

Summary
The Piétrain pig originates from the Belgian village Piétrain some time between 1920 and 1950. Owing to its superior conformation, the Piétrain has spread worldwide since the 1960s. As initial population sizes were limited and close inbreeding was commonplace, the breed’s genetic diversity has been questioned. Therefore, this study examines Piétrain breed substructure, diversity and selection signatures using SNP data in comparison with Duroc, Landrace and Large White populations. Principal component analysis indicated three subpopulations, and F ST analysis showed that US Piétrains differ most from European Piétrains. Average inbreeding based on runs of homozygosity (ROH) segments larger than 4 Mb ranged between 16.7 and 20.9%. The highest chromosomal inbreeding levels were found on SSC8 (42.7%). ROH islands were found on SSC8, SSC15 and SSC18 in all Piétrain populations, but numerous population‐specific ROH islands were also detected. Moreover, a large ROH island on SSC8 (34–126 Mb) appears nearly fixed in all Piétrain populations, with a unique genotype. Chromosomal ROH patterns were similar between Piétrain populations. This study shows that Piétrain populations are genetically diverging, with at least three genetically distinct populations worldwide. Increasing genetic diversity in local Piétrain populations by introgression from other Piétrain populations seems to be only limited. Moreover, a unique 90 Mb region on SSC8 appeared largely fixed in the Piétrain breed, indicating that fixation was already present before the 1960s. We believe that strong selection and inbreeding during breed formation fixed these genomic regions in Piétrains. Finally, we hypothesize that independent coat color selection may have led to large ROH pattern similarities on SSC8 between unrelated pig breeds.
Keywords: Duroc, effective population size, genetic diversity, inbreeding, Landrace, Large White, ROH islands, runs of homozygosity, selection signatures, single nucleotide polymorphism
Introduction
The Piétrain is a black spotted pig breed originating between 1920 and 1950 near the Belgian village Piétrain. The breed was founded by crossing local pigs (‘Indigenous White Pig’) with Berkshire, Large White and Bayeux pigs according to local sources (Departement Landbouw en Visserij et al. 2016). However, these sources also mention various other European breeds and even wild boars influencing the Piétrain breed (Porter 1993; Departement Landbouw en Visserij et al. 2016). Nevertheless, all sources agree that close inbreeding was commonplace during breed formation. This high degree of inbreeding fixed some key characteristics such as extreme muscularity and lean meat percentage (Porter 1993). Owing to its superior conformation and the rise of artificial insemination, the breed rapidly became Belgium’s most popular terminal boar breed. It conquered Europe from the 1960s, and now Piétrain populations are found worldwide (Porter 1993; Departement Landbouw en Visserij et al. 2016; FAO 2019; see Fig. S1).
Preserving local breed diversity is considered essential (FAO 2019; United Nations 2019). However, 34% of traditional local pig breeds in Europe are now extinct, 26% are at risk of extinction and only 3.2% are ‘safe’. Moreover the status of 38% of pig breeds worldwide is unknown (FAO 2019). Therefore, it is crucial to study the genetic diversity of our most important pig breeds. A loss of diversity will increase the incidence of hereditary diseases, jeopardize genetic improvement and decrease genetic adaptability to environmental changes (Lynch & Walsh 1998; Druet & Gautier 2017; Ceballos et al. 2018; Bosse et al. 2019). Owing to the assumed narrow genetic basis at the origin of the Piétrain and subsequent selective breeding, the genetic diversity of the breed is now questioned. Worldwide, commercial pig breeds like the Piétrain, but also Landrace, Large White and Duroc, are considered one breed, even though pigs are bred in closed populations with different selection criteria. As a consequence, populations may have diverged over time, giving rise to subpopulations.
Runs of homozygosity (ROH) are autozygous stretches in the genome that are considered to be homozygous‐by‐descent (HBD; Gibson et al. 2006; McQuillan et al. 2008; Druet & Gautier 2017; Ceballos et al. 2018). ROH analysis has become a standard method in genetic diversity studies both in humans and in livestock (Ferenčaković et al. 2013; Curik et al. 2014; Ceballos et al. 2018). ROH analysis can accurately estimate inbreeding (F ROH) at an individual level. Moreover, inbreeding history can be revealed and inbreeding can be genomically localized (Druet & Gautier 2017). The latter makes it possible to investigate highly inbred genomic regions within a population, also referred to as ROH islands (Nothnagel et al. 2010). These ROH islands are population‐specific selection signatures and allow the unraveling of genes that have been divergently selected for between populations, or can be used to link a specific genotype to phenotype (Lander & Botstein 1989; Ceballos et al. 2018).
The aim of this study was to unravel breed substructure, diversity and ROH in several Piétrain populations from Europe and USA in comparison with Duroc, Landrace and Large White populations. First, the breed substructure was analyzed. Second, diversity was further investigated by determining the F ROH and effective population size (N E). Last, ROH islands were identified and similarities in ROH patterns were analyzed to study selection signatures and founder effects within the Piétrain.
Materials and methods
Animal sampling and genotyping
Medium‐density SNP data from Piétrain populations from five countries (Belgium, France, Germany, The Netherlands and USA) were used. The Belgian population (PBE; n = 620), born between 2012 and 2017, was genotyped via the GGP porcine hd beadchip (70K). PBE samples were selected based on pedigree relationships to maximize genetic diversity. Genotypes and pedigree were provided by the herdbook Vlaamse Piétrain Fokkerij (VPF).
Illumina porcine snp60 beadchip data were obtained from the German (PGE; n = 992) and French (PFR; n = 173) Piétrain populations. PGE data were provided by the University of Hohenheim and originated from three different provinces (Baden–Württemberg, Nordrhein–Westfalen and Schleswig–Holstein); they refer to animals born between 1997 and 2009. PFR data were provided by INRA (Institut National de la Recherche Agronomique) and stem from three different breeding lines (P1, P2, P3), with birthdates between 2007 and 2012. More information on Piétrain subpopulations is provided in Table S1. Furthermore, Illumina 60K SNP data available online (http://dx.doi.org/10.5061/dryad.30tk6) were acquired for 2093 pigs originating from 146 different pig populations worldwide, comprising 20 Piétrains from both The Netherlands (PNL) and the USA (PUS) sampled in 2010 (Yang et al. 2017). These data also contain Landrace (LDR; n = 126; seven populations – Denmark, Norway, Finland, China, the USA, Spain and The Netherlands), Duroc (DUR; n = 79; four populations – Denmark, the USA, The Netherlands and China) and Large White (LWT; n = 96; five populations – Denmark, China, the USA, The Netherlands and the UK) pigs, hereafter denoted as ‘commercial’ breeds.
Genotype quality control
Quality control (QC) was performed per population via plink version 1.9 (Chang et al. 2015). Samples with a call rate of less than 91% were discarded and samples with outlying heterozygosity rate (>3 SD) and relatedness of greater than 95% were removed. Map files were updated to a reference map file using snpchimp v.3 to ensure that SNP names and locations were consistent across populations (Nicolazzi et al. 2015). Only autosomal SNPs were kept and SNPs with a call rate of less than 95% were removed. No MAF or HWE filtering was performed (Ferenčaković et al. 2013). Populations were merged for PCA and F ST analysis. The numbers of samples and SNPs retained after each step of QC are shown in Tables S1 and S2.
Breed substructure analysis
Breed substructure was investigated via two methods. First, PCA was performed using the ‐‐pca flag in plink v1.9 to investigate the clustering of populations. Hereafter, Weir & Cockerham (1984) F ST analysis was performed via the r package Hierfstat (Goudet 2005) to quantify genetic differentiation among populations.
Pedigree inbreeding
Pedigree data were available for PBE. Only genotyped individuals with a pedigree depth of more than 10 generations were considered (563 individuals). Pedigree was constructed via the r package pedigree (Coster 2008) and comprised 7041 individuals. Pedigree inbreeding (F PED) was calculated via the r package GeneticsPed (Gorjanc et al. 2007).
ROH analysis
Runs of homozygosity analysis was performed both by plink version 1.9 (Chang et al. 2015) and the RZooRoH package in r (Druet & Gautier 2017). plink uses a relatively simple rule‐based window approach to determine ROH‐segments, whereas RZooRoH uses a complex Bayesian full probabilistic modeling approach to identify ROH‐segments.
For plink, guidelines from Ferenčaković et al. (2013) were followed to set the allowed number of missing and heterozygous SNPs per ROH length category (Table 1). For every set of parameters, a separate ROH analysis was performed in plink. Total F ROH was calculated based on the most stringent criteria, i.e. no missing or heterozygote SNPs allowed.
Table 1.
Number of accepted missing (‐‐homozyg‐window‐mis) and heterozygote (‐‐homozyg‐window‐het and ‐‐homozyg‐het) SNPs in a runs of homozygosity (ROH) segment per ROH length category (Ferenčaković et al. 2013).
| ROH length category (in Mb) | |||||
|---|---|---|---|---|---|
| 1–2 Mb | 2–4 Mb | 4–8 Mb | 8–16 Mb | >16 Mb | |
| Allowed heterozygous SNPs | 0 | 0 | 0 | 0 | 1 |
| Allowed missing SNPs | 0 | 0 | 1 | 2 | 4 |
The following parameter specifications were used: a maximal gap of 1000 kb (‐‐homozyg‐gap), a minimum ROH length of 1000 kb (homozyg‐kb) and at least one SNP per 150 kb (‐‐homozyg‐density). The minimum numbers of SNPs (l) in both the sliding window (‐‐homozyg‐window‐snp) and in a final ROH segment (‐‐homozyg‐snp) were determined following Purfield et al. (2012), adapted from Lencz et al. (2007):
where α, the false‐positive ROH percentage, was set at 0.05, n s refers to the number of genotyped SNPs per individual, ni refers to the number of genotyped individuals and het denotes the mean heterozygosity level, calculated using the Hierfstat package in r (Goudet, 2005). Choosing a sliding window of appropriate length l eliminates short ROH probably caused by LD. In this study, l values were 53 (PBE), 57 (PGE), 55 (PFR), 47 (PNL) and 49 (PUS). Observed (H O) and expected (H E) heterozygosity levels were calculated per population using the Hierfstat package in r (Goudet, 2005).
Individual degree of inbreeding based on ROH analysis (F ROH) was calculated as:
(Purfield et al. 2012).where L ROH is the total ROH length of an individual i and L auto represents the total ROH length of a completely homozygous individual using identical parameter settings as the evaluated population. Genome coverage was calculated as the proportion of L auto to the reference pig genome length, being 2.5 Gb in the sscrofa11.1 assembly (GenBank accession no. GCA_000003025). The genome coverage denotes the proportion of the reference genome where ROH detection was possible given the map file and ROH parameter settings used in plink.
The model‐based approach via RZooRoH was then used to analyze ROH (Druet & Gautier 2017). We performed our ROH analysis using a mixKR model with fixed rates of ancestry changes Rk, because this model allows classification of ROH segments into K − 1 predefined age‐related HBD classes (Druet & Gautier 2017). In contrast to the method in plink, this model directly provides an estimate of inbreeding history and takes mutation rate, allele frequencies and mixing proportions of ROH segments into account. The optimal number of age‐related HBD classes (K) was determined by evaluating models from K = 3 to K = 10 with the Bayesian information criterion. The genotyping error rate ε was set to 0.25%, as suggested by Ferenčaković et al. (2013). An optimum was found for all populations at K = 8.
ROH islands and patterns
ROH islands were determined at population level. First, the proportion of SNP located within an ROH for a given population (Purfield et al. 2017) was calculated as
To determine ROH islands, standard normal z‐scores were calculated per population from the ROHincidence‐snp distribution. Based on these z‐scores, P‐values were calculated. The SNP with the highest ROHincidence‐snp was kept per scanning window of 1 Mb. Afterwards, only bins with a P‐value >0.99 were designated as ROH islands (adapted from Purfield et al. 2017).
Furthermore, we investigated whether ROH islands had the same underlying genotype. Using R, we performed a stepwise scan of the genome with a 50 SNP window, and for every step, frequencies of the most frequent homozygous and heterozygous genotypes were recorded as well as the total number of unique homozygous and heterozygous genotypes. ROH patterns between different populations were investigated by calculating pairwise Pearson correlations of ROHincidence‐snp both at the whole autosomal genome and at the chromosomal level.
Effective population size
LD‐based N E (Weir & Hill 1980) was calculated for all Piétrain populations via the snep 1.1 software (Barbato et al. 2015). N E was estimated on the autosomal genome level as well as on a chromosomal level. The same set of SNPs (34551) was used to compare populations. Parameters were sample size correction, no MAF‐pruning (Sved & Feldman 1973), recombination rate modifier, bin distance distribution of 3, 30 bins and maximum 10 Mb between evaluated SNPs. The recombination rate modifier was used to translate physical distance (δ) between two loci into linkage distance (d). Binning control flags were chosen in such a way that N E could be investigated for the more recent generations. This study aimed to investigate N E since population separation 50–60 years ago, therefore N E estimates were evaluated from five up to 21 generations ago.
Results
Breed substructure
Piétrains clearly clustered apart from DUR, LDR and LWT (Fig. 1). PCA on all Piétrain populations per country of sampling revealed three clusters on the first three principal components (Fig. S2). PBE clearly clustered separately from other Piétrain populations. PFR clustered separately from other populations on PC1 vs. PC2. Piétrains resulting from crossing different populations were situated between parent populations.
Figure 1.
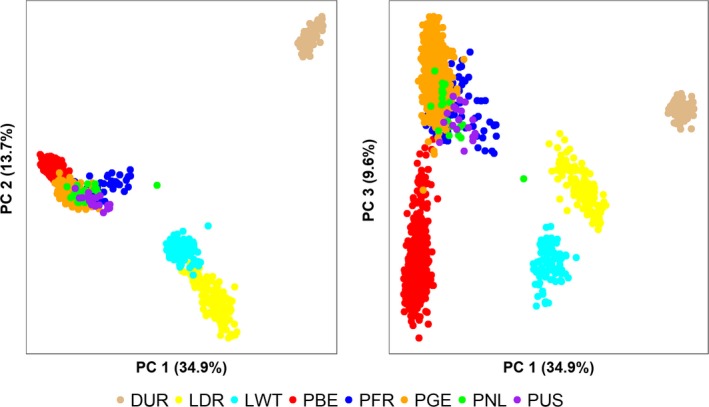
PCA shows that subclusters are present in Piétrain. DUR, Duroc; LDR, Landrace; LWT, Large White; PBE, Belgian Piétrain; PFR, French Piétrain; PGE, German Piétrain; PNL, Dutch Piétrain; PUS, USA Piétrain.
Pairwise Weir and Cockerham’s F ST values ranged between 0.03 and 0.10 within Piétrain populations (Fig. 2), representing little to moderate genetic differentiation (Hartl & Clark, 1997). PUS diverged most from the other Piétrain populations. F ST estimates were largest between Piétrain and DUR.
Figure 2.
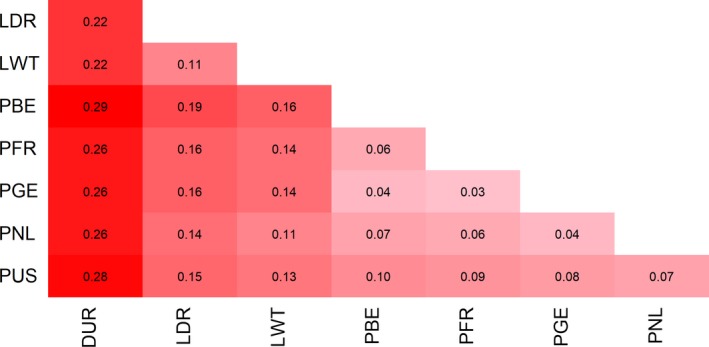
Weir and Cockerham F ST heatmap for all Piétrain and commercial populations. Values in red represent largest F ST. Breed abbreviations as in Fig. 1.
Inbreeding
For PBE, pedigree inbreeding (F PED) ranged between 0.0 and 26.7% and averaged 2.9% (SD = 3.2%). Although pedigree depth was 14–21 generations, F PED was only moderately correlated with F ROH estimated via plink (r = 0.54) and ZooRoH (r = 0.44).
Table 2 shows that the eight populations ranged from 18.0 to 26.1% F ROH using plink, whereas ZooRoH F ROH estimates were higher (from 25.7% to 34.1%). ZooRoH F ROH estimates were consistently larger than plink F ROH estimates for ROH segments between 4 and 16 Mb. F ROH>4Mb estimates were similar between both methods with high Pearson correlations (r = 0.96–1.00). Considering F ROH>4Mb, inbreeding was highest in PUS and lowest in PGE.
Table 2.
Observed (H O) and expected heterozygosity (H E) as well as inbreeding estimates based on runs of homozygosity (F ROH) in percentage per population and per runs of homozygosity length class.
| Population | H O | H E | Method | F ROH | F ROH>4Mb | F ROH 0–1Mb | F ROH 1–2Mb | F ROH 2–4Mb | F ROH 4–8Mb | F ROH 8–16Mb | F ROH>16Mb |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PBE | 0.32 | 0.32 | plink | 22.0 | 19.3 | – | 0.9 | 3.7 | 5.3 | 5.4 | 8.7 |
| ZooRoH | 28.0 | 19.7 | 1.5 | 2.7 | 4.2 | 5.0 | 4.9 | 9.8 | |||
| PGE | 0.34 | 0.34 | plink | 18.0 | 16.7 | – | 0.1 | 2.3 | 3.9 | 4.5 | 8.3 |
| ZooRoH | 25.1 | 17.8 | 1.3 | 2.4 | 3.6 | 3.9 | 4.2 | 9.8 | |||
| PFR | 0.33 | 0.34 | plink | 20.5 | 19.8 | – | 0.1 | 2.7 | 4.5 | 5.4 | 9.8 |
| ZooRoH | 26.9 | 19.8 | 1.2 | 2.2 | 3.6 | 4.3 | 4.4 | 11.1 | |||
| PNL | 0.34 | 0.35 | plink | 21.3 | 18.7 | – | 0.3 | 3.3 | 4.8 | 5.0 | 8.9 |
| ZooRoH | 25.7 | 18.6 | 1.2 | 2.2 | 3.7 | 4.4 | 4.5 | 9.7 | |||
| PUS | 0.33 | 0.33 | plink | 23.0 | 20.9 | – | 0.2 | 2.8 | 5.0 | 5.4 | 10.5 |
| ZooRoH | 26.2 | 20.7 | 0.8 | 1.7 | 3.0 | 4.4 | 5.3 | 11.0 | |||
| DUR | 0.28 | 0.31 | plink | 26.2 | 25.3 | – | 0.1 | 2.6 | 6.2 | 7.2 | 11.9 |
| ZooRoH | 34.1 | 25.9 | 1.1 | 2.5 | 4.6 | 6.0 | 6.3 | 13.6 | |||
| LDR | 0.33 | 0.36 | plink | 19.1 | 17.3 | – | 0.2 | 2.8 | 4.6 | 4.9 | 7.8 |
| ZooRoH | 26.8 | 18.1 | 1.6 | 2.9 | 4.2 | 4.5 | 4.7 | 8.9 | |||
| LWT | 0.33 | 0.37 | plink | 21.0 | 19.4 | – | 0.2 | 2.8 | 5.1 | 5.2 | 9.1 |
| ZooRoH | 32.3 | 22.2 | 1.9 | 3.4 | 4.7 | 5.6 | 5.3 | 11.3 |
For plink, total inbreeding (F ROH) was calculated using the most stringent parameter setting, not allowing any heterozygote or missing SNPs. Therefore, plink F ROH subclasses do not sum up to the total F ROH. DUR, Duroc; LDR, Landrace; LWT, Large White; PBE, Belgian Piétrain; PFR, French Piétrain; PGE, German Piétrain; PNL, Dutch Piétrain; PUS, USA Piétrain.
ZooRoH allows the investigation of inbreeding history (Fig. S3). In general, 40–50% of inbreeding in Piétrains was due to old inbreeding events, more than 32 generations ago (R K = 64 and R K = 128, with generations ago ≈ R K/2; Druet & Gautier 2017). In the most recent generations, cumulative inbreeding was lowest in PBE and highest in PUS.
Effective population size
was estimated for all Piétrain populations. PNL and PUS had the lowest , possibly owing to their limited sample size. PGE, PFR and PBE had similar estimates ranging between 85 and 92, five generations ago. Combining all Piétrain populations yielded an of 105. Results are shown in Fig. S4.
Chromosomal differences in F ROH and N E
Figure 3 shows differences in F ROH and N E at chromosomal level. The highest F ROH levels were found on SSC8 (42.7%), SSC15 (24.1%) and SSC18 (24.2%). Mean Pearson correlation was −0.29, indicating that high chromosomal levels of F ROH are associated with low N E.
Figure 3.
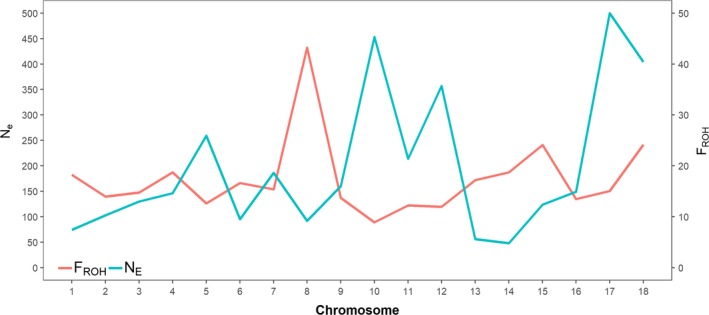
Chromosomal inbreeding (F ROH) and effective population size (N E) estimates for all Piétrains (n = 1632).
ROH islands and ROH patterns
Population‐specific ROH islands in Piétrains were detected on 10 different chromosomes (Table 3). Population‐specific ROHincidence‐snp are shown for all eight populations in Fig. S5. Combining all Piétrain samples, ROH islands were detected on SSC8, SSC15 and SSC18 (Fig. 4). The ROH island on SSC8 appeared in all sampled Piétrain subpopulations over a 90 Mb region, between 34 and 126 Mb, with ROHincidence‐snp estimates of up to 85% on a total of 1632 individuals. Further analysis showed that about 80% of all Piétrains across all subpopulations had identical homozygous genotypes on SSC8 from 50 to 70 Mb and from 90 to 105 Mb (Figs S6 and S7). These genotypes were also detected to a limited extent (up to 10%) in Landrace and Large White as well, but were absent in Duroc. Overlap in ROH islands between all Piétrain subpopulations, Landrace and Large White were found at 50–60 Mb on SSC8 (Tables 3 and S3).
Table 3.
Summary of runs of homozygosity islands per chromosome (SSC) per Piétrain population.
| SSC | Population | Location of detected ROH islands (Mb) |
|---|---|---|
| 3 | PBE | 92–98 |
| 4 | PBE | 127–128 |
| 6 | PBE | 139–141 |
| PUS | 59–62 | |
| 7 | PNL | 89–91; 97–99 2 |
| 8 | PBE | 39–46; 59–77 1 , 2 ; 86–108; 121–126 |
| PGE | 38–77 1 , 2 ; 81–126 | |
| PNL | 38–49; 55–74 1 , 2 ; 85–108; 111–113; 124–126 | |
| PFR | 38–110 1 , 2 ; 111–113; 121–126 | |
| PUS | 34–37; 38–77 1 , 2 ; 87–88; 121–124 | |
| 9 | PNL | 36–37 |
| 13 | PBE | 24–29 |
| PNL | 93–98 1 | |
| 14 | PNL | 73–77 |
| PUS | 37–41; 90–96 | |
| 15 | PBE | 76–88 |
| PGE | 73–86 | |
| PFR | 73–89; 96–104 | |
| PNL | 73–89 | |
| 18 | PBE | 9–26 |
| PGE | 10–13 | |
| PNL | 10–16 | |
| PUS | 10–19 | |
| 8 | All Piétrains | 38–44; 54–62; 63–77; 81–118; 122–126 |
| 15 | All Piétrains | 76–90 |
| 18 | All Piétrains | 10–16; 23–27 |
ROH island also found in LDR.
ROH island also found in LWT.
Figure 4.
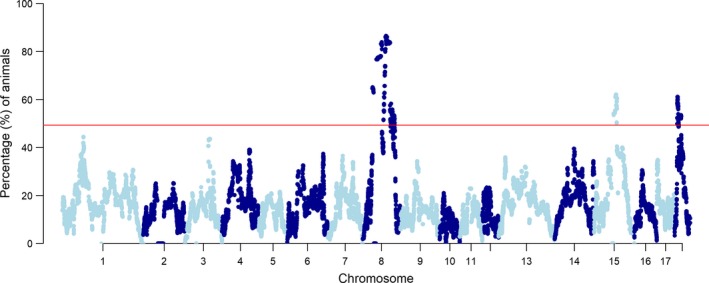
Percentage of animals with specific SNP in ROH (ROHincidence‐snp) for all Piétrains. In total, 1632 Piétrains were evaluated on 34551 SNPs. The horizontal line corresponds to the cutoff level for ROH island detection (49.3%).
Hereafter, a genome‐wide correlational analysis of ROHincidence‐snp was performed to analyze ROH pattern similarities (Fig. 5). Pearson correlations of ROHincidence‐snp between Piétrain populations were generally high, although PUS and PNL had lower correlations with PBE, PFR and PGE. Moderate pairwise correlations were found between DUR, LWT and LDR but these breeds had rather low correlations with the Piétrain populations. ROH pattern analysis on SSC8 (Fig. S8) showed high correlations (r > 0.93) between PBE, PNL, PGE and PFR whereas the PUS ROH pattern deviated more, with correlations ranging from 0.74 to 0.80.
Figure 5.
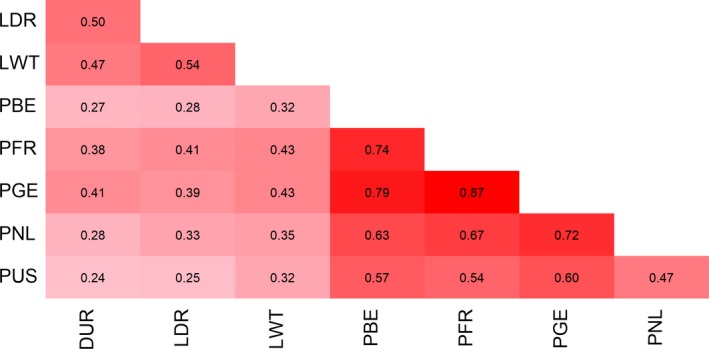
Correlational heatmap of ROHincidence‐snp shows high similarity in ROH patterns between Piétrain populations. Breed abbreviations as in Fig. 1.
Discussion
The Piétrain breed is known to originate from a small initial population and inbreeding was commonplace during breed formation. Furthermore, the Piétrain spread worldwide from the 1960s on, forming several closed breeding populations. Therefore, this study investigated breed substructure, diversity and ROH in Piétrain populations from Europe and USA and compared them with Duroc, Landrace and Large White populations.
Breed substructure
Analysis indicated that Piétrain populations are genetically diverging, probably because breeding populations were separated for more than 40 years and breeding goals differed. PGE, PFR and PNL clustered together in PCA and had low mutual F ST estimates, showing that these populations are genetically closely linked (Figs 1 and 2). PBE clustered separately from other Piétrain populations and had higher F ST estimates with PFR, PNL and PUS, suggesting that PBE has genetically diverged from these populations. The low F ST estimate of 0.04 between PBE and PGE denotes little genetic differentiation (Hartl & Clark, 1997) and is presumably due to the exchange of genetic material between both populations. PUS genetically diverged moderately from other Piétrain populations with F ST 0.07–0.10 (Fig. 2), possibly because originally only a limited number of founding animals could be exported to the USA and/or owing to a difference in breeding goals between the American and European populations.
Inbreeding
For PBE, correlations between genomic (F ROH) and pedigree (F PED) inbreeding were moderate (0.44 and 0.54), although pedigree depth was more than 14 generations. Peripolli et al. (2017) reported Pearson correlations between F ROH and F ped ranging from 0.39 to 0.81 in a review of 15 cattle and three pig populations, whereas correlations of 0.31, 0.32 and 0.53 were found respectively for Duroc, Landrace and Large White populations by Grossi et al. (2017). This shows that pedigree data have limited value in predicting actual degrees of inbreeding, possibly owing to Mendelian sampling, unknown remote relationships and pedigree errors (Howard et al. 2017; Druet & Gautier 2017).
Only large (>4 Mb) ROH segments were used to evaluate inbreeding, as proposed for medium‐density SNP data by Ferenčaković et al. (2013) and Purfield et al. (2012). Average F ROH>4Mb estimates were generally high for Piétrain populations, between 16.7 and 20.9%, but were similar to estimates in commercial populations, like LDR (F ROH>4Mb = 17.3%), LWT (F ROH>4Mb = 19.4%) and DUR (F ROH>4Mb = 25.3%). Based on the published data of Bosse et al. (2012), we computed F ROH estimates of 14.2% (LDR), 15.6% (LWT) and 20.8% (DUR). In our study, inbreeding comparison is rather qualitative, as generations are not discrete and sampling was in different time periods between populations. Remarkably, Piétrains originating from crosses between populations (PBE and PGE) on average had 15.7% F ROH>4Mb (plink), with estimates ranging between 7.9 and 21.8% (details not shown). Although these estimates are lower than the population averages, they are still substantial, indicating that large ROH similarities exist across these populations. Hence, F ROH can be decreased at population level by importing Piétrains from other subgroups, but only to a limited extent.
The high correlations and similarity of F ROH>4Mb estimates between plink and ZooRoH show that both methods detect ROH consistently. However, ZooRoH estimates HBD classes based on allele frequencies and mixing proportions of ROH segments and therefore allows for a more reliable estimation of inbreeding history (Druet & Gautier 2017). Using plink and the classical assumption that 1 cM = 1 Mb (Curik et al. 2014) seemed to overestimate recent inbreeding (<12.5 generations ≈ F ROH>4Mb in Table 2). Indeed, the Piétrain populations showed several large homozygous regions probably caused by founder effects. However, because of their large size, these regions would be considered as ‘recent’ inbreeding. For the different Piétrain populations, ZooRoH estimated F ROH at 14.3–18.9% for the most recent 16 generations (Fig. S3; cumulative proportion of inbreeding with R k ≤ 32). The F ROH for old inbreeding events >32 generations ago was estimated at 7.5–11.5%. Hence, 28–42% of total inbreeding is due to old inbreeding events, which is consistent with the fact that the Piétrain breed was formed by close inbreeding. Assuming a generation interval of 2–3 years, breed formation took place approximately 25–50 generations ago.
Effective population size N E
Piétrain N E estimates were well above the FAO guideline that suggests an N E ≥ 50 per generation (FAO 1998). Combining all available Piétrain populations (PBE, PGE, PFR, PNL and PUS) elevated the N E from 85 (median) to 105, an increase of 23.5%, indicating that importing Piétrains can enhance subpopulation diversity to some extent.
However, genetic diversity monitoring and controlled breeding remains advised, as a decline in population size and probably also N E is expected in Belgium. In PBE, breeding is mainly performed by independent, private breeders cooperating in a breed association. Most breeders of PBE are older than 55 years of age and succession is limited (Calus et al. 2008). Hence, active population sizes are expected to drop in the years to come. Also in an international context, the population sizes of pig breeds are under pressure. Owing to increased globalization, semen can be distributed beyond national borders, increasing competition. In addition, advances in artificial insemination techniques lead to more doses per boar, resulting in a need for fewer active boars (Knox, 2016).
Inbreeding and effective population size on a chromosomal level
Chromosomal F ROH differences in Piétrains were large (Fig. 3), with the highest ROH coverage on SSC8 (42.7%) and the lowest on SSC10 (6.3%). Bosse (2015) found the same pattern in 47 Dutch Piétrains. Likewise, N E estimates varied greatly between chromosomes and were negatively correlated (r = −0.29) with F ROH. This rather low correlation points out that average F ROH only explains a minor part of the correlational structure and recombination rates, upon which N E estimation is based (Barbato et al. 2015). However, Druet & Gautier (2017) suggest that individual F ROH estimates per HBD class can be related to the corresponding past N E. Recombination rate locally influences ROH patterns and N E (Bosse et al. 2019). A better understanding of this recombination rate might improve ROH and N E calculations. Breeding programs aiming to minimize inbreeding should not solely focus on average F ROH, but also take local inbreeding into account (Howard et al. 2017).
ROH islands and patterns
All Piétrain populations had numerous, overlapping ROH islands on a 90 Mb region on SSC8 between 34 and 126 Mb (Table 3 and Fig. 4), suggesting that SSC8 is largely fixed in the Piétrain breed. On SSC8, ROHincidence‐snp was high for all Piétrain populations – up to 100% for PFR and PUS – with a drop at 75–85 Mb (Fig. 4 and Figs S6 and S7). This drop is possibly caused by a local recombination hotspot at 70–80 Mb on SSC8 in pigs (Tortereau et al. 2012).
Bosse (2015) noted for 47 Dutch Piétrains that chromosomal regions on SSC8 and SSC15 were fixed, and found signatures for ongoing selection on SSC13 and SSC15 via extended haplotype homozygosity analysis. Our study confirms that a large region of SSC8 is almost completely fixed in Piétrains worldwide. However, the highly inbred region on SSC15 around 70–85 Mb was not found in PUS. Furthermore, our study only found ROH islands on SSC13 in PBE (24–29 Mb) and PNL (93–98 Mb), indicating that these selection signatures are population specific, rather than representative of the Piétrain breed. We did not find any relevant underlying genes which could explain these differences between populations.
Further analysis revealed that more than 75% of all sampled Piétrains had identical 50 SNP window homozygous genotypes on SSC8 at 50–70 and 90–105 Mb. These ROHs were also found in those Piétrains identified as crosses of PBE–PGE, proving that identical genotypes are present in different Piétrain populations. Hence, these genomic regions on SSC8 appear almost completely fixed in the Piétrain breed. It is remarkable that this 90 Mb region remains present over multiple subpopulations which have been largely separated for more than 40 years. Our hypothesis is that this region in SSC8 in Piétrains became fixed during breed formation owing to a strong selection on exterior, coat color and coat pattern combined with severe inbreeding. The KIT gene is located on SSC8 at 41.4–41.5 Mb and is known to affect coat color and pattern in pigs (Fontanesi et al. 2010). Fontanesi et al. (2010) found a selective sweep for Piétrains at the KIT gene. Within the KIT gene, the patch allele (I P) results into colored patches on a white background (Moller et al. 1996). Fontanesi et al. (2010) discovered that a duplication of the I P allele was uniquely found in Piétrains and they suggest that this duplication possibly occurred during breed formation. Furthermore, local Belgian sources mention a strong combined selection on carcass conformation and coat characteristics at the start of the Piétrain pigbook in 1945, including elimination of ‘white’ Piétrains (Departement Landbouw en Visserij et al. 2016). Unfortunately, coat color phenotypes were not available in this study. Although large fixed homozygous regions are documented in several highly inbred livestock breeds with limited population sizes, this is to our knowledge the first time this has been described in a commercial breed within several populations distributed worldwide. In any case, a similar large fixed region was not detected in the analyzed samples of Duroc, Landrace and Large White populations.
Further investigation into the SSC8 region using all available genotypes from the study of Yang et al. (2017) revealed highly similar ROH patterns between Piétrain populations (0.80 ≤ r ≤ 0.94; Fig. S8), but also with a dozen of other (unrelated) breeds, although the ROH genotypes were different (details not shown). For example, Piétrains had an ROH pattern correlation of 0.85 on SSC8 with the Chinese Rongchang pig, a breed known for its solid white coat color (Lai et al. 2006). Our hypothesis is that coat color selection may have led to this ROH pattern similarity on SSC8, owing to the presence of the KIT gene. Coat color was one of the first phenotypes man selected for in pigs and coat color alternations are regarded as the first sign of domestication (Fontanesi & Russo 2013). We believe that independent coat color selection may have resulted in similar ROH patterns on SSC8, yet with different phenotypes and genotypes. Indeed, a similar ROH pattern implies that selection/inbreeding took place at similar genomic regions, but it does not imply that selection took place in the same direction. It is reasonable that selecting individuals for opposite extremes – for example white vs. black coat color – will produce ROHs in similar genomic regions, but with different genotypes.
Including all chromosomes, ROH patterns (Fig. 5) were similar between PGE, PFR and PBE (r ≥ 0.74), and lower between the former and PNL and PUS (0.47 ≤ r ≤ 0.72). This indicates that breeding goals and selection methods between PGE, PFR and PBE are more similar compared with PUS and PNL.
Implications of the study
This study has shown that genetic subgroups exist within the Piétrain breed. Although genetic diversity of each of the subpopulations is considered as sufficient, a decrease in population size is expected in (some) subpopulations. Therefore, genetic diversity should be closely monitored in the years to come. Importing Piétrains from other subpopulations might be effective to only a limited extent, so we advise taking diversity into account in the breeding programs, for example by using optimal contribution selection. Because only Piétrain populations from Europe and USA were available, other Piétrain populations could contribute additional genetic variation within the breed. Furthermore, a large homozygous region on SSC8 appeared almost completely fixed in all Piétrain populations, possibly owing to coat color selection and/or founder effects. This should be taken into account when performing GWAS using Piétrain pigs.
Conclusions
Although the Piétrain is considered as one breed, this study shows that substantial genetic differences exist between some subpopulations from Europe and USA. We show that common genomic patterns are present in Piétrains, but our findings also suggest that Piétrain populations are genetically diverging. At least three genetically distinct subgroups within the Piétrain breed were found, with US Piétrains being most distinct from their European relatives. Average Piétrain F ROH estimates were high (18–23%), but in the same range as Duroc, Landrace and Large White populations (19–26%). We also found that a large part of SSC8 is fixed in Piétrain pigs worldwide, possibly owing to severe inbreeding and coat color selection during breed formation in the first half of the twentieth century. Moreover, we hypothesize that independent coat color selection may have led to large similarities in ROH patterns between unrelated breeds.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Table S1 Individual quality control statistics per Piétrain population.
Table S2 SNP quality control statistics per Piétrain population.
Table S3 Summary of runs of homozygosity (ROH) islands per chromosome (SSC) for Duroc, Landrace and Large White.
Figure S1 Presence of Piétrain pigs worldwide.
Figure S2 PCA on Piétrain populations shows three main clusters.
Figure S3 Inbreeding proportion (HBD, homozygosity by descent) as a function of rate parameter R K.
Figure S4 N E estimates for different Piétrain populations 5–21 generations ago.
Figure S5 ROHincidence‐snp per Piétrain population.
Figure S5 ROHincidence‐snp per Piétrain population.
Figure S6 Most frequently observed homozygous genotypes found in Piétrain (PIT) were also present at low frequencies in Landrace (LDR) and Large White (LWT), but absent in Duroc (DUR).
Figure S7 Identical ROH genotypes appear in all of the different Piétrain populations at high frequencies.
Figure S8 Correlational heatmap of ROHincidence‐snp on SSC8 shows high similarity in ROH patterns between Piétrain populations.
Acknowledgements
The authors would like to acknowledge the Vlaamse Piétrain Fokkerij vzw for the provision of samples and pedigree data of Belgian Piétrain pigs. Furthermore, we would like to acknowledge Professor Jörn Bennewitz and Dr Patrick Stratz from the University of Hohenheim for providing the German genotypes, and Dr Llibertat Tusell Palomero and Catherine Larzul from INRA (GenPhySE, Castanet‐Tolosan, France) for providing French genotypes. Belgian genotyping was funded by the Flemish Government: Department of Agriculture and Fisheries. French genotyping was funded by the UtOpIGe ANR‐10‐GENOM_BTV‐015 project.
Data availability
The 70K SNP dataset of Belgian Piétrains will be made accessible upon motivated request and was available upon request during the review process.
References
- Barbato M., Orozco‐terWengel P., Tapio M. & Bruford M.W. (2015) SNeP: a tool to estimate trends in recent effective population size trajectories using genome‐wide SNP data. Frontiers in Genetics 6, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosse M. (2015) The hybrid nature of pig genomes: unraveling the mosaic haplotype structure in wild and commercial Sus scrofa populations. Doctoral dissertation. Wageningen University.
- Bosse M., Megens H.J., Madsen O., Paudel Y., Frantz L.A., Schook L.B., Crooijmans R.P. & Groenen M.A. (2012) Regions of homozygosity in the porcine genome: consequence of demography and the recombination landscape. PLoS Genetics 8, e1003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosse M., Megens H.J., Derks M.F., de Cara Á.M. & Groenen M.A. (2019) Deleterious alleles in the context of domestication, inbreeding, and selection. Evolutionary Applications 12, 6–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calus M., Van Huylenbroeck G. & Van Lierde D. (2008) The relationship between farm succession and farm assets on Belgian farms. Sociologia Ruralis 48, 38–56. [Google Scholar]
- Ceballos F.C., Joshi P.K., Clark D.W., Ramsay M. & Wilson J.F. (2018) Runs of homozygosity: windows into population history and trait architecture. Nature Reviews Genetics 19, 220. [DOI] [PubMed] [Google Scholar]
- Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M. & Lee J.J. (2015) Second‐generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coster A. (2008) Pedigree: Pedigree Functions in R Package Version 1.1. R Foundation for Statisical Computing, Vienna. [Google Scholar]
- Curik I., Ferenčaković M. & Sölkner J. (2014) Inbreeding and runs of homozygosity: a possible solution to an old problem. Livestock Science 166, 26–34. [Google Scholar]
- Departement Landbouw en Visserij , Vettenburg N., Eskens J. & Pauwels H. (2016) Ontstaan en evolutie van het Piétrain ras.
- Druet T. & Gautier M. (2017) A model‐based approach to characterize individual inbreeding at both global and local genomic scales. Molecular Ecology 26, 5820–41. [DOI] [PubMed] [Google Scholar]
- FAO (1998) Secondary Guidelines for Development of National Farm Animal Genetic Resources Management Plans – Management of small populations at risk. Retrieved from http://www.fao.org/docrep/010/a1250e/a1250e00.htm.
- FAO (2019) Domestic Animal Diversity Information System (DAD‐IS). Retrieved November 18, 2018, from http://www.fao.org/dad-is/regional-national-nodes/efabis/en/.
- Ferenčaković M., Sölkner J. & Curik I. (2013) Estimating autozygosity from high‐throughput information: effects of SNP density and genotyping errors. Genetics Selection Evolution 45, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanesi L. & Russo V. (2013) Molecular genetics of coat colour in pigs. Acta Agric Slov 4, 16. [Google Scholar]
- Fontanesi L., D’Alessandro E., Scotti E., Liotta L., Crovetti A., Chiofalo V. & Russo V. (2010) Genetic heterogeneity and selection signature at the KIT gene in pigs showing different coat colours and patterns. Animal Genetics 41, 478–92. [DOI] [PubMed] [Google Scholar]
- Gibson J., Morton N.E. & Collins A. (2006) Extended tracts of homozygosity in outbred human populations. Human Molecular Genetics 15, 789–95. [DOI] [PubMed] [Google Scholar]
- Gorjanc G., Henderson D.A., Kinghorn B. & Percy A. (2007) Genetics‐Ped: pedigree and genetic relationship functions. R package version, 1(0).
- Goudet J. (2005) Hierfstat, a package for R to compute and test hierarchical F‐statistics. Molecular Ecology Notes 5, 184–6. [Google Scholar]
- Grossi D.A., Jafarikia M., Brito L.F., Buzanskas M.E., Sargolzaei M. & Schenkel F.S. (2017) Genetic diversity, extent of linkage disequilibrium and persistence of gametic phase in Canadian pigs. BMC Genetics 18, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl D.L., Clark A.G. (1997) Principles of Population Genetics, Vol. 3, pp. 117–20. Sinauer Associates, Sunderland, MA. [Google Scholar]
- Howard J.T., Pryce J.E., Baes C. & Maltecca C. (2017) Invited review: Inbreeding in the genomics era: inbreeding, inbreeding depression, and management of genomic variability. Journal of Dairy Science 100, 6009–24. [DOI] [PubMed] [Google Scholar]
- Knox R.V. (2016) Artificial insemination in pigs today. Theriogenology 85, 83–93. [DOI] [PubMed] [Google Scholar]
- Lai F., Ren J., Ai H., Ding N., Ma J., Zeng D., Chen C., Guo Y. & Huang L. (2006) Chinese white Rongchang pig does not have the dominant white allele of KIT but has the dominant black allele of MC1R. Journal of Heredity 98, 84–7. [DOI] [PubMed] [Google Scholar]
- Lander E.S. & Botstein D. (1989) Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121, 185–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lencz T., Lambert C., DeRosse P., Burdick K.E., Morgan T.V., Kane J.M., Kucherlapati R. & Malhotra A.K. (2007) Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proceedings of the National Academy of Sciences 104, 19942–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. & Walsh B. (1998) Genetics and Analysis of Quantitative traits, Vol 1, pp. 251–87. Sinauer, Sunderland, MA. [Google Scholar]
- McQuillan R., Leutenegger A.L., Abdel‐Rahman R. et al (2008) Runs of homozygosity in European populations. The American Journal of Human Genetics 83, 359–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller M.J., Chaudhary R., Hellmen E., Höyheim B., Chowdhary B. & Andersson L. (1996) Pigs with the dominant white coat color phenotype carry a duplication of the KIT gene encoding the mast/stem cell growth factor receptor. Mammalian Genome 7, 822–30. [DOI] [PubMed] [Google Scholar]
- Nicolazzi E.L., Caprera A., Nazzicari N. et al (2015) SNPchiMp v. 3: integrating and standardizing single nucleotide polymorphism data for livestock species. BMC Genomics 16, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothnagel M., Lu T.T., Kayser M. & Krawczak M. (2010) Genomic and geographic distribution of SNP‐defined runs of homozygosity in Europeans. Human Molecular Genetics 19, 2927–35. [DOI] [PubMed] [Google Scholar]
- Peripolli E., Munari D.P., Silva M.V.G.B., Lima A.L.F., Irgang R. & Baldi F. (2017) Runs of homozygosity: current knowledge and applications in livestock. Animal Genetics 48, 255–71. [DOI] [PubMed] [Google Scholar]
- Porter V. (1993) Pigs: A Handbook to the Breeds of the World, Vol. 1, pp. 129–30. Comstock Publishing Associates, Ithaca, NY. [Google Scholar]
- Purfield D.C., Berry D.P., McParland S. & Bradley D.G. (2012) Runs of homozygosity and population history in cattle. BMC Genetics 13, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purfield D.C., McParland S., Wall E. & Berry D.P. (2017) The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS ONE 12, e0176780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sved J.A. & Feldman M.W. (1973) Correlation and probability methods for one and two loci. Theoretical Population Biology 4, 129–32. [DOI] [PubMed] [Google Scholar]
- Tortereau F., Servin B., Frantz L. et al (2012) A high density recombination map of the pig reveals a correlation between sex‐specific recombination and GC content. BMC Genomics 13, 586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- United Nations (2019) Sustainable Development Goals. Retrieved February 20, 2019, from Goal 2: Zero Hunger website: https://www.un.org/sustainabledevelopment/hunger/.
- Weir B.S. & Cockerham C.C. (1984) Estimating F‐statistics for the analysis of population structure. Evolution 38, 1358–70. [DOI] [PubMed] [Google Scholar]
- Weir B.S., Hill W.G. (1980) Effect of mating structure on variation in linkage disequilibrium. Genetics 95, 477–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B., Cui L., Perez‐Enciso M. et al (2017) Genome‐wide SNP data unveils the globalization of domesticated pigs. Genetics Selection Evolution 49, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Individual quality control statistics per Piétrain population.
Table S2 SNP quality control statistics per Piétrain population.
Table S3 Summary of runs of homozygosity (ROH) islands per chromosome (SSC) for Duroc, Landrace and Large White.
Figure S1 Presence of Piétrain pigs worldwide.
Figure S2 PCA on Piétrain populations shows three main clusters.
Figure S3 Inbreeding proportion (HBD, homozygosity by descent) as a function of rate parameter R K.
Figure S4 N E estimates for different Piétrain populations 5–21 generations ago.
Figure S5 ROHincidence‐snp per Piétrain population.
Figure S5 ROHincidence‐snp per Piétrain population.
Figure S6 Most frequently observed homozygous genotypes found in Piétrain (PIT) were also present at low frequencies in Landrace (LDR) and Large White (LWT), but absent in Duroc (DUR).
Figure S7 Identical ROH genotypes appear in all of the different Piétrain populations at high frequencies.
Figure S8 Correlational heatmap of ROHincidence‐snp on SSC8 shows high similarity in ROH patterns between Piétrain populations.
Data Availability Statement
The 70K SNP dataset of Belgian Piétrains will be made accessible upon motivated request and was available upon request during the review process.