

Abstract
Alzheimer's disease (AD) is a common dementia affecting a vast number of individuals and significantly impairing quality of life. Despite extensive research in animal models and numerous promising treatment trials, there is still no curative treatment for AD. Astrocytes, the most common cell type of the central nervous system, have been shown to play a role in the major AD pathologies, including accumulation of amyloid plaques, neuroinflammation, and oxidative stress. Here, we show that inflammatory stimulation leads to metabolic activation of human astrocytes and reduces amyloid secretion. On the other hand, the activation of oxidative metabolism leads to increased reactive oxygen species production especially in AD astrocytes. While healthy astrocytes increase glutathione (GSH) release to protect the cells, Presenilin‐1‐mutated AD patient astrocytes do not. Thus, chronic inflammation is likely to induce oxidative damage in AD astrocytes. Activation of NRF2, the major regulator of cellular antioxidant defenses, encoded by the NFE2L2 gene, poses several beneficial effects on AD astrocytes. We report here that the activation of NRF2 pathway reduces amyloid secretion, normalizes cytokine release, and increases GSH secretion in AD astrocytes. NRF2 induction also activates the metabolism of astrocytes and increases the utilization of glycolysis. Taken together, targeting NRF2 in astrocytes could be a potent therapeutic strategy in AD.
Keywords: Alzheimer's disease, astrocytes, inflammation, NRF2, oxidative stress
Main points
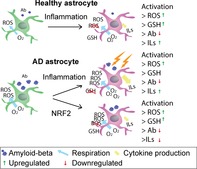
NRF2 induction ameliorates inflammatory and amyloid pathologies as well as antioxidant responses in AD astrocyte.
AD astrocytes fail to activate their antioxidant defense pathways upon inflammation.
Inflammation leads to metabolic activation of astrocytes and reduces their Aβ secretion.
1. INTRODUCTION
Alzheimer's disease (AD) is the most common dementia in the elderly population, significantly impairing cognitive functions and quality of life of the patients. Despite vast research, the exact pathophysiological mechanisms underlying disease progression are still unknown and no curative treatment exists for AD. The major hallmarks of the disease are episodic memory impairment and decline of cognitive functions. Typical neuropathological findings include intracellular neurofibrillary tangles and extracellular neurotic plaques, consisting of aggregates of β‐amyloid peptides (Aβ) (Blennow, de Leon, & Zetterberg, 2006). Most AD cases are sporadic and the underlying cause for the disease is unknown; however, a few percent of the cases are familial and causative mutations have been identified in amyloid precursor protein, and presenilin‐1, or −2 (PSEN1/2) genes (Waring & Rosenberg, 2008).
Astrocytes are active modulators of cognitive functions (Fields et al., 2014) and recent research has suggested that astrocytes are actively involved in AD disease progression (González‐Reyes, Nava‐Mesa, Vargas‐Sánchez, Ariza‐Salamanca, & Mora‐Muñoz, 2017). A commonly accepted amyloid cascade hypothesis suggests that increased extracellular levels of Aβ, especially its soluble forms, initiate AD pathology (Hardy & Selkoe, 2002). Astrocytes have been shown to contribute to both Aβ production and clearance (Kraft et al., 2013; Ries & Sastre, 2016) and also be significant targets themselves for Aβ toxicity. Accumulation of Aβ induces alterations in astrocytic calcium signaling, which is associated with increased reactive oxygen species (ROS) production (Askarova, Yang, Sheng, Sun, & Lee, 2011) and reduction in astrocytic glutathione (GSH), which impairs cellular defense mechanisms against oxidative stress (Abramov, Canevari, & Duchen, 2004a). This can also impact neurons, as astrocytic antioxidant defense pathways are thought to convey neuronal protection, which have low innate antioxidant defense mechanisms, and rely on astrocytic GSH metabolite CysGly for their GSH synthesis (Hirrlinger, Schulz, & Dringen, 2002). Thus, astrocytes have a role in both initiation and maintenance of oxidative stress in AD.
While not being considered inflammatory cells per se, astrocytes react to inflammatory responses by modifying their phenotype and by secreting inflammatory cytokines (Colombo & Farina, 2016; Sofroniew, 2014), and even become neurotoxic upon exposure to inflammatory mediators released by activated microglia (Liddelow et al., 2017). Further, accumulation of Aβ activates the nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NF‐κB) pathway, which is responsible for expression of astrocytic chemokines and pro‐inflammatory cytokines and thus promotes neuroinflammation (Shi et al., 2016). Hence, astrocytes are also important in neuroinflammation and can exert both pro‐ and anti‐inflammatory effects (Oksanen et al., 2019; Sofroniew, 2015). While neuroinflammation is thought to contribute to the neurodegeneration in AD (Heneka et al., 2015), it may also play an initial beneficial role, as it activates both astrocytes and microglia, and could thus increase clearance of Aβ. Chronic neuroinflammation, however, compromises the cellular microenvironment and leads to toxic effects and neurodegeneration (Heneka et al., 2015).
Transcription factor NF‐E2‐related factor 2 (NRF2), encoded by the NFE2L2 gene (Nuclear factor erythroid 2 like 2), is a key regulator of genes involved in antioxidant defense pathways. The E3 ligase adaptor Kelch‐like ECH‐associated protein 1 (KEAP1) targets NRF2 for proteasomal degradation and acts as a primary NRF2 repressor (Sun, Zhang, Chan, & Zhang, 2007). Modification of reactive cysteine residues of KEAP1 by electrophilic molecules inhibits proteolytic degradation of NRF2, leading to rapid accumulation of de novo synthesized NRF2 in the nucleus and induction of antioxidant response element (ARE) containing genes (Ma, 2013; Sun et al., 2007). Besides antioxidant proteins, the NRF2 target genes include anti‐inflammatory proteins, thus the NRF2‐KEAP1 pathway has been suggested to be critical in various diseases where oxidative stress or inflammation are involved in the disease progression. Therapeutic targeting of this pathway is of major interest in vast number of different pathologies, including many neurodegenerative diseases (Kanninen et al., 2015).
We have previously shown that Presenilin 1 mutated (PSEN1ΔE9) AD patient astrocytes, generated from induced pluripotent stem cells, display a toxic Aβ profile, manifest altered cytokine secretion upon inflammatory stimulation and use more oxidative metabolism, thus also producing more ROS than healthy control astrocytes (Oksanen et al., 2017). Here, we have further studied the effect of inflammation on both healthy and AD astrocytes and show that inflammation activates the metabolism of human astrocytes and also leads to decreased secretion of Aβ from both healthy and diseased cells. We further show that NRF2 activation has both anti‐inflammatory and anti‐oxidant effects on human astrocytes. Inducing the NRF2‐ARE pathway reduces Aβ and cytokine secretion, and increases GSH secretion from AD astrocytes, suggesting that targeting NRF2 in human astrocytes could be beneficial for AD patients.
2. METHODS
2.1. Human iPS cells
Previously established PSEN1ΔE9 mutant individual and their isogenic control iPS lines were used in this study (Oksanen et al., 2017). Two isogenic pairs were used in all experiments except the initial testing of NRFF2 induction which was done with one isogenic pair. The isogenic pairs included in this study were AD2B, AD2B iso, AD3B, and AD3B iso from Oksanen et al. (2017). iPS cells were generated under the ethical approval from the committee on Research Ethics of Northern Savo Hospital district (license no. 123/2016) and cultured in Essential 8™ Medium (E8; Thermo Fisher) as previously described (Oksanen et al., 2017).
2.2. Astroglial differentiation and characterization of their transcriptome
A previously described protocol was used for the astroglial differentiation of iPS cells (Oksanen et al., 2017). Briefly, dual SMAD inhibitors 10 μM SB431542 (Sigma‐Aldrich) and 200 nM LDN193189 (Selleckchem) were used to induce neural differentiation and rosette formation. Areas with rosettes were then mechanically lifted and cultured in suspension on ultra‐low attachment plates (Corning) to initiate sphere formation. Spheres were maintained in astrocyte differentiation medium (DMEM/F12 with N2, non‐essential amino acids, and 1 U/ml heparin from Leo Pharma and Glutamax) supplemented with 10 ng/ml bFGF and 10 ng/ml EGF (both from Peprotech). Medium was changed every 2–3 days and spheres were split manually once a week. Spheres were cultured in suspension for 5–7 months to ensure the purity of astrocyte cultures. For maturation, spheres were dissociated with Accutase (Stem Cell Technologies) and plated on Matrigel‐coated (Corning) dishes in astrocyte differentiation medium supplemented with 10 ng/ml CNTF and 10 ng/ml BMP4 (both from Peprotech) for 7 days prior to experiments.
For characterization of the iPSC‐derived astrocytes with whole transcriptome sequencing, we extracted total RNA from the iPSC lines of the two individuals with PSEN1ΔE9 mutation and the corresponding isogenic control iPSC lines using RNeasy Mini Kit (Qiagen). mRNA sequencing was done by Biomedicum Functional Genomics Unit (FuGu, University of Helsinki, Finland). The RNA quality was analyzed on the Agilent 2,100 Bioanalyzer™ using an RNA6000 assay. After ribosomal RNA depletion (RiboZero, Illumina), mRNA sequencing libraries were constructed with SureSelect Strand Specific RNA Seq kit (Agilent). Libraries were sequenced with Illumina NextSeq (76 bp, single‐end) resulting in 29–85 M reads per sample. Gene expression data is available in Gene Expression Omnibus (GEO; GSE138695) Reviewer link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138695 (token yxankeiqfjwldsn). Differential gene expression (DGE) analyses were conducted by GeneVia Technologies (Tampere, Finland). In short, reads were aligned to human genome GRCh38 and annotated to genes using STAR v2.5.2 (Dobin et al., 2013). DGE analysis of AD cell lines versus isogenic control samples was performed using R package DESeq2 (Love, Huber, & Anders, 2014). For comparing expression profiles of our iPSC‐derived astrocytes to other available data, we downloaded from GEO expression data of previously published iPSC‐derived astrocytes (Windrem et al., 2017, GSE86906) and three RNA‐sequencing samples of human cortical astrocytes, neurons, and oligodendrocytes (Zhang et al., 2016, GSE73721; GSM1901311, GSM1901333, and GSM1901334, respectively). To produce counts per million (CPM), RNA‐sequencing data of Zhang et al. (2016) was realigned to human genome GRCh38 using STAR 2.5.4b (Dobin et al., 2013) and annotated to gene exons with HTSeq v0.6.1 (Anders, Pyl, & Huber, 2015; GTF release 93). After merging these data with ours, we filtered the matrix for lowly expressed genes (<1 CPM) and normalized it using edgeR (Robinson, McCarthy, & Smyth, 2010). A clustering heatmap based on Euclidean distance was made with R package gplots 3.0.1.1 to compare similarity between samples. As seen in the heatmap with our iPSC‐derived astrocytes combined with other data sets (Windrem et al., 2017; Zhang et al., 2016), our samples form a uniform cluster, with replicate samples from the same cell lines clustering together (Figure S1). Our samples also display similarity with previously published iPSC‐derived astrocytes produced with the same culturing method (Windrem et al., 2017). In turn, samples collected from human brain fall outside these two clusters, the human astrocyte sample being most similar cell type to the cultured iPSC‐derived astrocytes when compared with samples of oligodendrocytes or neurons.
2.3. Lentiviral NRF2 induction
Cells were incubated overnight (approximately 16 hr) with the desired amount (Multiplicity of infection [MOI] of 0.5–2) of infectious particles produced from a lentiviral NRF2 vector, added to regular culture medium. After transduction, the cultures were washed and fresh medium was added. The cultures were used in experiments, 48 hr after transduction, to ensure adequate NRF2 expression. The control group was left untreated but went through the same media changes and washing steps.
2.4. Chemical stimulation of the cells
To optimize the chemical induction of NRF2 pathway in astrocytes, cells were treated with increasing concentrations (1–10 μM) of sulforaphane (Sfn, Sigma Aldrich) for different times (16–72 hr). For the actual experiments, a concentration of 5 μM for 72 hr was chosen. Fresh Sfn was added to cells daily. For inflammatory stimulation, cells were treated with 50 ng/ml hTNFα and 10 ng/ml hIL‐1β (both from Peprotech) for 48 hr. Control cells were maintained in the regular culture medium. For an additional experiment to determine possible changes in expression of genes and proteins regulating GSH synthesis and secretion, an 8‐hr time point was used in qPCR and 24‐hr time point in Western blots.
2.5. MTT assay
Prior to MTT assay, cells were cultured and treated as described above, similar to all experiments. Cells were incubated with MTT (0.5 mg/ml; Sigma) containing media until sufficient color formation (3–4 hr). Media were removed and crystals dissolved with adequate volume of DMSO. Absorbance was read at 595 nm.
2.6. Real‐time qPCR
RNA was extracted using RNeasy Mini Kit (Qiagen) followed by cDNA synthesis with Maxima reverse transcriptase (Life Technologies). The relative mRNA expression levels were determined by quantitative RT‐PCR (StepOne Plus™ Real‐Time PCR system, Life Technologies; or Bio‐Rad CFX96 Real‐Time System, Bio‐Rad) using the following TaqMan assay mixes (Life Technologies): NFE2L2 [Hs00232352_m1], HMOX1 [Hs01110250_m1], NQO1 [Hs01045993_g1], GCLC [Hs00155249_m1], GCLM [Hs00157694_m1], GSS [Hs00609286_m1], ABCC1 [Hs01561483_m1], and Universal Master Mix or Maxima Probe/ROX qPCR Master Mix (Life Technologies). Expression levels were normalized to β‐Actin (ACTB; Life Technologies) using Q‐gene program (Equation 2).
2.7. Immunofluorescence staining
Cells were first fixed with 4% paraformaldehyde followed by permeabilization with 0.2% Triton‐X. Cells were then treated with 6 M Guanidine HCl (Sigma Aldrich) for 10 min at room temperature (RT) for antigen retrieval and blocked with normal goat serum. NRF2 primary antibody (Santa Cruz; sc‐365,949) was diluted 1:100 and incubated for 2 hr at RT. For fluorescence detection, cells were then labeled with Alexa Fluor™ 488 Tyramide SuperBoost™ Kit (Thermo Fisher) according to manufacturer's instructions. Nuclei were counterstained with Hoechst 33258 (Sigma) and coverslips were mounted with Fluoromount‐G (Thermo Fisher). Images were acquired using Olympus BX51 fluorescence microscope. All images were acquired using the same settings.
2.8. Western blotting
Western blotting was done as previously described (Oksanen et al., 2017) with the following antibodies and dilutions: NRF2 (16396‐1‐AP, 1:5000, Proteintech), P‐NRF2 (ab76026, 1:1000, Abcam), Lamin B1 (ab133741, 1:2000, Abcam) and β‐Actin (A5441, 1:5000, Sigma). The GCLC and GCLM antibodies (both 1:100000) were a kind gift from Dr. Terrance J. Kavanagh, University of Washington, Seattle, USA.
For the subcellular fractionation, cells were cultured and matured as usual and washed once with cold PBS before harvesting. Cells were scraped in MNase buffer (10 mM Tris pH 7.4, 10 mM NaCl, 5 mM MgCl2, 0.1% NP‐40, and protease inhibitors), incubated on ice for 10 min, vortexed and centrifuged at 1500 ×g for 5 min. Supernatants were collected as cytoplasmic fraction, and pellets (i.e., nuclear fraction) were lysed with lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA, 1% Triton X‐100, 0.5% Na‐deoxycholate, 0.1% SDS, 10% glycerol, pH 7.5).
2.9. Analysis of cellular metabolism
Seahorse XF24 analyzer and Mito Stress Test were used to analyze cellular metabolism according to manufacturer's instructions (Agilent Technologies). The assay medium was supplemented with 1x Glutamax and 0.5 mM sodium pyruvate. Glucose (10 mM) was added as part of the assay. Final concentrations of oligomycin, FCCP, antimycin, and rotenone were 1 μM each. Results were normalized to total protein content in each well using Pierce BCA protein assay according to manufacturer's instructions (Thermo Fisher).
2.10. ROS measurement
To measure cellular ROS levels, cells were loaded with 1 μM CellROX green reagent (Molecular Probes) for 45 min. Before analysis, cells were washed with PBS, detached with Accutase and resuspended in PBS. Median fluorescence intensities of 10,000 single cell events from each sample were collected using FACSCalibur (BD Biosciences).
2.11. Glutathione secretion
Secreted and intracellular GSH levels were measured as described earlier (Liddell, Hoepken, Crack, Robinson, & Dringen, 2006). Briefly, conditioned medium was mixed with an equal volume of cold 1% sulfosalicylic acid (SSA, Sigma Aldrich) and cells were scraped directly with SSA. Samples were further diluted with water as needed (1:5–1:10) and GSH concentrations were determined from a standard curve following the rate of thionitrobenzoate production colorimetrically after addition of GSH reductase. Results were normalized to total protein amount using Pierce BCA protein assay.
2.12. Analysis of amyloid‐β levels
Aβ1‐40 and Aβ1‐42 concentrations were measured from cell culture media after conditioning for 72 hr with ELISA kits according to manufacturer's instructions (Invitrogen). For Aβ1‐42, an ultrasensitive kit was used. The results were normalized to the total protein concentration in the well (Pierce BCA protein assay).
2.13. Cytokine secretion
Cytokine concentrations were analyzed from conditioned media by CBA Flex Sets (BD) Human Soluble Protein (granulocyte‐macrophage colony‐stimulating factor [GM‐CSF] and IL‐6) and Human Enhanced Sensitivity (IL‐2 and IL‐10). Assays were done according to manufacturer's instructions with minor modifications. For Soluble Proteins, 20 μL of sample or cytokine standard and 20 μL of 1:50 bead mixture was used and for Enhanced Sensitivity, 25 μL of sample or cytokine standard and 10 μL of 1:20 bead mixture. Beads were analyzed on CytoFLEX Flow Cytometer (Beckman Coulter). More than 300 events per cytokine were measured. Data were analyzed using FCAP Array 2.0 (SoftFlow, Pécs, Hungary) and cytokine concentrations were calculated by regression analysis from known standard concentrations.
2.14. Statistical analysis
Statistical analyses were performed with GraphPad Prism 5.03 software (GraphPad Software Inc., San Diego, CA) and groups were compared with Student's t‐test or one‐way ANOVA with Tukey's post‐hoc test. Statistical significance was assumed if p < 0.05. All data are expressed as mean ± SEM.
3. RESULTS
3.1. Lentiviral or drug‐mediated activation of NRF2 induces expression of target genes and accumulation of NRF2 in the nucleus
To investigate putative differences in the activity of the NRF2 pathway between AD and control cells, we analyzed the expression of the NRF2 gene (Nuclear factor, erythroid 2 like 2, NFE2L2) and its target genes: Heme oxygenase 1 (HMOX1), NAD(P)H dehydrogenase quinone 1 (NQO1), Glutamate ‐cysteine ligase catalytic subunit (GCLC), Glutamate ‐cysteine ligase regulatory subunit (GCLM), and glutathione synthetase (GSS). In basal conditions, no significant differences were seen between AD and control astrocytes when the mRNA levels were analyzed by quantitative PCR (Figure S2).
Two alternative approaches were used to activate the NRF2 pathway: (a) the cells were transduced with lentiviral NRF2 (Kanninen et al., 2009); (b) chemical induction of the pathway was achieved by treatment with a dietary non‐nutrient, Sfn, an isothiocyanate present in cruciferous vegetables, and known to induce NRF2 (Houghton, Fassett, & Coombes, 2016). First, the conditions for the NRF2 induction were optimized by analyzing the expression of NFE2L2, its target genes (HMOX1, NQO1, GCLC, and GCLM) and the secretion of GSHfrom the astrocytes upon various conditions (Figure S3). For viral transduction, a MOI of 1 was chosen, and for Sfn, 5 μM concentration was used for 72 hr. With the chosen conditions, lentiviral NRF2 expression increased NFE2L2 transcript levels (Figure 1a) and both lentiviral transduction and Sfn treatment induced the transcription of NRF2 target genes (Figure 1b–e) as well as secretion of GSH from astrocytes (Figure 1f), verifying the induction of NRF2 activity. MTT assay verified that neither the lentiviral nor the Sfn treatment caused significant reductions in the MTT activity and thus, viability of the cells (Figure S3g).
Figure 1.
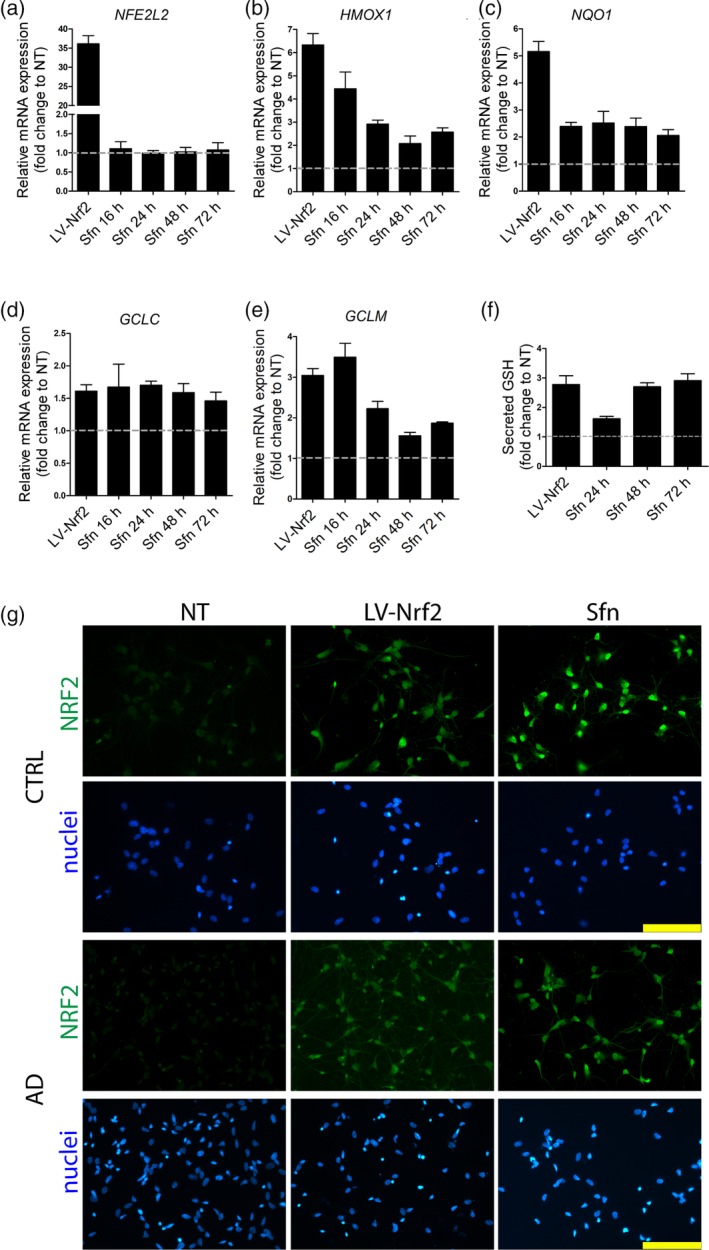
NRF2 induction in astrocytes by lentiviral transduction or Sfn treatment. (a) Relative mRNA expression of NRF2 (NFE2L2) after treatment shown as fold change to non‐treated (NT) astrocytes. Lentiviral transduction increases NRF2 expression, while Sfn treatment (5 μM) does not. (b–e) Relative mRNA expression of NRF2 target genes (HMOX1, NQO1, GCLC, and GCLM) after treatment. Both lentiviral transduction and Sfn treatment increase the expression of NRF2 target genes. (f) Glutathione secretion after treatment. Both lentiviral NRF2 and Sfn treatments increase the secretion of glutathione from astrocytes. (a–f) Experiments were performed with one isogenic pair and both AD and control cells behaved similarly. Representative data from one isogenic control line are shown as fold change to non‐treated astrocytes. Data are presented as mean ± SEM from three independent experiments. (g) Representative immunofluorescence images of NRF2 (green) staining show the accumulation of NRF2 in the nucleus upon both treatments in astrocytes of both genotypes. Nuclei were counterstained with Hoechst 33258 (blue). Scale bars 100 μm. (NT: non‐treated cells; GSH: glutathione; LV‐NRF2: lentiviral NRF2 transduction; Sfn: sulforaphane; CTRL: isogenic control astrocytes; AD: PSEN1 ΔE9 mutated Alzheimer's disease astrocytes) [Color figure can be viewed at http://wileyonlinelibrary.com]
Immunofluorescence staining of NRF2 in astrocytes in basal conditions and after induction revealed increased accumulation of NRF2 in the nucleus after lentiviral transduction or Sfn treatment (Figure 1h), further verifying the activation of the NRF2 pathway by these treatments. Western blotting after subcellular fractionation verified that the NRF2 was present only in the nuclear fraction (Figure S4).
These results show that both lentiviral transduction and Sfn treatment are able to activate the NRF2 pathway. Furthermore, they show that in basal conditions, NRF2 activity is not significantly altered between AD and control astrocytes.
3.2. Inflammatory stimulation and NRF2 induction activate astrocyte metabolism leading to increased ROS production
As inflammation is thought to play a significant role in AD pathology (Heneka et al., 2015), we further investigated the effect of inflammatory stimulation on astrocytes. To induce inflammatory pathways, AD and control astrocytes were stimulated with TNFα and IL‐1β for 48 hr. As inflammation is known to have an activating effect on astrocytes, we next analyzed the metabolic activity of the cells by Seahorse XF analyzer. Oxygen consumption and extracellular acidification were analyzed from untreated cells, from cells subjected to inflammatory stimulation and from cells where NRF2 was activated either by lentiviral transduction or Sfn treatment. As expected, inflammatory stimulation led to significant astrocytic activation and increased their metabolism significantly (Figure 2a–c, Figure S5). Interestingly, NRF2 activation also had a similar effect on astrocytes. Both AD and control cells showed increased oxygen consumption rates (OCRs) representing increased mitochondrial respiration upon inflammatory stimulation or NRF2 activation (Figure 2a, Figure S5a,b). Both inflammation and NRF2 induction also increased the extracellular acidification rate (ECAR) in both diseased and healthy cells (Figure 2b, Figure S5c,d), suggesting an increase also in glycolytic activity, as majority of the ECAR is due to the glycolytic conversion of pyruvate to lactate. Importantly, while in basal conditions, the AD cells were more respiratory than control cells, as shown by an increased OCR/ECAR ratio (Figure 2c); both inflammation and NRF2 activation normalized this to a similar level with the control cells. This suggests that the glycolytic activity increased more in AD than in control cells. Mitochondrial respiration produces ROS as a byproduct and in line with the increased metabolic activity and oxygen consumption, inflammatory stimulation also led to increased ROS production from both AD and control astrocytes (Figure 2d). Interestingly, AD astrocytes produced significantly more ROS than control cells in response to inflammation. Lentiviral induction of NRF2 resulted in similar increase in ROS production than inflammation did. However, treatment with Sfn did not increase cellular ROS production (Figure 2d), even though oxygen consumption was increased (Figure 2a, Figure S5a,b).
Figure 2.
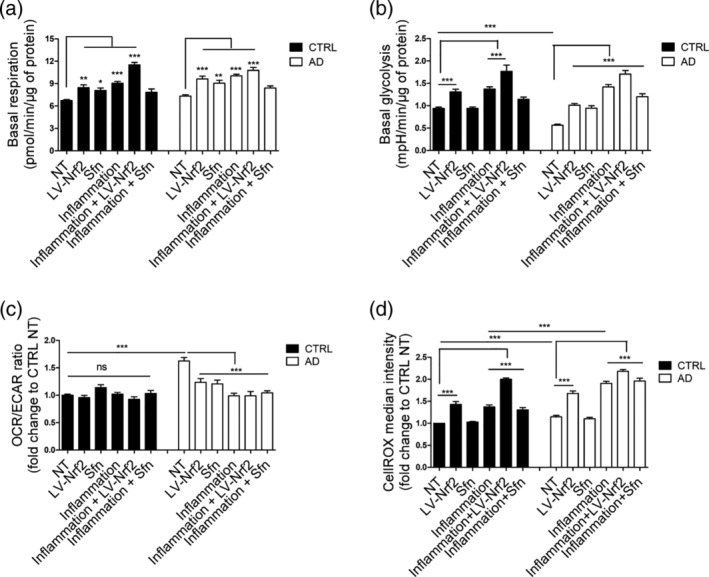
Both inflammatory stimulation and NRF2 induction lead to metabolic activation of astrocytes. (a) Quantification of the basal respiration rates from the oxygen consumption rate analyzed by Seahorse XF analyzer in the presence of glucose. Both inflammatory stimulation and NRF2 induction activate oxidative metabolism in both cell types. (b) Quantification of the basal glycolytic activity from extracellular acidification rate. Both inflammatory stimulation and LV‐NRF2 induction activate glycolytic metabolism in both cell types. (c) Relative OCR/ECAR ratio after glucose injection shown as fold change to non‐treated isogenic control astrocytes. Both inflammation and NRF2 induction reduce the ratio of oxidative to glycolytic metabolism in AD cells. (a–c) All data are presented as mean ± SEM, n = 4–6 independent experiments using two isogenic pairs with 3–4 technical replicates in each experiment. (d) Reactive oxygen species (ROS) production, measured by CellROX fluorogenic probe and FACS analysis. As expected, the increased respiratory activity upon inflammatory stimulation or NRF2 induction also leads to increased ROS production in both AD and isogenic control cells. However, in Sfn‐treated cells, ROS is not increased in either genotype. Relative results compared with non‐treated control cells shown as mean ± SEM, n = 3 independent experiments with two isogenic pairs. ***p < .001; **p < .01; *p < .05. ns, non‐significant
These results show that inflammatory stimulation leads to metabolic activation of both AD and control astrocytes. This metabolic activation increases ROS production, subjecting the cells to oxidative stress upon inflammation, and AD astrocytes are more vulnerable than control astrocytes. The results also show that induction of NRF2, either by lentiviral means or by Sfn treatment, leads to similar metabolic activation.
3.3. Secretion of glutathione from AD astrocytes is impaired but rescued by NRF2 activation
We previously showed that AD astrocytes rely more on oxidative metabolism than control cells and produce more ROS (Oksanen et al., 2017), thus subjecting the cells to oxidative stress. To analyze the activity of the cellular antioxidant defense pathways, we measured both cellular and secreted GSH levels in AD and control astrocytes upon basal conditions, inflammatory stimulation, and NRF2 induction using a previously established colorimetric assay (Liddell et al., 2006). Already in basal conditions, AD cells secreted 1.5‐fold less GSH than control cells (Figure 3a). Furthermore, upon inflammatory stimulation and induction of the respiratory activity, the control cells significantly increased their GSH secretion, whereas AD cells were not able to do so (Figure 3a). NRF2 is known to regulate GSH synthesis, and activation of NRF2 was able to drastically increase GSH secretion from both healthy and AD cells, with Sfn treatment resulting in a greater increase than the lentiviral NRF2 induction (Figure 3a; 1.5‐fold greater in control and twofold greater in AD astrocytes). This can explain why the Sfn treatment did not increase ROS production while lentiviral transduction did so (Figure 2g). No difference in cellular GSH levels were seen between AD and control astrocytes, but lentiviral induction of NRF2 increased this slightly in both genotypes (Figure 3b). In line with the results from the cellular GSH levels, no difference was seen in the expression of enzymes involved in GSH synthesis between AD and control cells, when untreated or when stimulated by inflammation or Sfn separately or in combination, even though inflammation and Sfn tended to increase the gene expression and the treatments had a synergic effect (Figure S6).
Figure 3.
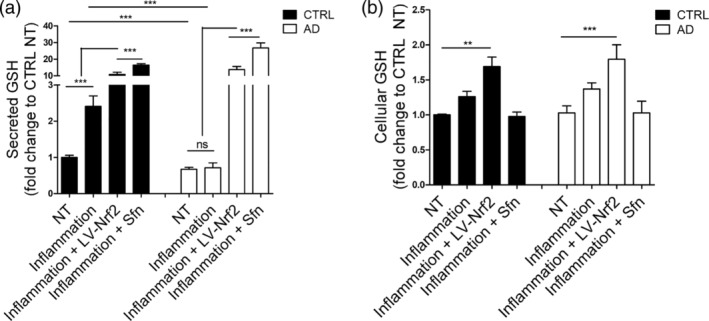
NRF2 activation rescues the reduced glutathione secretion in AD astrocytes. (a) Relative secreted GSH upon basal conditions and inflammatory stimulation shown as fold change to non‐treated isogenic control astrocytes. Healthy control cells secrete more GSH in basal conditions than AD cells and increase GSH secretion upon inflammatory stimulation, whereas AD cells do not. Activation of NRF2 drastically increases GSH secretion both in healthy and AD cells. (b) Relative cellular GSH levels shown as fold change to non‐treated isogenic control astrocytes. A slight increase is seen also in cellular GSH upon lentiviral induction of NRF2. No difference is seen in the cellular GSH levels between AD and control cells. All data are presented as mean ± SEM, n = 3–4 independent experiments with two isogenic pairs. ***p < .001; **p < .01. ns, non‐significant
These results show that secretion of GSH is impaired in AD cells both in basal conditions and upon inflammation, but activation of NRF2 is able to rescue the defect.
3.4. Astrocytic activation leads to decreased amyloid‐β secretion
Inflammation has been suggested to play a role in Aβ pathology with putative initial beneficial effects through activation of astrocytes and microglia (Meraz‐Ríos, Toral‐Rios, Franco‐Bocanegra, Villeda‐Hernández, & Campos‐Peña, 2013). We have also shown earlier that astrocytes are involved both in Aβ secretion and clearance (Oksanen et al., 2017). Thus, to analyze secreted Aβ levels upon inflammation and NRF2 induction, ELISA assays with antibodies detecting specifically either Aβ1‐40 or Aβ1‐42 peptides were used. Inflammation led to a similar approximately twofold decrease in both Aβ1‐40 and Aβ1‐42 peptides in AD and control cells (Figure 4a,b) with no significant difference in the ratio of the two peptides. However, induction of NRF2 led to a significant 1.4‐fold decrease specifically in Aβ1‐42 in AD cells (Figure 4b), whereas Aβ1‐40 levels were unaltered (Figure 4a). This normalized the toxic Aβ1‐42/Aβ1‐40 ratio in AD cells (Figure 4c).
Figure 4.
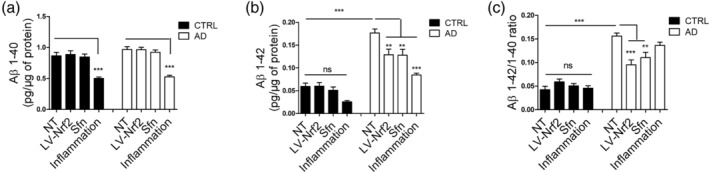
NRF2 activation ameliorates the amyloid secretion profile of AD astrocytes. (a) Secreted Aβ1‐40 levels. (b) Secreted Aβ1‐42 levels. (c) Aβ1‐40/Aβ1‐42 ratio. NRF2 induction specifically reduces the Aβ1‐42 secretion from AD astrocytes and thus improves the Aβ1‐40/Aβ1‐42 ratio while inflammatory stimulation leads to simultaneous reduction of both amyloid species. All data are presented as mean ± SEM, n = 3 independent experiments with two isogenic pairs. ***p < .001; **p < .01; ns, non‐significant
These data show that inflammatory stimulation reduces Aβ secretion from astrocytes. Activation of astrocytes through NRF2 induction, on the other hand, reduced the production of the toxic Aβ1‐42 peptide in AD cells, whereas it had no effect on the healthy astrocytes.
3.5. NRF2 activation has anti‐inflammatory effects on astrocytes
Besides antioxidant properties, induction of NRF2 also has anti‐inflammatory effects (Cuadrado et al., 2019). We have previously shown that AD astrocytes secrete more cytokines than control cells upon inflammatory stimulation (Oksanen et al., 2017). Thus, we finally analyzed the effect of NRF2 activation on the cytokine secretion from astrocytes. Upon inflammatory stimulation, AD astrocytes secrete significantly more Interleukins 2, 6, and 10 and GM‐CSF than healthy cells (Figure 5a–d). Induction of NRF2 by both lentiviral and Sfn treatment significantly reduced the cytokine secretion from cells of both genotypes (Figure 5a–d) resulting in normalized cytokine secretion from AD cells.
Figure 5.
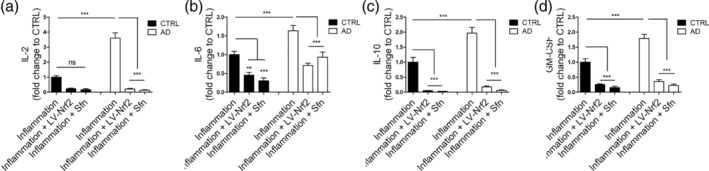
NRF2 induction shows anti‐inflammatory effects. Cytokine secretion from astrocytes upon inflammatory stimulation. (a) Interleukin‐2 (IL‐2). (b) Interleukin‐6 (IL‐6). (c) Interleukin‐10 (IL‐10). (d) Granulocyte‐macrophage colony‐stimulating factor (GM‐CSF). Secretion of inflammatory cytokines is increased in AD cells when compared with healthy control cells. Induction of NRF2 reduces cytokine secretion from cells of both genotypes. Relative levels are shown as fold change to inflammatory stimulated isogenic control astrocytes and presented as mean ± SEM, n = 3 independent experiments with two isogenic pairs. ***p < .001; **p < 0.01. ns, non‐significant
These data show that activation of the NRF2 pathway poses anti‐inflammatory effects on astrocytes upon inflammatory insult.
4. DISCUSSION
Despite vast research efforts, the detailed pathogenetic mechanisms underlying AD disease progression still remain unknown. Generally accepted underlying mechanisms include accumulation of soluble and deposited Aβ, oxidative stress and neuroinflammation. While familial AD has been traditionally considered a neuronal disease, the role of astrocytes in AD pathogenesis is becoming increasingly more evident, and astrocytes seem to play a role in all three of these processes. For example, exposure to Aβ leads to loss of mitochondrial membrane potential and increased ROS production specifically in astrocytes but not in neurons (Abramov, Canevari, & Duchen, 2004b). Also, Ca2+‐transients, one of the functional hallmarks of astrocytes, are impaired in AD patient brains and oxidative stress directly affects cellular Ca2+‐homeostasis (Lin et al., 2007). Inflammation in the central nervous system always involves astrocytes and microglia, the resident immune cells of the brain (Perry & Teeling, 2013), and is tightly linked to neurodegeneration (Chen, Zhang, & Huang, 2016). Aβ is involved in the inflammatory response, and astrocytic exposure to Aβ has been shown to induce a pro‐inflammatory profile and astrogliosis (Batarseh et al., 2016). Under physiological conditions, astrocytes are critical for maintaining a homeostatic balance in the brain (Vasile, Dossi, & Rouach, 2017). However, under a pathological inflammatory state, they can aggravate the disease pathology and maintain inflammatory stimuli through production of cytokines and interleukins (Glass, Saijo, Winner, Marchetto, & Gage, 2010) and through failure in stress resolution (Colombo & Farina, 2016). We showed previously that PSEN1 mutant AD patient astrocytes manifest hallmarks of AD, including increased Aβ secretion, altered inflammatory responses and increased oxidative metabolism with increased ROS production (Oksanen et al., 2017), thus further stressing the importance of astrocytes in AD pathology. Here we further investigated the role of inflammation in astrocyte function and metabolism.
Inflammatory stimulation activated astrocytes and led to an increased respiratory metabolism and ROS production. The metabolic activation was associated with increased antioxidant responses and GSH secretion in healthy astrocytes. However, PSEN1 mutant AD patient astrocytes were not able to increase GSH secretion, which was also low in basal conditions. Thus, chronic inflammation is likely to expose them and their nearby environment to chronic oxidative stress. This is also likely to impact neurons, as they have been shown to rely on astrocytes for antioxidant defenses (Hirrlinger et al., 2002). The impact of inflammatory stimulation on astrocytes, however, was not purely negative. As suggested earlier (Meraz‐Ríos et al., 2013), at least short‐term inflammatory activation leads to reduction of Aβ levels in both healthy and AD cells, most likely through increased clearance of Aβ by the reactive astrocytes.
No curative treatment exists for AD and only symptomatic treatment can be offered to patients. Numerous trials have been performed and several are currently ongoing with the aim of identifying treatment options. Most of the clinical trials have focused on targeting the Aβ deposits, but so far all of them have failed (Castello, Jeppson, & Soriano, 2014; Golde, Schneider, & Koo, 2011), thus alternative approaches are needed. Apart from targeting Aβ, oxidative stress and neuroinflammation are putative therapeutic targets in AD (Bronzuoli, Iacomino, Steardo, & Scuderi, 2016; Gella & Durany, 2009). As astrocytes play a role in both these processes, we investigated the effect of targeting these pathways in astrocytes.
The NRF2‐ARE pathway is emerging as a potent therapeutic target for various pathological conditions as it regulates both cellular redox balance and inflammatory processes (Cuadrado et al., 2019). NRF2 is the master regulator of around 250 genes, which contain the ARE‐sequence in their promoter region and are involved in various cellular processes including antioxidant response, cellular metabolism and inflammatory regulation (Hayes & Dinkova‐Kostova, 2014). Besides its appealing protective role against both oxidative stress and inflammation, another attractive feature of NRF2 is that it can be activated by pharmaceutical means. A vast number of different NRF2 activators and modulators are currently being developed by pharmaceutical companies with the aim of treating various diseases ranging from psoriasis to multiple sclerosis and breast cancer (Cuadrado et al., 2019). Dimethyl fumarate, a potent NRF2 activator, has already shown encouraging safety and tolerability results in two clinical phase III trials (Fernández et al., 2017; Fox et al., 2017). Sulforaphane (Sfn), a naturally occurring isothiocyanate, has been the primary choice in neurological conditions, as it has been shown to be able to penetrate the blood–brain barrier (Jazwa et al., 2011) and it has exhibited beneficial effects in numerous preclinical trials for neurological diseases (Holmström, Kostov, & Dinkova‐Kostova, 2016). Activation of the NRF2 pathway has been shown to confer protection also in AD (Johnson & Johnson, 2015). Interestingly, the NRF2‐ARE pathway is particularly weak in neurons, while astrocytes show a stronger response to NRF2 activation and have higher expression of ARE‐containing genes than neurons (Baxter & Hardingham, 2016). Furthermore, studies on mice with astrocyte specific overexpression of NRF2 show that the NRF2‐induced neuronal protection is dependent on astrocytes (Chen et al., 2009; Vargas, Johnson, Sirkis, Messing, & Johnson, 2008). Thus, we hypothesized that activation of the NRF2 pathway could also have beneficial effects on the human AD patient astrocytes. We induced NRF2 in AD and control astrocytes by two alternative approaches; lentiviral mediated NRF2 overexpression and activation of NRF2 with Sfn. Induction of the NRF2 pathway exerted anti‐oxidative, anti‐inflammatory, and anti‐amyloid effects on AD cells, suggesting that it could target all these three processes. Besides regulating cellular redox balance and inflammatory reactions, NRF2 has also been shown to modulate cellular metabolism and regulate metabolic remodeling during stress (Hayes & Dinkova‐Kostova, 2014). In line with this, NRF2‐induced astrocytic activation and led to increased metabolic activity. Both oxidative and glycolytic metabolisms were increased; however, AD cells shifted their metabolism more toward glycolysis, as is typical for the healthy control cells. Most importantly, all the beneficial effects were achieved also by pharmaceutical activation of NRF2 with Sfn, a drug already in preclinical trials. Sfn further increased GSH levels more than lentiviral induction of NRF2, resulting in unaltered ROS levels and thus a better safety profile.
While the novel NRF2 activators are promising therapeutic agents, challenges still remain with their use. Activation of the NRF2 pathway is tissue‐ and cell‐specific and dependent on dose, timing, and duration of the treatment. The half‐life of NRF2 is very short even after drug‐mediated activation (Cuadrado et al., 2019). Furthermore, many of the NRF2 target genes are much more stable and have fairly long half‐lives, complicating the dosing of NRF2 modulating drugs (Cuadrado et al., 2019). NRF2 activity is known to decline upon age (Suh et al., 2004). This decline, however, seems to be reversible and at least in aged rats, NRF2 can be re‐activated by pharmaceutical means (Suh et al., 2004), suggesting that it can also be activated in human patients with age‐associated diseases like AD. However, at least one clinical trial has shown that the level of NRF2 induction correlated negatively with the age of the patient (Hammer et al., 2018). Thus, the dosing of NRF2 activating drugs likely needs adjusting depending on the age and disease status of the patients. However, we did not detect any alterations in the NRF2 pathway activity between AD and control cells in the basal conditions, suggesting that there are no underlying defects in the pathway that would prevent its activation in the patient cells. Our data should be exploited by caution, as iPSC‐derived astrocytes, including ours, resemble more closely to fetal rather than adult human astrocytes (Windrem et al., 2017; Zhang et al., 2016), even though iPSC‐derived astrocytes show all the key morphological and functional characteristics of adult human astrocytes (Oksanen et al., 2017).
In conclusion, our data shows the beneficial effects of NRF2 activation on amyloidogenic, inflammatory and anti‐oxidant properties in AD patient astrocytes and suggests that targeting NRF2 in astrocytes could be beneficial in AD patients.
CONFLICT OF INTEREST
The authors report no conflict of interest.
Supporting information
Appendix S1: Supporting information
ACKNOWLEDGEMENTS
We wish to thank L. Kaskela, E. Korhonen, V. Keksa‐Goldsteine, and J. Voutilainen for technical assistance and A.J. Petersen and S.‐C. Zhang for their invaluable help with isogenic control lines. This work was supported by University of Eastern Finland, Sigrid Juselius Foundation, Academy of Finland and through the Academy of Finland under the aegis of JPND ‐ http://www.jpnd.eu. This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No. 643417 (L.R. and J.K.)
Oksanen M, Hyötyläinen I, Trontti K, et al. NF‐E2‐related factor 2 activation boosts antioxidant defenses and ameliorates inflammatory and amyloid properties in human Presenilin‐1 mutated Alzheimer's disease astrocytes. Glia. 2020;68:589–599. 10.1002/glia.23741
Riikka H. Hämäläinen and Jari Koistinaho contributed equally to this study.
Funding information Academy of Finland; Academy of Finland and through the Academy of Finland under the aegis of JPND ‐ http://www.jpnd.eu. This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 643417; Sigrid Juselius Foundation; Horizon 2020; University of Eastern Finland
REFERENCES
- Abramov, A. Y. , Canevari, L. , & Duchen, M. R. (2004a). Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture. Biochimica et Biophysica Acta, 1742, 81–87. [DOI] [PubMed] [Google Scholar]
- Abramov, A. Y. , Canevari, L. , & Duchen, M. R. (2004b). Beta‐amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. The Journal of Neuroscience, 24, 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders, S. , Pyl, P. T. , & Huber, W. (2015). HTSeq—A python framework to work with high‐throughput sequencing data. Bioinformatics, 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askarova, S. , Yang, X. , Sheng, W. , Sun, G. Y. , & Lee, J. C.‐M. (2011). Role of Aβ‐receptor for advanced glycation endproducts interaction in oxidative stress and cytosolic phospholipase A2 activation in astrocytes and cerebral endothelial cells. Neuroscience, 199, 375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batarseh, Y. S. , Duong, Q.‐V. , Mousa, Y. M. , Al Rihani, S. B. , Elfakhri, K. , & Kaddoumi, A. (2016). Amyloid‐β and astrocytes interplay in amyloid‐β related disorders. International Journal of Molecular Sciences, 17, 1‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter, P. S. , & Hardingham, G. E. (2016). Adaptive regulation of the brain's antioxidant defences by neurons and astrocytes. Free Radical Biology & Medicine, 100, 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blennow, K. , de Leon, M. J. , & Zetterberg, H. (2006). Alzheimer's disease. Lancet, 368, 387–403. [DOI] [PubMed] [Google Scholar]
- Bronzuoli, M. R. , Iacomino, A. , Steardo, L. , & Scuderi, C. (2016). Targeting neuroinflammation in Alzheimer's disease. Journal of Inflammation Research, 9, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello, M. A. , Jeppson, J. D. , & Soriano, S. (2014). Moving beyond anti‐amyloid therapy for the prevention and treatment of Alzheimer's disease. BMC Neurology, 14, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, P. C. , Vargas, M. R. , Pani, A. K. , Smeyne, R. J. , Johnson, D. A. , Kan, Y. W. , & Johnson, J. A. (2009). Nrf2‐mediated neuroprotection in the MPTP mouse model of Parkinson's disease: Critical role for the astrocyte. Proceedings of the National Academy of Sciences of the United States of America, 106, 2933–2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, W. W. , Zhang, X. , & Huang, W. J. (2016). Role of neuroinflammation in neurodegenerative diseases (review). Molecular Medicine Reports, 13, 3391–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo, E. , & Farina, C. (2016). Astrocytes: Key regulators of Neuroinflammation. Trends in Immunology, 37, 608–620. [DOI] [PubMed] [Google Scholar]
- Cuadrado, A. , Rojo, A. I. , Wells, G. , Hayes, J. D. , Cousin, S. P. , Rumsey, W. L. , … Dinkova‐Kostova, A. T. (2019). Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nature Reviews. Drug Discovery, 18, 295–317. [DOI] [PubMed] [Google Scholar]
- Dobin, A. , Davis, C. A. , Schlesinger, F. , Drenkow, J. , Zaleski, C. , Jha, S. , … Gingeras, T. R. (2013). STAR: ultrafast universal RNA‐seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández, Ó. , Giovannoni, G. , Fox, R. J. , Gold, R. , Phillips, J. T. , Potts, J. , … Marantz, J. L. (2017). Efficacy and safety of delayed‐release dimethyl Fumarate for relapsing‐remitting multiple sclerosis in prior interferon users: An integrated analysis of DEFINE and CONFIRM. Clinical Therapeutics, 39, 1671–1679. [DOI] [PubMed] [Google Scholar]
- Fields, R. D. , Araque, A. , Johansen‐Berg, H. , Lim, S.‐S. , Lynch, G. , Nave, K.‐A. , … Wake, H. (2014). Glial biology in learning and cognition. The Neuroscientist, 20, 426–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox, R. J. , Gold, R. , Phillips, J. T. , Okwuokenye, M. , Zhang, A. , & Marantz, J. L. (2017). Efficacy and tolerability of delayed‐release dimethyl Fumarate in black, Hispanic, and Asian patients with relapsing‐remitting multiple sclerosis: Post hoc integrated analysis of DEFINE and CONFIRM. Neurology and Therapy, 6, 175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gella, A. , & Durany, N. (2009). Oxidative stress in Alzheimer disease. Cell Adhesion & Migration, 3, 88–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass, C. K. , Saijo, K. , Winner, B. , Marchetto, M. C. , & Gage, F. H. (2010). Mechanisms underlying inflammation in neurodegeneration. Cell, 140, 918–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde, T. E. , Schneider, L. S. , & Koo, E. H. (2011). Anti‐abeta therapeutics in Alzheimer's disease: The need for a paradigm shift. Neuron, 69, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González‐Reyes, R. E. , Nava‐Mesa, M. O. , Vargas‐Sánchez, K. , Ariza‐Salamanca, D. , & Mora‐Muñoz, L. (2017). Involvement of astrocytes in Alzheimer's disease from a neuroinflammatory and oxidative stress perspective. Frontiers in Molecular Neuroscience, 10, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer, A. , Waschbisch, A. , Kuhbandner, K. , Bayas, A. , Lee, D.‐H. , Duscha, A. , … Linker, R. A. (2018). The NRF2 pathway as potential biomarker for dimethyl fumarate treatment in multiple sclerosis. Annals of Clinical Translational Neurology, 5, 668–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy, J. , & Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science, 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Hayes, J. D. , & Dinkova‐Kostova, A. T. (2014). The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends in Biochemical Sciences, 39, 199–218. [DOI] [PubMed] [Google Scholar]
- Heneka, M. T. , Carson, M. J. , El Khoury, J. , Landreth, G. E. , Brosseron, F. , Feinstein, D. L. , … Kummer, M. P. (2015). Neuroinflammation in Alzheimer's disease. Lancet Neurology, 14, 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirrlinger, J. , Schulz, J. B. , & Dringen, R. (2002). Glutathione release from cultured brain cells: Multidrug resistance protein 1 mediates the release of GSH from rat astroglial cells. Journal of Neuroscience Research, 69, 318–326. [DOI] [PubMed] [Google Scholar]
- Holmström, K. M. , Kostov, R. V. , & Dinkova‐Kostova, A. T. (2016). The multifaceted role of Nrf2 in mitochondrial function. Current Opinion in Toxicology, 1, 80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghton, C. A. , Fassett, R. G. , & Coombes, J. S. (2016). Sulforaphane and other Nutrigenomic Nrf2 activators: Can the Clinician's expectation be matched by the reality? Oxidative Medicine and Cellular Longevity [Internet, 2016 Available from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4736808/, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazwa, A. , Rojo, A. I. , Innamorato, N. G. , Hesse, M. , Fernández‐Ruiz, J. , & Cuadrado, A. (2011). Pharmacological targeting of the transcription factor Nrf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism. Antioxidants & Redox Signaling, 14, 2347–2360. [DOI] [PubMed] [Google Scholar]
- Johnson, D. A. , & Johnson, J. A. (2015). Nrf2—a therapeutic target for the treatment of neurodegenerative diseases. Free Radical Biology & Medicine, 88, 253–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanninen, K. , Heikkinen, R. , Malm, T. , Rolova, T. , Kuhmonen, S. , Leinonen, H. , … Koistinaho, J. (2009). Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America, 106, 16505–16510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanninen, K. M. , Pomeshchik, Y. , Leinonen, H. , Malm, T. , Koistinaho, J. , & Levonen, A.‐L. (2015). Applications of the Keap1‐Nrf2 system for gene and cell therapy. Free Radical Biology & Medicine, 88, 350–361. [DOI] [PubMed] [Google Scholar]
- Kraft, A. W. , Hu, X. , Yoon, H. , Yan, P. , Xiao, Q. , Wang, Y. , … Lee, J.‐M. (2013). Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. The FASEB Journal, 27, 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddell, J. R. , Hoepken, H. H. , Crack, P. J. , Robinson, S. R. , & Dringen, R. (2006). Glutathione peroxidase 1 and glutathione are required to protect mouse astrocytes from iron‐mediated hydrogen peroxide toxicity. Journal of Neuroscience Research, 84, 578–586. [DOI] [PubMed] [Google Scholar]
- Liddelow, S. A. , Guttenplan, K. A. , Clarke, L. E. , Bennett, F. C. , Bohlen, C. J. , Schirmer, L. , … Barres, B. A. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, D. T. , Wu, J. , Holstein, D. , Upadhyay, G. , Rourk, W. , Muller, E. , & Lechleiter, J. D. (2007). Ca2+ signaling, mitochondria and sensitivity to oxidative stress in aging astrocytes. Neurobiology of Aging, 28, 99–111. [DOI] [PubMed] [Google Scholar]
- Love, M. I. , Huber, W. , & Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Q. (2013). Role of nrf2 in oxidative stress and toxicity. Annual Review of Pharmacology and Toxicology, 53, 401–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraz‐Ríos, M. A. , Toral‐Rios, D. , Franco‐Bocanegra, D. , Villeda‐Hernández, J. , & Campos‐Peña, V. (2013). Inflammatory process in Alzheimer's disease. Frontiers in Integrative Neuroscience, 7, 1–15. Available from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3741576/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen, M. , Lehtonen, S. , Jaronen, M. , Goldsteins, G. , Hämäläinen, R. H. , & Koistinaho, J. (2019). Astrocyte alterations in neurodegenerative pathologies and their modeling in human induced pluripotent stem cell platforms. Cellular and Molecular Life Sciences, 76, 2739–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen, M. , Petersen, A. J. , Naumenko, N. , Puttonen, K. , Lehtonen, S. , Gubert Olive, M. , … Koistinaho, J. (2017). PSEN1 mutant iPSC‐derived model reveals severe astrocyte pathology in Alzheimer's disease. Stem Cell Reports, 9, 1885–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry, V. H. , & Teeling, J. (2013). Microglia and macrophages of the central nervous system: The contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Seminars in Immunopathology, 35, 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries, M. , & Sastre, M. (2016). Mechanisms of abeta clearance and degradation by glial cells. Frontiers in Aging Neuroscience, 8, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, M. D. , McCarthy, D. J. , & Smyth, G. K. (2010). edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, Z.‐M. , Han, Y.‐W. , Han, X.‐H. , Zhang, K. , Chang, Y.‐N. , Hu, Z.‐M. , … Hong, W. (2016). Upstream regulators and downstream effectors of NF‐κB in Alzheimer's disease. Journal of the Neurological Sciences, 366, 127–134. [DOI] [PubMed] [Google Scholar]
- Sofroniew, M. V. (2014). Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. The Neuroscientist, 20, 160–172. [DOI] [PubMed] [Google Scholar]
- Sofroniew, M. V. (2015). Astrocyte barriers to neurotoxic inflammation. Nature Reviews. Neuroscience, 16, 249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh, J. H. , Shenvi, S. V. , Dixon, B. M. , Liu, H. , Jaiswal, A. K. , Liu, R.‐M. , & Hagen, T. M. (2004). Decline in transcriptional activity of Nrf2 causes age‐related loss of glutathione synthesis, which is reversible with lipoic acid. Proceedings of the National Academy of Sciences of the United States of America, 101, 3381–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Z. , Zhang, S. , Chan, J. Y. , & Zhang, D. D. (2007). Keap1 controls postinduction repression of the Nrf2‐mediated antioxidant response by escorting nuclear export of Nrf2. Molecular and Cellular Biology, 27, 6334–6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas, M. R. , Johnson, D. A. , Sirkis, D. W. , Messing, A. , & Johnson, J. A. (2008). Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. The Journal of Neuroscience, 28, 13574–13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasile, F. , Dossi, E. , & Rouach, N. (2017). Human astrocytes: Structure and functions in the healthy brain. Brain Structure & Function, 222, 2017–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waring, S. C. , & Rosenberg, R. N. (2008). Genome‐wide association studies in Alzheimer disease. Archives of Neurology, 65, 329–334. [DOI] [PubMed] [Google Scholar]
- Windrem, M. S. , Osipovitch, M. , Liu, Z. , Bates, J. , Chandler‐Militello, D. , Zou, L. , … Goldman, S. A. (2017). Human iPSC glial mouse chimeras reveal glial contributions to schizophrenia. Cell Stem Cell, 21, 195–208.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Sloan, S. A. , Clarke, L. E. , Caneda, C. , Plaza, C. A. , Blumenthal, P. D. , … Barres, B. A. (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron, 89, 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information