

Abstract
Viruses can inhibit host autophagy through multiple mechanisms, and evasion of autophagy plays an important role in immune suppression and viral oncogenesis. Merkel cell polyomavirus (MCPyV) T‐antigens are expressed and involved in the pathogenesis of a large proportion of Merkel cell carcinoma (MCC). Yet, how MCPyV induces tumorigenesis is not fully understood. Herein, we show that MCPyV T‐antigens induce miR‐375, miR‐30a‐3p and miR‐30a‐5p expressions, which target multiple key genes involved in autophagy, including ATG7, SQSTM1 (p62) and BECN1. In MCC tumors, low expression of ATG7 and p62 are associated with MCPyV‐positive tumors. Ectopic expression of MCPyV small T‐antigen and truncated large T‐antigen (LT), but not the wild‐type LT, resulted in autophagy suppression, suggesting the importance of autophagy evasion in MCPyV‐mediated tumorigenesis. Torin‐1 treatment induced cell death, which was attenuated by autophagy inhibitor, but not pan‐caspase inhibitor, suggesting a potential role of autophagy in promoting cell death in MCC. Conceptually, our study shows that MCPyV oncoproteins suppress autophagy to protect cancer cells from cell death, which contribute to a better understanding of MCPyV‐mediated tumorigenesis and potential MCC treatment.
Keywords: Merkel cell polyomavirus, Merkel cell carcinoma, autophagy, microRNA, cell survival
Short abstract
What's new?
About four‐fifths of Merkel cell carcinomas harbor the Merkel cell polyomavirus (MCPyV) genome, though mutations in viral T antigens generally render MCPyV replication‐deficient. Here, the authors describe a network by which MCPyV oncoproteins and viral‐regulated miRNAs hijack autophagy machinery in MCC. In cells, expression of MCPyV T‐antigens induced miR‐375, miR‐30a‐3p, and miR‐30a‐5p expression. Subsequent experiments validated these miRNAs as autophagy regulators that specifically target the genes ATG7, SQSTM1, and BECN1. Autophagy suppression protected MCC cell survival. This novel insight into the role of MCPyV and autophagy regulation in MCC may be relevant in the generation of future therapeutic strategies.
Abbreviations
- LT
large T‐antigen
- MCC
Merkel cell carcinoma
- MCPyV
Merkel cell polyomavirus
- miRNA
microRNA
- mut
mutated‐type
- shRNA
short hairpin RNA
- sT
small T‐antigen
- UTR
untranslated region
- wt
wild‐type
Introduction
Merkel cell carcinoma (MCC) is an aggressive skin cancer with poor prognosis, typically occurring after 65 years of age and in immunosuppressed individuals.1 About 80% of MCCs harbor integrated Merkel cell polyomavirus (MCPyV) genome with a mutation in the large T (LT) gene,2, 3 leading to expression of a truncated LT that retains the retinoblastoma protein (RB) binding but eliminates viral replication capacity. The viral LT–RB interaction is required for RB sequestration, which releases E2F for cell growth activation.4 Besides the truncated LT, most MCC tumors also express intact small T‐antigen (sT). The LT‐stabilization domain (LSD) of MCPyV sT can inhibit FBW7 ubiquitin ligase and thus stabilizing its substrates, including LT, c‐MYC and cyclin E.5 The LSD also targets the translation regulator 4E‐BP1, which activates mitotic‐dependent protein translation for cell cycle progression.6, 7 The protein phosphatase 4C (PP4C) domain of the sT interacts directly with the NF‐κB essential modulator (NEMO) protein and disrupts host cell inflammatory signaling mediated by NF‐κB.8 The PP4C domain is also important for microtubule destabilization that promotes cell motility and migration.9 Additionally, MCPyV sT can also promote oncogenic activities through recruitment of MYCL to the EP400 complex,10 perturbation of metabolic pathways11 and induction of ADAM metalloproteinases12 and chloride channels.13 Moreover, the sT can contribute to tumor development in transgenic mouse models.14, 15, 16 Thus, both truncated LT and sT are key players in the pathogenesis of MCC. However, the detailed network for involvement of these viral T‐antigens in MCC development is not completely understood.
We previously identified a subset of microRNAs (miRNAs) associated with MCPyV status in MCC tumors, including miR‐203, miR‐30a‐3p, miR‐769‐5p, miR‐34a, miR‐30a‐5p and miR‐375.17 Higher expressions of miR‐375, miR‐30a and miR‐34a have also been observed in MCPyV‐positive (MCPyV+) compared to MCPyV‐negative (MCPyV−) MCC tumors or cell lines by other groups.18, 19, 20 Importantly, miR‐375 is specific for MCC and its serum level correlates with tumor burden.21 To date, only a few miRNAs have been functionally characterized in MCC. miR‐203 was found to target BIRC5 and regulate cell growth and cell cycle progression in MCPyV−, but not in MCPyV+ MCC cells.17 miR‐375 was shown to promote neuroendocrine differentiation and act as a tumor suppressor in MCPyV− MCC cell lines,18, 22 but function as an oncogene in MCPyV+ MCC cell lines.22
Given that the identified MCPyV‐associated miR‐375 and miR‐30a‐5p are known to be involved in autophagy,23, 24 we investigated whether MCPyV T‐antigens regulate autophagy in MCCs. Indeed, we show that MCPyV T‐antigens and the MCPyV‐regulated miRNAs miR‐375, miR‐30a‐3p and miR‐30a‐5p suppress autophagy by targeting multiple autophagy genes.
Materials and Methods
MCC cell lines
The MCPyV− cell lines MCC13, MCC14/2 and MCC26 were available from Cell Bank Australia (Westmead, NSW, Australia). The MCPyV+ cell lines WaGa and MKL‐1 were kindly provided by Drs J.C. Becker (Medical University of Graz) and N.L. Krett (Northwestern University), respectively. Cells were cultured at 37°C with 5% CO2 in RPMI‐1640 medium supplemented with 15% (MCC13, MCC14/2 and MCC26) or 10% (WaGa and MKL‐1) fetal bovine serum. All cell lines were genotyped for short tandem repeats (STRs) at Bio‐Synthesis, Inc. (Lewisville, TX) and the STR‐genotypes are detailed in Supporting Information Table S1. The authenticity of the cell lines was confirmed by comparing the genotypes from Daily et al.25 and Cellosaurus (https://web.expasy.org/cellosaurus/). These MCPyV− cell lines were chosen for gain‐of‐function studies due to their low levels of endogenous miR‐375, miR‐30a‐3p and miR‐30a‐5p.
Clinical samples
Forty‐five formalin‐fixed paraffin‐embedded MCC tumor samples were collected from Karolinska University Hospital and Stockholm South General Hospital. The MCPyV status of 38 samples were characterized and included in our previous studies,17, 26 and seven samples were characterized in this study using the same approaches, that is, PCR detection of MCPyV DNA in tumor samples and immunohistochemical detection of MCPyV LT expression using CM2B4 (Santa Cruz Biotechnology, Dallas, TX) or Ab3 (kindly provided by Dr. J.A. DeCaprio) antibody. The study was approved by the Ethics Committee of Karolinska Institutet.
Plasmids
MCPyV wild‐type (wt; LTco, #40200) and truncated (LT339, #28193) LT expression vectors were purchased from Addgene (Cambridge, MA). sTco and LTcoD44N plasmids were gifts from Drs Y. Chang and P. Moore (University of Pittsburgh). LTco and sTco are codon‐optimized vectors with deleted splicing donor and acceptor sites in the sequence, therefore only express LT or sT, respectively.6 shTA, short hairpin RNA (shRNA) vector targeting the common exon 1 of MCPyV T‐antigens, was generated in our previous study.17 The autophagy mRFP‐GFP‐LC3 reporter was kindly provided by Dr. B. Joseph (Karolinska Institutet).
shRNA vectors targeting the MCPyV sT (shsTA) and miRNA expression vectors (miR‐30a, miR‐30a‐3p and miR‐30a‐5p) were cloned into pcDNA3‐U6M2, between BglII and KpnI sites. miRNA sponge sequences (miR‐375sp, miR‐30a‐3psp and miR‐30a‐5psp) containing five or four tandem miRNA binding sites with bulged site at miRNA position 9–12 were cloned into pcDNA3 vector at BamHI and XhoI sites (Supporting Information Fig. S1). All luciferase reporters were generated using pmirGLO dual‐luciferase miRNA target expression vector (Promega Corporation, Madison, WI). Wild‐type (wt) and mutated‐type (mut) of the putative miRNA binding sites of ATG7, SQSTM1 and BECN1 were cloned into 3′‐UTR downstream of luc2 firefly luciferase gene at PmeI and XbaI sites. Sequences for all oligonucleotides used are given in Supporting Information Table S2. All constructs were confirmed by sequencing at KI Gene. The efficacy of miRNA inhibition for all three miRNA sponges was evaluated using their respective pmirGLO luciferase reporter containing the reverse complementary sequence of miRNA, while miRNA overexpression was quantified by RT‐qPCR (Supporting Information Fig. S2). Nonspecific effects of miRNA sponges on MCPyV LT were excluded by Western blotting (Supporting Information Fig. S3).
Transfections, bafilomycin A1 and actinomycin D treatments
For silencing of MCPyV T‐antigens or miRNAs, 2 μg of shRNA or miRNA sponge plasmid DNA was transfected into 3 × 106 WaGa and MKL‐1 cells using Nucleofector kit V (program D‐24 and A‐24, respectively; Lonza, Basel, Switzerland). For overexpression of MCPyV T‐antigen(s) or miRNAs, 2 μg of T‐antigen or miRNA expression plasmid DNA was transfected into 3 × 105 cells of MCC13, MCC14/2 and MCC26 cell lines using Lipofectamine LTX with plus reagent (Invitrogen, Carlsbad, CA). For transfection of miR‐375 mimics (MC10327; Ambion) or miRNA mimic negative control (NC, AM17110; Ambion), 10 nM of miRNA mimic was transfected into cells using Lipofectamine RNAiMAX Reagent (Invitrogen).
For inhibition of autophagy flux, 40 nM bafilomycin A1 (B1793; Sigma‐Aldrich) was added in the growth medium and incubated for 2 hr prior to analysis. Cells treated with dimethyl sulfoxide (DMSO) alone (1:1,000 dilution; Sigma‐Aldrich, St. Louis, MO) were used as a control. For inhibition of transcription, 2.5 μg/μl actinomycin D (A1410; Sigma‐Aldrich) was added in the growth medium for 0, 6 and 24 hr.
Reverse‐transcription quantitative PCR
Total RNA was isolated by mirVana miRNA isolation kit (Ambion) and the concentrations were measured with a NanoDrop ND‐1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). RT‐qPCR was performed using the StepOnePlus™ Real‐Time PCR system (Life Technologies). TaqMan assays for miR‐375, miR‐30a‐3p, miR‐30a‐5p, miR‐125a‐3p, miR‐16, pri‐mir‐375, pri‐mir‐30a, RNU6B and 18S rRNA were purchased from Applied Biosystems (Foster City, CA). cDNA was synthesized from 120 ng of total RNA using TaqMan MicroRNA Reverse Transcription Kit or RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA). Detection of MCPyV LT and sT transcripts were performed using SYBR Green detection system (Applied Biosystems) and previously described gene‐specific primers,27 which are listed in Supporting Information Table S2. All reactions were performed in triplicate. RNU6B was used as an endogenous control for miRNA quantification, while pri‐mir‐375 and pri‐mir‐30a were normalized to 18S rRNA. MCPyV LT or sT transcripts were normalized to GAPDH mRNA.
Protein isolation and Western blotting
Total protein was isolated using NP‐40 lysis buffer (Life Technologies), supplemented with 1 mM of phenylmethanesulfonyl fluoride (Sigma‐Aldrich) and complete protease inhibitor cocktail (Roche Diagnostics GmbH, Mannheim, Germany). Protein concentrations were measured using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). Protein lysates (40 μg) were run in 12% NuPAGE SDS or 4–12% Bis‐Tris gels (Invitrogen) and transferred to polyvinylidene fluoride or nitrocellulose membranes. Antibodies are listed in Supporting Information Table S3. Detection was performed using the Novex ECL HRP chemiluminescent substrate reagent (Life Technologies) and LAS‐1000 Image Analyzer (Fujifilm, Tokyo, Japan). GAPDH, α‐ or β‐tubulin were used as endogenous controls for quantification purposes. Quantification was done using LAS‐1000 Image Analyzer (Fujifilm) and Image J.
Transmission electron microscopy
After 72 hr of transfection, cells were collected and fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer. Electron microscopy was performed at the electron microscopy unit, Karolinska Institutet.
LC3 reporter assay
The mRFP‐GFP‐LC3 tandem reporter was used to monitor the autophagic flux. Transfected cells were fixed with 4% paraformaldehyde in PBS (USB Corporation/Affymetrix, Santa Clara, CA), and counter‐stained with 4′,6‐diamidino‐2‐phenylindole (Sigma‐Aldrich) in Vectorshield® mounting medium (Vector Laboratories, Burlingame, CA). Detection of LT was performed using CM2B4 antibody (sc‐136172, Santa Cruz Biotechnology; 1:50). All images were captured in a confocal microscope Leica TCS SP5 (Leica Microsystems GmbH, Wetzlar, Germany). Autophagy was determined by counting the number of cells with LC3‐positive puncta. A minimum of 20 cells was captured and scored per condition using particle analysis tool of ImageJ software (https://imagej.net/Particle_Analysis).
pmirGLO reporter assays
Fifty nanograms of pmirGLO reporter plasmid and 5 pmol of miR‐375 mimic or 50 ng of miR‐30a‐3p expression plasmid were cotransfected into MCC14/2 cells (2.5 × 104) using Lipofectamine 2000 transfection reagent (Invitrogen). All reporter assays were evaluated at 24 hr posttransfection. Dual‐GLO Luciferase Assay system (Promega Corporation, Madison, WI) was used to determine the firefly and Renilla luciferase luminescence signals using a microplate luminometer (Berthold Technologies, Bad Wildbad, Germany). All experiments were performed in 4–5 wells for each condition and repeated at least three times independently. Data were analyzed by dividing firefly luciferase activity with Renilla luciferase activity for each well and normalized to the empty pmirGLO vector control.
WST‐1 and apoptosis assays
Cells were treated with bafilomycin A1 (10 nM; Sigma‐Aldrich), z‐VAD‐FMK (20 μM; R&D Systems, Minneapolis, MN) and/or Torin‐1 (5 or 10 μM; Tocris/Bio‐Techne Ltd, Abingdon, OX, UK). All treatments were performed for 24 hr, except bafilomycin A1 for 6 hr. As control experiments, we included cells treated with DMSO or camptothecin (100 μM; Sigma‐Aldrich) and a combination of camptothecin (100 μM) and z‐VAD‐FMK (20 μM). For cell viability assay, 10 μl of WST‐1 reagent (Roche Diagnostics, Indianapolis, IN) was added into each well and incubated for 2 hr. Measurements were recorded at wavelengths 450 nm (WST‐1 cleavage products) and 690 nm (reference) using VERSAmax microplate reader (Molecular Devices, Sunnyvale, CA) and analyzed with SoftMax Pro 7 (Molecular Devices). Five replicate wells were included for each treatment group. Relative cell viability in each treatment was normalized to DMSO‐treated group. All experiments were repeated three times independently. Cell apoptosis was evaluated using Annexin V‐FITC apoptosis kit (#640905; BioLegend, San Diego, CA). All staining conditions were performed according to the manufacturer's instructions and analyzed by NovoCyte flow cytometer (ACEA Biosciences, San Diego, CA). All experiments were repeated four times independently.
Immunohistochemistry
Immunohistochemistry was performed in sections from 45 MCC tumor samples according to previously described methodology,17 using ATG7 and p62 antibodies (Supporting Information Table S4). The immunoreactivity was classified as negative, weak (1+), moderate (2+) or strong (3+) based on the scoring of the cytoplasmic intensity.
Statistical analysis
Paired Student's t‐test (Microsoft Office Excel) was used to compare two groups in transfection experiments. Multiple comparisons between treatment groups were evaluated using One‐way ANOVA with post hoc Tukey test (http://astatsa.com/OneWay_Anova_with_TukeyHSD/). Associations of immunohistochemical staining and MCPyV status were evaluated using Fisher's exact test. All the analyses were two‐tailed, and p values <0.05 were considered as statistically significant.
Results
MCPyV T‐antigens posttranscriptionally induce miR‐375, miR‐30a‐3p and miR‐30a‐5p
Three of the MCPyV‐associated miRNAs (miR‐375, miR‐30a‐3p and miR‐30a‐5p) identified in our previous screen in clinical samples17 were further investigated concerning their biological roles in MCC. First, we determined whether these miRNAs are regulated by MCPyV T‐antigens. In MCPyV+ cell lines (WaGa and MKL‐1), we silenced LT and/or sT using shTA or shsTA, respectively. In MCPyV− MCC14/2 cells, we ectopically expressed sT, wt or truncated LT using sTco, LTco or LT339, respectively. Vector constructs are illustrated in Figure 1 a. The specificity of shTA and shsTA was verified by RT‐qPCR and Western blot analyses of the LT and sT transcripts and protein, respectively (Fig. 1 b and Supporting Information Fig. S4a ). Additionally, no off‐target effects were observed in the MCPyV− cell lines (Supporting Information Fig. S4b ). Ectopic expressions of viral T‐antigens in MCPyV− cell lines were verified by Western blotting (Fig. 1 c). In line with their associations to MCPyV status in MCC tumors, we observed decreased expressions of miR‐375, miR‐30a‐3p and miR‐30a‐5p in WaGa and MKL‐1 cells upon silencing of LT and/or sT (Fig. 1 b). Similarly, miR‐375, miR‐30a‐3p and miR‐30a‐5p expressions were increased in cells expressing sTco, LTco or LT339, which were relatively lower than their endogenous levels in MCPyV+ cell lines (Fig. 1 c). As negative controls, we included two miRNAs (miR‐125a‐3p and miR‐16), which were not differentially expressed between MCPyV+ and MCPyV− MCC tumors in our previous study.17 Concordantly, these control miRNAs did not show significant changes in cells with expression or silencing of T‐antigens (Figs. 1 b and 1c).
Figure 1.
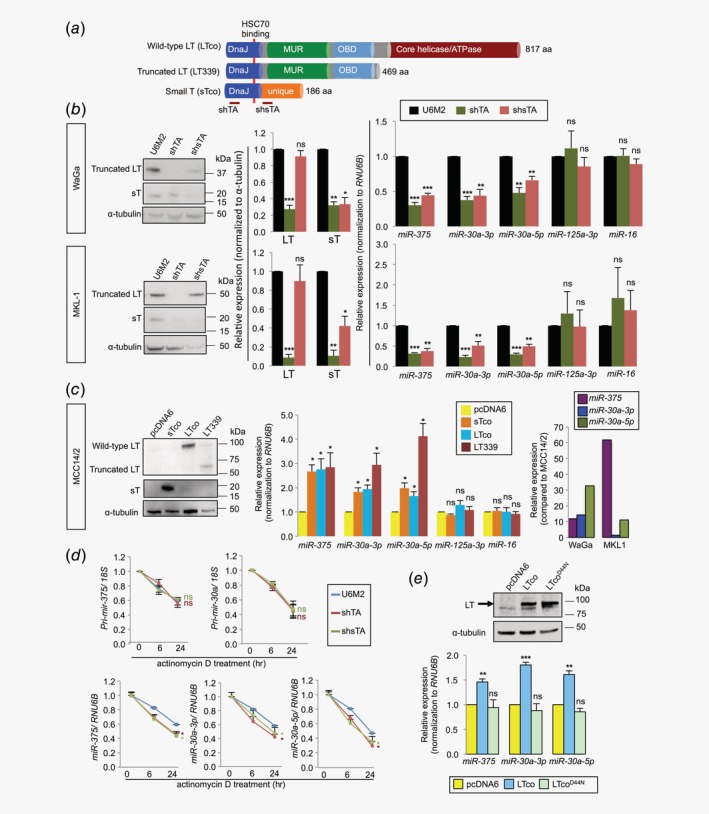
MCPyV T‐antigens regulate miRNA expressions at the posttranscriptional level. (a) Overview of plasmids expressing different MCPyV T‐antigens and short‐hairpin RNAs (shRNAs) targeting T‐antigens. LTco and LT339 plasmids express wild‐type and truncated large T‐antigen (LT), respectively; sTco plasmid expresses only small T‐antigen (sT). shTA is a shRNA vector targeting both LT and sT, while shsTA silences only sT. Both LT and sT share a common DnaJ domain, which interacts with HSC70 (42–47 aa), as indicated by red line. (b, c) Validation of MCPyV T‐antigen‐regulated miRNAs. Cells were transfected with pcDNA3‐U6M2 (U6M2), shTA or shsTA in MCPyV+ MCC cell lines (b), and pcDNA6, sTco, LTco or LT339 in the MCPyV− MCC14/2 cells (c). Cells were harvested for RNA extraction and RT‐qPCR after 72 hr (WaGa and MKL‐1) or 48 hr (MCC14/2) of transfection. Representative images showing the silencing effects on MCPyV LT and/or sT by Western blotting (left). Quantification of the Western blot data from three independent experiments in (b) (middle). miRNA expressions were quantified by RT‐qPCR and normalized to RNU6B. miR‐125a‐3p and miR‐16 were included as negative controls (b, right and c, middle). The relative expression of each miRNA in each treatment group was compared to the corresponding vector control‐transfected cells. Data represent mean ± SEM of four independent experiments. As comparison, miRNA levels in WaGa and MKL‐1 cells were shown in the right panel (c) after normalization to the mean expression levels of respective miRNAs in MCC14/2 cells. (d) WaGa cells were transfected with shTA, shsTA or U6M2 for 72 hr, followed by actinomycin D treatment. Cells were harvested at 0, 6 and 24 hr after actinomycin D treatment for RT‐qPCR analysis of primary and mature miRNAs. The treatment of actinomycin D at 0 hr was set as 1. Expressions of primary miRNAs were normalized to 18S rRNA and mature miRNAs were normalized to RNU6B. Red and green * refer to significant p values for shTA‐ and shsTA‐transfected cells, respectively, as compared to vector control (blue line). Error bars indicate SEM of mean from three independent biological replicates. (e) Wild‐type LT, D44N mutant (LTcoD44N) or vector control (pcDNA6) was transfected into MCC26 cell line. Western blotting and RT‐qPCR analyses were performed after 48 hr of transfection. Upper: Immunoblot images showing the transfection efficiency of LT. Lower: Quantification of miRNAs in cells expressing LTco, LTcoD44N and pcDNA6. Data represent mean ± SEM of four independent experiments. (b–e) Differences between the treatment groups and the vector control were calculated by paired Student's t‐test. *p < 0.05, **p < 0.01, ***p < 0.001, ns = not significant.
To determine whether MCPyV T‐antigens regulate miRNAs at the transcriptional or posttranscriptional level, we first quantified primary transcripts of miR‐375 and miR‐30a using RT‐qPCR. We observed no difference in pri‐mir‐375 or pri‐mir‐30a expressions upon silencing of LT and/or sT (Supporting Information Fig. S4c ). We next determined the stability of primary miRNA transcripts in WaGa cells with and without silencing of LT and/or sT upon inhibition of transcription using actinomycin D. Consistently, silencing of MCPyV T‐antigens had no effect on primary miRNA transcripts (Fig. 1 d). On the other hand, we observed reductions of all three mature miRNAs in actinomycin D‐treated cells upon silencing of LT and/or sT (Fig. 1 d). The data suggest that MCPyV T‐antigens regulate miRNAs at the posttranscriptional level.
A mutation in the DnaJ domain of LT abolishes MCPyV‐induced miRNA expressions
To address which domain of the viral T‐antigens is responsible for miRNA regulation, we focused on the DnaJ domain, which is the only common domain shared between the LT and sT (Fig. 1 a). This domain is known to interact with heat shock protein HSC70.28 HSC70 is a part of the miRNA‐processing complex and is required for loading miRNA duplexes into the silencing complex.29 We therefore investigated whether this domain is required for miRNA upregulation using wt (LTco) and DnaJ mutant D44N (LTcoD44N) LT, which abolishes the interaction with HSC70.28 Consistently, we observed increased expression of miR‐375, miR‐30a‐3p and miR‐30a‐5p in both MCC14/2 and MCC26 cell lines expressing wt LT; however, the DnaJ mutant completely abolished the effect from the LT (Fig. 1 e and Supporting Information Fig. S5), suggesting that the MCPyV DnaJ domain is required for miRNA regulation.
MCPyV T‐antigens regulate autophagy in MCC cells
We next investigated whether MCPyV T‐antigens can regulate autophagy. As demonstrated by Western blotting, we observed an increase of LC3‐II in both WaGa and MKL‐1 cells transfected with shTA or shsTA (Figs. 2 a and 2b; Supporting Information, Fig. S4d ). The LC3‐II level is a hallmark of autophagy, which correlates with the number of autophagosomes and/or autophagic degradation.30 To assess whether the changes of LC3‐II were due to autophagosome formation or degradation, we quantified LC3‐II levels in the presence of bafilomycin A1, a lysosomal protease inhibitor that blocks autophagosome‐lysosome fusion and LC3‐II degradation. Upon bafilomycin A1 treatment, we observed further increase of LC3‐II in cells with silencing of T‐antigens (Figs. 2 a and 2b), suggesting perturbations of autophagy formation. Similarly, we found decrease of LC3‐II levels in cells with ectopic expression of sT or truncated LT, however, the effect of wt LT was subtle or no difference (Figs. 2 a and 2b). It was noted that the wt LT was more abundant than the truncated LT, indicating that lack of effect in the wt LT‐transfected cells was unlikely due to low transfection efficiency. On the other hand, the observation suggests that wt LT does not suppress autophagy.
Figure 2.
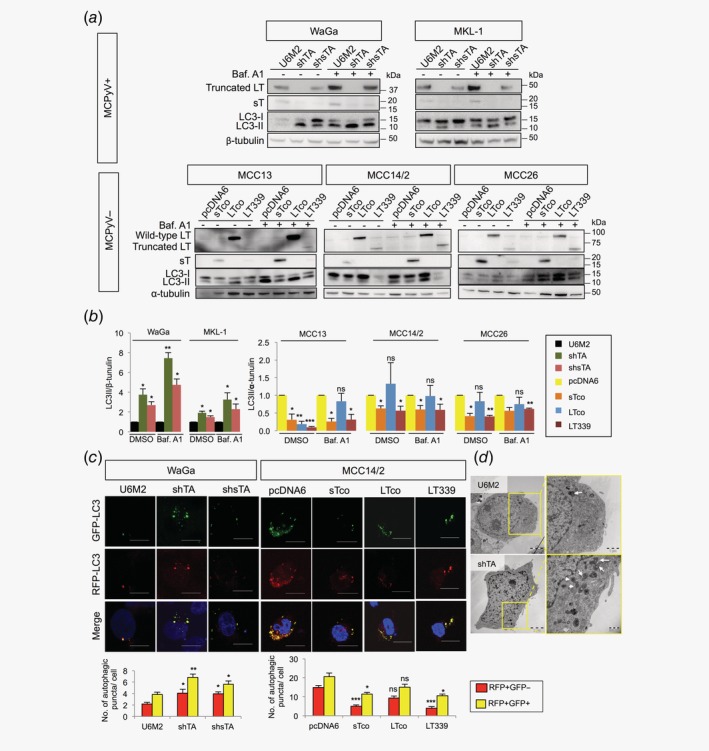
MCPyV T‐antigens inhibit autophagy in MCC cells. (a) Representative Western blots of LC3 protein levels in MCPyV+ cell lines (WaGa and MKL‐1) transfected with shTA, shsTA or pcDNA3‐U6M2 and in MCPyV− cell lines (MCC13, MCC14/2 and MCC26) transfected with sTco, LTco, LT339 or pcDNA6 with bafilomycin A1 (Baf. A1, 40 nM) or DMSO (control) treatment for 2 hr. β‐tubulin (for MCPyV+ cells) and α‐tubulin (for MCPyV− cells) were used as loading controls for quantification. (b) Quantification of the Western blot data described in Figure 2 a (n = 3). (c) WaGa cells were cotransfected with mRFP‐GFP‐LC3 and pcDNA3‐U6M2, shTA or shsTA, and MCC14/2 cells were cotransfected with mRFP‐GFP‐LC3 and pcDNA6, sTco, LTco or LT339. After 72 hr (WaGa) or 48 hr (MCC14/2), cells were fixed and analyzed by confocal microscopy. Representative images are shown. Scale bars represent 10 μm. Number of autophagosomes (RFP+/GFP+, yellow puncta) and autolysosomes (RFP+/GFP−, red puncta) were counted for 22 cells from three independent experiments. Representative lower magnification images are shown in Supporting Information Figure S6. (d) Representative images from transmission electron microscopy of WaGa cells transfected with shTA or pcDNA3‐U6M2 for 72 hr. Areas within yellow boxes (left) are shown at higher magnification (right). Arrows indicate autophagosomes or autolysosomes. (b, c) Error bars indicate SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ns = not significant by paired Student's t‐test.
To further validate the effect of MCPyV T‐antigens on autophagy, we quantified the number of autophagosomes and autolysosomes using a tandem mRFP‐GFP‐LC3 reporter. Silencing of T‐antigens or sT only in the MCPyV+ WaGa cells resulted in an increase of both autophagosomes (RFP+/GFP+) and autolysosomes (RFP+/GFP−), while expression of sT or truncated LT in the MCPyV− MCC14/2 cells decreased the number of autophagosomes and autolysosomes (Fig. 2 c and Supporting Information Fig. S6). Similar to the results from the LC3 immunoblotting, expression of wt LT had a subtle effect on the number of autophagosomes and autolysosomes, but not statistically significant (Fig. 2 c). Using transmission electron microscopy, we also observed an increase of autophagosomes and autolysosomes upon silencing of T‐antigens (Fig. 2 d), which corroborated with the findings from LC3 reporter assay. Together, our data support that MCPyV oncoproteins suppress autophagy in MCC, and we hypothesized that these viral oncoproteins may regulate autophagy through miRNAs.
Validation of miR‐375 and miR‐30a‐5p as regulators of autophagy in MCC cells
miR‐375 and miR‐30a‐5p are known to inhibit autophagy by targeting ATG7 23 and BECN1,24 respectively. We investigated whether miR‐375 and miR‐30a‐5p also regulate autophagy in MCC. Inhibition of miR‐375 in MCPyV+ MCC cell lines increased LC3‐II, which was further increased in the presence of bafilomycin A1 (Fig. 3 a). On the other hand, overexpression of miR‐375 in MCPyV− cell lines reduced LC3‐II in both conditions with and without bafilomycin A1 (Fig. 3 a). Using LC3 reporter assay, we observed an increase of autophagosomes and autolysosomes upon inhibition of miR‐375, and a decrease upon overexpression of miR‐375 (Figs. 3 b and 3c; Supporting Information Fig. S7a ). Similar results were observed using electron microscopy (Fig. 3 d).
Figure 3.
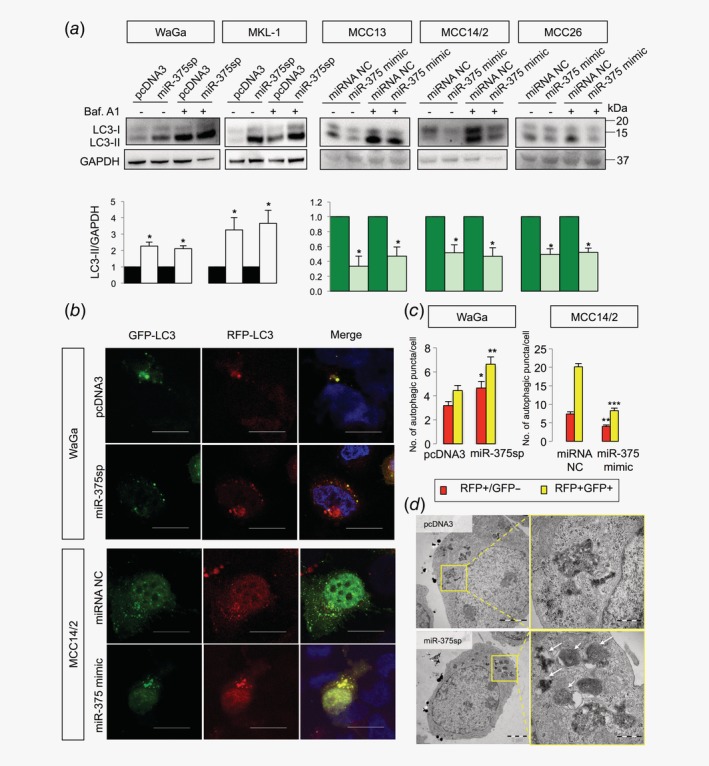
miR‐375 regulates autophagy in MCC cells. (a) MCPyV+ cell lines were transfected with miR‐375 sponge (miR‐375sp) or pcDNA3 as a control, and MCPyV− cell lines were transfected with miR‐375 mimic or miRNA negative control (NC) in the presence of bafilomycin A1 (Baf. A1, 40 nM for 2 hr) or DMSO. After 72 or 48 hr of transfection, cells were harvested for Western blot analysis of LC3 and GAPDH. The quantification data of LC3‐II/GAPDH are shown below the Western blot images (n = 3). (b) WaGa cells were cotransfected with mRFP‐GFP‐LC3 and miR‐375sp or pcDNA3, while MCC14/2 cells were cotransfected with mRFP‐GFP‐LC3 and miR‐375 mimic or miRNA NC. Cells were then fixed and analyzed by confocal microscopy. Scale bars represent 10 μm. (c) Number of autophagosomes (RFP+/GFP+, yellow puncta) and autolysosomes (RFP+/GFP−, red puncta) were counted for 30 cells in each transfection. Representative lower magnification images are shown in Supporting Information, Figure S7a . (d) Representative transmission electron microscopy images showing autophagosomes and autolysosomes in WaGa cells transfected with pcDNA3 or miR‐375sp. Areas within yellow boxes are shown at higher magnification on the right panels. Arrows indicate autophagosomes or autolysosomes. (a, c) Error bars indicate SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
To validate the involvement of miR‐30a‐5p in autophagy in MCC, we first constructed an expression vector by amplifying the miR‐30a precursor sequence (designated as miR‐30a) and cloning into pcDNA3‐U6M2. We found that the miR‐30a plasmid expressed both miR‐30a‐5p and miR‐30a‐3p (Supporting Information Figs. S2d and S2e ). This led us to design strand‐specific miRNA expression vector by cloning the miR‐30a‐5p or miR‐30a‐3p mature sequence into the pcDNA3‐U6M2 shRNA vector (designated as miR‐30a‐5p and miR‐30a‐3p, respectively) and their respective expressions were validated by RT‐qPCR (Supporting Information Figs. S2d and S2e ). As expected, cells expressing miR‐30a‐5p showed a decrease of LC3‐II in all three MCPyV− cell lines, while inhibition of miR‐30a‐5p increased LC3‐II levels in MCPyV+ cells, in the presence or absence of bafilomycin A1 (Figs. 4 a and 4b). Unexpectedly, we noted that modulation of miR‐30a‐3p expression had similar effects as miR‐30a‐5p on LC3‐II expression in both MCPyV+ and MCPyV− cell lines (Figs. 4 a and 4b). The effect of miR‐30a‐3p on autophagy was further supported by the mRFP‐GFP‐LC3 reporter assay (Fig. 4 c and Supporting Information Fig. S7b ). Inhibition of miR‐30a‐3p and miR‐30a‐5p in WaGa cells led to an increase in autophagosomes and autolysosomes. Conversely, overexpression of miR‐30a‐3p and miR‐30a‐5p reduced the number of autophagosomes and autolysosomes in MCC14/2 cells.
Figure 4.
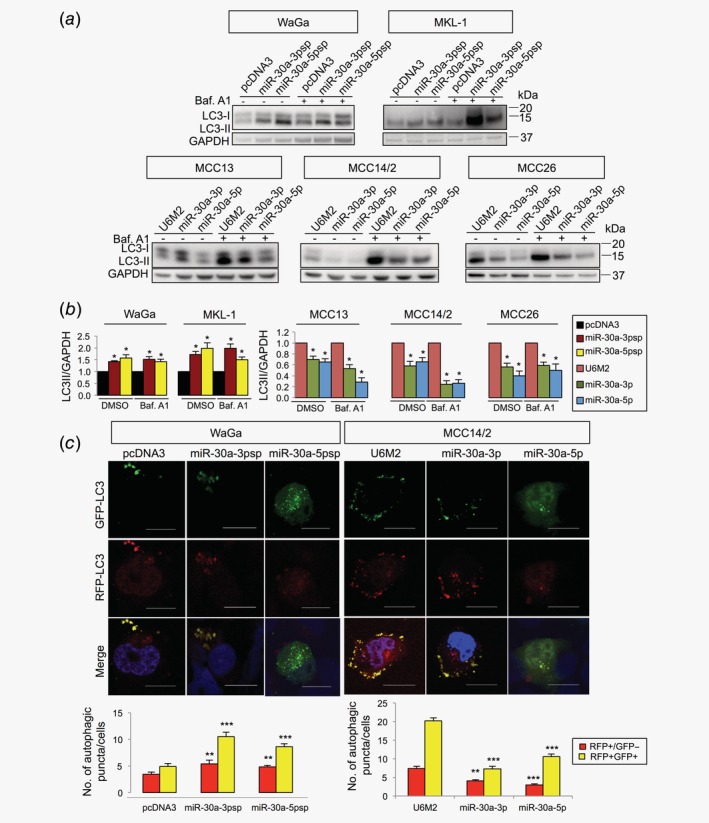
Both miR‐30a‐3p and miR‐30a‐5p regulate autophagy in MCC cells. (a) MCPyV+ MCC cell lines (WaGa and MKL‐1) were transfected with miRNA sponges for miR‐30a‐3p (miR‐30a‐3psp), miR‐30a‐5p (miR‐30a‐5psp) or vector control (pcDNA3) for 72 hr, while the MCPyV− MCC cell lines (MCC13, MCC14/2 and MCC26) were transfected with pcDNA3‐U6M2 (vector control), miR30a‐3p or miR‐30a‐5p expression vectors for 48 hr and treated with bafilomycin A1 (Baf. A1, 40 nM) or DMSO (1:1,000) for 2 hr. Representative Western blot images are shown. (b) Quantification of the LC3‐II protein levels described in Figure 4 a (n = 3). GAPDH was used as endogenous control. (c) WaGa cells were cotransfected with mRFP‐GFP‐LC3 reporter together pcDNA3, miR‐30a‐3p or miR‐30a‐5p sponges, while MCC14/2 cells were cotransfected with mRFP‐GFP‐LC3 reporter and miR‐30a‐3p, miR‐30a‐5p or pcDNA3‐U6M2. Cells were fixed and analyzed with confocal microscopy for autophagosome (yellow puncta) and autolysosome (red puncta). Scale bars represent 10 μm. The related quantification data are shown at the bottom panel (n = 30). (b, c) Error bars indicate SEM. **p < 0.01, ***p < 0.001. Representative lower magnification images are shown in Supporting Information Figure S7b .
miR‐375 and miR‐30a‐5p target ATG7 and BECN1, respectively, in MCC
We next investigated the consequence of miR‐375 regulation on ATG7 expression. ATG7 protein expression was found increased (23–54%) in MCPyV+ cells upon inhibition of miR‐375, and reduced (20–40%) in MCPyV− cells upon overexpression of miR‐375 (Figs. 5 a and 5b). To validate ATG7 as a direct target of miR‐375, we cloned the miR‐375 binding site of ATG7 into the 3′‐untranslated region (3′‐UTR) of pmirGLO luciferase reporter (ATG7 wt) and cotransfected the reporter with miR‐375 mimic or miRNA mimic negative control (NC). We observed a ~30% reduction of luciferase activity in the cells overexpressing miR‐375 compared to the miRNA mimic NC (Fig. 5 c). The ATG7 mutant construct (ATG7 mut) with three mismatches in the seed interaction sites of miR‐375 partly rescued the miR‐375‐mediated suppression of luciferase activity (Fig. 5 c).
Figure 5.
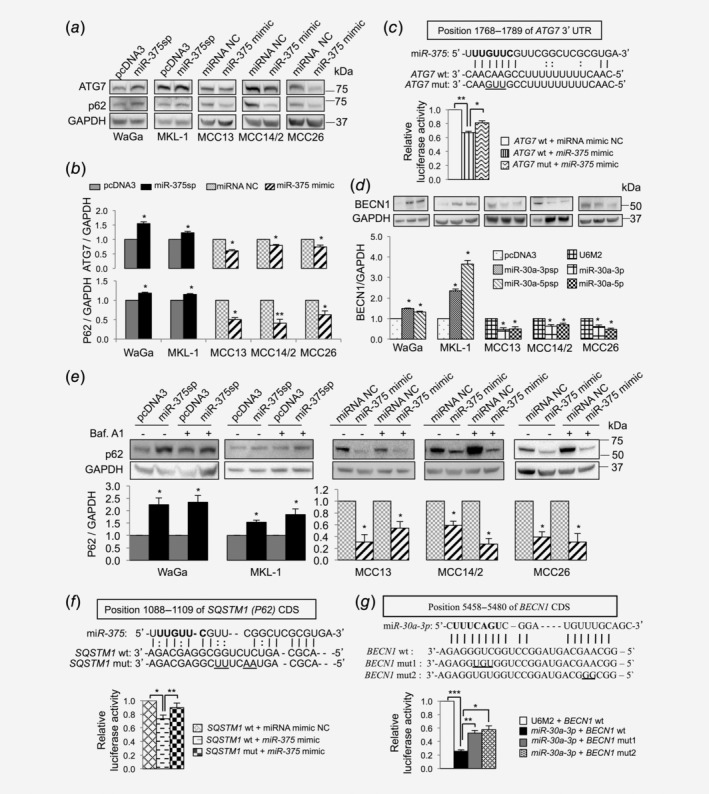
miR‐375, miR‐30a‐3p and miR‐30a‐5p target multiple autophagy genes. (a, b) miR‐375 regulates ATG7 and p62 expression. (a) Representative Western blots showing the effect of miR‐375 silencing (miR‐375sp) or overexpression (miR‐375 mimic) on ATG7 and p62 expression in MCPyV+ and MCPyV− MCC cell lines. (b) Quantification of ATG7 and p62 protein levels in MCPyV+ MCC cell lines upon inhibition of miR‐375 expression and in MCPyV− MCC cell lines upon overexpression of miR‐375. The expression was normalized to GAPDH, and their respective vector controls were set to 1 (n = 3). (c) Verification of ATG7 as a direct target of miR‐375 in MCC. Upper: Illustrations of sequence alignment of miR‐375 and the wild‐type (wt) and the mutated (mut) target sequence of ATG7. The seed sequence of miR‐375 is highlighted in bold and the mutated sequence is underlined. Lower: The effect of miR‐375 on luciferase activity was evaluated 24 hr after cotransfection of miR‐375 mimic or NC together with the wt or mut of ATG7 reporter constructs in MCC14/2 cells. (d) Effect of miR‐30a‐3p and miR‐30a‐5p regulation on BECN1 expression. Top: Representative Western blot images showing the effect of miR‐30a‐3p and miR‐30a‐5p on BECN1 expression. Bottom: Quantification of BECN1 expression normalized to GAPDH and vector control‐treated cells were set to 1 (n = 4). (e) Experiments described in (a) were repeated with DMSO or bafilomycin A1 treatment (40 nM for 2 hr). Top: Representative Western blot images showed the effect on p62 expression in the presence or absence of bafilomycin A1. Bottom: Quantification of p62 levels normalized to GAPDH (n = 3). (f, g) Validation of SQSTM1 and BECN1 as novel targets of miR‐375 and miR‐30a‐3p, respectively, by luciferase reporter assays (n = 3). The seed sequence is in bold and the mutated sequences are underlined. (b–g) Error bars refer to SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by paired Student's t‐test.
We also evaluated whether BECN1 could be a target of miR‐30a‐5p in MCC. Inhibition of miR‐30a‐5p increased BECN1 expression in MCPyV+ MCC cell lines, and its overexpression reduced BECN1 expression in MCPyV− MCC cell lines (Fig. 5 d). In line with its effect on autophagy assays, miR‐30a‐3p overexpression had similar suppression of BECN1 as miR‐30a‐5p (Fig. 5 d), suggesting that BECN1 could also be a putative target of miR‐30a‐3p.
SQSTM1 (p62) and BECN1 are novel targets of miR‐375 and miR‐30a‐3p, respectively
When we investigated the effect of miR‐375 regulation, we also noted consistent changes of p62 (encoded by the SQSTM1 gene) expression in cells transfected with miR‐375 sponge or mimic (Figs. 5 a and 5b). p62 is frequently used as another marker for autophagy in addition to LC3‐II, which can bind to LC3 and be degraded by autophagy.31 To distinguish whether the effect of miR‐375 regulation on p62 expression was a direct effect of miR‐375 regulation or a consequence of autophagy‐modulatory effects, we repeated the experiments with and without bafilomycin A1 treatment. The results were similar with and without bafilomycin A1 (Fig. 5 e) indicating that the effect of miR‐375 regulation on p62 expression is unlikely caused by consequences from deregulation of autophagic degradation. The results also suggest that SQSTM1 can be a putative target of miR‐375.
To search for potential miR‐375 target site(s) of SQSTM1, we extracted the coding and 3′‐UTR sequences of SQSTM1 and predicted the interaction with miR‐375 sequence using RNA22.32 The analysis revealed a potential target site in the coding region of SQSTM1 (Fig. 5 f). To verify the miR‐375 target site of SQSTM1, we cotransfected a luciferase reporter containing the miR‐375 target site of SQSTM1 and miR‐375 mimic or NC. The results showed ~30% reduction of luciferase activity in the cells transfected with miR‐375 mimic (p < 0.05). Notably, a mutation in the seed interaction site of miR‐375 did not abolish the repression of luciferase activity (data not shown), however, mutations in the central region of miR‐375 pairing completely abolished the repression activity (Fig. 5 f). Using a similar approach, we searched for the potential miR‐30a‐3p binding site of BECN1, which revealed a putative binding site in the coding sequence of BECN1. We constructed two different mutant constructs: one with mismatches in the seed pairing region (designated as BECN1 mut1), and the other with mismatches in the 3′ of miR‐30a‐3p pairing region (designated as BECN1 mut2; Fig. 5 g). Overexpression of miR‐30a‐3p reduced luciferase activity of the reporter containing miR‐30a‐3p target site of BECN1 (BECN1 wt), and the two mutant constructs partially rescued the miR‐30a‐3p suppression effect. Together, our data provide evidence that miR‐375, miR‐30a‐3p and miR‐30a‐5p regulate multiple target genes involved in autophagy.
ATG7 and p62 expressions are lower in MCPyV+ than MCPyV− MCC tumors
We also assessed ATG7 and p62 expressions in 45 MCC tumors using immunohistochemistry. For both proteins, we scored the intensity of cytoplasmic immunoreactivity in each specimen; examples of immunostaining are given in Supporting Information Figure S8a and the results are detailed in Supporting Information Table S4. For ATG7, 23 tumors showed moderate (2+) immunostaining and one had strong expression (3+), while 19 tumors had weak expression (1+) and two were negative. To assess whether ATG7 expression was associated with MCPyV status, we divided the tumors into two groups: moderate/strong (≥2+) and weak/negative (≤1+) expression. We noted that weak/negative ATG7 expression was associated with MCPyV+ MCCs (p = 0.028; Supporting Information Fig. S8b ). For p62, moderate (2+) immunostaining was only observed in 11 MCCs and strong (3+) staining in one case, while 20 of the remaining tumors showed weak immunostaining (1+) and 13 were negative (Supporting Information Table S4). Similar to the ATG7 expression pattern, most MCPyV+ MCCs had weak/negative p62 immunostaining (p = 0.004; Supporting Information Fig. S8b ). The results indicate that these autophagic proteins are lower in MCPyV+ than MCPyV− MCC tumors.
Autophagy inhibitor bafilomycin A1 rescues cell death induced by Torin‐1
Autophagy can regulate cell viability and apoptosis, depending on cellular context.33 Here we speculated that autophagy suppression might protect MCC cells from apoptosis. To address this issue, we treated WaGa cells with 5 or 10 μM of Torin‐1 (an mTOR inhibitor that is known to induce autophagy)34 or a combination of Torin‐1 and bafilomycin A1 (autophagy inhibitor) or z‐VAD‐FMK (pan‐caspase inhibitor), followed by evaluation of cell viability using WST‐1 assay. As shown in Figure 6 a, Torin‐1 treatment reduced cell viability to 0.66 ± 0.04 (5 μM) and 0.63 ± 0.10 (10 μM) as compared to DMSO‐treated cells. A combination of Torin‐1 and bafilomycin A1, but not the combination of Torin‐1 and z‐VAD‐FMK, was able to completely or partially rescue the cytotoxicity induced by Torin‐1 (5 and 10 μM, respectively; Fig. 6 a). Similar effects were also observed in MKL‐1 (Fig. 6 a) and MCPyV− MCC cell lines (Supporting Information Fig. S9).
Figure 6.
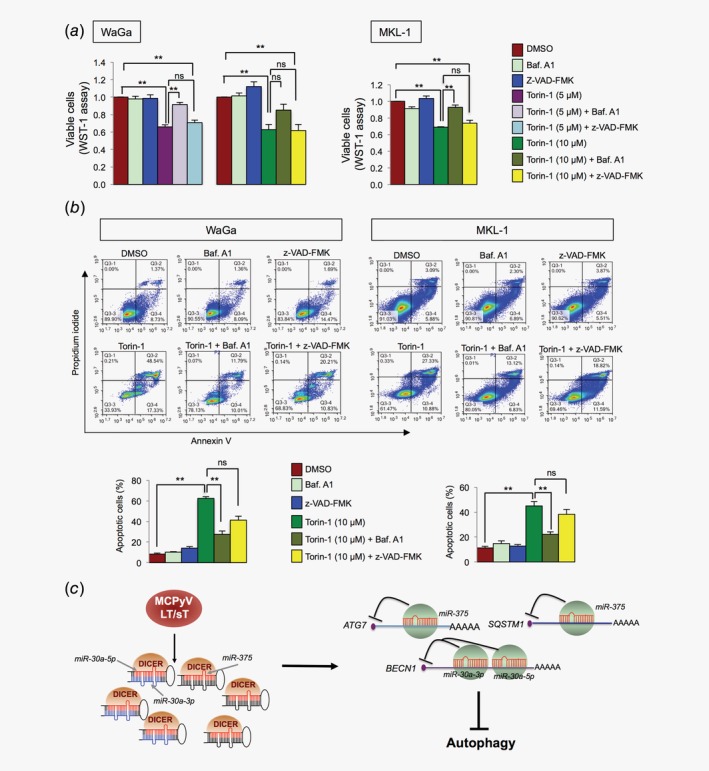
Inhibition of autophagy rescues Torin‐1 mediated cytotoxicity and model for MCPyV‐mediated suppression of autophagy. (a, b) MCPyV+ MCC cells were treated with DMSO, Baf. A1 (10 nM for 6 hr), z‐VAD‐FMK (20 μM, 24 hr) or Torin‐1 (5 or 10 μM for 24 hr), and a combination of Torin‐1 and Baf. A1 or z‐VAD‐FMK. (a) WST‐1 assay was used to evaluate cell viability. Mean ± SEM (n = 3). (b) Annexin V apoptosis assay was used to evaluate cell apoptosis in different treatments described in (a) for 10 μM Torin‐1. Representative flow cytometric images showing cells costained with Annexin V‐FITC and propidium iodide. The early and late apoptotic cells are presented by Annexin V+/PI− (lower right) and Annexin V+/PI+ (upper right) cells, respectively. Quantification of total apoptotic cells (Annexin V+) is shown below the images (n = 4). Error bars refer to SEM. (a, b) *p < 0.05, **p < 0.01 and ns = not significant by one‐way ANOVA with post hoc Tukey test. (c) Model for MCPyV T‐antigens mediated regulation of autophagy through miRNAs. MCPyV T‐antigens induce miR‐375, miR‐30a‐3p and miR‐30a‐5p expressions, which target multiple autophagy genes that lead to autophagy suppression in MCC.
To further support the effect on cell death, we evaluated the effects using Annexin V assay. As shown in Figure 6 b, the Torin‐1 treatment increased the proportion of apoptotic cells to 62% in WaGa and 45% in MKL‐1 from DMSO‐treated cells (8% and 11% in WaGa and MKL‐1, respectively). A combination of Torin‐1 and bafilomycin A1 treatment significantly rescued the number of apoptotic cells, while the effect of combined Torin‐1 and z‐VAD‐FMK treatment was not significant. Together, the results indicate that inhibition of autophagy, but not pan‐caspases, could rescue cell death induced by Torin‐1.
Discussion
Here we uncover a novel role for MCPyV T‐antigens in autophagy, in which the viral oncoproteins induce miRNA expressions that target multiple autophagy genes. Autophagy is a regulatory pathway of cellular degradation for proteins and organelles, which is critical in recycling macromolecules in response to nutrient and environmental stress. Deregulation of autophagy is involved in virus infection and tumorigenesis.33, 35 Although the involvement of autophagy has not been reported in MCPyV infection or MCPyV‐induced tumorigenesis, viral evasion of autophagy has been described for several human tumor viruses, including human Kaposi's sarcoma‐associated herpesvirus (KSHV), Epstein–Barr virus (EBV) and human papillomavirus (HPV),35 which contributes to immune escape and facilitates viral replication.
The role of autophagy is complex and tissue dependent. In normal cells, autophagy generally acts as a protective mechanism. In cancer cells, autophagy can promote or inhibit cell growth or tumorigenesis depending on cellular context.33 For example, autophagy is required to promote tumor growth in KRAS‐driven pancreatic cancer and glioblastoma mouse models.36, 37 Similarly, autophagy is essential for limiting oxidative stress in BRAF‐driven melanoma and lung tumors.38, 39 Tumor suppressive function of autophagy has been demonstrated in various tumor models. For example, autophagy deficiency through heterozygous disruption of Becn1 promotes development of lymphomas, liver and lung tumors,40 and mice with homozygous deletion of Atg5 or Atg7 develop multiple liver tumors.41
In MCC, two pieces of evidence indicate a link of autophagy to MCPyV‐induced tumorigenesis. First, Shuda et al. demonstrated that MCPyV sT could activate mTOR signaling through phosphorylation of two downstream molecules, 4E‐BP1 and S6K.6 mTOR is a master regulator of cellular metabolism and promotes cell growth in response to environmental cues. This signaling also plays an essential role in autophagy regulation. Its activation can lead to inhibition of autophagy through phosphorylation of multiple autophagy‐related proteins.34 Second, Lin et al. revealed that mTOR activation and suppressed autophagy is common in MCC tumors.42 Here, we provide direct evidence that MCPyV sT and truncated LT, but not wt LT, can suppress autophagy, supporting the importance of autophagy evasion in MCC development. The difference between the wt and truncated LT is the carboxyl‐terminal of LT protein containing viral replication elements, which is consistently deleted in MCC tumors. This carboxyl‐end of LT can activate DNA damage response, leading to cell cycle arrest and decreased cell growth.43 Given that DNA damage response generally induces autophagy as a protective survival mechanism, we would expect that the carboxyl‐terminal of LT could counter the autophagy suppression effect mediated by the amino‐terminal of LT. Additionally, we further demonstrated that inhibition of autophagy could rescue cytotoxicity induced by the mTOR inhibitor Torin‐1 in MCC cells, suggesting that autophagy suppression is a protective mechanism for cell survival in MCC.
In general, tumor viruses have evolved two different mechanisms to modulate host autophagy. First, some viruses encode viral homologs of Bcl‐2 and FLIP, which directly target the autophagy. The viral Bcl‐2 homologs lack a phosphorylation site‐containing regulatory loop that constitutively associates with Beclin 1 to inhibit autophagosome formation.44 The viral homolog of FLIP can directly interact with the ATG3 protein and inhibits autophagosome elongation.45 Alternatively, viral oncoproteins can target autophagy‐regulating signaling pathways, such as mTOR, PI3K‐AKT, ERK, AMPK, p53 and NF‐κB.35 Based on previous reports and our findings, autophagy suppression in MCC is likely contributed by both direct targeting of autophagy factors and activation of the PI3K/AKT/mTOR pathway. In MCPyV− tumors, the activation is likely caused by mutation or deregulation of the effectors involved in the PI3K/AKT/mTOR pathway,46 whereas, in MCPyV+ tumors, MCPyV T‐antigens are responsible for activation of the pathway or direct regulation of autophagy genes, which lead to suppression of autophagy.
One of the most noteworthy findings in this study is the identification of MCPyV‐regulated miRNAs. Although we have not demonstrated the exact mechanism of how MCPyV T‐antigens regulate miRNAs, our results indicate that the DnaJ domain of the T‐antigen is required for the miRNA regulation. One possible mechanism would be the direct or indirect regulation of miRNA processing factors by MCPyV T‐antigens, which affect miRNA biogenesis in the host cells. Such a mechanism has been observed for HPV16 E6 and E7 oncoproteins, in which the viral oncoproteins induce DICER and DROSHA expressions that modulate miRNA expressions.47 Alternatively, MCPyV T‐antigens may regulate enzymes that alter miRNA stability or turnover. In human, at least two terminal uridylyl transferases (TUTase‐2 and TUTase‐3) have been identified to regulate mature miRNA stability,48 and three miRNA degrading enzymes (XRN1, PTPase and RRP41).49 Although it remains to be determined whether MCPyV T‐antigens can regulate these enzymes, hepatitis C virus core protein has been demonstrated to inhibit TUTase‐2 that destabilizes miR‐122.50
In conclusion, we provide evidence that MCPyV oncoproteins regulate miRNAs and autophagy in MCC. MCPyV T‐antigens increase miRNA expressions and several of these miRNAs can directly target multiple autophagy genes that lead to suppression of autophagy in MCC (Fig. 6 c). We therefore propose that induction of autophagy could become an effective therapeutic strategy for MCC patients.
Author contributions
Experimental concept and design: Kumar S, HX and W‐OL. Experimental analysis: Kumar S, HX, HS, JG and LL. Data analysis and interpretation of results: Kumar S, HX, Shi H, Gao J, CCJ, VB, LL, AH and W‐OL. Article writing: Kumar S, HX, CL and W‐OL.
Supporting information
Figure S1. Illustration of miRNA sponge vectors design.
Figure S2. Evaluation of the efficacy of miRNA inhibition or overexpression.
Figure S3. miRNA sponges do not affect MCPyV LT expression.
Figure S4. RT‐qPCR analysis of primary and mature miRNAs in cells transfected with shTA, shsTA or vector control.
Figure S5. Evaluation of the effect of the LT DnaJ mutation D44N on miRNA regulation.
Figure S6. Evaluation of the effect of MCPyV T‐antigens expression on autophagosomes and autolysosomes by mRFP‐GFP‐LC3 reporter and confocal microscopy.
Figure S7. Effect of miRNA inhibition on autophagosomes and autolysosomes by mRFP‐GFP‐LC3 reporter and confocal microscopy.
Figure S8. Weak/negative ATG7 and p62 immunohistochemical stainings are associated with MCPyV+ MCC tumors.
Figure S9. Evaluation of the effect of Torin‐1 and a combination of Torin‐1 and bafilomycin A1 or pan‐caspase inhibitor z‐VAD‐FMK in MCPyV− MCC cell lines
Table S1. Short tandem repeat (STR) profiles of the five MCC cell lines used in this study.
Table S2. List of the oligonucleotides used in the study.
Table S3. The source and working concentrations of the antibodies used for Western blotting and immunohistochemistry.
Table S4. Immunohistochemical analysis of ATG7 and p62 in 45 MCC tumors.
Acknowledgements
We thank Drs N.L. Krett and J.C. Becker for providing MCC cell lines, Drs P. Moore and Y. Chang for plasmids, Dr J.A. DeCaprio for the Ab3 antibody, Drs J. Füllgrabe and B. Joseph for suggestions and reagents related to autophagy experiments, Dr K. Hultenby for the advice in electron microscopy work, Ms L. Ånfalk for processing tumor specimens and the members of the sRNA group for their helps and suggestions. This work was supported by Swedish Research Council, Cancer Research Funds of Radiumhemmet, Swedish Cancer Society, Karolinska Institutet, Stockholm County Council, National Natural Science Foundation of China (81773002) and the Natural Science Foundation of Tianjin (16JCYBJC42400). S.K. is supported by the Karolinska Institutet PhD program (KID), H.S. and J.G. by the China Scholarship Council training grants.
Conflict of interest: The authors disclose no potential conflicts of interest.
Data Availability Statement:The data that support the findings of this study are available from the corresponding author upon reasonable request.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Coggshall K, Tello TL, North JP, et al. Merkel cell carcinoma: an update and review: pathogenesis, diagnosis, and staging. J Am Acad Dermatol 2018;78:433–42. [DOI] [PubMed] [Google Scholar]
- 2. Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008;319:1096–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shuda M, Feng H, Kwun HJ, et al. T antigen mutations are a human tumor‐specific signature for Merkel cell polyomavirus. Proc Natl Acad Sci USA 2008;105:16272–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Houben R, Adam C, Baeurle A, et al. An intact retinoblastoma protein‐binding site in Merkel cell polyomavirus large T antigen is required for promoting growth of Merkel cell carcinoma cells. Int J Cancer 2012;130:847–56. [DOI] [PubMed] [Google Scholar]
- 5. Kwun HJ, Shuda M, Feng H, et al. Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe 2013;14:125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shuda M, Kwun HJ, Feng H, et al. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E‐BP1 translation regulator. J Clin Invest 2011;121:3623–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shuda M, Velasquez C, Cheng E, et al. CDK1 substitutes for mTOR kinase to activate mitotic cap‐dependent protein translation. Proc Natl Acad Sci USA 2015;112:5875–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Griffiths DA, Abdul‐Sada H, Knight LM, et al. Merkel cell polyomavirus small T antigen targets the NEMO adaptor protein to disrupt inflammatory signaling. J Virol 2013;87:13853–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Knight LM, Stakaityte G, Wood JJ, et al. Merkel cell polyomavirus small T antigen mediates microtubule destabilization to promote cell motility and migration. J Virol 2015;89:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheng J, Park DE, Berrios C, et al. Merkel cell polyomavirus recruits MYCL to the EP400 complex to promote oncogenesis. PLoS Pathog 2017;13:e1006668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Berrios C, Padi M, Keibler MA, et al. Merkel cell polyomavirus small T antigen promotes pro‐glycolytic metabolic perturbations required for transformation. PLoS Pathog 2016;12:e1006020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nwogu N, Boyne JR, Dobson SJ, et al. Cellular sheddases are induced by Merkel cell polyomavirus small tumour antigen to mediate cell dissociation and invasiveness. PLoS Pathog 2018;14:e1007276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stakaityte G, Nwogu N, Lippiat JD, et al. The cellular chloride channels CLIC1 and CLIC4 contribute to virus‐mediated cell motility. PLoS Pathog 2018;293:4582–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shuda M, Guastafierro A, Geng X, et al. Merkel cell polyomavirus small T antigen induces cancer and embryonic Merkel cell proliferation in a transgenic mouse model. PLoS One 2015;10:e0142329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Verhaegen ME, Mangelberger D, Harms PW, et al. Merkel cell polyomavirus small T antigen initiates Merkel cell carcinoma‐like tumor development in mice. Cancer Res 2017;77:3151–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Verhaegen ME, Mangelberger D, Harms PW, et al. Merkel cell polyomavirus small T antigen is oncogenic in transgenic mice. J Invest Dermatol 2015;135:1415–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xie H, Lee L, Caramuta S, et al. MicroRNA expression patterns related to Merkel cell polyomavirus infection in human Merkel cell carcinoma. J Invest Dermatol 2014;134:507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Abraham KJ, Zhang X, Vidal R, et al. Roles for miR‐375 in neuroendocrine differentiation and tumor suppression via notch pathway suppression in Merkel cell carcinoma. Am J Pathol 2016;186:1025–35. [DOI] [PubMed] [Google Scholar]
- 19. Renwick N, Cekan P, Masry PA, et al. Multicolor microRNA FISH effectively differentiates tumor types. J Clin Invest 2013;123:2694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Veija T, Sahi H, Koljonen V, et al. miRNA‐34a underexpressed in Merkel cell polyomavirus‐negative Merkel cell carcinoma. Virchows Arch 2015;466:289–95. [DOI] [PubMed] [Google Scholar]
- 21. Fan K, Ritter C, Nghiem P, et al. Circulating cell‐free miR‐375 as surrogate marker of tumor burden in Merkel cell carcinoma. Clin Cancer Res 2018;24:5873–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kumar S, Xie H, Scicluna P, et al. MiR‐375 regulation of LDHB plays distinct roles in polyomavirus‐positive and ‐negative Merkel cell carcinoma. Cancers (Basel) 2018;10:443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chang Y, Yan W, He X, et al. miR‐375 inhibits autophagy and reduces viability of hepatocellular carcinoma cells under hypoxic conditions. Gastroenterology 2012;143:177–87. e8. [DOI] [PubMed] [Google Scholar]
- 24. Zhu H, Wu H, Liu X, et al. Regulation of autophagy by a beclin 1‐targeted microRNA, miR‐30a, in cancer cells. Autophagy 2009;5:816–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Daily K, Coxon A, Williams JS, et al. Assessment of cancer cell line representativeness using microarrays for Merkel cell carcinoma. J Invest Dermatol 2015;135:1138–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xie H, Liu T, Wang N, et al. TERT promoter mutations and gene amplification: promoting TERT expression in Merkel cell carcinoma. Oncotarget 2014;5:10048–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sastre‐Garau X, Peter M, Avril MF, et al. Merkel cell carcinoma of the skin: pathological and molecular evidence for a causative role of MCV in oncogenesis. Cancers (Basel) 2009;218:48–56. [DOI] [PubMed] [Google Scholar]
- 28. Kwun HJ, Guastafierro A, Shuda M, et al. The minimum replication origin of Merkel cell polyomavirus has a unique large T‐antigen loading architecture and requires small T‐antigen expression for optimal replication. J Virol 2009;83:12118–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Iwasaki S, Kobayashi M, Yoda M, et al. Hsc70/Hsp90 chaperone machinery mediates ATP‐dependent RISC loading of small RNA duplexes. Mol Cell 2010;39:292–9. [DOI] [PubMed] [Google Scholar]
- 30. Tanida I, Minematsu‐Ikeguchi N, Ueno T, et al. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005;1:84–91. [DOI] [PubMed] [Google Scholar]
- 31. Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007;282:24131–45. [DOI] [PubMed] [Google Scholar]
- 32. Miranda KC, Huynh T, Tay Y, et al. A pattern‐based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 2006;126:1203–17. [DOI] [PubMed] [Google Scholar]
- 33. White E. The role for autophagy in cancer. J Clin Invest 2015;125:42–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest 2015;125:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Silva LM, Jung JU. Modulation of the autophagy pathway by human tumor viruses. Semin Cancer Biol 2013;23:323–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gammoh N, Fraser J, Puente C, et al. Suppression of autophagy impedes glioblastoma development and induces senescence. Autophagy 2016;12:1431–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rosenfeldt MT, O'Prey J, Morton JP, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013;504:296–300. [DOI] [PubMed] [Google Scholar]
- 38. Strohecker AM, Guo JY, Karsli‐Uzunbas G, et al. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E‐driven lung tumors. Cancer Discov 2013;3:1272–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xie X, Koh JY, Price S, et al. Atg7 overcomes senescence and promotes growth of BrafV600E‐driven melanoma. Cancer Discov 2015;5:410–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003;112:1809–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Takamura A, Komatsu M, Hara T, et al. Autophagy‐deficient mice develop multiple liver tumors. Genes Dev 2011;25:795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lin Z, McDermott A, Shao L, et al. Chronic mTOR activation promotes cell survival in Merkel cell carcinoma. Cancer Lett 2014;344:272–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li J, Wang X, Diaz J, et al. Merkel cell polyomavirus large T antigen disrupts host genomic integrity and inhibits cellular proliferation. J Virol 2013;87:9173–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wei Y, Pattingre S, Sinha S, et al. JNK1‐mediated phosphorylation of Bcl‐2 regulates starvation‐induced autophagy. Mol Cell 2008;30:678–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee JS, Li Q, Lee JY, et al. FLIP‐mediated autophagy regulation in cell death control. Nat Cell Biol 2009;11:1355–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wong SQ, Waldeck K, Vergara IA, et al. UV‐associated mutations underlie the etiology of MCV‐negative Merkel cell carcinomas. Cancer Res 2015;75:5228–34. [DOI] [PubMed] [Google Scholar]
- 47. Harden ME, Munger K. Perturbation of DROSHA and DICER expression by human papillomavirus 16 oncoproteins. Virology 2017;507:192–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Burns DM, D'Ambrogio A, Nottrott S, et al. CPEB and two poly(a) polymerases control miR‐122 stability and p53 mRNA translation. Nature 2011;473:105–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Das SK, Sokhi UK, Bhutia SK, et al. Human polynucleotide phosphorylase selectively and preferentially degrades microRNA‐221 in human melanoma cells. Proc Natl Acad Sci USA 2010;107:11948–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kim GW, Lee SH, Cho H, et al. Hepatitis C virus core protein promotes miR‐122 destabilization by inhibiting GLD‐2. PLoS Pathog 2016;12:e1005714. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Illustration of miRNA sponge vectors design.
Figure S2. Evaluation of the efficacy of miRNA inhibition or overexpression.
Figure S3. miRNA sponges do not affect MCPyV LT expression.
Figure S4. RT‐qPCR analysis of primary and mature miRNAs in cells transfected with shTA, shsTA or vector control.
Figure S5. Evaluation of the effect of the LT DnaJ mutation D44N on miRNA regulation.
Figure S6. Evaluation of the effect of MCPyV T‐antigens expression on autophagosomes and autolysosomes by mRFP‐GFP‐LC3 reporter and confocal microscopy.
Figure S7. Effect of miRNA inhibition on autophagosomes and autolysosomes by mRFP‐GFP‐LC3 reporter and confocal microscopy.
Figure S8. Weak/negative ATG7 and p62 immunohistochemical stainings are associated with MCPyV+ MCC tumors.
Figure S9. Evaluation of the effect of Torin‐1 and a combination of Torin‐1 and bafilomycin A1 or pan‐caspase inhibitor z‐VAD‐FMK in MCPyV− MCC cell lines
Table S1. Short tandem repeat (STR) profiles of the five MCC cell lines used in this study.
Table S2. List of the oligonucleotides used in the study.
Table S3. The source and working concentrations of the antibodies used for Western blotting and immunohistochemistry.
Table S4. Immunohistochemical analysis of ATG7 and p62 in 45 MCC tumors.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.