

Abstract
Purpose
Diffuse large B‐cell lymphoma (DLBCL), the most common non‐Hodgkin lymphoma, is a heterogeneous lymphoma with different clinical manifestations and molecular alterations, and several markers are currently being measured routinely for its diagnosis, subtyping, or prognostication by immunohistochemistry (IHC). Here, the utility of a reverse‐phase‐protein‐array (RPPA) as a novel supportive tool to measure multiple biomarkers for DLBCL diagnosis is validated.
Experimental design
The expression of seven markers (CD5, CD10, BCL2, BCL6, MUM1, Ki‐67, and C‐MYC) is analyzed by RPPA and IHC using 37 DLBCL tissues, and the correlation between the two methods is determined. To normalize tumor content ratio in the tissues, the raw RPPA values of each marker are adjusted by that of CD20 or PAX‐5.
Results
The CD20‐adjusted data for CD5, MUM1, BCL2, Ki‐67, and C‐MYC has better correlation with IHC results than PAX‐5‐adjusted data. Receiver operating characteristic (ROC) analysis reveals that CD5, MUM1, BCL2, and C‐MYC exhibit a better sensitivity and specificity >0.750. Furthermore, the CD20‐adjusted C‐MYC value strongly correlates with that of IHC, and has a particularly high specificity (0.882).
Conclusions and clinical relevance
Although further investigation using a large number of DLBCL specimens needs to be conducted, these results suggest that RPPA could be applicable as a supportive tool for determining lymphoma prognosis.
Keywords: C‐MYC, diffuse large B‐cell lymphoma (DLBCL), immunohistochemistry (IHC), lymphoma prognosis, reverse‐phase‐protein‐array (RPPA)
1. Introduction
Diffuse large B‐cell lymphoma (DLBCL) is the most common non‐Hodgkin lymphoma in adults, accounting for one‐third of cases.1 DLBCL is regarded as a heterogeneous group of lymphomas with varying clinical manifestations and underlying molecular alterations, and prognostic subtyping is currently based on immunohistochemical (IHC) algorithms, gene expression profiling, and fluorescence in‐situ hybridization. Hans et al. developed an IHC algorithm that classified DLBCL into germinal center B cell (GCB) and non‐GCB subtypes by analyzing expression patterns of CD10, BCL6, and MUM1,2 and patients with the GCB subtype have generally favorable outcomes compared with those with the non‐GCB subtype.2, 3, 4 DLBCL with CD5 expression accounts for approximately 10% of DLBCL,5 and is characterized by aggressive clinical features and frequent central nervous system relapses.6, 7, 8 Previous studies have investigated the protein abundance and/or gene translocation of C‐MYC, BCL2, and BCL6, which may also have predictive prognostic potential.9, 10, 11, 12, 13, 14, 15, 16, 17, 18 Flow cytometry has been used for the evaluation of cell surface markers to diagnose malignant lymphomas. IHC is especially important for pathological diagnosis and prognostication of DLBCL. However, IHC specimens occasionally show heterogeneous, weak, or nonspecific staining, and such specimens make it difficult to evaluate protein abundance in a reproducible manner. Moreover, IHC is a semi‐quantitative method for measuring protein abundance in vivo, and cannot provide quantitative results. For clinical DLBCL diagnosis, additional quantitative methods are required to improve the false positive rate, sensitivity, and specificity.
In the present study, we focused on reverse phase protein arrays (RPPA), which are a high‐throughput clinical proteomics technique, and evaluated whether RPPA can provide quantitative diagnostic information for DLBCL diagnosis. An RPPA analytical system using clinical specimens was previously established by Liotta's group.19 RPPA analysis is a proteomic technology providing quantitative, multiplexed analysis of protein abundance and posttranslational modifications in limited clinical specimens such as cells, tissues, and body fluids. RPPA uses an approach similar to western blotting (antibody‐based), but the methodology is different, as there is no separation based on weight. This makes it very important to use highly validated antibodies that do not show any non‐specificity upon western blotting. An RPPA microarray can blot more than 1000 protein spots on a single glass slide over a nitrocellulose membrane, allowing for the detection of target proteins with specific antibodies. The greatest advantage of RPPA analysis is that about 10 nL of sample is enough for printing each spot, and a wider range of protein concentrations are acceptable for accurate quantification by RPPA analysis than by western blotting. RPPA analysis is therefore well suited for the detection and quantitation of proteins in clinical samples.
The aim of the present study is to validate the utility of RPPA as a novel complementary diagnostic method for quantitative detection of multiple biomarkers for DLBCL alongside IHC. We analyzed and compared the expression of seven biomarkers by RPPA and IHC using tumor tissue specimens from 37 DLBCL cases, and investigated the correlation between the two methods. We attempted to normalize the tumor cell ratios of the tissue samples used for RPPA. We also determined the specificity and sensitivity of the signal intensities following RPPA analysis. To our knowledge, this is the first comparison of RPPA and IHC using malignant lymphoma tissue samples in the clinical context.
2. Experimental Section
2.1. Case Selection
The ethics committee of the Kanagawa Cancer Center approved the experimental design. The cohort comprised of 37 cases diagnosed as DLBCL from 2007 to 2014 in Kanagawa Cancer Center. Cases were selected for inclusion in the cohort based on the availability of formalin‐fixed paraffin‐embedded (FFPE) and frozen tissue obtained from biopsy, with DLBCL diagnosed according to 2017 World Health Organization (WHO) classification criteria.20 The clinical data were retrieved from medical records. Frozen tissue specimens were stored at –80 °C at Kanagawa Cancer Center Biobank. Cases that had received chemotherapy prior to biopsy were excluded.
Clinical Relevance
Diffuse large B‐cell lymphoma (DLBCL), the most common non‐Hodgkin lymphoma, is a heterogeneous lymphoma with different clinical manifestations and molecular alterations, and several markers are currently being measured routinely for its diagnosis, subtyping, or prognostication by immunohistochemistry (IHC). However, IHC specimens occasionally show heterogeneous, weak, or nonspecific staining, making it difficult to evaluate protein abundance in a reproducible manner. Moreover, IHC is a semi‐quantitative method for measuring protein abundance in vivo, and cannot provide quantitative results. For clinical DLBCL diagnosis, additional quantitative methods are required to improve the false positive rate, sensitivity, and specificity. By contrast, reverse phase protein array (RPPA) can automatically analyze and quantify protein abundance, and can provide high reproducibility for high‐throughput analysis. Moreover, RPPA enables quantitative analysis for many biomarkers that are expressed in small amount of cancer tissue.
Here, RPPA analysis revealed four biomarkers that may have clinical utility in diagnosing and classifying DLBCL. In particular, CD20‐adjusted RPPA expression of C‐MYC strongly correlated with IHC data, and had high specificity. Since RPPA can analyze and simultaneously quantify multiple proteins in many biological samples, it could be suitable for molecular analysis of lymphoma including DLBCL.
2.2. Antibodies
Monoclonal antibodies (mAbs) used for IHC and RPPA against CD10 (ab208778), BCL6 (ab172610), MUM1 (ab133590), Ki‐67 (ab92742), C‐MYC (ab32072), CD20 (ab9475), and PAX5 (ab109443) were obtained from Abcam (Cambridge, UK). Anti‐CD5 mAb (MA5‐13308) was obtained from Thermo Fisher Scientific (Waltham, MA). Anti‐BCL2 mAb (M0887) was obtained from Agilent (Santa Clara, CA). Detailed antibody information is presented in Table 1a.
Table 1a.
Antibodies used for the immunohistochemistry and RPPA
| Antibody | Clone | Type | Source | Dilution/IHC | Dilution/RPPA |
|---|---|---|---|---|---|
| CD20 | L26 | Mouse mAb | Abcam | 1:100 | 1:50 |
| PAX5 | EPR3730 (2) | Rabbit mAb | Abcam | 1:1000 | 1:1000 |
| CD5 | 4C7 | Mouse mAb | Thermo Fisher Scientific | 1:50 | 1:100 |
| CD10 | EPR5904‐110 | Rabbit mAb | Abcam | 1:1000 | 1:250 |
| BCL6 | EPR11410‐43 | Rabbit mAb | Abcam | 1:250 | 1:250 |
| MUM1 | EP5699 | Rabbit mAb | Abcam | 1:500 | 1:500 |
| BCL2 | 124 | Mouse mAb | Dako | 1:100 | 1:50 |
| Ki‐67 | EPR3610 | Rabbit mAb | Abcam | 1:500 | 1:1000 |
| C‐MYC | Y69 | Rabbit mAb | Abcam | 1:250 | 1:500 |
mAb, monoclonal antibody.
2.3. Western Blot Analysis
Total cell lysates (15 µg per lane) were separated by SDS‐PAGE on a 5–20% gradient gel under reducing conditions. After electrophoresis, the proteins were transferred electrophoretically onto an Immobilon membrane (Millipore). Nonspecific sites were blocked with 5% dry milk in TBS‐T (TBS containing 0.05% Tween‐20) at 37 °C for 1 h, and the membrane was then incubated overnight at 4 °C with mAbs at the concentrations indicated in Table 1a. After washing with TBS‐T, the membrane was incubated for 1 h with peroxidase‐conjugated anti‐rabbit IgG or anti‐mouse IgG (GE Healthcare UK Ltd., UK) The resulting bands were detected by chemiluminescence according to the manufacturer's instructions (PerkinElmer, Waltham, MA). An ImmunoStar LD western blotting detection system (FUJIFILM Wako Pure Chemical Co., Tokyo, Japan) was used for detection.
2.4. Immunohistochemical Analysis
IHC staining was performed on FFPE sections (4 µm thick) using an automated immunostainer (HISTOSTAINER 48A; Nichirei, Tokyo, Japan) with primary antibodies listed in Table 1a. The expression of each marker was detected using commercially available detection kits (Histofine Simple Stain MAX‐PO kit; Nichirei, Tokyo, Japan). Appropriate positive and negative control tissues were used in each case. Staining for CD5, CD10, BCL2, BCL6, MUM1, PAX5, and CD20 were classified by a score from 1 to 6 based on the proportion of immunopositive tumor cells (1: <1%, 2: 1–9%, 3: 10–29%, 4: 30–49%, 5: 50–79%, 6: 80–100%). Staining for C‐MYC and Ki‐67 were evaluated in increments of 10%.
2.5. Reverse Phase Protein Array
Frozen DLBCL tissue was homogenized using a bead homogenizer (Yasui Kikai, Osaka, Japan). Tumor tissue and cell lysate were extracted from the respective specimens using T‐PER protein extraction reagent (Thermo Fisher Scientific, Waltham, MA) supplemented with inhibitors (100 mm NaF, 1 mm Na3VO4, 10 mm NaPPi, 1 mm EDTA, PhosSTOP; Sigma–Aldrich, Missouri, MO) and protease inhibitor cocktail (1.04 mm AEBSF, 800 nm aprotinin, 40 µm bestatin, 14 µm E‐64, 20 µm leupeptin, 15 µm pepstatin A; (Sigma–Aldrich). The lysates were centrifuged at 15 000 × g at 4 °C for 20 min, and the supernatant was subjected to following analyses. After adjustment of protein concentration to about 1.5 mg mL–1 according to Bradford protein assay, the lysates were manually diluted in twofold serial dilutions with extraction buffer. The diluted lysates were boiled with 2% SDS and 2.5% β‐mercaptoethanol, and printed onto nitrocellulose‐coated slides in four replicates (Grace Bio‐Labs, Bend, OR) using an Aushon Biosystems 2470 arrayer (Burlington, MA). After blocking with an odyssey blocking buffer (LI‐COR biosciences, Lincoln, NE) supplemented with 0.1% Tween‐20, the blotted slides were probed with validated primary antibodies, followed by secondary antibodies conjugated to infrared dyes, IRDye 680RD and 800CW (LI‐COR biosciences). Slides were scanned using an ODYSSEY scanner (LI‐COR biosciences). The signal intensity of each spot was quantified using Image Studio (LI‐COR biosciences) according to the manufacturer's instructions.
2.6. Cells and Culture Conditions
Human hepatoblastoma, HepG2, pancreatic epithelioid carcinoma, Panc‐1, normal embryonic kidney epithelium, HEK293T, endocervical adenocarcinoma, HeLa, astrocytoma, U‐251 MG (KO), KHM‐10B and Daudi, Burkitt's lymphoma, and T acute lymphoblastic leukemia, CCRF‐CEM cells were purchased from JCRB cell bank (National Institute of Biomedical Innovation, Health and Nutrition, Osaka Japan). Human colorectal carcinoma, HCT‐116, and ovarian serous adenocarcinoma, OVCAR‐8, were purchased from ATCC as a part of the NCI‐60 cancer cell line panel. HepG2, Panc‐1, HEK293T, HeLa, and U‐251 MG (KO) were cultured in DMEM (Thermo Fisher Scientific) supplemented with 10% FBS at 37 °C and 5% CO2. HCT‐116, OVCAR‐8, KHM‐10B, and CCRF‐CEM were cultured in RPMI 1640 medium (Thermo Fisher Scientific) supplemented with 10% FBS. Daudi cells were cultured in RPMI 1640 medium supplemented with 20% FBS at 37 °C and 5% CO2.
2.7. Statistical Analysis
The results acquired from immunohistochemical staining and RPPA analysis were compared using the Spearman's rank correlation method. Receiver operating characteristic (ROC) curves, plotted as true positive fraction against false positive fraction, were used to analyze diagnostic accuracy. Statistical analysis was conducted using SPSS version 23 (IBM, NY, USA).
3. Results
3.1. Clinicopathological Features
The clinicopathological features of the 37 cases comprising the present cohort are summarized in Table 1b. The ages of these cases ranged from 19 to 86 years (median 64 years). Nineteen patients were female (51%), and 18 were male (49%). The biopsy site was the cervical lymph node in 16 (43%) cases, the axillary lymph node in three (8%), the inguinal lymph node in three (8%), the abdominal lymph node in three (8%), the retroperitoneal lymph node in two (5%), the subcutaneous tissue in four (11%), the thyroid gland in three (8%), the testis in two (5%), and the mammary gland in one (3%) case. Histologically, all cases showed diffuse proliferation of large neoplastic lymphoid cells with prominent nucleoli. Except for a case of methotrexate‐associated DLBCL, 36 cases were designated as DLBCL, not otherwise specified, and two were transformed cases from follicular lymphomas.
Table 1b.
Clinicopathological features of diffuse large B‐cell lymphoma cases
| Total number | 37 |
|---|---|
| Age (year) | |
| Median | 64 |
| Range | 19–86 |
| Sex (n) | |
| Female | 19 |
| Male | 18 |
| LDH | |
| Normal | 15 |
| Elevated | 22 |
| PS | |
| 0–1 | 31 |
| 2–4 | 6 |
| Ann Arbor stage | |
| I | 6 |
| II | 11 |
| III | 7 |
| IV | 13 |
| IPI score | |
| 0–1 | 12 |
| 2 | 8 |
| 3 | 8 |
| 4–5 | 8 |
| Unknown | 1 |
PS, performance status; IPI, International Prognostic Index.
3.2. Validation of Antibodies for IHC and RPPA Analysis
To select mAbs for IHC and RPPA analysis, we checked the specificity of mAbs using cell lysates obtained from normal and cancer cells, including DLBCL cell lines (KHM‐10B and Daudi). Cell lysates from ten cell lines were subjected to western blot to select highly specific mAbs against CD10, CD5, BCL6, BCL2, Ki‐67, MUM‐1, C‐MYC, PAX5, and CD20. The mAbs against eight of these (CD10, CD5, BCL6, BCL2, MUM‐1, C‐MYC, PAX5, and CD20) detected a single or a major band with the predicted molecular weight, while the mAb against Ki‐67 showed multiple smeared bands with broad range of molecular weights (Figure 1). We next confirmed positive correlations between western blotting and RPPA. RPPA slides were prepared using the same set of cell lysates and the screened antibodies were used as probes. Comparison of signal intensities obtained from RPPA and western blotting are shown in Figure S1, Supporting Information. RPPA signal intensity obtained with all nine mAbs was positively correlated with the results of western blot analysis, with correlation coefficients ranging from 0.842 to 1.000. These nine mAbs were therefore used for IHC and RPPA analysis using lymphoma tissues.
Figure 1.
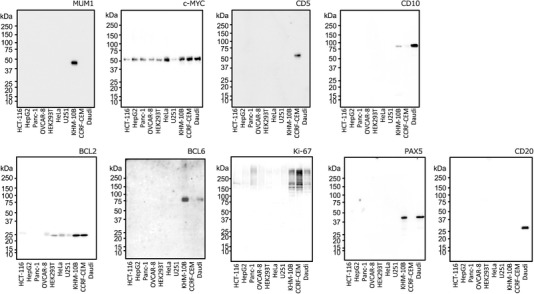
Validation of monoclonal antibodies (mAbs) by western blot analysis. Total cell lysates (15 µg per lane) prepared from ten cell lines were analyzed by western blot using mAbs against nine biomarkers for DLBCL.
3.3. Immunohistochemical Features
Lymphoma cells obtained from patient samples were positive for prognostic markers (Table 2a,b and Figure 2). These markers were often expressed even in background cells, such as T‐cells positive for CD5 and BCL2, and interstitial cells positive for CD10 (Figure S3, Supporting Information). In all cases, lymphoma cells showed diffuse strong positive immunoreactivity for CD20. Almost all cases showed diffuse expression for PAX5, but some cases showed heterogeneous and/or weak expression for PAX5.
Table 2a.
Immunohistochemical analysis of CD5, CD10, BCL6, MUM1, BCL2, PAX5, and CD20
| Markers | Proportion of immunopositive tumor cells | ||||||
|---|---|---|---|---|---|---|---|
| Score 1 | Score 2 | Score 3 | Score 4 | Score 5 | Score 6 | ||
| <1% | 1–9% | 10–29% | 30–49% | 50–79% | >80% | Total | |
| CD5 | 28 | 1 | 3 | 0 | 4 | 1 | 37 |
| CD10 | 20 | 1 | 2 | 0 | 3 | 11 | 37 |
| BCL6 | 5 | 3 | 4 | 4 | 14 | 7 | 37 |
| MUM1 | 4 | 1 | 10 | 5 | 9 | 8 | 37 |
| BCL2 | 4 | 2 | 0 | 2 | 5 | 24 | 37 |
| PAX5 | 0 | 0 | 0 | 0 | 3 | 34 | 37 |
| CD20 | 0 | 0 | 0 | 0 | 0 | 37 | 37 |
Table 2b.
Immunohistochemical analysis of Ki‐67 and C‐MYC
| Markers | Proportion of immunopositive tumor cells | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 10–19% | 20–29% | 30–39% | 40–49% | 50–59% | 60–69% | 70–79% | 80–89% | >90% | Total | |
| Ki‐67 | 0 | 0 | 1 | 0 | 3 | 3 | 18 | 7 | 5 | 37 |
| C‐MYC | 3 | 9 | 5 | 1 | 3 | 3 | 8 | 3 | 2 | 37 |
Figure 2.
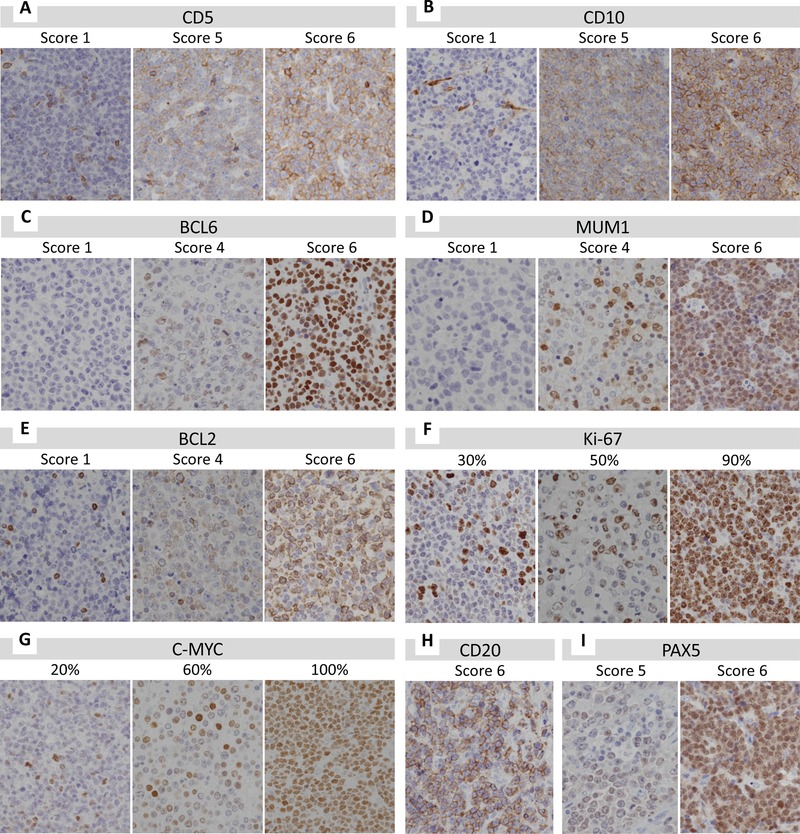
Immunohistochemistry analysis of each marker in DLBCL cells. A–G) DLBCL cells were positive for CD5 (A), CD10 (B), BCL6 (C), MUM1 (D), BCL2 (E), Ki‐67 (F), and C‐MYC (G). H) All cases showed diffuse positive staining for CD20. I) Most cases showed diffuse positive for PAX5, but some cases showed heterogeneous and/or weak PAX5 expression.
3.4. Quantitative Analysis of Nine DLBCL Biomarker Expressions by RPPA
To measure nine kinds of biomarker expressions in 37 DLBCL tissues quantitatively, the RPPA were performed using validated mAbs as shown in Figure 1. The detection images of RPPA obtained by an IR image scanner are shown in Figure S2, Supporting Information. The average expression values are shown in Table S1, Supporting Information and plotted on Figure 3a.
Figure 3.
a) Correlation between the results of reverse phase protein array (RPPA) and immunohistochemistry (IHC) for each marker. A) For CD5, the correlation coefficient with IHC data was highest in CD20‐adjusted data and lowest in PAX5‐adjusted data. B) For CD10, the correlation coefficient with IHC data was highest in the raw data and lowest in CD20‐adjusted data. C) For BCL6, the correlation coefficient with IHC data was highest in the raw data and lowest in CD20‐adjusted data. D) For MUM1, the correlation coefficient with IHC data was highest in CD20‐adjusted data and lowest in PAX5‐adjusted data. E) For BCL2, the correlation coefficient with IHC data was highest in CD20‐adjusted data and lowest in the raw data. F) For Ki‐67, the correlation coefficient with IHC data was highest in CD20‐adjusted data and lowest in the raw data. G) For C‐MYC, the correlation coefficient with IHC data was highest in CD20‐adjusted data and lowest in PAX5‐adjusted data. R = correlation coefficient. b) Receiver operating characteristic (ROC) analysis of the signal intensities obtained by reverse phase protein array (RPPA) analysis. A) ROC curves of the raw and CD20‐adjusted RPPA data for CD5. B) ROC curves of the raw and CD20‐adjusted RPPA data for CD10. C) ROC curves of the raw and CD20‐adjusted RPPA data for BCL6. D) ROC curves of the raw and CD20‐adjusted RPPA data for MUM1. E) ROC curves of the raw and CD20‐adjusted RPPA data for BCL2. F) ROC curves of the raw and CD20‐adjusted RPPA data for Ki‐67. G) ROC curves of the raw and CD20‐adjusted RPPA data for C‐MYC.
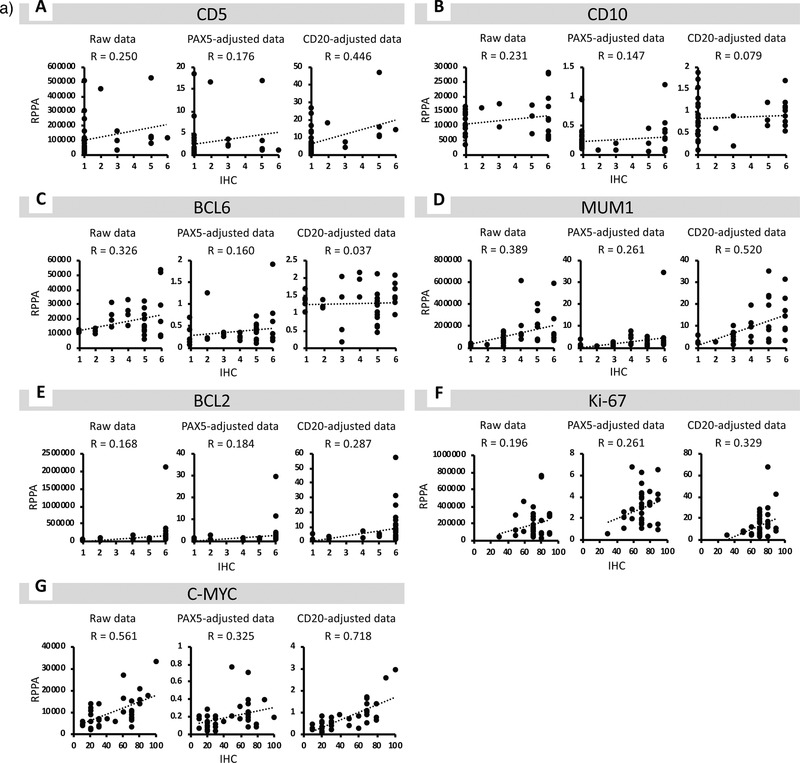
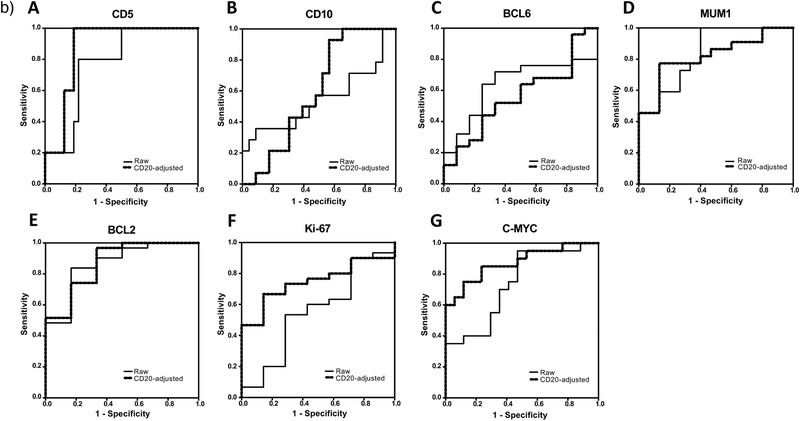
3.5. Correlation of RPPA and IHC Diagnostic Values
The correlation between diagnostic values obtained from RPPA and IHC analysis is shown in Figure 3a. To normalize the tumor cell content in each tissue, the raw RPPA values of CD5, CD10, BCL6, MUM1, BCL2, Ki‐67, and C‐MYC were adjusted to that of PAX5 or CD20. CD20‐adjusted results had correlation coefficients of 0.446, 0.079, 0.037, 0.520, 0.287, 0.329, and 0.718 for CD5, CD10, BCL6, MUM1, BCL2, Ki‐67, and C‐MYC, respectively. In contrast, PAX5‐adjusted results had correlation coefficients of 0.176, 0.147, 0.160, 0.261, 0.184, 0.261, and 0.325 for of CD5, CD10, BCL6, MUM1, BCL2, Ki‐67, and C‐MYC, respectively. CD20‐adjusted RPPA values for CD5, MUM1, BCL2, Ki‐67, and C‐MYC had better correlation with IHC results than raw or PAX‐5‐adjusted RPPA values.
3.6. ROC Analysis
ROC analysis was carried out using RPPA analysis results (Figure 3b and Table 2c). For CD5, CD10, BCL2, BCL6, and MUM1, an IHC score of 1 to 3 was classified as IHC‐negative, while a score of 4 to 6 was considered as IHC‐positive. ICH cutoff values for Ki‐67 and C‐MYC were set at 69% and 39%, respectively. The ROC AUC with RPPA raw data for CD5, CD10, BCL6, MUM1, BCL2, Ki‐67, and C‐MYC was 0.775, 0.543, 0.640, 0.839, 0.866, 0.552, and 0.738, respectively. In contrast, the ROC AUC with CD20‐adjusted RPPA data for CD5, CD10, BCL6, MUM1, BCL2, Ki‐67, and C‐MYC was 0.875, 0.599, 0.563, 0.818, 0.871, 0.748, and 0.874, respectively. The CD20‐adjusted AUC values for CD5, CD10, BCL2, Ki‐67, and C‐MYC were better than those of their raw data. Furthermore, four biomarkers (CD5, MUM1, BCL2, and C‐MYC) had both high sensitivity (1.000, 0.783, 0.750, and 0.750, respectively) and specificity (0.788, 0.867, 0.833, and 0.882, respectively).
Table 2c.
Results of ROC analysis
| Markers | Data | AUC | Sensitivity | Specificity |
|---|---|---|---|---|
| CD5 | Raw data | 0.775 | 0.800 | 0.788 |
| PAX5‐adjusted data | 0.612 | 0.600 | 0.606 | |
| CD20‐adjusted data | 0.875 | 1.000 | 0.788 | |
| CD10 | Raw data | 0.543 | 0.571 | 0.542 |
| PAX5‐adjusted data | 0.565 | 0.571 | 0.708 | |
| CD20‐adjusted data | 0.599 | 0.929 | 0.417 | |
| BCL6 | Raw data | 0.640 | 0.720 | 0.692 |
| PAX5‐adjusted data | 0.580 | 0.560 | 0.692 | |
| CD20‐adjusted data | 0.563 | 0.520 | 0.615 | |
| MUM1 | Raw data | 0.839 | 0.739 | 0.733 |
| PAX5‐adjusted data | 0.752 | 0.609 | 0.733 | |
| CD20‐adjusted data | 0.818 | 0.783 | 0.867 | |
| BCL2 | Raw data | 0.866 | 0.813 | 0.833 |
| PAX5‐adjusted data | 0.731 | 0.750 | 0.667 | |
| CD20‐adjusted data | 0.871 | 0.750 | 0.833 | |
| Ki‐67 | Raw data | 0.552 | 0.533 | 0.750 |
| PAX5‐adjusted data | 0.667 | 0.633 | 0.875 | |
| CD20‐adjusted data | 0.748 | 0.667 | 0.750 | |
| C‐MYC | Raw data | 0.738 | 0.700 | 0.647 |
| PAX5‐adjusted data | 0.756 | 0.750 | 0.647 | |
| CD20‐adjusted data | 0.874 | 0.750 | 0.882 |
4. Discussion
To examine the utility of RPPA in cancer diagnosis, we compared the correlation between prognostic biomarker expression in DLBCL tissue using both RPPA and conventional IHC analyses. Notably, CD20‐adjusted RPPA values were positively correlated with IHC expression in CD5, MUM1, BCL2, Ki‐67, and C‐MYC. ROC analysis of CD20‐adjusted RPPA values showed that CD5, MUM1, BCL2, and C‐MYC had high sensitivity (0.750) and specificity (0.882). RPPA C‐MYC expression was strongly correlated with IHC expression, and had particularly high specificity. According to IHC analysis, lymphoma cells often had heterogeneous C‐MYC expression, which may confound the use of C‐MYC expression as a prognostic biomarker. In contrast to IHC, RPPA provides highly quantitative, reproducible measurements for protein abundance in clinical tissue, and therefore could be used to determine protein abundance in clinical tissue that is complementary to IHC.21
Although DLBCL is a clinicopathologically heterogeneous group of lymphomas, DLBCL with CD5 expression is characterized by aggressive clinical features.5, 6, 7, 8 The prognostic significance of C‐MYC expression has been controversial, but some studies suggest that C‐MYC expression is associated with poor prognosis in patients with DLBCL.11, 14, 17 Co‐expression of C‐MYC and BCL2 may also contribute to poor prognosis in DLBCL cases.9, 10, 13 Our RPPA data for CD5, BCL2, and C‐MYC were positively correlated with IHC results, and also showed high sensitivity and specificity. RPPA enables simultaneous quantification of multiple protein markers in a large number of biological samples, and has previously been used for biomarker analysis in tumors.13, 21, 22, 23, 24, 25, 26, 27 Advantages of using RPPA over conventional biochemical assays, such as western blotting and ELISAs, include RPPA's ability to provide high‐throughput quantification and the requirement of less material for analysis.
As RPPA can be used to analyze many samples at once, it is suitable for use in a research setting but may not be appropriate for clinical use with a few samples. However, it may offer advantages for clinical diagnosis, subtyping, and prognostic classification of lymphoma cases, as it can be used to evaluate a number of biomarkers and could be applied for prediction of DLBCL outcomes. Unfortunately, the RPPA results did not correlate with those of IHC for CD10 and BCL6. Although DLBCL cases can be divided into two groups (GCB and non‐GCB subtype) based on the expression of a combination of the markers CD10, BCL6, and MUM1,2 it was difficult to divide the DLBCL cases into these subtypes using RPPA analysis alone. As the cause, in addition to DLBCL, CD10 is also expressed in many types of cells such as background lymphocytes, neutrophils, and interstitial cells, and BCL6 often shows heterogeneous expression in lymphoma cells (Figure S3, Supporting Information). Therefore, RPPA analysis without morphological information may not be suitable for classification of the GCB and non‐GCB subtyping.
To our knowledge, this is the first study that compared the results of RPPA and IHC using tissue samples from malignant lymphoma cases. IHC enables the identification of cells expressing biomarkers for cancer. However, RPPA detects protein abundance in cell lysates, which include both neoplastic and non‐neoplastic cells. A previous study quantified multiple biomarkers using formalin‐fixed paraffin‐embedded breast cancer tissues, and compared the results of RPPA and IHC.21 RPPA expression data for macro‐dissected tumor samples was able to separate the cases into the respective IHC‐based groups for seven of eight biomarkers. On the other hand, RPPA data obtained for full‐face tissue samples were discriminative for four of eight biomarkers. In the present study, the raw RPPA expression value for each marker was normalized to that of CD20 or PAX5 to account for the tumor cell ratios of the tissue samples. The CD20‐adjusted RPPA values generally had better correlation with IHC scores than the raw RPPA data. In contrast, the PAX5‐adjusted RPPA values had worse correlation with IHC scores. CD20 is therefore suitable for tumor content ratio normalization, and all cases exhibited diffuse strong positive immunoreactivity for CD20. We found that cases that received chemotherapy with rituximab had fewer CD20‐positive lymphoma cells, and the RPPA data did not correlate with that of IHC in these cases (data not shown). Therefore, the use of CD20 RPPA values to normalize for tumor content ratio should be restricted to untreated DLBCL cases. Based on these results, it appears that PAX5 is not a suitable marker for determining tumor content, possibly due to heterogeneous and/or weak expression in tumor cells. As PAX‐5 is a transcription factor in the nucleus, it may have resistance to solubility of a non‐ionic T‐PER Lysis buffer in RPPA analysis.
The specificity of each marker for tumor cells may also be an important factor in DLBCL diagnosis. There were only a few background cells with C‐MYC expression according to IHC, and this may explain why the CD20‐adjusted C‐MYC data had the highest correlation coefficient with the IHC data in the present study. In contrast, expression of other markers was immunohistochemically detected in background cells in some cases. Heterogeneous expression of BCL6 was often observed in lymphoma cells. CD10 and BCL6 expression by RPPA did not correlate with that of IHC, and further evaluation of these antibodies is therefore required.
In conclusion, we demonstrated that RPPA analysis using four biomarkers may have clinical utility in diagnosing and classifying DLBCL. CD20‐adjusted RPPA expression of C‐MYC strongly correlated with IHC data, and had high specificity (0.882). RPPA can analyze and simultaneously quantify multiple proteins in many biological samples, and is therefore suitable for molecular analysis of lymphoma including DLBCL. These findings strongly suggest that combination of quantitative and morphological biomarker information from RPPA and IHC could be a powerful tool for the diagnosis and prognosis of lymphomas. Further investigation into the correlation between RPPA data for relevant biomarkers and the clinical outcomes of each patient is needed to determine the clinical utility of this analysis.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
M.S. and A.M. contributed equally to this work. The authors are grateful to Yoshiyasu Nakamura and Mitsuyo Yoshihara, Kanagawa Cancer Center, for their expert technical assistance. This work was supported in part by Kanagawa Cancer Center Hospital‐Research Institute Joint Study, and by a Grant‐in‐Aid for Scientific Research from a Grant‐in‐Aid for Scientific Research on Innovative Areas (17H06329; N.K. and A.M.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT), a Grant‐in‐Aid for Scientific Research (C) 17K09027; N.K.), and a JSPS Core‐to Core program (Establishing International Research Network of Mathematical Oncology) from the Japan Society for the Promotion Science (N.K.).
Suzuki M., Muroi A., Nojima M., Numata A., Takasaki H., Sakai R., Yokose T., Miyagi Y., Koshikawa N., Utility of a Reverse Phase Protein Array to Evaluate Multiple Biomarkers in Diffuse Large B‐Cell Lymphoma. Prot. Clin. Appl. 2020, 14, 1900091 10.1002/prca.201900091
References
- 1. The Non‐Hodgkin's Lymphoma Classification Project , Blood 1997, 89, 3909. [PubMed] [Google Scholar]
- 2. Hans C. P., Weisenburger D. D., Greiner T. C., Gascoyne R. D., Delabie J., Ott G., Muller‐Hermelink H. K., Campo E., Braziel R. M., Jaffe E. S., Pan Z., Farinha P., Smith L. M., Falini B., Banham A. H., Rosenwald A., Staudt L. M., Connors J. M., Armitage J. O., Chan W. C., Blood 2004, 103, 275. [DOI] [PubMed] [Google Scholar]
- 3. Berglund M., Thunberg U., Amini R.‐M., Book M., Roos G., Erlanson M., Linderoth J., Dictor M., Jerkeman M., Cavallin‐Ståhl E., Sundström C., Rehn‐Eriksson S., Backlin C., Hagberg H., Rosenquist R., Enblad G., Mod. Pathol. 2005, 18, 1113. [DOI] [PubMed] [Google Scholar]
- 4. Alizadeh A. A., Eisen M. B., Davis R. E., Ma C., Lossos I. S., Rosenwald A., Boldrick J. C., Sabet H., Tran T., Yu X., Powell J. I., Yang L., Marti G. E., Moore T., Hudson J. Jr, Lu L., Lewis D. B., Tibshirani R., Sherlock G., Chan W. C., Greiner T. C., Weisenburger D. D., Armitage J. O., Warnke R., Levy R., Wilson W., Grever M. R., Byrd J. C., Botstein D., Brown P. O., et al., Nature 2000, 403, 503. [DOI] [PubMed] [Google Scholar]
- 5. Taniguchi M., Oka K., Hiasa A., Yamaguchi M., Ohno T., Kita K., Shiku H., Blood 1998, 91, 1145. [PubMed] [Google Scholar]
- 6. Yamaguchi M., Seto M., Okamoto M., Ichinohasama R., Nakamura N., Yoshino T., Suzumiya J., Murase T., Miura I., Akasaka T., Tamaru J., Suzuki R., Kagami Y., Hirano M., Morishima Y., Ueda R., Shiku H., Nakamura S., Blood 2002, 99, 815. [DOI] [PubMed] [Google Scholar]
- 7. Miyazaki K., Yamaguchi M., Suzuki R., Kobayashi Y., Maeshima A. M., Niitsu N., Ennishi D., Tamaru J. I., Ishizawa K., Kashimura M., Kagami Y., Sunami K., Yamane H., Nishikori M., Kosugi H., Yujiri T., Hyo R., Katayama N., Kinoshita T., Nakamura S., Ann. Oncol. 2011, 22, 1601. [DOI] [PubMed] [Google Scholar]
- 8. Yamaguchi M., Nakamura N., Suzuki R., Kagami Y., Okamoto M., Ichinohasama R., Yoshino T., Suzumiya J., Murase T., Miura I., Ohshima K., Nishikori M., Tamaru J., Taniwaki M., Hirano M., Morishima Y., Ueda R., Shiku H., Nakamura S., Haematologica 2008, 93, 1195. [DOI] [PubMed] [Google Scholar]
- 9. Green T. M., Young K. H., Visco C., Xu‐Monette Z. Y., Orazi A., Go R. S., Nielsen O., Gadeberg O. V., Mourits‐Andersen T., Frederiksen M., Pedersen L. M., Moller M. B., J. Clin. Oncol. 2012, 30, 3460. [DOI] [PubMed] [Google Scholar]
- 10. Johnson N. A., Slack G. W., Savage K. J., Connors J. M., Ben‐Neriah S., Rogic S., Scott D. W., Tan K. L., Steidl C., Sehn L. H., Cha W. C., Iqbal J., Meyer P. N., Lenz G., Wright G., Rimsza L. M., Valentino C., Brunhoeber P., Grogan T. M., Braziel R. M., Cook J. R., Tubbs R. R., Weisenburger D. D., Campo E., Rosenwald A., Ott G., Delabie J., Holcroft C., Jaffe E. S., Staudt L. M., et al., J. Clin. Oncol. 2012, 30, 3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kluk M. J., Chapuy B., Sinha P., Roy A., Cin P. D., Neuberg D. S., Monti S., Pinkus G. S., Shipp M. A., Rodig S. J., PLoS One 2012, 7, e33813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Horn H., Ziepert M., Becher C., Barth T. F. E., Bernd H‐W., Feller A. C., Klapper W., Hummel M., Stein H., Hansmann M‐L., Schmelter C., Möller P., Cogliatti S., Pfreundschuh M., Schmitz N., Trümper L., Siebert R., Loeffler M., Rosenwald A., Ott G., Blood 2013, 121, 2253. [DOI] [PubMed] [Google Scholar]
- 13. Hu S., Xu‐Monette Z. Y., Tzankov A., Green T., Wu L., Balasubramanyam A., Liu W. M., Visco C., Li Y., Miranda R. N., Montes‐Moreno S., Dybkaer K., Chiu A., Orazi A., Zu Y., Bhagat G., Richards K. L., Hsi E. D., Choi W. W., Zhao X., van Krieken J. H., Huang Q., Huh J., Ai W., Ponzoni M., Ferreri A. J., Zhou F., Slack G. W., Gascoyne R. D., Tu M., et al., Blood 2013, 121, 4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Perry A. M., Alvarado‐Bernal Y., Laurini J. A., Smith L. M., Slack G. W., Tan K. L., Sehn L. H., Fu K., Aoun P., Greiner T. C., Chan W. C., Bierman P. J., Bociek R. G., Armitage J. O., Vose J. M., Gascoyne R. D., Weisenburger D. D., Br. J. Haematol. 2014, 165, 382. [DOI] [PubMed] [Google Scholar]
- 15. Yan L. X., Liu Y. H., Luo D. L., Zhang F., Cheng Y., Luo X. L., Xu J., Cheng J., Zhuang H. G., PLoS One 2014, 9, e104068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou K., Xu D., Cao Y., Wang J., Yang Y., Huang M., PLoS One 2014, 9, e95020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou M., Wang J., Ouyang J., Xu J. Y., Chen B., Zhang Q. G., Zhou R. F., Yang Y. G., Shao X. Y., Xu Y., Chen Y. M., Fan X. S., Wu H. Y., Tumor Biol. 2014, 35, 6757. [DOI] [PubMed] [Google Scholar]
- 18. Kawamoto K., Miyoshi H., Yoshida N., Nakamura N., Ohshima K., Sone H., Takizawa J., Cancer Sci. 2016, 107, 853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Charboneau L., Tory H., Chen T., Winters M., Petricoin E. F., Liotta L. A., Paweletz C. P., Briefings Funct. Genomics Proteomics 2002, 1, 305. [DOI] [PubMed] [Google Scholar]
- 20. Swerdlow S. H., Campo E., Harris N. L., Jaffe E. S., Pileri S. A., Stein H., Thiele J. (eds), WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, IARC, Lyon, France, 2017. [Google Scholar]
- 21. Negm O. H., Muftah A. A., Aleskandarany M. A., Hamed M. R., Ahmad D. A., Nolan C. C., Diez‐Rodriguez M., Tighe P. J., Ellis I. O., Rakha E. A., Green A. R., Breast Cancer Res. Treat. 2016, 155, 25. [DOI] [PubMed] [Google Scholar]
- 22. Sewell A., Brown B., Biktasova A., Mills G. B., Lu Y., Tyson D. R., Issaeva N., Yarbrough W. G., Clin. Cancer Res. 2014, 20, 2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Du D., Ma W., Yates M. S., Chen T., Lu K. H., Lu Y., Weinstein J. N., Broaddus R. R., Mills G. B., Liu Y., Oncotarget 2018, 9, 19704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Patil V., Mahalingam K., Gene 2019, 685, 85. [DOI] [PubMed] [Google Scholar]
- 25. Spreafico F., Bongarzone I., Pizzamiglio S., Magni R., Taverna E., De Bortoli M., Ciniselli C. M., Barzano E., Biassoni V., Luchini A., Liotta L. A., Zhou W., Signore M., Verderio P., Massimino M., Oncotarget 2017, 8, 46177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Clarke C. N., Lee M. S., Wei W., Manyam G., Jiang Z. Q., Lu Y., Morris J., Broom B., Menter D., Vilar‐Sanchez E., Raghav K., Eng C., Chang G. J., Simon I., Bernards R., Overman M., Mills G. B., Maru D., Kopetz S., Ann. Surg. Oncol. 2017, 24, 4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Han G., Zhao W., Song X., Ng P. K. S., Karam J. A., Jonasch E., Mills G. B., Zhao Z., Ding Z., Jia P., BMC Genomics 2017, 18, 678. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information