

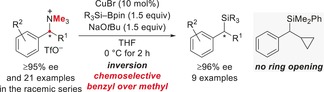
Abstract
A method for the synthesis of benzylsilanes starting from the corresponding ammonium triflates is reported. Silyl boronic esters are employed as silicon pronucleophiles, and the reaction is catalyzed by copper(I) salts. Enantioenriched benzylic ammonium salts react stereospecifically through an SN2‐type displacement of the ammonium group to afford α‐chiral silanes with inversion of the configuration. A cyclopropyl‐substituted substrate does not undergo ring opening, thus suggesting an ionic reaction mechanism with no benzyl radical intermediate.
Keywords: copper, nucleophilic substitution, silanes, stereospecificity, transmetalation
SiR, the NMe has surrendered: Benzylic ammonium salts can be transformed into the corresponding silanes by a copper‐catalyzed SN2‐type displacement. The enantioenrichment of the precursors is completely retained in the α‐chiral silanes. A cyclopropyl group at the benzylic carbon atom remains intact, thereby supporting an ionic reaction mechanism.
Catalytic methods for the asymmetric synthesis of α‐chiral silanes using silicon nucleophiles have gone through a tremendous development in recent years.1 However, enantioselective C(sp3)−Si bond formation by cross‐coupling of either the aforementioned silicon nucleophiles or, in a reverse approach, silicon electrophiles with aliphatic coupling partners is still essentially unknown.2, 3 As shown for benzylic precursors in the racemic series, our group4 as well as Watson and co‐workers5 have developed mechanistically different cross‐coupling reactions (Scheme 1, top).2, 6 Our efforts to render these radical reactions enantioconvergent have been fruitless so far.7 We therefore turned away from alkyl halides and redox‐active carboxylic acid derivatives and considered the use of quaternary ammonium salts as electrophiles.8 These are stable towards light and oxygen and easily accessible in high enantiomeric purity.9 Watson and co‐workers have demonstrated their stereospecific displacement10, 11, 12 for C(sp3)−C(sp2)10 and C(sp3)−B11 bond formation (Scheme 1, bottom left). A related C(sp3)−Si cross‐coupling of these ammonium salts is unprecedented and would offer an alternative to Markovnikov hydrosilylation of styrenes.13 We disclose here a copper‐catalyzed deaminative silylation of those benzylic quaternary ammonium salts with inversion of the configuration (Scheme 1, bottom right).
Scheme 1.
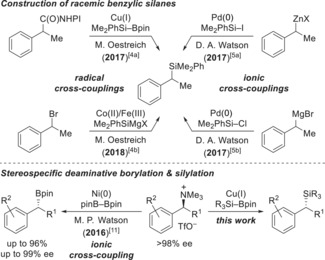
Synthesis of racemic benzylsilanes and stereospecific deaminative cross‐couplings. NHPI=N‐hydroxyphthalimide.
We began our investigation by testing the typical protocol of our earlier ionic silylation3 in the reactions of racemic ammonium triflate 1 a and iodide 2 a with silyl boronic ester14 Me2PhSi‐Bpin (3 a). After a systematic screening of reaction conditions (see the Supporting Information for details), an excellent yield was obtained at 0 °C for triflate 1 a with 10 mol % CuBr as the catalyst and NaOtBu as the base (Table 1, entry 1). The corresponding iodide 2 a was less reactive,10a affording the benzylsilane 4 aa in moderate yield (entry 2). As expected, the alkoxide base had a profound effect on the reaction outcome (entries 3–5), and without base, there was no reaction (entry 6). Both higher and lower loadings of Me2PhSi‐Bpin/NaOtBu resulted in diminished yields (entries 7 and 8). Reactions were sluggish at temperatures other than 0 °C (entries 9 and 10). The yield dropped with 5.0 mol % CuBr (entry 11), and there was an alkoxide‐promoted background reaction15 in the absence of the copper catalyst but conversion was low (entry 12). The use of other silicon nucleophiles4b, 16 led to inferior yields (entries 13 and 14).
Table 1.
Selected examples from the optimization of the reaction conditions.[a]

|
Entry |
X |
Variation |
Yield [%][b] |
|---|---|---|---|
|
1 |
OTf (1 a) |
none |
92 |
|
2 |
I (2 a) |
18 h instead of 2 h |
64 |
|
3 |
OTf (1 a) |
NaOMe instead of NaOtBu |
46 |
|
4 |
OTf (1 a) |
LiOtBu instead of NaOtBu |
64 |
|
5 |
OTf (1 a) |
KOtBu instead of NaOtBu |
0 |
|
6 |
OTf (1 a) |
w/o NaOtBu, 18 h |
0 |
|
7 |
OTf (1 a) |
1.1 equiv of Me2PhSiBpin/NaOtBu |
59 |
|
8 |
OTf (1 a) |
2.0 equiv of Me2PhSiBpin/NaOtBu |
41 |
|
9 |
OTf (1 a) |
25 °C, 2 h |
65 |
|
10 |
OTf (1 a) |
−78 °C, 20 h |
34 |
|
11 |
OTf (1 a) |
w/ 5.0 mol % CuBr, 2 h |
45 |
|
12 |
OTf (1 a) |
w/o CuBr, 18 h |
9 |
|
13 |
OTf (1 a) |
w/ (Me2PhSi)2Zn |
35 |
|
14 |
OTf (1 a) |
w/ Me2PhSiMgX |
66 |
[a] All reactions performed on a 0.25 mmol scale. [b] Determined by GLC analysis with 1,3,5‐trimethoxybenzene as an internal standard.
With the optimized setup in hand, we examined the scope of the nucleophilic substitution for secondary as well as primary and tertiary benzylic ammonium triflates in the racemic series (Scheme 2). The reaction proceeded well with representative silyl boronic esters 3 a–c (1 a→4 aa–ac). Methyl substitution at the aryl group was tolerated in the para (as in 1 b), meta (as in 1 c), and ortho (as in 1 d) positions but the yield was rather low for 1 d→4 da. A broad range of electronically modified aryl groups was compatible, but we could not identify any particular trend (1 e–k→4 ea–ka). Bromine substitution in the para position was an exception since this substrate underwent hydrodebromination (not shown); we had observed this before under similar conditions.3, 17 Benzylic substrates containing naphthyl groups (as 1 l and 1 m) as well as heteroaryl groups (as 1 n and 1 o) reacted in good yields. A longer aliphatic chain at the benzylic position resulted in lower yield (1 p→4 pa); the same applied to a cyclic substrate (1 q→4 qa). Importantly, cyclopropyl‐substituted 1 r yielded 4 ra in 42 % with no ring opening (gray box); this supports an ionic mechanism and excludes the intermediacy of a benzyl radical. However, increasing the steric demand of the alkyl substituent further (isopropyl in 1 s and tert‐butyl in 1 t) thwarted the nucleophilic displacement and defunctionalization was observed. The parent primary substrate reacted in good yield (1 u→4 ua) but, with regard to the assumed SN2‐type mechanism, the tertiary benzylic ammonium salt 1 v decomposed and only furnished traces of 4 va. For completion, diaryl‐substituted 1 w converted into 4 wa in 50 % yield. Conversely, dialkyl‐substituted 1 x, that is, an unactivated ammonium triflate, only formed the desired silane 4 xa in trace amounts. Me2PhSi‐Me (=Me3PhSi) was found to be the major product, which is proof that a primary site (methyl) reacts preferentially over an unactivated secondary position. This is indeed the current limitation of the method. The formation of that byproduct was also seen to a much lesser extent in the reactions of the benzylic substrates and accounts for the lower yields for a few substrates.
Scheme 2.
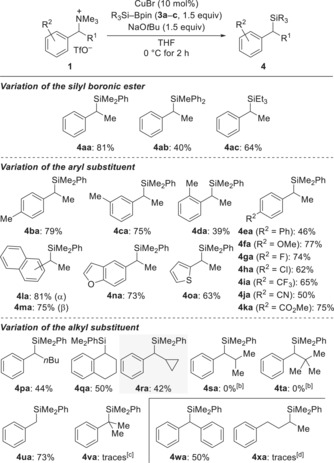
Copper‐catalyzed nucleophilic substitution of benzylic ammonium triflates.[a] [a] Yields are isolated material after purification by flash chromatography on silica gel. Me2PhSi‐SiPhMe2 is always obtained as a byproduct but can be selectively oxidized18 for easier separation from the less polar benzylsilanes. [b] Mainly defunctionalization was observed. [c] Defunctionalization and decomposition was observed. [d] Me3PhSi formed as the major product.
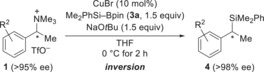
We next studied the stereospecificity of this nucleophilic substitution (Table 2). A number of highly enantioenriched benzyl ammonium triflates 1 were transformed into the desired benzyl silanes 4 with essentially no loss of enantiomeric purity. Substitution of the aryl ring with electron‐donating (entry 4) or electron‐withdrawing (entries 5 and 7) groups had no effect on the stereospecificity. The absolute configuration of 4 aa was assigned as S by Tamao–Fleming oxidation and comparison of the optical rotation of the resulting (S)‐1‐phenylethanol with the reported value (see the Supporting Information for details).19 Hence, the stereochemical course of the transformation of (R)‐1 a into (S)‐4 aa is inversion, which is in agreement with our previously reported ionic nucleophilic silylation of α‐triflyloxy nitriles and esters.3
Table 2.
Stereospecific copper‐catalyzed nucleophilic substitution of benzylic ammonium triflates with a silicon nucleophile.[a]
|
Entry |
Ammonium triflate |
ee [%] |
Benzylsilane |
Yield [%][b] |
ee [%][c] |
|---|---|---|---|---|---|
|
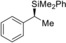
1 |
(R)‐1 a |
>99[d] |
|
79 |
>99 |
|
(S)‐4 aa | |||||
|
|
|
|
|
|
|
|
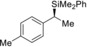
2 |
(R)‐1 b |
98[e] |
|
77 |
98 |
|
(S)‐4 ba | |||||
|
|
|
|
|
|
|
|
3 |
(R)‐1 c |
98[e] |
|
76 |
98 |
|
(S)‐4 ca | |||||
|
|
|
|
|
|
|
|
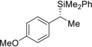
4 |
(S)‐1 f |
98[d] |
|
75 |
97 |
|
(R)‐4 fa | |||||
|
|
|
|
|
|
|
|
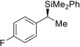
5 |
(R)‐1 g |
98[d] |
|
71 |
98 |
|
(S)‐4 ga | |||||
|
|
|
|
|
|
|
|
6 |
(S)‐1 h |
98[d] |
|
64 |
98 |
|
(R)‐4 ha | |||||
|
|
|
|
|
|
|
|
7 |
(R)‐1 j |
>95[e,f] |
|
48 |
97[g] |
|
(S)‐4 ja | |||||
|
|
|
|
|
|
|
|
8 |
(R)‐1 l |
99[d] |
|
82 |
99 |
|
(S)‐4 la | |||||
|
|
|
|
|
|
|
|
9 |
(R)‐1 m |
99[d] |
|
77 |
97 |
|
(S)‐4 ma |


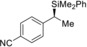
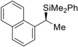
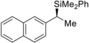
[a] All reactions performed on a 0.25 mmol scale. [b] Yield of isolated product after purification by flash chromatography on silica gel. [c] Determined by HPLC analysis on chiral stationary phases after Tamao–Fleming oxidation. [d] Starting amine is commercially available in enantiomerically enriched form. [e] Determined by HPLC analysis on a chiral stationary phase at the stage of the free amine after amide formation with 4‐nitrobenzoic acid chloride (see the Supporting Information for details). [f] Racemization during amide formation; the enantiomeric excess is derived from the diastereomeric excess (de) of the highly diastereoenriched precursor10b (Ellman's tert‐butanesulfinyl amines9a). [g] The enantiomeric excess was determined at the stage of the silane; no Tamao–Fleming oxidation required.
We then moved to other activated ammonium triflate salts. Since the synthesis of unknown secondary allylic ammonium salts failed in our hands, a primary derivative was tested successfully (see the Supporting Information for details). In turn, a secondary propargylic ammonium triflate could be prepared with high enantiomeric excess, and both linear‐ and branched‐selective stereospecific substitution reactions with Grignard reagents have been reported.20 We subjected (S)‐5 to the usual procedure and obtained allene γ‐6 in reasonable yield, noting the chemical instability of (S)‐5 (Scheme 3). In contrast to Tortosa's work20b and our own method21 with a phosphate leaving group, the enantiomeric excess of (R)‐γ‐6 was significantly eroded; several runs of the reaction showed that the ee value varied, probably as result of the partial decomposition of (S)‐5.
Scheme 3.
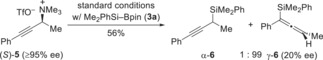
Copper‐catalyzed silylation of a propargylic ammonium salt.
In summary, we have developed a nucleophilic silylation of benzylic ammonium triflates, which represents the first example of a deaminative silylation. Our procedure offers an alternative pathway for the synthesis of racemic benzylsilanes and exhibits good functional‐group tolerance. The reaction is stereospecific and likely to proceed by an SN2‐type mechanism. The extension of this approach to allylic or propargylic ammonium salts will require further optimization and is currently under investigation in our laboratory.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was supported by the Deutsche Forschungsgemeinschaft (Oe 249/15‐1). M.O. is indebted to the Einstein Foundation Berlin for an endowed professorship.
J. Scharfbier, B. M. Gross, M. Oestreich, Angew. Chem. Int. Ed. 2020, 59, 1577.
Contributor Information
Jonas Scharfbier, http://www.organometallics.tu-berlin.de.
Prof. Dr. Martin Oestreich, Email: martin.oestreich@tu-berlin.de.
References
- 1.
- 1a. Wilkinson J., Nuyen C., Carpenter T., Harruff S., van Hoveln R., ACS Catal. 2019, 9, 8961–8979; [Google Scholar]
- 1b. Hensel A., Oestreich M., Top. Organomet. Chem. 2016, 58, 135–167; [Google Scholar]
- 1c. Sawamura M., Ito H. in Copper-Catalyzed Asymmetric Synthesis (Eds.: A. Alexakis, N. Krause, S. Woodward), Wiley-VCH, Weinheim, 2014, pp. 157–177. [Google Scholar]
- 2.A comprehensive summary:
- 2a. Bähr S., Xue W., Oestreich M., ACS Catal. 2019, 9, 16–24 and cited references; see also [Google Scholar]
- 2b. Korch K. M., Watson D. A., Chem. Rev. 2019, 119, 8192–8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Scharfbier J., Hazrati H., Irran E., Oestreich M., Org. Lett. 2017, 19, 6562–6565. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Xue W., Oestreich M., Angew. Chem. Int. Ed. 2017, 56, 11649–11652; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11808–11811; [Google Scholar]
- 4b. Xue W., Shishido R., Oestreich M., Angew. Chem. Int. Ed. 2018, 57, 12141–12145; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12318–12322; related methods without examples of benzylic electrophiles: [Google Scholar]
- 4c. Chu C. K., Liang Y., Fu G. C., J. Am. Chem. Soc. 2016, 138, 6404–6407; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4d. Xue W., Qu Z.-W., Grimme S., Oestreich M., J. Am. Chem. Soc. 2016, 138, 14222–14225. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Cinderella A. P., Vulovic B., Watson D. A., J. Am. Chem. Soc. 2017, 139, 7741–7744; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Vulovic B., Cinderella A. P., Watson D. A., ACS Catal. 2017, 7, 8113–8117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loh and co-workers recently developed a palladium-catalyzed silylation of benzyl halides, focusing on primary substrates: Huang Z.-D., Ding R., Wang P., Xu Y.-H., Loh T.-P., Chem. Commun. 2016, 52, 5609–5612. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a.W. Xue, H. Yi, M. Oestreich, unpublished results, 2017/2018; for an alternative approach, see:
- 7b. Yi H., Mao W., Oestreich M., Angew. Chem. Int. Ed. 2019, 58, 3575–3578; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3613–3616; [Google Scholar]
- 7c. Schwarzwalder G. M., Matier C. D., Fu G. C., Angew. Chem. Int. Ed. 2019, 58, 3571–3574; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3609–3612; in contrast, the enantioconvergent borylation of benzyl halides was recently accomplished: [Google Scholar]
- 7d. Wang Z., Bachman S., Dudnik A. S., Fu G. C., Angew. Chem. Int. Ed. 2018, 57, 14529–14532; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14737–14740; [Google Scholar]
- 7e. Iwamoto H., Endo K., Ozawa Y., Watanabe Y., Kubota K., Imamoto T., Ito H., Angew. Chem. Int. Ed. 2019, 58, 11112–11117; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 11229–11234. [Google Scholar]
- 8.A recent review on C−N bond cleavage, including ammonium salts: Ouyang K., Hao W., Zhang W.-X., Xi Z., Chem. Rev. 2015, 115, 12045–12090. [DOI] [PubMed] [Google Scholar]
- 9.Preparation of enantioenriched amines by diastereoselective reduction of tert-butanesulfinyl ketimines:
- 9a. Borg G., Cogan D. A., Ellman J. A., Tetrahedron Lett. 1999, 40, 6709–6712; racemization-free dimethylation of amines: [Google Scholar]
- 9b. Icke R. N., Wisegarver B. B., Alles G. A., Org. Synth. 1945, 25, 89–90. [Google Scholar]
- 10.
- 10a. Maity P., Shacklady-McAtee D. M., Yap G. P. A., Sirianni E. R., Watson M. P., J. Am. Chem. Soc. 2013, 135, 280–285; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Shacklady-McAtee D. M., Roberts K. M., Basch C. H., Song Y.-G., Watson M. P., Tetrahedron 2014, 70, 4257–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Basch C. H., Cobb K. M., Watson M. P., Org. Lett. 2016, 18, 136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stereospecific C(sp3)−S bond formation: Jiang W., Li N., Zhou L., Zeng Q., ACS Catal. 2018, 8, 9899–9906. [Google Scholar]
- 13.Recent examples with di- or trihydrosilanes:
- 13a. Cheng B., Lu P., Zhang H., Cheng X., Lu Z., J. Am. Chem. Soc. 2017, 139, 9439–9442; [DOI] [PubMed] [Google Scholar]
- 13b. Cheng B., Liu W., Lu Z., J. Am. Chem. Soc. 2018, 140, 5014–5017. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Delvos L. B., Oestreich M. in Science of Synthesis Knowledge Update 2017/1 (Ed.: M. Oestreich), Thieme, Stuttgart, 2017, pp. 65–176; [Google Scholar]
- 14b. Oestreich M., Hartmann E., Mewald M., Chem. Rev. 2013, 113, 402–441; preparation of Si−B reagents: [DOI] [PubMed] [Google Scholar]
- 14c. Suginome M., Matsuda T., Ito Y., Organometallics 2000, 19, 4647–4649 [Me2PhSi-Bpin (3 a) and MePh2Si-Bpin (3 b)]; [Google Scholar]
- 14d. Boebel T. A., Hartwig J. F., Organometallics 2008, 27, 6013–6019 [Et3Si-Bpin (3 c)]. [Google Scholar]
- 15.
- 15a. Kawachi A., Minamimoto T., Tamao K., Chem. Lett. 2001, 30, 1216–1217; [Google Scholar]
- 15b. Ito H., Horita Y., Yamamoto E., Chem. Commun. 2012, 48, 8006–8008. [DOI] [PubMed] [Google Scholar]
- 16. Weickgenannt A., Oestreich M., Chem. Eur. J. 2010, 16, 402–412 and cited references. [DOI] [PubMed] [Google Scholar]
- 17. Yamamoto E., Ukigai S., Ito H., Chem. Sci. 2015, 6, 2943–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gryparis C., Stratakis M., Chem. Commun. 2012, 48, 10751–10753. [DOI] [PubMed] [Google Scholar]
- 19. Chan T. H., Pellon P., J. Am. Chem. Soc. 1989, 111, 8737–8738. [Google Scholar]
- 20.
- 20a. Guisán-Ceinos M., Martín-Heras V., Tortosa M., J. Am. Chem. Soc. 2017, 139, 8448–8451; [DOI] [PubMed] [Google Scholar]
- 20b. Guisán-Ceinos M., Martín-Heras V., Soler-Yanes R., Cárdenas D. J., Tortosa M., Chem. Commun. 2018, 54, 8343–8346. [DOI] [PubMed] [Google Scholar]
- 21.A related stereospecific allene formation: Vyas D. J., Hazra C. K., Oestreich M., Org. Lett. 2011, 13, 4462–4465. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary