

Abstract
The SMYD2 protein lysine methyltransferase methylates various histone and non‐histone proteins and is overexpressed in several cancers. Using peptide arrays, we investigated the substrate specificity of the enzyme, revealing a recognition of leucine (or weaker phenylalanine) at the −1 peptide site and disfavor of acidic residues at the +1 to +3 sites. Using this motif, novel SMYD2 peptide substrates were identified, leading to the discovery of 32 novel peptide substrates with a validated target site. Among them, 19 were previously reported to be methylated at the target lysine in human cells, strongly suggesting that SMYD2 is the protein lysine methyltransferase responsible for this activity. Methylation of some of the novel peptide substrates was tested at the protein level, leading to the identification of 14 novel protein substrates of SMYD2, six of which were more strongly methylated than p53, the best SMYD2 substrate described so far. The novel SMYD2 substrate proteins are involved in diverse biological processes such as chromatin regulation, transcription, and intracellular signaling. The results of our study provide a fundament for future investigations into the role of this important enzyme in normal development and cancer.
Keywords: enzyme specificity, peptide array, protein lysine methyltransferase, SMYD2
SMYD2 methylates different histone and non‐histone proteins. We investigated its substrate specificity and discovered several novel peptide and protein substrates, many of which are methylated more strongly than the p53 protein, the best SMYD2 substrate described so far. Our data will aid in understanding the role of SMYD2 in development and cancer.
Introduction
In recent years, it has been discovered that lysine methylation, which was initially identified on histone proteins, occurs on a wide range of non‐histone proteins, where it plays essential regulatory roles in various cellular processes.1, 2, 3, 4 Lysine residues can be mono‐, di‐ and trimethylated and the biological outcome of the modification depends on the site and degree of methylation in target proteins. Lysine methylation is introduced by protein lysine methyltransferases (PKMTs)5, 6 including enzymes of the SET and MYND‐containing protein (SMYD) family.6, 7, 8 The biological function of SMYD PKMTs is diverse including gene regulation, chromatin remodeling, transcription, signal transduction, cell cycle control, and DNA damage response.6, 7, 8 The SMYD PKMT family consists of five members, called SMYD1 to 5. They share their structural domain architecture with other SET domain PKMTs, where a catalytically active SET domain containing a SET‐I insertion involved in target peptide interaction is followed by a Post‐SET domain. Different from other SET domain PKMTs, the SET domain of SMYD family enzymes is further split by the insertion of a MYND (Myeloid‐Nervey‐DEAF‐1) domain between the initial part of the SET domain (the S‐sequence) and the SET‐I part. The MYND domain is a zinc‐finger domain that mediates protein‐protein interactions. Different from other SET‐domain PKMTs, the SMYD family enzymes do not contain a Pre‐SET domain, but they carry an additional SMYD family specific C‐terminal domain (CTD).
The SMYD2 enzyme (also called KMT3C) was initially discovered as an H3K36 dimethyltransferase and SMYD2 activity had been shown to lead to repression of transcribed genes.9 Later it was found that SMYD2 also methylates H3K4 in the presence of the HSP90 protein.10 In addition to the methylation of histone targets, SMYD2 was reported to methylate several non‐histone proteins like p5311 and the retinoblastoma protein (RB).12 TP53 is one of the most studied tumor suppressor genes and it has important biological functions in the regulation of cell cycle arrest, apoptosis and the DNA damage response. Four lysine residues in the C‐terminal part of p53 can be methylated by different PKMTs leading to distinct biological outputs.13 Methylation studies with p53 lysine to arginine mutants revealed that K370 is the main methylation site of SMYD2 on p53.11 Methylation of K370 results in repression of p53 regulated transcription and p53 mediated apoptosis in cancer cells due to decreased occupancy of methylated p53 at its target genes.11, 12 Different studies reported that methylation of p53 by SMYD2 was higher than methylation of the other identified histone targets.14, 15, 16 The retinoblastoma tumor suppressor protein (RB) involved in the regulation of cell cycle progression and apoptosis is another non‐histone substrate of SMYD2 with a strong cancer connection. SMYD2 methylation of RB occurs at lysine 860, leading to the progression of the cell cycle via different molecular pathways.17 Moreover, SMYD2 also methylates the estrogen receptor alpha (ERα) protein at lysine 266 repressing ERα transactivation activity.18 Details of the substrate peptide recognition of SMYD2 have been identified by two structures of the enzyme with the p53 and the ERα peptide substrates.14, 19 In addition, several other non‐histone targets were identified in the last couple of years, including PARP1,20 MAPKAPK321 and PTEN.22 SMYD2 is essential for normal organismal development6, 7, 8 and dysregulation of SMYD2 was found in cardiovascular disease and cancer.6, 7, 8 For example, SMYD2 is significantly overexpressed in many cancers including esophageal squamous cell carcinoma (ESCC), bladder and gastric cancer,23, 24 and in triple negative breast cancer it was shown to have a tumor promoting effect.8 In some cases, the molecular mechanisms connecting the methylation of SMYD2 non‐histone substrates and oncogenic effects have been discovered and, based on these, inhibitors of SMYD2 have been tested as therapeutic agents.24
Better understanding of the biology of the SMYD2 protein lysine methylation signaling network critically depends on the identification of the substrate profile of SMYD2, but in spite of progress in this direction, the identification of PKMT substrates is not trivial.25 General proteome‐wide screenings were undertaken aiming to identify SMYD2 substrates in an unbiased way. By combining proteomic lysine methylation data with SMYD2 knock‐down, 34 SMYD2 dependent methylation events were discovered in human cell lines, two of which were validated by in vitro methylation experiments (AHNAK and AHNAK2).26 In an approach combining enzyme specificity and modelling, four novel SMYD2 substrates (SIX1, SIX2, SIN3B and DHX15) were discovered and validated.27 Another bioinformatic approach combining different data sources like known substrates, proteomic data, protein interaction, gene ontology and structural data has led to the identification of six novel substrates (MAPT, CCAR2, EEF2, NCOA3, STUB1 and UTP14A).28 In the current study, we also applied an unbiased search for novel SMYD2 substrates. In our workflow, the substrate specificity of SMYD2 is analyzed by methylation of peptide arrays to identify additional non‐histone targets in a systematic manner. Different studies already showed that the analysis of the specificity profiles of PKMTs is a powerful method to investigate their substrate recognition and based on this discover novel substrates.25, 29, 30, 31, 32, 33, 34, 35 With this approach, we identified 32 novel peptide substrates of SMYD2 with validated methylation sites. Among them, 14 novel non‐histone protein substrates of SMYD2 were discovered, six of which were more strongly methylated than p53, the best SMYD2 substrate described so far. The novel SMYD2 substrates are involved in diverse cellular processes like chromatin interaction and modification, gene regulation, DNA repair, actin filament dynamics, and Ras signaling.
Results and Discussion
As described above, it is a big challenge to link protein lysine methyltransferases with lysine methylation events of specific substrates. In this study, we wanted to analyze the substrate sequence specificity of SMYD2 in detail using a peptide array approach. Afterward, we used the specificity information to search for and validate additional non‐histone substrates of SMYD2 that will give deeper insight into the wide substrate spectrum of SMYD2 and the connected biological functions.
Protein expression and catalytic activity of SMYD2 on p53‐K370
Full‐length SMYD2 and p53 were expressed as GST‐fusion proteins in Escherichia coli cells and purified by affinity chromatography (Figure 1 A). The circular dichroism (CD) spectrum of the purified SMYD2 showed minima around 208 and 220 nm characteristic for a folded protein (Figure S1 A in the Supporting Information) and the obtained melting temperature of 55.9 °C (Figure S1 B) documented stable folding. SMYD2 activity was tested using peptide SPOT arrays probing several well‐studied histone methylation marks together with p53‐K370. To this end, 20‐residue peptides were synthesized on a cellulose membrane using the SPOT technology.36, 37 The resulting SPOT peptide arrays were incubated with SMYD2 using radioactively labelled AdoMet as cofactor and the transfer of radioactively labelled methyl groups to p53 was detected by autoradiography (Figure 1 B). Note that the different peptide sequences could yield slightly different background signal levels due to unspecific binding of AdoMet. This experiment confirmed the methylation of H3K36 as shown by the clear methylation signal of the H3 28–48 peptide, which was completely lost after the K36A exchange. In the case of the H3 1–20 peptide, a methylation signal was observed, which was decreased after the H3K4 exchange but not affected by the K9A exchange, indicating methylation of H3K4. However, the strongest methylation was observed on the p53 peptide, which was almost completely lost after the K370A exchange demonstrating a strong and specific methylation of p53‐K370 by SMYD2. The methylation activity of SMYD2 was next tested on the p53 protein using radioactively labeled AdoMet. The methylated reaction mixture was separated using sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and the transfer of radioactively labelled methyl groups to p53 was detected by autoradiography (Figure 1 C). The result revealed a strong methylation of p53, whereas no signal could be detected in the absence of p53.
Figure 1.
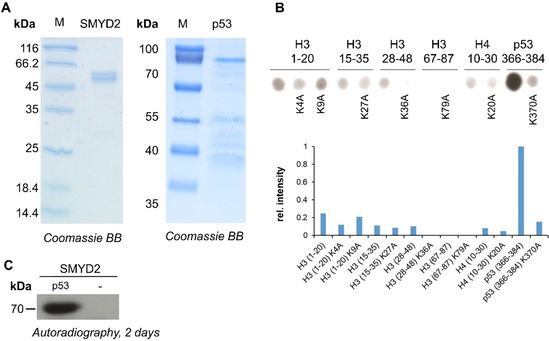
Protein purification and activity validation of SMYD2. A) Full‐length SMYD2 and p53 were cloned as GST‐fusion proteins and purified by affinity chromatography. B) Methylation of peptide SPOT arrays containing the previously identified methylation sites of SMYD2 on H3, some additional well studied histone methylation sites and the p53 methylation substrate. K‐to‐A peptides were included to identify the lysine residues that were methylated in the corresponding wild‐type peptide. The lower part shows a quantification of the spot intensities. C) Methylation of the purified p53 protein with radioactively labeled AdoMet. Methylated samples were separated by SDS‐PAGE and the methyl group transfer was detected by autoradiography. After two days of exposure methylation of p53 could be detected, whereas in absence of p53 no methylation was observed.
Substrate specificity analysis of SMYD2
For specificity analysis, larger SPOT peptide libraries were synthesized by using the p53 sequence (363–377) as template, which was selected based on the strong methylation signal observed in Figure 1 B. The peptide arrays consisted of 300 individual peptides in each of which one amino acid of the original sequence was exchanged against one of the 20 natural amino acids generating all possible single amino acid exchanges of the template sequence. With this approach, the influence of each single amino acid at every position of the substrate peptide on the enzymatic activity of SMYD2 can be investigated in detail. The peptide arrays were incubated with methylation buffer containing SMYD2 and radioactively labeled AdoMet as cofactor. The transfer of methyl groups to the immobilized peptides was detected by autoradiography (Figure 2 A). The experiment was conducted two times. Afterward, each array was quantified and the results of the two independent experiments were normalized and averaged (Figure 2 B). Based on these data, the standard deviations (SD) were calculated for the methylation activity on each single spot to evaluate the quality of the peptide array methylation experiments.
Figure 2.
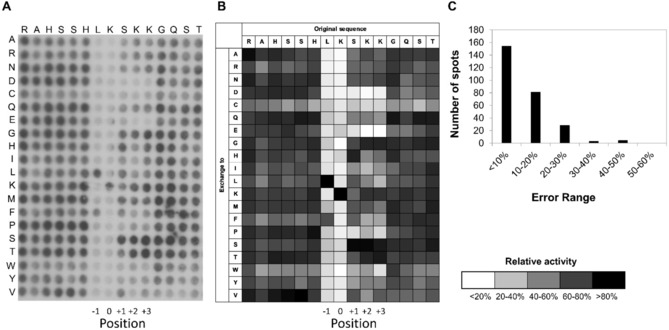
Substrate sequence specificity analysis of SMYD2. A) Example of a substrate sequence specificity SPOT array methylated by SMYD2. Arrays of 15‐residue peptides were synthesized by using p53 (363–377) as template sequence, represented in the horizontal axis. Each residue was exchanged against all 20 natural amino acid residues, shown in the vertical axis. For methylation, the membrane was incubated with SMYD2 in the presence of radioactively labelled AdoMet and the transfer of methyl groups visualized by autoradiography. The exposition time of the film was three days. B) Two independent peptide array methylation experiments were performed and the data were normalized, averaged and the methylation signals presented as greyscale heatmap. Black color represents strong methylation, while light grey represents weak methylation. C) Distribution of the standard deviations of SMYD2 activity on all peptides tested in two repetitions.
As shown in Figure 2 C, the SPOT peptide array methylation data were highly reproducible, because 87 % of the peptides showed an SD smaller than 20 %, around 97 % of the peptides showed an SD smaller than 30 % and only seven peptides had an SD larger than 30 % (Figure 2 C). Our data show that SMYD2 is specific toward a leucine at position −1 of the substrate peptide sequence (considering the target lysine as 0). It only weakly tolerates phenylalanine at the same site. SMYD2 did not exhibit specificity for amino acids N‐terminal to position −1. At the +1 to +3 positions, polar uncharged and basic residues are preferred, but acidic residues (aspartate and glutamate), cysteine and large hydrophobic as well as aromatic residues were not tolerated to a different degree leading to the following specificity profile: [LF]‐K‐[ARNGHLKFSTV]‐[ARNGKST]‐[ARNGKSTV].
Several of the known non‐histone targets, including p53 (SHLK370SKK, the template sequence of the peptide array), ERα (RMLK266HKR), Rb (RVLK860RSA), PARP1 (LTLK528GGA) and MAPKAPK3 (KDLK355TSN) fit to the derived sequence motif, as well as the K36 methylation site (GGVK36KPH) on histone H3. The preference of SMYD2 for leucine and phenylalanine at the −1 site of the target peptide can be explained by the structure of the enzyme in complex with the p53 (PDB ID: https://www.rcsb.org/structure/3S7F)14 or ERα peptides (PDB ID: https://www.rcsb.org/structure/4O6F).19 In both structures, the corresponding target peptide leucine residue at the −1 site is in close proximity to methyl groups of SMYD2 residues T105, L108, V179, and T185 and further contacted by the Cα of N180 and G183 and Cβ of S196, altogether creating a hydrophobic pocket large enough to accommodate a leucine residue (Figure 3 A). The disfavor for acidic residues at the +1 to +3 sites in the target peptide can be explained by the close proximity of two acidic side chains from SMYD2 (E187 and D242) and the general acidic nature of the peptide binding pocket (Figure 3 B). Overall SMYD2 showed a relatively weak peptide sequence specificity with surprisingly little sequence specific readout. This finding suggests that the methylation specificity of this enzyme very likely is also controlled by substrate binding specificity at the protein level.
Figure 3.
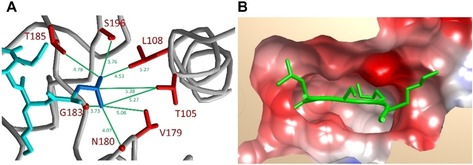
Structural details of the SMYD2⋅p53 complex (3SF7).14 A) Residues in the vicinity of the Leu at the −1 position. The peptide is shown in cyan with the side chain of the Leu residue in blue. SMYD2 is shown in ribbon view (grey) with the interacting residues in red. Distances are indicated in Å. B) Electrostatic surface view of SMYD2 illustrating the dominance of acidic residues in the binding pocket. The bound peptide is shown in green.
Methylation of non‐histone peptide substrates by SMYD2
Since we were interested to discover novel biological relevant substrates of SMYD2, the newly identified sequence motif was used to screen for potential novel non‐histone targets in the human proteome using Scansite (https://scansite4.mit.edu/).38 Among the almost 8000 hits, 124 proteins with nuclear localization and important biological functions were selected for further analysis (Table S1). To check if SMYD2 can methylate these predicted targets, 15‐residue peptides were synthesized on a cellulose membrane using the SPOT synthesis method. The p53 peptide and an artificial peptide with an optimized SMYD2 target sequence were included as positive controls and their corresponding K‐to‐A mutants were used as negative controls. To compare the SMYD2 activity with the activity at the histone methylation sites, H3K4, H3K36 and the corresponding K‐to‐A mutant peptides were included on the array as well. Figure 4 A shows that SMYD2 can methylate about 40 peptides with same or higher activity than p53 indicating that about of the predicted targets were methylated. The other peptides were either weakly or not methylated at all. Out of these 40 substrates, 14 were selected for further investigation based on their high methylation intensity.
Figure 4.

Screening of SMYD2 non‐histone peptide substrates. A) SPOT array methylation of candidate SMYD2 targets taken from the Scansite search database (Table S1). B) SPOT array methylation of candidate SMYD2 targets taken from the PhosphoSite Plus database (Table S2). C) Selected peptides found to be methylated in panel A and B were synthesized on an additional SPOT peptide array together with their corresponding K‐to‐A mutants (Table 1). On each array, the artificial peptide and p53 peptide were included as positive controls. As negative controls, the corresponding K‐to‐A mutants of the artificial peptide and p53 peptide were synthesized next to the corresponding wild‐type spots. Spots marked with red circles in (C) were selected for further investigation of protein methylation.
To further develop the detection of possible novel substrates, the PhosphoSite Plus database (https://www.phosphosite.org/)39 was used, which allows to identify proteins with known methylation within the broadened SMYD2 sequence motif [LF]‐[K]. With this approach, 155 methylation sites in non‐histone proteins were identified which are methylated in cells at potential SMYD2 target sites (Table S2). All potential targets were synthesized as 15 amino acid long peptides with the target lysine in the center on a SPOT peptide array and treated as explained previously (Figure 4 B). As before, this array contained the sequence of p53 and the artificial peptide together with their K‐to‐A mutants as controls. Out of the 155 potential non‐histone targets, 19 peptides were methylated equally strong as p53 or even stronger and were selected for further investigation. Interestingly, the more global search applied here, which did not include the information about the +1 to +3 sites, led to a significantly lower hit rate of only 12 % as compared to 33 % found in the screen based on the full motif.
The 33 selected non‐histone targets from both screens were synthesized on an additional peptide SPOT array together with their K‐to‐A mutants to confirm that the predicted target lysine is methylated by SMYD2 (Figure 4 C). Strikingly, for 32 out of the 33 tested peptides a complete loss or strong decrease in the methylation signal was detected with the alanine mutant relative to the wild‐type peptides (Table 1). This shows that the target lysine is indeed methylated by SMYD2 indicating an excellent power of the previous screens. Among the peptides with validated methylation sites, 24 were selected for further protein work based on their strong methylation and the known important biological functions of the corresponding proteins.
Table 1.
List of potential novel non‐histone targets identified in Scansite search (Figure 4 A) and PhosphoSite Plus database (Figure 4 B) for which target site methylation was investigated in the SPOT array shown in Figure 4 C. The target lysine residue is highlighted in boldface. Column 1: Swissprot protein number; column 2: abbreviation; column 3: target lysine; column 4: original screen leading to the discovery of this methylation site (Figure 4 A or B); column 5: position of the spot in Figure 4 C; column 6: position of K‐to‐A variant spot in Figure 4 C; column 7: target site methylation validated; column 8: methylation sites studied at protein level are indicated by “+”.
|
1 |
2 |
Name |
Sequence |
3 |
4 |
5 |
6 |
7 |
8 |
|---|---|---|---|---|---|---|---|---|---|
|
|
|
artificial peptide |
RNEPPKLKRSRGAFT |
8 |
|
1A |
2A |
|
|
|
POLR3B |
DNA‐directed RNA polymerase III subunit RPC2 |
PVYYQKLKHMVLDKM |
1013 |
B |
3A |
4A |
+ |
|
|
|
POLR2B |
DNA‐directed RNA polymerase II subunit RPB2 |
PTYYQRLKHMVDDKI |
1052 |
B |
5A |
6A |
+ |
+ |
|
|
H1F0 |
histone H1.0 |
PKKSVAFKKTKKEIK |
107 |
B |
7A |
8A |
+ |
|
|
|
RECQL4 |
ATP‐dependent DNA helicase Q4 |
PDYGQRLKANLKGTL |
110 |
B |
9A |
10A |
+ |
|
|
|
AHNAK |
neuroblast differentiation‐associated protein AHNAK |
KLKGPKFKMPEMHFK |
806 |
B |
11A |
12A |
− |
|
|
|
FGD5 |
FYVE, RhoGEF and PH domain‐containing protein 5 |
DGCFGELKKRGRAVP |
1301 |
B |
13A |
14A |
+ |
+ |
|
|
RAPH1 |
Ras‐associated and pleckstrin homology domains‐containing protein 1 |
TLKHGTLKGLSSSSN |
134 |
B |
15A |
16A |
+ |
+ |
|
|
LIN9 |
TGS2 |
HRGGQPLKKRRGSSK |
143 |
B |
17A |
18A |
+ |
|
|
|
MYH11 |
myosin‐11 |
GREVNALKSKLRRGN |
1925 |
B |
21A |
1B |
+ |
|
|
|
ETFB |
electron transfer flavoprotein subunit β |
ATLPNIMKAKKKKIE |
200 |
B |
2B |
3B |
+ |
|
|
|
NCBP1 |
nuclear cap‐binding protein subunit 1 |
ANTESYLKRRQKTHV |
204 |
B |
4B |
5B |
+ |
|
|
|
CAP1 |
adenylyl cyclase‐associated protein 1 |
THKNPALKAQSGPVR |
286 |
B |
6B |
7B |
+ |
+ |
|
|
PI4KB |
phosphatidylinositol 4‐kinase β |
ISLSSNLKRTASNPK |
290 |
B |
8B |
9B |
+ |
|
|
|
OR6C74 |
olfactory receptor 6C74 |
KQVKDVFKHTVKKIE |
300 |
B |
10B |
11B |
+ |
|
|
|
CENPU |
centromere protein U |
SQMLTNLKRKNAKMI |
303 |
B |
12B |
13B |
+ |
|
|
|
p53 |
cellular tumor antigen p53 |
RAHSSHLKSKKGQST |
370 |
B |
14B |
15B |
+ |
+ |
|
|
SETD1B |
histone‐lysine N‐methyltransferase SETD1B |
LMIDPALKKGHHKLY |
41 |
B |
16B |
17B |
+ |
|
|
|
TRIM71 |
E3 ubiquitin‐protein ligase TRIM71 |
KATGDGLKRALQGKV |
495 |
B |
18B |
19B |
+ |
|
|
|
PARD3B |
partitioning defective 3 homologue B |
AGLGVSLKGNKSRET |
514 |
B |
20B |
21B |
+ |
|
|
|
MADD |
MAP kinase‐activating death domain protein |
ATPFPSLKGNRRALV |
884 |
B |
1C |
2C |
+ |
|
|
|
NFKBID |
NF‐κB inhibitor δ |
EGLRQLLKRSRVAPP |
454 |
A |
3C |
4C |
+ |
|
|
|
AFF1 |
AF4/FMR2 family member 1 |
KPAKPALKRSRREAD |
883 |
A |
5C |
6C |
+ |
+ |
|
|
CENPW |
centromere protein W |
HVLAAAKVILKKSRG |
84 |
A |
7C |
8C |
+ |
+ |
|
|
CHD3 |
chromodomain‐helicase‐DNA‐binding protein 3 |
PVRTKKLKRGRPGRK |
348 |
A |
9C |
10C |
+ |
+ |
|
|
AGAP2 |
Arf‐GAP with GTPase, ANK repeat and PH domain‐containing protein 2 |
EPPAPGLKRGREGGR |
329 |
A |
11C |
12C |
+ |
+ |
|
|
UHRF2 |
E3 ubiquitin‐protein ligase UHRF2 |
SRGKTPLKNGSSCKR |
166 |
A |
13C |
14C |
+ |
+ |
|
|
UHRF2 |
E3 ubiquitin‐protein ligase UHRF2 |
VVKAGERLKMSKKKA |
407 |
A |
15C |
16C |
+ |
|
|
|
TMUB1 |
transmembrane and ubiquitin‐like domain‐containing protein 1 |
HDTIGSLKRTQFPGR |
129 |
A |
17C |
18C |
+ |
+ |
|
|
PHF2 |
lysine‐specific demethylase PHF2 |
AGKRLLKRAKNSVDL |
847 |
A |
19C |
20C |
+ |
+ |
|
|
RRN3 |
RNA polymerase I‐specific transcription initiation factor RRN3 |
PFDPCVLKRSKKFID |
567 |
A |
21C |
1D |
+ |
+ |
|
|
CUL3 |
cullin‐3 |
LFIDDKLKKGVKGLT |
397 |
A |
2D |
3D |
+ |
+ |
|
|
TAF1 |
transcription initiation factor TFIID subunit 1 |
SKKESSLKKSRILLG |
557 |
A |
4D |
5D |
+ |
+ |
|
|
PHF20 |
PHD finger protein 20 |
KGCEVPLKRPRLDKN |
299 |
A |
6D |
7D |
+ |
+ |
|
|
ELAC1 |
zinc phosphodiesterase ELAC protein 1 |
QLMKSQLKAGRITKI |
51 |
A |
8D |
9D |
+ |
+ |
Methylation of non‐histone protein substrates by SMYD2
After confirming the methylation of 32 peptide substrates, further methylation analysis of the validated substrates followed at protein level. Methylation of the targets at the protein level can differ from peptide level, because the target lysine may not be accessible in the context of the folded protein. The selected targets were cloned as GST fusion proteins, overexpressed and purified by affinity chromatography. For 18 of them purification was possible at sufficient quality for the follow‐up work (Figure S2). Sequencing results of NFKBID revealed mutations and this target was excluded from further analysis. Roughly equal protein amounts were incubated with SMYD2 in methylation buffer containing radioactively labeled AdoMet. After methylation, the samples were separated on a 16 % SDS gel and the methyl group transfer was detected by autoradiography. As positive control, the p53 protein was included. The results shown in Figure 5 demonstrate that six proteins (AFF1, CENPW, TAF1, PHF20, FGD5 and RAPH1, marked with a red asterisk) were stronger methylated than p53. The methylation signals of three proteins (UHRF2, RAD18 and CAP1, marked with blue asterisk) were similar to p53. For five proteins (TMUB1, CHD3, AGAP2, PHF2, and ELAC1, marked with black asterisk) methylation was detected, but weaker than for p53. Only three of the proteins did not show detectable methylation (RRN3, CLU3 and PolR2B), indicating an excellent overall success rate of the validation of peptide substrates in this analysis of 82 %.
Figure 5.

Methylation of SMYD2 non‐histone targets at protein level. The selected candidate non‐histone proteins were purified by affinity chromatography and similar protein amounts (Figure S2) were incubated with radioactively labeled AdoMet and SMYD2. As positive control, p53 was included in each experiment. After methylation, the samples were separated by SDS‐PAGE and the transfer of methyl groups was detected by autoradiography. A) Methylated non‐histone target protein domains coming from the Scansite search. B) Methylated non‐histone target protein domains coming from the PhosphoSite Plus database search for methylated proteins. Red asterisks indicate substrates with stronger methylation than p53. Targets marked with blue or black asterisks were methylated similarly to p53 or weaker than p53, correspondingly.
Because we were mostly interested in targets with strong methylation, verification of the predicted target lysine methylation was performed for nine selected protein substrates (AFF1, CENPW, UHRF2, RAD18, TAF1, PHF20, CAP1, FGD5 and RAPH1; Table 2). To this end, site‐directed mutagenesis was performed to exchange the target lysine against arginine. The mutated proteins were overexpressed, purified by affinity chromatography and methylated as described before (Figure 6). All K‐to‐R mutant non‐histone targets (except RAPH1) exhibited a loss or strong decrease in methylation relative to their corresponding wild‐type proteins. In the case of FGD5, mutation of the predicated target lysine only led to a gradual decrease in methylation signal. The sequence of this protein contains two additional LK motif in close proximity to the predicted target lysine, but mutation of these two lysine residues did not lead to a further decrease in the methylation signal (data not shown). Further investigation is necessary to detect which additional lysine residue of FGD5 is methylated by SMYD2. In the case of RAPH1, the initial mutation of the target lysine K134 did not lead to a decrease in methylation. This protein contains additional LK motifs and mutation of K129 to arginine led to a decrease in methylation. Complete loss of methylation was achieved by combination of the K129R and K134R mutations indicating that SMYD2 methylates the predicted target lysine K134 but also K129.
Table 2.
List of newly discovered SMYD2 protein substrates with validated target lysine methylation. The target lysine residue is highlighted in boldface. Column 1: Swissprot protein number; column 2: abbreviation; column 3: target lysine position; column 4: boundaries of the cloned domains used in this study; column 5: approximate methylation level estimated from the autoradiographic images in Figure 5 indicated by ++ or +, if the methylation was stronger than p53 or similar to p53.
|
1 |
2 |
Name |
Sequence |
3 |
4 |
5 |
|---|---|---|---|---|---|---|
|
FGD5 |
FYVE, RhoGEF and PH domain‐containing protein 5 |
DGCFGELKKRGRAVP |
1301 |
1207–1462 |
++ |
|
|
RAPH1 |
Ras‐associated and pleckstrin homology domains‐containing protein 1 |
TLKHGTLKGLSSSSN |
129, 134 |
1–260 |
++ |
|
|
PHF20 |
PHD finger protein 20 |
KGCEVPLKRPRLDKN |
299 |
267–452 |
++ |
|
|
TAF1 |
transcription initiation factor TFIID subunit 1 |
SKKESSLKKSRILLG |
557 |
414–665 |
++ |
|
|
AFF1 |
AF4/FMR2 family member 1 |
KPAKPALKRSRREAD |
883 |
668–916 |
++ |
|
|
CENPW |
centromere protein W |
HVLAAAKVILKKSRG |
84 |
9–88 |
++ |
|
|
CAP1 |
adenylyl cyclase‐associated protein 1 |
THKNPALKAQSGPVR |
286 |
1–320 |
+ |
|
|
RAD18 |
E3 ubiquitin‐protein ligase RAD18 |
ASRQSLKQGSRLMDN |
127 |
64–232 |
+ |
|
|
UHRF2 |
E3 ubiquitin‐protein ligase UHRF2 |
SRGKTPLKNGSSCKR |
166 |
81–350 |
+ |
Figure 6.
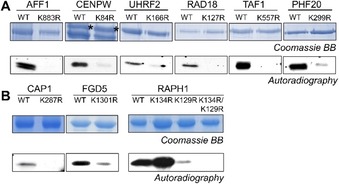
Validation of target lysine methylation of the novel SMYD2 protein substrates. Site directed mutagenesis was performed to create lysine to arginine mutants of the selected non‐histone protein substrates. Similar protein amounts of the wild‐type and K‐to‐R proteins were used for methylation experiments shown in the autoradiography images. For CENPW, black asterisks indicate the protein bands that correspond to methylation signal. A) Non‐histone target protein domains and their K‐to‐R mutants identified in the Scansite search. B) Non‐histone target protein domains and their K‐to‐R mutants identified in the PhosphoSite Plus database search of methylated proteins. Exposure of the autoradiography films was 8 h for all targets, except RAPH1 (1 day) and CAP1, FGD5 (3 days).
In summary, methylation of the lysine residue predicted from peptide studies was validated for all nine tested proteins. Three of them (CAP1, FGD5 and RAPH1) had been shown previously in proteomics screens to be methylated in cells at the SMYD2 target lysine identified here, but the PKMT responsible to introduce these methylations was not known. Our data strongly suggest that these methylation events are catalyzed by SMYD2. The newly discovered SMYD2 substrates all have important biological functions in chromatin regulation (CENPW, PHF20, UHRF2, and RAD18), transcription (AFF1, PHF20, TAF1), and intracellular signaling via ubiquitylation (UHRF2, RAD18), the Ras pathway (FGD5, RAPH1) or other pathways (CAP1, AGAP2). Future studies will need to validate the SMYD2 dependent methylation of these proteins in cells and study its biological consequences.
Conclusions
It was the aim of this work to identify novel substrates of the SMYD2 PKMT. Using peptide arrays, we investigated the substrate sequence specificity profile of the enzyme, revealing a rather low level of specificity, because only at the −1 site a real recognition of leucine (or at weaker level phenylalanine) was detected. At the +1 to +3 sites, some less specific additional effects were observed including the disfavor for acidic residues. Using the derived sequence specificity motif, novel SMYD2 peptide substrates were identified and, in a second step, methylation at the target lysine was validated. Strikingly, high fractions of predicted SMYD2 peptide substrates were indeed found to be methylated, 33 % in the case of the full motif and still 12 % in the case of the reduced [LF]‐[K] motif. Our study has led to the discovery of 32 novel peptide substrates with validated target sites. Among them, 19 were already reported to be methylated at the target lysine in human cells, strongly suggesting that SMYD2 is the PKMT responsible for these methylation events. Next, methylation of the novel substrates was tested at the protein level leading to the identification of 14 novel protein substrates of SMYD2. However, as validation of protein methylation was very efficient for those proteins which could be successfully cloned, expressed and purified, it is very likely that many of the additional novel peptide substrates will also be methylated by SMYD2 at protein level. Six of the discovered protein substrates were methylated more strongly than p53, the best SMYD2 substrate described so far. The novel SMYD2 substrate proteins are involved in diverse biological processes like chromatin regulation, transcription and different intracellular signaling pathways. Overall, our study boosts the number of known SMYD2 substrates considerably providing a fundament for future studies to investigate the function of the methylation of these novel SMYD2 substrates in cells.
Experimental Section
Cloning, expression, and purification of the proteins: SMDY2 was cloned as described.14 The domains of the non‐histone substrates and p53 were amplified by PCR using cDNA isolated from HEK293 cells as template. The amplified DNA was inserted into the pGEX‐6p2 bacterial expression vector for protein expression. Protein domains of the non‐histone substrates were predicted with the Scooby domain prediction tool (http://www.ibi.vu.nl/programs/scoobywww/).40 The different mutations were introduced by site‐directed mutagenesis.41 All cloning steps were confirmed by sequencing. Protein overexpression was performed in BL21 DE3 Codon Plus cells (Novagen) in ampicillin containing lysogeny broth at 37 °C till the main culture reached an OD600 of 0.6. Afterward, protein expression was induced by the addition of 1 mm isopropyl β‐d‐thiogalactopyranoside (IPTG) at 22 °C for 12 h. Thereafter, the cells were centrifuged (4000 g, 20 min, 4 °C) and stored at −20 °C. For protein purification, the pellet was resuspended in 25 mL sonication buffer (50 mm Tris⋅HCl pH 7.4, 150 mm NaCl, 1 mm DTT, 5 % glycerol) and sonicated (13 rounds, 30 % power, 40‐second intervals). Following centrifugation (20 000 g, 80 min, 4 °C), the obtained supernatant was loaded onto a column containing pre‐equilibrated glutathione‐Sepharose 4B beads (GE Healthcare). The column was washed once with sonication buffer and twice with wash buffer (50 mm Tris⋅HCl pH 8, 500 mm NaCl, 1 mm DTT, 5 % glycerol). Afterward, the protein was eluted from the column with wash buffer supplemented with 40 mm reduced glutathione (pH 8). The pooled fractions were dialyzed against dialysis buffer I (20 mm Tris⋅HCl pH 7.4, 100 mm KCl, 0.5 mm DTT, 10 % glycerol) for 3 h and overnight in dialysis buffer II (20 mm Tris⋅HCl pH 7.4, 100 mm KCl, 0.5 mm DTT, 60 % glycerol). The proteins were stored at −20 °C.
Peptide array synthesis and methylation: The peptide arrays were synthesized with an Autospot peptide array synthesizer (Intavis AG, Köln, Germany) using the SPOT synthesis method.36, 37 The arrays contain 15 or 20 amino acid long peptides immobilized on a cellulose membrane. After synthesis, the membrane was pre‐incubated in methylation buffer (50 mm Tris⋅HCl pH 9, 100 mm NaCl, 5 mm DTT) for 5 min. Thereafter, the membrane was incubated in methylation buffer supplemented with 0.76 μm radioactively labeled AdoMet (PerkinElmer) and 0.6 μm SMYD2 for 1 h at 25 °C on a shaker. Afterward, the membrane was washed five times for 5 min with wash buffer (100 mm NH4HCO3, 1 % SDS), followed by the incubation in amplify NAMP100V (GE Healthcare) for 10 min. Then, the membrane was exposed to a Hyperfilm high‐performance autoradiography film (GE Healthcare) at −80 °C in the dark for various periods of time and developed with a developing machine.
Protein methylation assay: Protein methylation was performed using 5–10 μg of the substrate proteins in a total volume of 40 μL methylation buffer (50 mm Tris⋅HCl pH 9, 100 mm NaCl, 5 mm DTT) supplemented with 0.76 μm radioactive labeled AdoMet (PerkinElmer) and 0.6 μm SMYD2 for 3–8 h at 25 °C. The reactions were stopped by the addition of SDS loading buffer and incubation at 95 °C for 5 min. Thereafter, the samples were separated by 12 or 16 % SDS‐PAGE. This step was followed by the incubation of the gel in amplify NAMP100V (GE Healthcare) for 45 min and drying of the gel in vacuum at 60 °C for 90 min. The dried SDS gel was then exposed to a Hyperfilm high performance autoradiography film as described above.
Circular dichroism analysis: Circular dichroism (CD) spectra of SMYD2 were measured using a J‐815 circular dichroism spectrophotometer (Jasco) using 7 μm protein diluted in a buffer containing 100 mm KCl. The spectra were measured in a wavelength range between 190 nm and 240 nm by using a 0.1 mm cuvette. The measurement was performed with an accumulation of 120 scans and a scan speed of 200 nm min−1. The spectrum obtained with the dialysis buffer II was used as baseline. For the determination of the melting curve, 16 μm protein was used. The measurement was performed at a wavelength of 210 nm in the temperature range of 20 and 80 °C using a heating rate of 1 °C min−1. The obtained data was analyzed with Microsoft Excel as described.42
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by Deutsche Forschungsgemeinschaft (DFG) grant JE 252/7‐4.
S. Weirich, M. K. Schuhmacher, S. Kudithipudi, C. Lungu, A. D. Ferguson, A. Jeltsch, ChemBioChem 2020, 21, 256.
References
- 1. Lanouette S., Mongeon V., Figeys D., Couture J. F., Mol. Syst. Biol. 2014, 10, 724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Clarke S. G., Trends Biochem. Sci. 2013, 38, 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang X., Huang Y., Shi X., Cell. Mol. Life Sci. 2015, 72, 4257–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Biggar K. K., Li S. S., Nat. Rev. Mol. Cell Biol. 2015, 16, 5–17. [DOI] [PubMed] [Google Scholar]
- 5. Del Rizzo P. A., Trievel R. C., Biochim. Biophys. Acta Gene Regul. Mech. 2014, 1839, 1404–1415. [DOI] [PubMed] [Google Scholar]
- 6. Boriack-Sjodin P. A., Swinger K. K., Biochemistry 2016, 55, 1557–1569. [DOI] [PubMed] [Google Scholar]
- 7. Spellmon N., Holcomb J., Trescott L., Sirinupong N., Yang Z., Int. J. Mol. Sci. 2015, 16, 1406–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tracy C., Warren J. S., Szulik M., Wang L., Garcia J., Makaju A., Russell K., Miller M., Franklin S., Curr. Opin. Physiol. 2018, 1, 140–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brown M. A., R. J. Sims III , Gottlieb P. D., Tucker P. W., Mol. Cancer 2006, 5, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abu-Farha M., Lambert J. P., Al-Madhoun A. S., Elisma F., Skerjanc I. S., Figeys D., Mol. Cell. Proteomics 2008, 7, 560–572. [DOI] [PubMed] [Google Scholar]
- 11. Huang J., Perez-Burgos L., Placek B. J., Sengupta R., Richter M., Dorsey J. A., Kubicek S., Opravil S., Jenuwein T., Berger S. L., Nature 2006, 444, 629–632. [DOI] [PubMed] [Google Scholar]
- 12. Saddic L. A., West L. E., Aslanian A., J. R. Yates, 3rd , Rubin S. M., Gozani O., Sage J., J. Biol. Chem. 2010, 285, 37733–37740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marouco D., Garabadgiu A. V., Melino G., Barlev N. A., Oncotarget 2013, 4, 1556–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ferguson A. D., Larsen N. A., Howard T., Pollard H., Green I., Grande C., Cheung T., Garcia-Arenas R., Cowen S., Wu J., Godin R., Chen H., Keen N., Structure 2011, 19, 1262–1273. [DOI] [PubMed] [Google Scholar]
- 15. Wang L., Li L., Zhang H., Luo X., Dai J., Zhou S., Gu J., Zhu J., Atadja P., Lu C., Li E., Zhao K., J. Biol. Chem. 2011, 286, 38725–38737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu J., Cheung T., Grande C., Ferguson A. D., Zhu X., Theriault K., Code E., Birr C., Keen N., Chen H., Biochemistry 2011, 50, 6488–6497. [DOI] [PubMed] [Google Scholar]
- 17. Hamamoto R., Saloura V., Nakamura Y., Nat. Rev. Cancer 2015, 15, 110–124. [DOI] [PubMed] [Google Scholar]
- 18. Zhang X., Tanaka K., Yan J., Li J., Peng D., Jiang Y., Yang Z., Barton M. C., Wen H., Shi X., Proc. Natl. Acad. Sci. USA 2013, 110, 17284–17289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jiang Y., Trescott L., Holcomb J., Zhang X., Brunzelle J., Sirinupong N., Shi X., Yang Z., J. Mol. Biol. 2014, 426, 3413–3425. [DOI] [PubMed] [Google Scholar]
- 20. Piao L., Kang D., Suzuki T., Masuda A., Dohmae N., Nakamura Y., Hamamoto R., Neoplasia 2014, 16, 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reynoird N., Mazur P. K., Stellfeld T., Flores N. M., Lofgren S. M., Carlson S. M., Brambilla E., Hainaut P., Kaznowska E. B., Arrowsmith C. H., Khatri P., Stresemann C., Gozani O., Sage J., Genes Dev. 2016, 30, 772–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nakakido M., Deng Z., Suzuki T., Dohmae N., Nakamura Y., Hamamoto R., Neoplasia 2015, 17, 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Weirich S., Jeltsch A. in Encyclopedia of Cancer, 3rd ed (Eds.: P. Boffetta, P. Hainaut), Elsevier, Amsterdam, 2019, pp. 538–550. [Google Scholar]
- 24. Yi X., Jiang X. J., Fang Z. M., Clin. Epigenetics 2019, 11, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weirich S., Kudithipudi S., Jeltsch A., J. Mol. Biol. 2016, 428, 2344–2358. [DOI] [PubMed] [Google Scholar]
- 26. Olsen J. B., Cao X. J., Han B., Chen L. H., Horvath A., Richardson T. I., Campbell R. M., Garcia B. A., Nguyen H., Mol. Cell. Proteomics 2016, 15, 892–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lanouette S., Davey J. A., Elisma F., Ning Z., Figeys D., Chica R. A., Couture J. F., Structure 2015, 23, 206–215. [DOI] [PubMed] [Google Scholar]
- 28. Ahmed H., Duan S., Arrowsmith C. H., Barsyte-Lovejoy D., Schapira M., J. Proteome Res. 2016, 15, 2052–2059. [DOI] [PubMed] [Google Scholar]
- 29. Rathert P., Dhayalan A., Murakami M., Zhang X., Tamas R., Jurkowska R., Komatsu Y., Shinkai Y., Cheng X., Jeltsch A., Nat. Chem. Biol. 2008, 4, 344–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dhayalan A., Kudithipudi S., Rathert P., Jeltsch A., Chem. Biol. 2011, 18, 111–120. [DOI] [PubMed] [Google Scholar]
- 31. Kudithipudi S., Dhayalan A., Kebede A. F., Jeltsch A., Biochimie 2012, 94, 2212–2218. [DOI] [PubMed] [Google Scholar]
- 32. Kudithipudi S., Lungu C., Rathert P., Happel N., Jeltsch A., Chem. Biol. 2014, 21, 226–237. [DOI] [PubMed] [Google Scholar]
- 33. Schuhmacher M. K., Kudithipudi S., Kusevic D., Weirich S., Jeltsch A., Biochim. Biophys. Acta Gene Regul. Mech. 2015, 1849, 55–63. [DOI] [PubMed] [Google Scholar]
- 34. Kudithipudi S., Schuhmacher M. K., Kebede A. F., Jeltsch A., ACS Chem. Biol. 2017, 12, 958–968. [DOI] [PubMed] [Google Scholar]
- 35. Schuhmacher M. K., Rolando M., Brohm A., Weirich S., Kudithipudi S., Buchrieser C., Jeltsch A., J. Mol. Biol. 2018, 430, 1912–1925. [DOI] [PubMed] [Google Scholar]
- 36. Kudithipudi S., Kusevic D., Weirich S., Jeltsch A., J. Vis. Exp. 2014, e52203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Frank R., J. Immunol. Methods 2002, 267, 13–26. [DOI] [PubMed] [Google Scholar]
- 38. Obenauer J. C., Cantley L. C., Yaffe M. B., Nucleic Acids Res. 2003, 31, 3635–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hornbeck P. V., Kornhauser J. M., Latham V., Murray B., Nandhikonda V., Nord A., Skrzypek E., Wheeler T., Zhang B., Gnad F., Nucleic Acids Res. 2019, 47, D433–D441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. George R. A., Lin K., Heringa J., Nucleic Acids Res. 2005, 33, W160–W163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jeltsch A., Lanio T., Methods Mol. Biol. 2002, 182, 85–94. [DOI] [PubMed] [Google Scholar]
- 42. Greenfield N. J., Nat. Protoc. 2006, 1, 2527–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary