untreated cell control samples were cleaned (reads lengths exceeding Myr: > 300 bp, PPYP: > 400 bp removed) and clustered using cd‐hit (v4.6.8, ‐n 11 ‐c 0.97 ‐A 0.95). Clusters with more than 0.3% of members were retained, and a consensus sequence for each cluster was created based on 100 random sequences (mafft v7.310; ‐‐globalpair ‐‐maxiterate 1000 and cons from EMBOSS suite v6.6.0). Multiple sequence alignments were performed using these consensus sequences (mafft; ‐‐globalpair ‐‐maxiterate 1000) and translated into weighted profiles with pfmake (pftools suite 2.3.5.d; −0 ‐G 3 and a search‐like scoring matrix). Profiles were calibrated using scores from pfsearch against scrambled sequences (60 bp window).
The calibrated profiles were used to search both the parental cell control reads and CRISPR‐treated cell sample reads with pfsearch, generating a psa output. Modifications observed in comparison to the profile were determined and further analyzed in R (v3.4.2). Alignment differences also observed in parental control samples were removed in CRISPR‐treated sample reads, and only CRISPR‐unique events were kept. Events which appeared in less than 0.1% of total reads were removed and reads from identical events clustered using mafft (<1000 members: ‐‐globalpair ‐‐maxiterate 1000; >1000 members: ‐‐globalpair ‐‐retree 1 ‐‐maxiterate 0) to generate one consensus sequence per observed CRISPR mutation. For all identified CRISPR‐derived mutations, Illumina raw reads were clustered using the Jukes‐Cantor genetic distance model under the UPGMA tree building method to test for ERV locus‐specific genetic variations in the mutation flanking region.
2.11. Whole genome sequencing of ERV‐mutated CHO cell clone
To further identify the mutated ERV loci in the whole CHO genome, high‐molecular‐weight DNA was extracted from the sgRNA PPYP6‐treated E10 clone using the Blood & Cell culture DNA kit (Qiagen). DNA quality and quantity were verified using Fragment Analyzer (Advanced Analytical) and Qubit (Thermo Fisher Scientific), respectively. Five SMRT cells were used for sequencing on a PacBio Sequel system (Pacific Biosciences) at the Genomic Technologies Facility of the University of Lausanne (Switzerland). Each SMRT cell resulted in 5.9–7.2 Gbp in subreads and a N50 of approximately 18.5 kbp. This yielded in total 34.15 Gbp of sequenced DNA in subreads, corresponding to an approximately 14× theoretical coverage over the CHO genome. The consensus sequence describing the CRISPR‐derived mutation at the PPYP site in clone E10 and the PPYP parental control cluster consensus sequences were used as reference for this analysis. PacBio reads from the E10 clone were aligned against these reference sequences (minimap2 v.2.8; ‐x map‐pb–secondary = no; samtools v.1.8) and 2 subreads with the E10 PPYP mutation were identified (lengths of subreads: 4.3 kbp and 15 kbp). The latter contained sufficient genomic non‐ERV sequence to be successfully mapped onto the NCBI CHO‐K1 reference genome using minimap2.
2.12. Analysis of therapeutic protein expression from genome‐edited CHO cells
To assess the therapeutic protein production capacity of ERV‐mutated cells, polyclonal cell populations and cell clones previously treated with ERV‐specific or empty sgRNA expression plasmids were electroporated with a trastuzumab immunoglobulin G1 (IgG1) heavy and light chain expression vector bearing a puromycin resistance gene (Le Fourn, Girod, Buceta, Regamey, & Mermod, 2014). Untreated CHO‐K1 cells were also transfected with this IgG1 expression vector in parallel, as control. Two days after transfection, cells were transferred to culture medium containing 5 μg/ml puromycin and stable antibiotic‐resistant cells were selected for 3 weeks. Immunoglobulin titers from cultures of the stable trastuzumab‐expressing polyclonal cell populations were quantified during 10‐day fed‐batch cultures as previously described (Le Fourn et al., 2014). Briefly, cells were seeded at 0.3 × 106 cells/ml in 5 ml initial culture volume without puromycin selection. Cell cultures were fed with HyClone Cell boost 5 Supplement (GE Healthcare) at 16% of the initial culture volume on Days 0, 2, 3, and 6–8 of the cell batch culture. Cell density and viability were assessed at Days 3, 6, 8, and 10, whereas immunoglobulin secretion in the culture supernatant was measured on Days 6, 8, and 10 by sandwich ELISA.
3. RESULTS
3.1. Characterization of ERV elements in the CHO‐K1 cell genome
CHO cell genome databases were searched for sequences bearing homologies to the previously reported ML2G murine retroviral sequence present in CHO cell genomes (Lie et al., 1994). ERV sequence profiles were then built from the ERV sequences identified by sequence similarity, and these profiles were then used to screen the CHO‐K1 genome assembly to generate a database of ERV elements and to identify their genomic locus of integration. Short read Illumina sequencing alone did not allow to assemble and to locate correctly the ERVs within the CHO genome, because of their high sequence similarity and high copy number. Thus, we also relied on single‐molecule long‐read PacBio sequencing, and the residual remaining sequencing errors were algorithmically polished and manually curated. A total of 260 potential ERVs were thereby identified, sharing a minimum of 60% sequence homology with conserved motifs of the ML2G gag sequence. All these ERV sequences appeared to be related to Gammaretrovirus species and they could be clustered in several distinct phylogenetic groups. Representative sequences of each group were thus used to determine their phylogenetic positions within the Gammaretrovirus classification (Figure 1a). To compare Gammaretrovirus from different species, we relied on the pol sequence, as it is the most conserved gene among the retroviral families. This allowed the identification of three groups of CHO ERV sequences, termed groups 1, 2, and 3, which are most related to the sequence ML2G described by Lie et al. (1994). Interestingly, these ERVs are close relatives to infectious type‐C retroviral elements such as the Feline leukemia virus (FeLV) and the Murine leukemia virus (MLV). We thus concluded that these three groups encompass the CHO type‐C ERVs. Interestingly, other CHO ERVs were identified as relatives of sequences found in different mammalian species, such as the Porcine ERV, or of the Gibbon ape leukemia virus.
Among the 260 identified ERVs, 173 were classified as members of the three groups of CHO type‐C provirus sequences, among which 131 consisted of full‐length ERVs (Table 1). We then relied on the gag, pol, env and LTR sequences for the phylogenetic analysis of these complete and highly conserved CHO type‐C provirus sequences, to increase resolution (Figure 1b). This analysis indicated that group 1 ERVs were less diverse than groups 2 and 3. However, the overall high conservation of these sequences and the frequency of residual sequencing errors in the available CHO cell genomes hampered the direct identification of which of these group 1, 2 and 3 ERVs might be functional and potentially active. Although the identified number of type‐C proviruses is in line with previous estimations (Dinowitz et al., 1992), we noticed that some ERVs could not be successfully placed in available genome assemblies, suggesting that these 173 copies are likely an underestimate of the total reservoir of type‐C elements in CHO cells. Overall, group 1, 2, and 3 type‐C ERVs formed the predominant and functionally most conserved sequence clusters, with complete 5′LTR‐gag‐pol‐env‐3′LTR proviral structures for 131 elements, and they also shared most similarity to MLV elements, which are known to produce VPs infecting primate cell lines (Donahue et al., 1992). This revealed the ERVs of groups 1, 2, and 3 as the most likely candidates for viral particle formation.
Table 1.
Number of distinct type‐C ERV sequences detected at the genome, transcriptome and viral particle levels for CHO‐K1 cells and number of corresponding genomic loci
| Group 1 | Group 2 | Group 3 | Total | ||||
|---|---|---|---|---|---|---|---|
| Detection level | # sequences | # loci | # sequences | # loci | # sequences | # loci | # loci |
| Genomic DNA | 75 | 75 | 25 | 25 | 31 | 31 | 173 a |
| Cellular RNA | 1 | 1–30 | 1 | 1 | 1 | 1 | 3–32 |
| Particle RNA | 1 | 1–30 | 0 | 0 | 0 | 0 | 1–30 |
Among the 173 detected type‐C ERVs, 42 could not be phylogenetically linked to the different groups as they are incomplete.
3.2. Characterization of expressed ERV elements in CHO‐K1 cells
To complement the genomic CHO ERV characterization as well as to identify expressed sequences, the total cellular polyadenylated mRNA was sequenced using Illumina sequencing technology. Type‐C ERV mRNAs were among the top 70 most abundant transcripts in CHO cells, and the RNA levels of the env gene were three‐fold higher than those of gag and pol, as previously observed for other proviral elements (Tokuyama et al., 2018), while those of the unique 5′ (U5) part of the LTR were underrepresented (Figure 2a).
Figure 2.
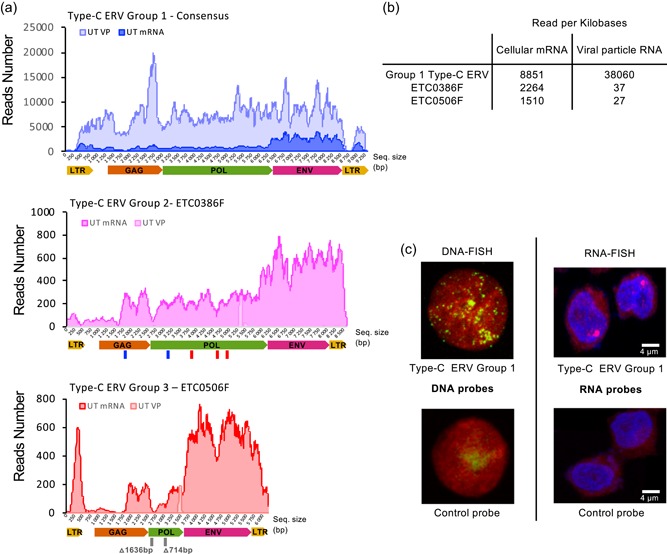
Characterization of expressed type‐C endogenous retrovirus (ERV) sequences in parental CHO‐K1 cells. (a) Illumina sequencing reads of the total cellular mRNA (UT mRNA, dark colors), or of the viral genomic RNA extracted from viral particles (UT VP, light colors) obtained from CHO‐K1 cell cultures, were mapped on group 1, 2, and 3 type‐C ERV sequences. Reads were mapped to a consensus of the possibly expressed group 1 sequences and on two distinct loci for group 2 (ETC386F) and group 3 (ETC506F) sequences. The ERV sequence sizes are represented by the x‐axis in base pairs. The lines under the schematic representation of group 2 and 3 ERVs loci indicate probable loss of function mutations occurring in these ERV sequences, blue for frameshift mutations, red for stop codon mutations and gray for deletions, with the deletion size indicated in base pairs. (b) Quantification of the average number of reads per kb of proviral sequence that mapped to the three different ERVs presented in panel A, from a total of 25 Gb and 208 Mb of mappable genomic sequences for cellular mRNA and VP RNA, respectively. (c) Confocal pictures of interphase CHO‐K1 cells subjected to DNA‐FISH (left pictures) or RNA‐FISH (right pictures), using a probe specifically targeting group 1 type‐C ERV (top pictures) or a nonspecific negative control probe (bottom pictures). Pictures were pseudocolored for visualization purposes, chromosomal DNA being represented in red and the DNA fluorescence in situ hybridization (FISH) signals of integrated retroviral sequences shown as green dots. For the RNA‐FISH, DNA staining is shown in blue, whereas ERV type‐C group 1 RNA signals are shown in red. The bright purple dot represents the nascent group 1 RNA signal at the transcription locus. Complete FISH analysis is presented in Figure S1 [Color figure can be viewed at wileyonlinelibrary.com]
A high proportion of the ERV‐derived reads could be mapped to approximately 30 group 1 ERV sequences. The high sequence identity, between 99% and 99.5% identity between distinct group 1 ERVs, and the residual sequencing error rate did not allow unambiguous attribution of these reads to specific group 1 loci (Table 1). The transcribed group 1 ERV sequences appeared to encode full‐length and in‐frame gag, pol, and env transcripts flanked by two LTRs and might thus produce functionally active viruses. Interestingly, the retrieved RNA sequencing reads mapping to group 1 did not contain any SNPs, as would be expected from the expression of multiple group 1 ERV sequences, suggesting that most if not all group 1 RNA‐derived reads originated from a single expressed ERV sequence.
Among the remaining mRNA reads corresponding to group 2 or to group 3 ERV sequences, 70% and 69% of the reads from each group mapped without mismatch uniquely to one type‐C ERV sequence, termed ETC386F (group 2) and ETC506F (group 3), respectively. This transcribed ETC386F and ETC506F sequences contained interrupted ORFs and/or missing coding sequences, as confirmed by PCR amplification and Sanger sequencing of these genomic loci (Figure 2a). While ETC386F had one frameshift in the gag and pol genes, as well as three‐stop codon mutations in pol, ETC506F possessed two deletions for a total of 2350 bp in the pol gene (Figure 2a). As these two sequences bear specific mutations that distinguish them from other members of their respective ERV sequence group, this suggested that ETC386F and ETC506F are the sole group 2 and 3 ERVs that are detectably transcribed, although this should not result in the production of functional Pol and/or Gag proteins.
The possible presence of viral genomes within VPs was assessed by extracting RNA from the particles released in the CHO cell culture supernatant, followed by their characterization by Illumina sequencing.
Analysis of the viral genome reads revealed that they mapped mostly to the group 1 ERV sequences, covering all sequences from the 5′ to the 3′ LTR, but not to the group 2 and 3 ERV sequences found to be transcribed in CHO cells (Figure 2a). This indicated that CHO cells are able to shed retroviral particles containing viral genomic RNA into the cell supernatant and that the genomic RNA present in the released VPs originated mostly from group 1 ERVs (Figure 2b). Moreover, the group 1 ERV sequences isolated from VPs were identical to the previously obtained group 1 mRNA sequences, implying that the transcribed group 1 ERV sequences are responsible for the release of VPs by CHO cells (Table 1).
To further characterize the functional group 1 type‐C ERV sequences, we designed group 1‐specific probes for fluorescent in‐situ hybridization (FISH) experiments. Using these probes, we detected approximately 80–110 group 1 ERV integration loci in the CHO‐K1 genome (Figure 2c; Figure S1A,B). However, when staining for group 1 nascent RNAs, we observed a unique highly transcribed site, supporting the notion that only a single group 1 ERV locus might be transcriptionally active (Figure 2c; Figure S1C).
Altogether, systematic ERV characterization at the genome, transcriptome and VP RNA levels allowed the identification of several groups 1 type‐C ERVs as strong candidates for the expression and release of retroviral particles by CHO‐K1 cells. Although the high sequence identity among the type‐C ERV sequences did not allow to determine the exact number of expressed type‐C ERV loci, these data implied that mutating few or possibly even just one group 1 ERV loci by genome editing might suffice to prevent ERV‐derived VP release.
3.3. Targeting group 1 ERV sequences by CRISPR‐Cas9 genome editing
To inhibit the release of potentially infective VPs from CHO cells, we aimed to disrupt conserved ERV sequence motifs critical for VP release. The Gag protein plays a pivotal role during retrovirus budding, and, consistently, its coding sequence is conserved among all type‐C ERVs in CHO cells (92.4% identity). However, the gag sequences are sufficiently different to distinguish group 1 from group 2 and group 3 type‐C ERV sequences, allowing to specifically target group 1 ERVs (96.4% identity between the related group 1 and 2 ERV gag sequences, vs. 98.2% identity for pol). Furthermore, two highly conserved gag sequences were shown to be required for viral budding, the myristoylation (Myr) and the PPxY motifs (Morikawa et al., 1996; Segura‐Morales et al., 2005), which were thus chosen as targets for CRISPR‐Cas9‐mediated mutagenesis. The N‐terminal Myr motif locates at a glycine residue at position 2 downstream of the translation initiation codon (Figure S2A). The myristoylation of Gag is generally considered as essential for targeting the protein to the host plasma membrane (Morikawa et al., 1996). Dominant‐negative mutations that directly interfere with Gag myristoylation, or indel mutations that block translation from the physiological start site or that create a loss‐of‐function gag transcript will perturb proper viral particle assembly at the plasma membrane, and hence block retroviral particle budding (Morikawa et al., 1996; Wapling, Srivastava, Shehu‐Xhilaga, & Tachedjian, 2007). In addition to Myr, the conserved proline‐rich PPxY motif also contributes to retrovirus budding, likely by interacting with the ESCRT machinery (Henzy, Gifford, Johnson, & Coffin, 2014), and its mutation strongly inhibits viral particle release (Segura‐Morales et al., 2005). The PPxY motif overlapped with a PPYP motif that is conserved in type‐C CHO ERVs, which is termed PPYP hereafter to refer to this CHO‐specific PPxY‐related budding motif.
Two sgRNAs were designed to target conserved sequences next to either the Myr motif (Myr2 sgRNA) or the PPYP motif (PPYP6 sgRNA) of potentially expressed group 1 gag sequences (Figure S2A). These two sgRNA were predicted to perfectly match over 50 type‐C ERV sequences, but to target up to 147 ERVs when allowing a maximum of three mismatches and noncanonical protospacer adjacent motif (PAM) sites (Table 1). Importantly, all these potential cleavage sites map specifically to various types of ERV sequences, while off‐target sites were not detected in the CHO genome. Although these sgRNA sequences contain a multitude of predicted target sites, we hypothesized that expressed ERVs might be preferentially cleaved by the CRISPR‐Cas9 nuclease, due to the more efficient cleavage of open chromatin (Daer, Cutts, Brafman, & Haynes, 2017).
To mutate the Gag budding motifs, CHO‐K1 cells were transiently transfected with CRISPR‐Cas9 and Myr or PPYP sgRNA expression vectors together with a dsRed transfection control plasmid. For CRISPR control samples, the gag‐specific sgRNA expression plasmids were replaced with a nontargeting empty sgRNA expression vector (empty vector), or cells were left untreated (UT). Transfected dsRed positive (dsRed+) cells were bulk‐sorted to enrich for cells containing mutations in the target motifs (Lee, Kallehauge, Pedersen, & Kildegaard, 2015). Following treatments with ERV‐specific sgRNAs, we noted an overall reduced frequency of transfected dsRed+ cells, as well as a significant drop in dsRed fluorescence intensity of positive cells when compared to control samples, suggesting that the most highly transfected cells might not survive because of a high frequency of genome cleavage (Figure S2B,C). High cell granularity is a marker of proapoptotic and/or dying cell populations (Gosselin et al., 2009). Consistently, granularity was also increased following the ERV‐specific CRISPR treatments, which inversely correlated with the frequency and expression intensity of dsRed + cells (Figure S2D,E). Altogether, this indicated that CRISPR‐mediated ERV cleavage impedes cell survival, especially in highly transfected cells, implying that the ERV‐specific sgRNAs mediated the efficient introduction of DSBs at multiple target sites in the CHO genome.
3.4. Isolation and characterization of ERV‐mutated CHO‐K1 clones
Single CHO cell clones were isolated from bulk‐sorted Myr2‐ or PPYP6‐edited cell pools and screened for those having mutations in the expressed group 1 ERV mRNA sequences. 18 out of 95 screened Myr2 sgRNA‐treated clones (18%) and 14 out of 181 screened PPYP6 sgRNA‐treated clones (8%) contained group 1 ERV mutations at the RNA level (Tables S3 and S4). Among the Myr2 sgRNA‐treated and mutated clones, the majority possessed an identical 1 bp insertion upstream of the ATG start codon (Tables S4 and S5), which likely resulted from staggered CRISPR‐Cas9 cleavage (Lemos et al., 2018). No clone treated with the PPYP6 sgRNA acquired a mutation disrupting the PPYP motif. Nevertheless, two Myr2‐ and 11 PPYP6‐derived clones contained mutations either blocking translation or frameshifting the gag transcripts, hence making them promising candidates for reduced VP release. The Sanger sequencing chromatogram of group 1 ERV mRNA of all clones consisted of a single clearly mutated sequence, without any other mixed sequence pattern, further supporting the earlier hypothesis that only a single group 1 ERV locus may be prominently transcribed.
To further investigate the CRISPR‐derived mutations at the genome level, we deep‐sequenced the Myr and PPYP flanking regions of type‐C ERVs in a subset of CHO cell clones. We selected two Myr2‐ and four PPYP6‐edited clones with Gag loss‐of‐function mutations in the group 1 type‐C ERV mRNA sequences (clones CO2 and D12 for Myr2; A02, E10, K03. and K14 for PPYP6) as well as one Myr2‐derived clone with a mutation in the 5′‐untranslated mRNA sequence (G09) and genotyped them, along with empty vector‐treated and untreated control cells (Table S5). To detect CRISPR‐derived mutations and distinguish them from sequence variations naturally occurring at each target site, we clustered the reads from untreated parental CHO cells and used these cluster consensus sequences to create diversity profiles (Figure 3a). This approach identified 34Myr and 28 PPYP clusters representing the natural sequence diversity within the Myr and PPYP flanking regions of CHO cell ERVs (Figure 3b,c; Figure S3). Despite the observed sequence diversity, the Myr and PPYP motifs themselves were highly conserved, as expected from their biological significance for viral budding. Overall, the identified clusters correlated well with the type‐C ERV groups previously characterized from the CHO genome reference assembly as well as with their predicted frequencies, suggesting that the captured sequencing reads represent well the ERV sequences previously characterized at the whole genome level (Figure 3b,c; Table S6). However, the high gag sequence similarity and short amplicon size prevented to fully distinguish group 1 and group 2 type‐C ERVs.
Figure 3.
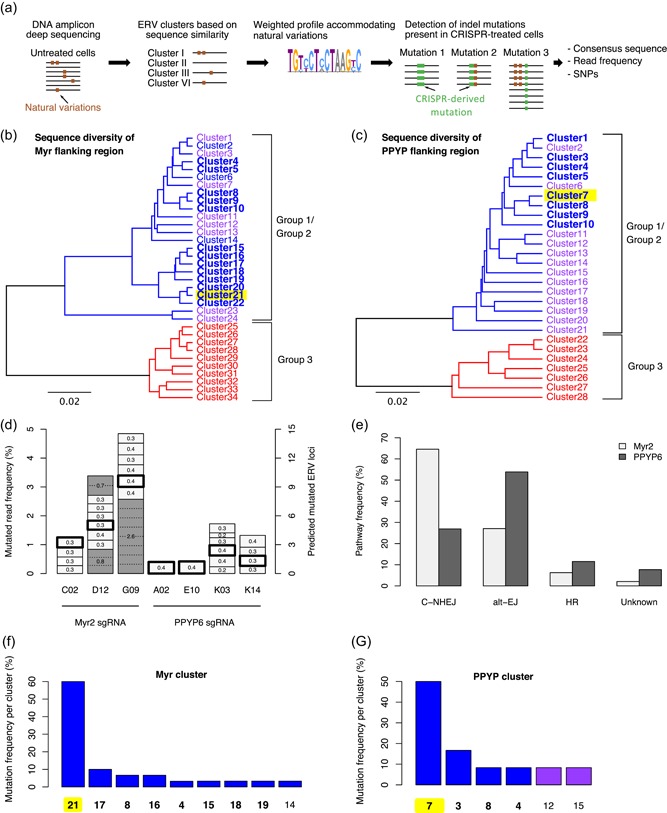
Assessment of the diversity of Myr and PPYP flanking sequences and CRISPR‐derived mutations by DNA deep sequencing. (a) Schematic illustration of the pipeline established to identify CRISPR‐derived indel mutations in type‐C endogenous retrovirus (ERV) sequences from targeted DNA amplicon sequencing. Type‐C ERV specific primers were used to amplify approximately 300 bp surrounding the Myr or PPYP CRISPR target sites of the gag genes from untreated and CRISPR‐treated cells, and amplicons were analyzed by Illumina sequencing. Untreated reads were clustered as based on 97% sequence similarity to establish weighted profiles. Profiles were used to distinguish between natural ERV variations and indel mutations in CRISPR‐treated cells. (b, c) Clusters of Myr (panel B) or PPYP (panel C) deep sequencing reads of untreated parental CHO‐K1 cells. Clusters consisting of group 1, group 2 and group 3 type‐C ERV sequences are indicated in blue, purple and red lettering, respectively, according to the phylogenetic groups depicted in Figure 1. Clustered sequences expected to be targeted by CRISPR‐Cas9, as they contain the Myr2 sgRNA or PPYP6 sgRNA recognition sites and an adjacent PAM sequence, are shown in bold. The cluster representing the expressed group 1 type‐C ERV sequence is highlighted in yellow. (d) Number of distinct mutations and their corresponding read frequencies in seven clones (C02, D12, G09, A02, E10, K03, K14) isolated from Myr2 or PPYP6 sgRNA‐treated polyclonal populations, as indicated. Mutations of the expressed group 1 ERV, as previously detected in the mRNA in each clone, are indicated with a bold frame. Gray shaded boxes represent mutations occurring at a frequency higher than 0.4% (left‐hand side axis), thus implying the occurrence of the same mutation in more than one ERV locus, where the distinct ERV loci are separated by dotted lines. The estimated total number of mutated ERV loci of each clone is indicated by the right‐hand side axis. (e) Frequency of Myr2 or PPYP6 sgRNA‐induced repair junctions compatible with C‐NHEJ, alt‐EJ or HR DSB repair mechanisms. Repair junctions incompatible with these three main DSB repair mechanisms are grouped as Unknown. A total of 67 DNA repair junctions (nMyr = 45, nPPYP = 22) obtained from both Sanger cDNA and Illumina deep DNA sequencing were analyzed. (f, g) Proportion of the various mutations detected in each of the ERV sequence clusters shown in panels B and C, respectively. Clusters containing the Myr2 or PPYP6 sgRNA recognition sites including an adjacent PAM site are shown in bold letters as in panels B and C, while clusters with sgRNA possessing mismatches at position 13 or 15 in the sgRNA recognition site mismatches are shown in normal letters. The cluster representing the expressed group 1 type‐C ERV sequence is highlighted in yellow, as for panels B and C [Color figure can be viewed at wileyonlinelibrary.com]
Using these sequence clusters and the corresponding diversity profiles, we found between 1 and 7 distinct CRISPR‐derived ERV mutations per clone at the genomic DNA level (depicted by the number of boxes of Figure 3d; Table S4). Some of the identified sequence alterations were identical to the mutations previously detected at the mRNA level (Figure 3d; bold frames). Other mutations, for instance, a 1 bp insertion occurring within all three genotyped Myr2‐treated clones, were absent in the PPYP6 clones, as expected from sgRNA‐specific repair outcomes (Bae, Kweon, Kim, & Kim, 2014). Consistently, control CHO cells treated with the empty sgRNA expression plasmid lacked additional mutations in the CRISPR target sites.
Typically, a given mutation was detected at a read frequency of approximately 0.2–0.4%, which must thus represent a single ERV locus in the CHO genome (Figure 3d). However, three Myr2‐derived mutations occurred at a read frequency well above 0.4%, as illustrated by the 1 bp insertion present in 2.6% of all G09 clone reads (Figure 3d; gray shaded boxes). This implied that the same mutation may have occurred more than once in distinct ERVs within the same clone. In support for this hypothesis, analysis of the mutation flanking regions allowed to distinguish five ERV groups among the G09 reads with the same 1 bp insertion, with one group having four‐times more reads than the others, indicating that this mutation should have occurred at eight distinct ERV loci in the G09 clone (Figure S4). Therefore, we concluded that each clone acquired between 1 and 14 ERV mutations following transient CRISPR transfection (Figure 3d). Characterization of the A02 and E10 clones having distinct mutations in a single mutated ERV at the DNA level, together with the finding that their mutation was identical to the single mutation detected at the cellular mRNA level, further substantiated that only one single group 1 type‐C ERV locus is transcribed, and that this single ERV is thus most likely responsible for the release of type‐C retroviral particles from CHO cells.
The majority of Myr2‐ and PPYP6‐derived repair junctions were compatible with classical nonhomologous end‐joining (C‐NHEJ) and alternative end‐joining (alt‐EJ) repair activities (Figure 3e). C‐NHEJ typically leads to small insertion and deletions, whereas alternative end‐joining (alt‐EJ) repair activities relying on microhomologies at the DSB site to anneal broken ends often results in larger and more complex mutations. Although alt‐EJ repair is considered to be a backup pathway in most mammalian cells, we detected between 25% and 55% alt‐EJ compatible junctions when targeting the gag gene, supporting our previous conclusions of intrinsically elevated alt‐EJ activities in CHO cells (Bosshard, Duroy, & Mermod, 2019; Kostyrko & Mermod, 2015; Kostyrko et al., 2017). Interestingly, approximately 10% of all analyzed repair junctions contained insertions templated from other ERV loci, or from the same ERV locus but using a distant sequence, while others manifested apparent duplications devoid of microhomologies, as required by alt‐EJ mechanisms. These latter junctions are consistent with homologous recombination (HR)‐related repair activities at Myr2‐ and PPYP6 target sites following CRISPR cleavage (Figure 3e).
Next, we assessed whether mutations occurred more frequently in some type‐C ERV clusters, indicating a preferential cleavage of certain ERV loci. The majority of mutations were observed in the Myr cluster 21 and in the PPYP cluster 7 (Figure 3f,g; yellow highlights). Interestingly, the consensus sequence of these clusters was identical to the actively transcribed group 1 type‐C ERV sequence present in VPs. Additional mutations were observed in other clusters, which contained a Myr2 or PPYP6 sgRNA recognition sites adjacent to a PAM sequence (Figure 3f,g; bold font). We also witnessed CRISPR cleavage in Myr and PPYP clusters containing a one base pair mismatch to the sgRNA target site (Figure 3f,g; normal font), supporting previous reports of CRISPR‐Cas9 tolerating small mismatches during target recognition (Lin et al., 2014). Altogether, these observations indicated that the expressed type‐C group 1 ERV was preferentially mutated and thus more likely cleaved by CRISPR‐Cas9.
We next assessed whether transient CRISPR/Cas9 expression and the resulting ERV editing in selected clones might be accompanied by gross chromosomal rearrangements. This was performed by determining the karyotypes of two representative clones, namely D12 and E10. The karyotype of these clones was similar to that of the untreated parental cells, with just one difference for a short acrocentric chromosome of clone E10, which appeared to have been subjected to a translocation (Figure S5). While retroviral integrations were dispersed throughout the CHO‐K1 genome, the presence of at least one ERV on this chromosome, as assessed by FISH analysis, supported the possibility that this rearrangement may have resulted from a Cas9 nuclease‐induced DSB. Nevertheless, we concluded that the editing of expressed ERV was not accompanied by major genomic alterations, despite the occurrence of a high number of CRISPR‐Cas9 target sites.
3.5. Identification of a unique VP‐producing ERV locus
The Sanger chromatograms, as well as the read frequencies of gag mutations observed during RNA and targeted DNA amplicon sequencing, corroborated the assumption that a single group 1 type‐C ERV locus is transcribed and may, therefore, mediate VP production by CHO cells. To further substantiate this assumption, the genome of the E10 clone was fully sequenced using a PacBio approach, so as to obtain sufficiently long reads for the unambiguous determination of ERV integration sites. This clone was selected as it appeared to contain only a single mutated ERV, allowing to correlate its unique mutation at the mRNA level with a potentially unique ERV genomic locus (Figure 3d). Analysis of the E10 clone genome sequence led to the identification of a single ERV locus bearing a mutation that was identical to the one detected at the mRNA level (Figure 4a). However, this ERV sequence was not present in the publicly available CHO or Chinese hamster genome scaffolds, which thus correspond to the allele lacking the ERV integration. The group 1 type‐C ERV integrated into this locus, named ETC109F, was found to lie between two moderately expressed CHO cell genes, and thus likely within permissive chromatin structures. The predicted ETC109F integration site was validated by PCR amplification and DNA Sanger sequencing using locus‐specific primers located outside of the ERV sequence, in parallel to a similar analysis of the unedited parental CHO cell line. Other deep‐sequenced mutated clones were analyzed similarly, as well as clones devoid of detectable mutations in the expressed group 1 ERV sequence (B01 for Myr2, B03 for PPYP6) as additional controls. All deep‐sequenced clones containing CRISPR‐derived mutations at the mRNA level possessed an identical mutation at the ETC109F genomic locus (Figure 4b). This indicated that the ETC109F genomic locus harbors the single expressed type‐C group 1 ERV element in CHO‐K1 cells.
Figure 4.
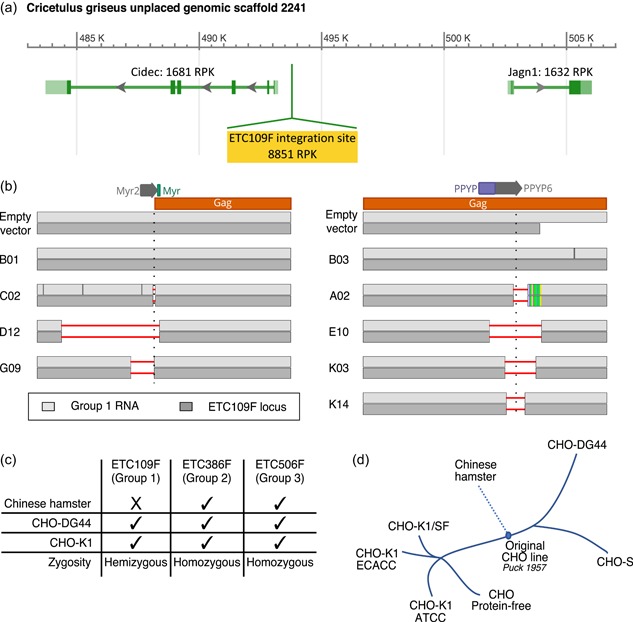
Identification of a unique functionally active group 1 type‐C endogenous retrovirus (ERV) locus and assessment of ERV conservation and zygosity in CHO cells and Chinese hamster genome. (a) Predicted group 1 type‐C ETC109F ERV integration site (highlighted in yellow) in the publicly available NCBI CHO‐K1 genome (NW_003613637.1 assembly). The genomic region surrounding the ERV integration site contains two annotated protein‐coding genes (Cidec, Jagn1) encoding the cell death‐inducing DFFA‐like effector protein involved in lipid metabolism and an endoplasmic reticulum protein involved in the early secretory pathway, respectively (Boztug et al., 2014; Puri et al., 2007). The predicted RNA expression levels for each gene in CHO‐K1 cells were estimated by RNA sequencing data and are expressed as Reads per Kilobase (RPK). (b) Sanger sequencing results of the Myr2 (left‐hand side) and PPYP6 (right‐hand side) sgRNA flanking regions of the indicated ERV‐mutated CHO cell clones. Sanger sequencing was performed on polymerase chain reaction (PCR) amplicons obtained from reverse‐transcribed total cellular mRNA using group 1‐specific primers (in light gray), or from genomic DNA using primers specific to the ETC109F genomic locus (in the dark gray). Clones C02, D12, G09, A02, E10, K03, K14 contain deletions in the functionally active group 1 type‐C ERV locus (horizontal red lines), unlike the B01 and B03 control clones as well as the empty vector‐treated control cells. The predicted Myr2 and PPYP6 sgRNA‐mediated DNA cleavage sites are indicated with a vertical dotted line. (c) Genomic sequences of the expressed ETC109F, ETC386F, and ETC506F and flanking sequences were PCR amplified from CHO‐K1, CHO‐DG44, and Chinese hamster genomic DNA using primers specific to the left and right DNA borders or the ERV extremities, and the amplified DNA was analyzed by gel electrophoresis. Summary of the detection (tick) or absence (cross) of stably integrated ERVs into their corresponding genomic locus as well as their zygosity in the indicated genomes for the three expressed group 1, 2, or 3 ERVs (d) Description of the history of CHO cell lines, adapted from Lewis et al. (2013). CHO, Chinese hamster ovary [Color figure can be viewed at wileyonlinelibrary.com]
Next, we assessed the conservation and zygosity of the three expressed ERVs in various CHO cell populations. We thus analyzed genomic DNA obtained from CHO‐K1 and CHO‐DG44 cells, as well as from the original Chinese hamster colony from which CHO cell lines were derived (Lewis et al., 2013), for which the three transcribed ERV loci were PCR amplified and sequenced. The expressed group 2 ETC386F and group 3 ETC506F ERV sequences were conserved and present in a homozygous state in all three tested Chinese hamster and CHO cell populations (Figure 4c). Interestingly, the expressed group 1 ETC109F ERV that encodes the viral genome of released VPs was observed in both the CHO‐K1 and CHO‐DG44 cell lines, but it was not detected in the Chinese hamster cell genome. Moreover, ETC109F integration in CHO‐K1 and CHO‐DG44 cells was found to be hemizygous, as the other allele was found to be devoid of a corresponding ERV integration. These observations were also validated from the PacBio sequencing of CHO‐K1, which yielded two alternative contig assemblies at the ETC109F integration locus, one with the ERV and one without it, thus validating the hemizygous state of this locus. This implies a possible recent appearance of ETC109F in CHO cells, for instance during an early cultivation step before the establishment of the original CHO cell line in 1957 (Figure 4d).
3.6. Quantification of VP production by ERV‐mutated CHO‐K1 cell clones
We next tested whether Gag loss‐of‐function mutations in ETC109F may lead to the anticipated inhibition of VP budding. Besides the previously characterized clones, we analyzed in parallel their corresponding bulk‐sorted polyclonal populations, as negative controls. To do so, the viral genomic RNA was extracted from the VPs released in the cell culture supernatants, followed by Illumina sequencing analysis and mapping of the reads to their corresponding ERV DNA sequences. Besides group 1 ERV sequences, we were unable to detect other endogenous viral RNA sequences in the cell culture supernatants that could be used as a normalization reference, when comparing edited to control cells. However, we could detect a significant amount of reads mapping to the 45S ribosomal RNA sequence, as the trace amounts of ribosomes shed by spontaneously lysing cells copurify with the VPs (Figure S6). Thus, the 45S ribosomal RNA was used as a reference for further analysis, as the variability to the mean between the 45S RNA values of the 13 sequenced samples was small (8%).
Interestingly, the number of reads mapping to the ETC109F ERV was drastically reduced for the clones having the largest deletions within the ERV gag sequences, namely clones D12 and E10, yielding three orders of magnitude reduction of the genome‐loaded VP release (Figures 4b, 5a,b). The release of viral RNA released within VPs was also strongly reduced for clones A02 and C02, whereas it was more moderate for other mutated clones. These sequencing results were also validated by q‐PCR assays of the LTR‐containing viral genome, providing consistent results (Figure 5c).
Figure 5.
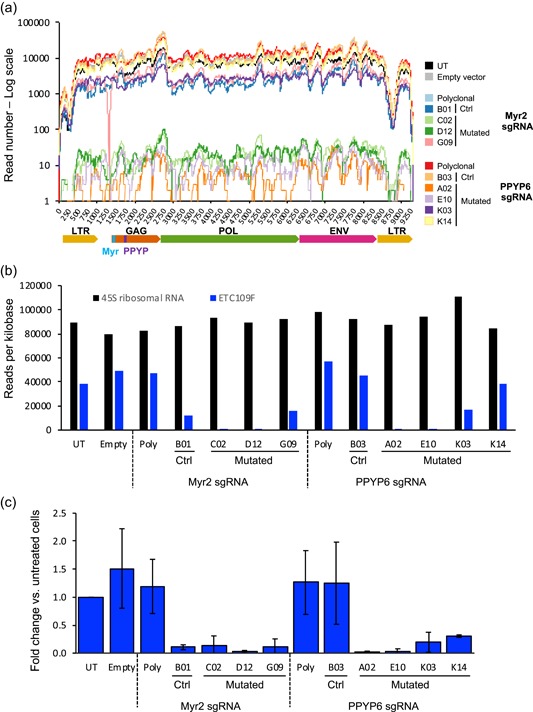
Assessment of viral RNA amounts released in VP by ERV‐mutated CHO cells. The retroviral RNA genomes were isolated from viral particles present within the supernatants of five‐day cultures of untreated cells (UT), empty sgRNA vector‐treated cells (Empty), bulk‐sorted polyclonal CRISPR‐treated cells (Poly), as well as clones containing mutations in the expressed type‐C group 1 ERV locus (C02, D12, G09, A02, E10, K03, and K14) or without a detected ERV mutation (B01, B03). (a) The RNA was processed for Illumina sequencing and the obtained reads were mapped onto the group 1 type‐C ERV locus ETC109F sequence. The ERV sequence coordinates are shown by the x‐axis in base pairs and the numbers of reads beginning at each ERV sequence position are shown by the log scale y‐axis. (b) The reads as analyzed in panel A were mapped to the sequences of the expressed group 1 ERV locus ETC109F (blue bars) and to the 45S ribosomal RNA (black bars) sequences of CHO cells used as a control. The y‐axis represents the number of reads per kilobases for each sequencing reaction. (c) Quantitative polymerase chain reaction (qPCR) analysis of the reverse‐transcribed total RNA isolated from VP released in cell culture supernatants. Reverse transcription and qPCR analysis were performed in triplicates from samples obtained from three independent CHO cell cultures. The genomic retroviral sequences were quantified using group 1 ERV LTR‐specific primers. Data were normalized to the number of analyzed cells and are represented as the average and standard deviation of the fold change relative to those of UT cells. CHO, Chinese hamster ovary; ERV, endogenous retrovirus [Color figure can be viewed at wileyonlinelibrary.com]
Interestingly, B01, one of the control clones that did not yield a detectable mutation in the expressed ERVs, was associated with an approximately 10‐fold reduction of viral particle release. A similar reduction was also observed for the G09 mutated clone, which has a point mutation in the ETC109F locus that is not expected to impact either transcription or translation. Since B01 and G09 were generated using the Myr2 sgRNA expected to cause possibly dominant‐negative mutations, we hypothesize that this effect may result from a dominant effect due to the mutation of the gag sequence from another ERV family member. However, we cannot rule out that this partial VP reduction phenotype may result from as yet unidentified genomic mutations that would inhibit viral‐particle release or from epigenetic effects on expressed ERV resulting from the DNA cleavage and repair processes.
Interestingly, CRISPR mutagenesis and/or the reduction of VP release did not affect the ETC109F, ETC386F, and ETC506F mRNA levels of the ERV‐mutated CHO cell clones (Figure S7). Overall, this indicated that mutations in the ETC109F sequence that remove the translation initiation sequences (D12, C02) or introduce a frameshift in the gag gene downstream of the PPYP motif (A02, E10) are sufficient to severely reduce the budding of complete VPs without affecting ERV transcription.
3.7. Characterization of edited CHO cell lines displaying reduced VP release
Having observed that CRISPR mutagenesis had efficiently prevented VP release by some cell clones, we next tested whether ERV inactivation may affect other CHO cell properties, such as cell growth or size, and therapeutic protein production. ERV‐edited clones were found to proliferate at comparable rates as polyclonal populations, empty vector‐treated and untreated cell controls, with a density reaching approximately 12.5×106 cells/ml after 5 days in culture (Figure S8A). Such a cell density concords with the expected CHO‐K1 doubling time of roughly 20 hr (Byrne et al., 2018). However, two Myr2‐edited clones (C02, D12) and one PPYP6‐mutated clone (K14) showed slightly modified cell cycle durations, and the maximal viable cell density of C02 was reduced, although this effect was not statistically significant. In addition, cell sizes appeared to be somewhat elevated in ERV‐edited cells, notably in the C02 clone, but they did not differ significantly when compared to the empty vector control cells (Figure S8B).
Finally, we assessed the capacity of ERV‐edited CHO cells to produce therapeutic proteins, a key property for the pharmaceutical use of CHO cells. We used the previously characterized ERV‐mutated cells to generate polyclonal populations stably expressing a humanized therapeutic immunoglobulin (IgG) and quantified the IgG secretion during ten‐days fed‐batch cultures. ERV‐edited clones and polyclonal populations expressing the IgG protein demonstrated cell growth and cell viability properties similar to those of untreated and empty vector‐treated control cells, except for the C02 clone, as observed without therapeutic protein expression (Figure 6a,b). IgG titers in the cell culture supernatants increased over the course of the fed‐batch experiment, as expected from the accumulation of the secreted IgG protein, reaching around 300–400 mg/l at the end of the fed‐batch for control cells and most ERV‐edited cell clones (Figure 6c). While the Myr2 sgRNA‐treated clone C02 secreted significantly fewer immunoglobulins, likely reflecting its reduced growth and increased cell size, ERV mutagenesis did not globally impair the ability of CHO cells to produce IgG proteins. Interestingly, two of the four clones generated with the PPYP6 sgRNA (E10 and K03) produced 50% more IgG relative to the empty vector control. Overall, this indicated that CHO clones that were exposed to multi‐locus ERV editing generally maintain normal CHO characteristics, whereas some clones with mutations in the PPYP region might have acquired a higher metabolic capacity to produce therapeutic proteins. As E10 and K03 have the largest gag sequence deletions of the four analyzed clones, this apparently augmented metabolism capacity suggests a possible correlation between particular PPYP deletions and the observed clonal fitness.
Figure 6.
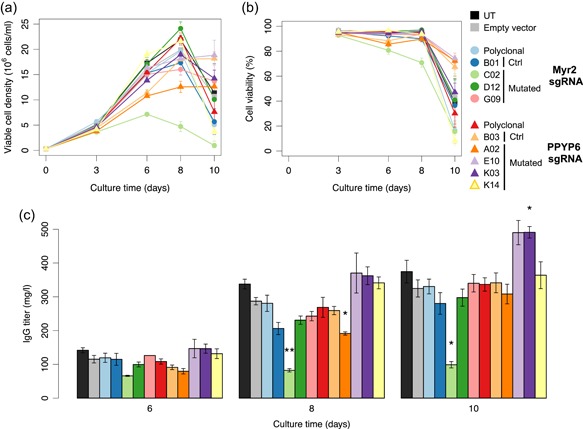
Analysis of cell growth and therapeutic immunoglobulin protein production by ERV‐mutated CHO cell clones. Untreated (UT) or empty sgRNA vector‐treated (Empty) control cells, bulk‐sorted polyclonal CRISPR‐treated cells (Poly), as well as cells from isolated clones containing mutations in the expressed ERV locus (C02, D12, G09, A02, E10, K03, and K14) or not (B01, B03) were stably transfected to express the trastuzumab immunoglobulin (IgG), and the stable polyclonal cell pools were assessed for cell density (a), cell viability (b) and IgG production (c) during 10‐days fed‐batch cultures. Statistical significance relative to the empty vector control was calculated using the two‐tailed unpaired Student's t‐test with Benjamini and Hochberg false discovery rate correction (n = 3 for all samples, except for A02 for which n = 2, error bars represent standard error of mean, *p < .05; **p < .01). CHO, Chinese hamster ovary; ERV, endogenous retrovirus [Color figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
CHO cells are a widely used expression host to produce therapeutic proteins, but also a recognized source of endogenous VLPs for more than 40 years (Anderson et al., 1991; Gould & Borisy, 1977; Heine et al., 1979; Manly et al., 1978). Although these particles were never shown to be infectious, their genomic origin and possible evolution have remained mostly unknown. Thus, potential safety concerns have persisted, and ample precautions must be taken when producing therapeutic proteins using such mammalian cell lines. Thus, the engineering of cells that would not release potentially functional VP together with the therapeutic protein, but without modifying their intrinsic protein production ability, should be advantageous. Moreover, the presence of incompletely characterized retroviral nucleic acids in CHO culture supernatants might lead to false positives when using next‐generation sequencing for the detection of viral adventitious agents. Therefore, further improvements in our knowledge of CHO cells endogenous viral elements may contribute to the establishment of next‐generation sequencing as a method of choice for adventitious agent detection in cell cultures.
The analysis of the Gammaretrovirus ERVs present in the CHO‐K1 genome revealed a wide diversity of viral sequences that may have integrated in the Chinese hamster genome during evolution. This diversity is coherent with the ERV diversity present in other rodent species such as mice, where different type of virus integration are associated with various levels of pseudogenization (Hayward, Grabherr, & Jern, 2013). This ERV sequence diversity and the identification of multiple LTR sequences corroborated the fact that the Chinese hamster species have faced multiple retrovirus integration periods during their evolutionary history. Among all identified CHO cell ERVs, group 1 of the type‐C ERVs appears to be most recent in terms of sequence conservation, and it was the sole evolutionary group identified to have full‐length ERV sequences bearing complete retroviral gag, pol, env genes, and thus to possibly produce functional viral proteins and particles.
The characterization of CHO endogenous retroviral elements at the genome, transcriptome, and viral particle levels indicated that CHO cells are able to release VP loaded with viral RNA genomes of the group 1 type‐C ERVs, while group 2 and 3 ERV sequences were below detection level in RNA‐loaded VPs. From these genomic and RNA analyses, we curtailed the number of possible ERV loci responsible for the expression and release of CHO viral particles to a group of up to 30 well‐conserved group 1 type‐C ERV sequences in the CHO genome. Whether these expressed ERV sequences are replication‐competent and produce retroviral particles that can infect CHO and/or other cells remains to be further characterized. Nevertheless, this possibility prompted us to generate ERV‐mutated CHO cells that would no longer release RNA‐loaded VPs, for safer therapeutic protein production.
The introduction and characterization of distinct mutations into defined ERV sequences allowed the identification of a single group 1 type‐C ERV sequence—ETC109F—as the origin of viral type‐C particle formation by CHO cells. Indeed, various types of ETC109F gag loss‐of‐function mutations strongly reduced VP release down to detection limits. This indicated that the other ERVs present in the CHO genome may be unable to complement the Gag loss‐of‐function and that ETC109F is the main source of VP release by these CHO cells.
The finding that the ETC109F ERV is present as a single allele in the CHO‐K1 and CHO‐DG44 genomes but not in cells obtained from the parental Chinese hamster strain suggests that the ERV integration at this locus occurred during or just after the isolation of the original CHO cell line by T. Puck and collaborators in the late 1950s. A recent occurrence of this event is further supported by the high degree of sequence conservation of this retroviral group, and also by the high expression level and ability to mediate VP release of ETC109F.
Interestingly, two clones that did not to contain a frameshift mutation or a large deletion in the ETC109F gag gene were also associated with a small decrease of VP production, although this ERV remained actively transcribed. This suggests that other as yet unidentified genomic mutations may have inhibited VP release, such as dominant‐negative mutations of other expressed but truncated ERVs. For instance, work in FeLV and HIV showed that nonmyristoylated Gag proteins can acquire dominant‐negative properties, leading to severe inhibition of VP release (Kawada, Goto, Haraguchi, Ono, & Morikawa, 2008; Manrique, Celma, González, & Affranchino, 2001). Further work will be required to evaluate the possibility that such ERV mutants might also inhibit the potential infection of CHO cells by other MLV‐related adventitious agents.
A common technical challenge for multi‐locus genome editing is the possible occurrence of extensive DNA damage elicited by the multiple Cas9‐induced DSBs, which usually activate p53 signaling and may cause cell death. The sgRNAs designed in this study were predicted to perfectly match roughly 60 distinct group 1 type‐C ERV loci in the CHO genome, and CRISPR‐Cas9 treated clones acquired up to 14 different mutated loci following a single transient transfection. This implies that CHO cells are able to handle the DNA damage response and repair of multiple DSBs, as might be expected from immortalized cell lines that encounter and tolerate high levels of endogenous DNA damage, in contrast to primary cells where a single DSB break might cause cell death (Ihry et al., 2018; O'Connor, 2015). This may explain why a recent study reporting the isolation of primary porcine cells containing mutations in up to 62 endogenous viral elements required the persistent expression of CRISPR‐Cas9 as well as antiapoptotic treatments to suppress cell death (Niu et al., 2017), unlike the transient CRISPR‐Cas9 expression used in the present study to inhibit CHO cell VP release.
Clones mutated in the expressed group 1 ETC109F sequence showed unchanged RNA levels for group 1, 2, and 3 ERVs, while being strongly impaired in releasing encapsulated viral RNA. In addition, ERV‐mutated clones did not consistently differ in cell growth, the cell size or therapeutic protein production compared to control samples. However, 2 of the 4 clones bearing deletions near the gag PPYP‐encoding motif yielded higher amounts of the therapeutic protein, raising the possibility that Gag expression might limit protein secretion by CHO cells. Clonal variability is a common phenomenon when isolating clones from polyclonal populations, or after subcloning individual cells from a monoclonal population (Lee et al., 2018; Pilbrough, Munro, & Gray, 2009), which was attributed to the acquisition of random mutations, stochastic protein level fluctuations and/or epigenetic effects (Aguirre et al., 2016; Pilbrough et al., 2009; Sigal et al., 2006). However, given the role of Gag in assembling at the cell membrane to release viral particles, it will be interesting to determine if specific mutations in this ERV protein might affect the secretion efficacy of endogenous cellular or therapeutic proteins. In this respect, the viral envelope gene is within the 100 most highly transcribed CHO cell genes. This together with the finding that the expressed ETC109F locus is present in CHO but not in primary Chinese hamster cells, suggests a possible evolutionary advantage elicited by high viral gene expression during the selection of the most efficient CHO cell line derivatives, such as the ability of retroviruses to counter the unfolded protein response (UPR; Yoshimura, Luo, Zhao, Gerard, & Hudson, 2008). Thus, the inactivation of the Gag protein, which associates with the cell plasma membrane and may thereby affect therapeutic protein secretion, without impairing the potential positive effect of high env expression, might explain the occurrence of engineered clones with increased ability to secrete a therapeutic protein. Further work will be needed to assess the interesting possibility that cell hosts engineered to be unable to release viral particles with the secreted therapeutic protein, and thus with potentially safer properties, might concomitantly acquire the capability to produce therapeutic proteins at higher levels.
CONFLICTS OF INTEREST
Some of the authors are employed by and/or are consultants of Selexis SA, a company generating therapeutic protein‐producing CHO cell lines. P.O.D. and S.B. university positions were funded in part by the Swiss government, the University of Lausanne and Selexis SA.
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENT
The authors want to thank Dr. Mojtaba Samoudi from the Nathan Lewis Lab (University of California, San Diego) for providing Chinese hamster DNA. The authors thank Alexander Kuhn for help with the sgRNA design and CRISPR off‐target analysis and Yves Dusserre for assistance with the vector constructions. We also thank the Genomic Technologies Facility at the University of Lausanne for support with library preparation and next‐generation sequencing, and the flow cytometry core facility at EPFL for assistance during cell sorting. This study was supported by the Swiss Government Commission for Technology and Innovation [grant number 17196.1 PFLS‐LS], Selexis SA and University of Lausanne. Funding for open access charge: University of Lausanne.
Duroy P‐O, Bosshard S, Schmid‐Siegert E, et al. Characterization and mutagenesis of Chinese hamster ovary cells endogenous retroviruses to inactivate viral particle release. Biotechnology and Bioengineering. 2020;117:466–485. 10.1002/bit.27200
REFERENCES
- Aguirre, A. J. , Meyers, R. M. , Weir, B. A. , Vazquez, F. , Zhang, C. Z. , Ben‐David, U. , & Hahn, W. C. (2016). Genomic copy number dictates a gene‐independent cell response to CRISPR/Cas9 targeting. Cancer Discovery, 6(8), 914–929. 10.1158/2159-8290.CD-16-0154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S. F. , Gish, W. , Miller, W. , Myers, E. W. , & Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215(3), 403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Anderson, K. P. , Lie, Y. S. , Low, M. A. , Williams, S. R. , Fennie, E. H. , Nguyen, T. P. , & Wurm, F. M. (1990). Presence and transcription of intracisternal A‐particle‐related sequences in CHO cells. Journal of Virology, 64(5), 2021–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, K. P. , Low, M.‐A. , Lie, Y. S. , Keller, G.‐A. , & Dinowitz, M. (1991). Endogenous origin of defective retroviruslike particles from a recombinant Chinese hamster ovary cell line. Virology, 181(1), 305–311. 10.1016/0042-6822(91)90496-x [DOI] [PubMed] [Google Scholar]
- Bae, S. , Kweon, J. , Kim, H. S. , & Kim, J. (2014). Microhomology‐based choice of Cas9 nuclease target sites. Nature Methods, 11(7), 705–706. 10.1038/nmeth.3015 [DOI] [PubMed] [Google Scholar]
- Bantignies, F. , Grimaud, C. , Lavrov, S. , Gabut, M. , & Cavalli, G. (2003). Inheritance of Polycomb‐dependent chromosomal interactions in Drosophila. Genes & Development, 17(19), 2406–2420. 10.1101/gad.269503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berting, A. , Farcet, M. R. , & Kreil, T. R. (2010). Virus susceptibility of Chinese hamster ovary (CHO) cells and detection of viral contaminations by adventitious agent testing. Biotechnology and Bioengineering, 106(4), 598–607. 10.1002/bit.22723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosshard, S. , Duroy, P. ‐O. , & Mermod, N. (2019). A role for alternative end‐joining factors in homologous recombination and genome editing in Chinese hamster ovary cells. DNA Repair, 82, 102691 10.1016/j.dnarep.2019.102691 [DOI] [PubMed] [Google Scholar]
- Boztug, K. , Järvinen, P. M. , Salzer, E. , Racek, T. , Mönch, S. , Garncarz, W. , & Klein, C. (2014). JAGN1 deficiency causes aberrant myeloid cell homeostasis and congenital neutropenia. Nature Genetics, 46(9), 1021–1027. 10.1038/ng.3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne, G. , O'Rourke, S. M. , Alexander, D. L. , Yu, B. , Doran, R. C. , Wright, M. , & Berman, P. W. (2018). CRISPR/Cas9 gene editing for the creation of an MGAT1‐deficient CHO cell line to control HIV‐1 vaccine glycosylation. PLOS Biology, 16(8), e2005817 10.1371/journal.pbio.2005817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chari, R. , Mali, P. , Moosburner, M. , & Church, G. M. (2015). Unraveling CRISPR‐Cas9 genome engineering parameters via a library‐on‐library approach. Nature Methods, 12(9), 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, C.‐S. , Peluso, P. , Sedlazeck, F. J. , Nattestad, M. , Concepcion, G. T. , Clum, A. , & Schatz, M. C. (2016). Phased diploid genome assembly with single‐molecule real‐time sequencing. Nature Methods, 13(12), 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daer, R. M. , Cutts, J. P. , Brafman, D. A. , & Haynes, K. A. (2017). The impact of chromatin dynamics on Cas9‐mediated genome editing in human cells. ACS Synthetic Biology, 6(3), 428–438. 10.1021/acssynbio.5b00299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinowitz, M. , Lie, Y. S. , Low, M. A. , Lazar, R. , Fautz, C. , Potts, B. , & Anderson, K. (1992). Recent studies on retrovirus‐like particles in Chinese hamster ovary cells. Developments in Biological Standardization, 76, 201–207. [PubMed] [Google Scholar]
- Donahue, R. E. , Kessler, S. W. , Bodine, D. , McDonagh, K. , Dunbar, C. , Goodman, S. , & Moen, R. (1992). Helper virus induced T cell lymphoma in nonhuman primates after retroviral mediated gene transfer. The Journal of Experimental Medicine, 176, 1125–1135. 10.1084/jem.176.4.1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanoil‐Ravier, R. , Hojman, F. , Servenay, M. , Lesser, J. , Bernardi, A. , & Peries, J. (1991). Biological and molecular studies of endogenous retrovirus‐like genes in Chinese hamster cell lines. Developments in Biological Standardization, 75, 113–122. [PubMed] [Google Scholar]
- Fadrosh, D. W. , Ma, B. , Gajer, P. , Sengamalay, N. , Ott, S. , Brotman, R. M. , & Ravel, J. (2014). An improved dual‐indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome, 2(1), 6 10.1186/2049-2618-2-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Fourn, V. , Girod, P. A. , Buceta, M. , Regamey, A. , & Mermod, N. (2014). CHO cell engineering to prevent polypeptide aggregation and improve therapeutic protein secretion. Metabolic Engineering, 21, 91–102. 10.1016/j.ymben.2012.12.003 [DOI] [PubMed] [Google Scholar]
- Fu, Y. , Foden, J. A. , Khayter, C. , Maeder, M. L. , Reyon, D. , Joung, J. K. , & Sander, J. D. (2013). High‐frequency off‐target mutagenesis induced by CRISPR‐Cas nucleases in human cells. Nature Biotechnology, 31(9), 822–826. 10.1038/nbt.2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin, K. , Deruy, E. , Martien, S. , Vercamer, C. , Bouali, F. , Dujardin, T. , & Abbadie, C. (2009). Senescent keratinocytes die by autophagic programmed cell death. The American Journal of Pathology, 174(2), 423–435. 10.2353/ajpath.2009.080332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould, R. R. , & Borisy, G. G. (1977). The pericentriolar material in Chinese hamster ovary cells nucleates microtubule formation. The Journal of Cell Biology, 73(3), 601–615. 10.1083/jcb.73.3.601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley, J. W. , & Rowe, W. P. (1976). Naturally occurring murine leukemia viruses in wild mice: Characterization of a new « amphotropic » class. Journal of Virology, 19(1), 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward, A. , Grabherr, M. , & Jern, P. (2013). Broad‐scale phylogenomics provides insights into retrovirus‐host evolution. Proceedings of the National Academy of Sciences, 110(50), 20146–20151. 10.1073/pnas.1315419110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine, U. I. , Kramarsky, B. , Wendel, E. , & Suskind, R. G. (1979). Enhanced proliferation of endogenous virus in Chinese hamster cells associated with microtubules and the mitotic apparatus of the host cell. Journal of General Virology, 44(1), 45–55. [DOI] [PubMed] [Google Scholar]
- Henzy, J. E. , Gifford, R. J. , Johnson, W. E. , & Coffin, J. M. (2014). A novel recombinant retrovirus in the genomes of modern birds combines features of avian and mammalian retroviruses. Journal of Virology, 88(5), 2398–2405. 10.1128/JVI.02863-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojman, F. , Emanoil‐Raivier, R. , Lesser, J. , & Périès, J. (1989). Biological and molecular characterization of an endogenous retrovirus present in CHO/HBs‐A Chinese hamster cell line. Developments in Biological Standardization, 70, 195–202. [PubMed] [Google Scholar]
- Ihry, R. J. , Worringer, K. A. , Salick, M. R. , Frias, E. , Ho, D. , Theriault, K. , & Kaykas, A. (2018). P53 inhibits CRISPR‐Cas9 engineering in human pluripotent stem cells. Nature Medicine, 24(7), 939–946. 10.1038/s41591-018-0050-6 [DOI] [PubMed] [Google Scholar]
- Kaminski, R. , Chen, Y. , Fischer, T. , Tedaldi, E. , Napoli, A. , Zhang, Y. , & Khalili, K. (2016). Elimination of HIV‐1 genomes from human T‐lymphoid cells by CRISPR/Cas9 gene editing. Scientific Reports, 6, 22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawada, S. , Goto, T. , Haraguchi, H. , Ono, A. , & Morikawa, Y. (2008). Dominant negative inhibition of human immunodeficiency virus particle production by the nonmyristoylated form of Gag. Journal of Virology, 82(9), 4384–4399. 10.1128/JVI.01953-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostyrko, K. , & Mermod, N. (2015). Assays for DNA double‐strand break repair by microhomology‐based end‐joining repair mechanisms. Nucleic Acids Research, 44(6), e56 10.1093/nar/gkv1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostyrko, K. , Neuenschwander, S. , Junier, T. , Regamey, A. , Iseli, C. , Schmid‐Siegert, E. , & Mermod, N. (2017). MAR‐mediated transgene integration into permissive chromatin and increased expression by recombination pathway engineering. Biotechnology and Bioengineering, 114(2), 384–396. 10.1002/bit.26086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. S. , Kallehauge, T. B. , Pedersen, L. E. , & Kildegaard, H. F. (2015). Site‐specific integration in CHO cells mediated by CRISPR/Cas9 and homology‐directed DNA repair pathway. Scientific Reports, 5, 8572 10.1038/srep08572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. S. , Park, J. H. , Ha, T. K. , Samoudi, M. , Lewis, N. E. , Palsson, B. O. , & Lee, G. M. (2018). Revealing key determinants of clonal variation in transgene expression in recombinant CHO cells using targeted genome editing. ACS Synthetic Biology, 7(12), 2867–2878. 10.1021/acssynbio.8b00290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos, B. R. , Kaplan, A. C. , Bae, J. E. , Ferrazzoli, A. E. , Kuo, J. , Anand, R. P. , & Haber, J. E. (2018). CRISPR/Cas9 cleavages in budding yeast reveal templated insertions and strand‐specific insertion/deletion profiles. Proceedings of the National Academy of Sciences, 115(9), E2040–E2047. 10.1073/pnas.1716855115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, N. E. , Liu, X. , Li, Y. , Nagarajan, H. , Yerganian, G. , O'Brien, E. , & Palsson, B. O. (2013). Genomic landscapes of Chinese hamster ovary cell lines as revealed by the Cricetulus griseus draft genome. Nature Biotechnology, 31(8), 759–765. 10.1038/nbt.2624 [DOI] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S. , Cha, S. W. , Heffner, K. , Hizal, D. B. , Bowen, M. , Chaerkady, R. , & Lewis, N. (2019). Proteogenomic annotation of the Chinese hamster reveals extensive novel translation events and endogenous retroviral elements. Journal of Proteome Research, 18(6), 2433–2455. 10.1101/468181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie, Y. S. , Penuel, E. M. , Low, M. A. , Nguyen, T. P. , Mangahas, J. O. , Anderson, K. P. , & Petropoulos, C. J. (1994). Chinese hamster ovary cells contain transcriptionally active full‐length type C proviruses. Journal of Virology, 68(12), 7840–7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber, M. M. , Benveniste, R. E. , Livingston, D. M. , & Todaro, G. J. (1973). Mammalian cells in culture frequently release type C viruses. Science, 182(4107), 56–59. 10.1126/science.182.4107.56 [DOI] [PubMed] [Google Scholar]
- Lin, Y. , Cradick, T. J. , Brown, M. T. , Deshmukh, H. , Ranjan, P. , Sarode, N. , & Bao, G. (2014). CRISPR/Cas9 systems have off‐target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Research, 42(11), 7473–7485. 10.1093/nar/gku402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manly, K. F. , Givens, J. F. , Taber, R. L. , & Zeigel, R. F. (1978). Characterization of virus‐like particles released from the hamster cell line CHO‐K1 After Treatment with 5‐Bromodeoxyuridine. Journal of General Virology, 39(3), 505–517. 10.1099/0022-1317-39-3-505 [DOI] [PubMed] [Google Scholar]
- Manrique, M. L. , Celma, C. C. P. , González, S. A. , & Affranchino, J. L. (2001). Mutational analysis of the feline immunodeficiency virus matrix protein. Virus Research, 76(1), 103–113. 10.1016/S0168-1702(01)00249-0 [DOI] [PubMed] [Google Scholar]
- Morikawa, Y. , Hinata, S. , Tomoda, H. , Goto, T. , Nakai, M. , Aizawa, C. , & Mura, S. (1996). Complete inhibition of human immunodeficiency virus Gag myristoylation is necessary for inhibition of particle budding. Journal of Biological Chemistry, 271(5), 2868–2873. 10.1074/jbc.271.5.2868 [DOI] [PubMed] [Google Scholar]
- Nakamura, T. , Yamada, K. D. , Tomii, K. , & Katoh, K. (2018). Parallelization of MAFFT for large‐scale multiplesequence alignments. Bioinformatics, 34(14), 2490–2492. 10.1093/bioinformatics/bty121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu, D. , Wei, H.‐J. , Lin, L. , George, H. , Wang, T. , Lee, I.‐H. , & Yang, L. (2017). Inactivation of porcine endogenous retrovirus in pigs using CRISPR‐Cas9. Science, 357(6357), 1303–1307. 10.1126/science.aan4187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor, M. J. (2015). Targeting the DNA damage response in cancer. Molecular Cell, 60(4), 547–560. 10.1016/j.molcel.2015.10.040 [DOI] [PubMed] [Google Scholar]
- Pilbrough, W. , Munro, T. P. , & Gray, P. (2009). Intraclonal protein expression heterogeneity in recombinant CHO cells. PLOS One, 4(12), e8432 10.1371/journal.pone.0008432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puck, T. T. (1957). The genetics of somatic mammalian cells. Advances in Biological and Medical Physics, 5, 75–101. 10.1016/B978-1-4832-3111-2.50006-7 [DOI] [PubMed] [Google Scholar]
- Puri, V. , Konda, S. , Ranjit, S. , Aouadi, M. , Chawla, A. , Chouinard, M. , & Czech, M. P. (2007). Fat‐specific protein 27, a novel lipid droplet protein that enhances triglyceride storage. Journal of Biological Chemistry, 282(47), 34213–34218. 10.1074/jbc.m707404200 [DOI] [PubMed] [Google Scholar]
- Ran, F. A. , Hsu, P. D. , Wright, J. , Agarwala, V. , Scott, D. A. , & Zhang, F. (2013). Genome engineering using the CRISPR‐Cas9 system. Nature Protocols, 8(11), 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuss, F. U. (1992). Expression of intracisternal A‐particle‐related retroviral element‐encoded envelope proteins detected in cell lines. Journal of Virology, 66(4), 1915–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander, J. D. , Maeder, M. L. , Reyon, D. , Voytas, D. F. , Joung, J. K. , & Dobbs, D. (2010). ZiFiT (zinc finger targeter): An updated zinc finger engineering tool. Nucleic Acids Research, 38, 462–468. 10.1093/nar/gkq319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander, J. D. , Zaback, P. , Joung, J. K. , Voytas, D. F. , & Dobbs, D. (2007). Zinc finger targeter (ZiFiT): An engineered zinc finger/target site design tool. Nucleic Acids Research, 35, 599–605. 10.1093/nar/gkm349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuepbach, T. , Pagni, M. , Bridge, A. , Bougueleret, L. , Xenarios, I. , & Cerutti, L. (2013). pfsearchV3: A code acceleration and heuristic to search PROSITE profiles. Bioinformatics, 29(9), 1215–1217. 10.1093/bioinformatics/btt129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura‐Morales, C. , Pescia, C. , Chatellard‐Causse, C. , Sadoul, R. , Bertrand, E. , & Basyuk, E. (2005). Tsg101 and alix interact with murine leukemia virus gag and cooperate with Nedd4 ubiquitin ligases during budding. Journal of Biological Chemistry, 280(29), 27004–27012. 10.1074/jbc.M413735200 [DOI] [PubMed] [Google Scholar]
- Semaan, M. , Ivanusic, D. , & Denner, J. (2015). Cytotoxic effects during knock out of multiple porcine endogenous retrovirus (PERV) sequences in the pig genome by zinc finger nucleases (ZFN). PLOS One, 10(4), e0122059 10.1371/journal.pone.0122059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd, A. J. , Wilson, N. J. , & Smith, K. T. (2003). Characterisation of endogenous retrovirus in rodent cell lines used for production of biologicals. Biologicals, 31(4), 251–260. 10.1016/S1045-1056(03)00065-4 [DOI] [PubMed] [Google Scholar]
- Sigal, A. , Milo, R. , Cohen, A. , Geva‐Zatorsky, N. , Klein, Y. , Liron, Y. , & Alon, U. (2006). Variability and memory of protein levels in human cells. Nature, 444(7119), 643–646. 10.1038/nature05316 [DOI] [PubMed] [Google Scholar]
- Tamura, K. , & Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Molecular Biology and Evolution, 10(3), 512–526. 10.1093/oxfordjournals.molbev.a040023 [DOI] [PubMed] [Google Scholar]
- Tihon, C. , & Green, M. (1973). Cyclic AMP‐amplified replication of RNA tumour virus‐like particles in Chinese hamster ovary cells. Nature: New Biology, 244, 227 10.1038/newbio244227a0 [DOI] [PubMed] [Google Scholar]
- Tokuyama, M. , Kong, Y. , Song, E. , Jayewickreme, T. , Kang, I. , & Iwasaki, A. (2018). ERVmap analysis reveals genome‐wide transcription of human endogenous retroviruses. Proceedings of the National Academy of Sciences, 115(50), 12565–12572. 10.1073/pnas.1814589115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urlaub, G. , Käs, E. , Carothers, A. M. , & Chasin, L. A. (1983). Deletion of the diploid dihydrofolate reductase locus from cultured mammalian cells. Cell, 33(2), 405–412. 10.1016/0092-8674(83)90422-1 [DOI] [PubMed] [Google Scholar]
- Urnovitz, H. B. , & Murphy, W. H. (1996). Human endogenous retroviruses: Nature, occurrence, and clinical implications in human disease. Clinical Microbiology Reviews, 9(1), 72–99. 10.1128/cmr.9.1.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapling, J. , Srivastava, S. , Shehu‐Xhilaga, M. , & Tachedjian, G. (2007). Targeting human immunodeficiency virus type 1 assembly, maturation and budding. Drug Target Insights, 2, 159–182. [PMC free article] [PubMed] [Google Scholar]
- De Wit, C. , Fautz, C. , & Xu, Y. (2000). Real‐time quantitative PCR for retrovirus‐like particle quantification in CHO cell culture. Biologicals, 28(3), 137–148. 10.1006/biol.2000.0250 [DOI] [PubMed] [Google Scholar]
- Wurm, F. (2013). CHO quasispecies—Implications for manufacturing processes. Processes, 1(3), 296–311. 10.3390/pr1030296 [DOI] [Google Scholar]
- Xu, H. , Xiao, T. , Chen, C.‐H. , Li, W. , Meyer, C. A. , Wu, Q. , & Liu, X. S. (2015). Sequence determinants of improved CRISPR sgRNA design. Genome Research, 25(8), 1147–1157. 10.1101/gr.191452.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X. , Nagarajan, H. , Lewis, N. E. , Pan, S. , Cai, Z. , Liu, X. , & Wang, J. (2011). The genomic sequence of the Chinese hamster ovary (CHO)‐K1 cell line. Nature Biotechnology, 29(8), 735–741. 10.1038/nbt.1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, L. , Güell, M. , Niu, D. , George, H. , Lesha, E. , Grishin, D. , & Church, G. (2015). Genome‐wide inactivation of porcine endogenous retroviruses (PERVs). Science, 350(6264), 1101–1104. 10.1126/science.aad1191 [DOI] [PubMed] [Google Scholar]
- Yoon, H. , & Leitner, T. (2015). PrimerDesign‐M: A multiple‐alignment based multiple‐primer design tool for walking across variable genomes. Bioinformatics, 31(9), 1472–1474. 10.1093/bioinformatics/btu832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura, F. K. , Luo, X. , Zhao, X. , Gerard, H. C. , & Hudson, A. P. (2008). Up‐regulation of a cellular protein at the translational level by a retrovirus. Proceedings of the National Academy of Sciences, 105(14), 5543–5548. 10.1073/pnas.0710526105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, L. J. , Holmes, B. R. , Aronin, N. , & Brodsky, M. H. (2014). CRISPRseek: A Bioconductor package to identify target‐specific guide RNAs for CRISPR‐Cas9 genome‐editing systems. PLOS One, 9(9), e108424 10.1371/journal.pone.0108424 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information