

Abstract
Objective
Drug‐resistant seizures are common in patients with leucine‐rich, glioma‐inactivated 1 (LGI1)‐IgG associated and contactin‐associated protein‐like 2 (CASPR2)‐IgG associated encephalitis. We performed the first randomized double‐blind placebo‐controlled trial to evaluate efficacy of intravenous immunoglobulin (IVIG) in reducing seizure frequency.
Methods
Our enrollment goal was 30 LGI1/CASPR2‐IgG–seropositive adult patients with ≥2 seizures per week. Patients were randomized to receive IVIG (0.5g/kg day 1, 1g/kg day 2, 0.6g/kg weeks 3 and 5) or volume‐matched intravenous normal saline. Following the blinded phase, the nonresponders in the placebo group received IVIG. The primary clinical outcome was 50% reduction in seizure frequency from baseline to 5 weeks.
Results
After enrollment of 17 patients (LGI1‐IgG, 14; CASPR2‐IgG, 3) over 34 months, the study was terminated due to slow enrollment. Six of 8 patients in the IVIG group were responders, compared to 2 of 9 in the placebo group (p = 0.044, odds ratio = 10.5, 95% confidence interval = 1.1–98.9). For the LGI1‐IgG seropositive subgroup, 6 of 8 patients in the IVIG group were responders, compared to zero of 6 in the placebo group. Two LGI1‐IgG–seropositive patients receiving IVIG, but none receiving placebo, were seizure‐free at the end of the blinded phase. Four of the 6 patients entering the open‐label IVIG arm reported ≥50% reduction in seizure frequency. There were no correlations with LGI1/CASPR2‐IgG1–4 subclasses.
Interpretation
Superiority of IVIG to placebo reached statistical significance for the primary endpoint for all patients and the subset with LGI1‐IgG. These results have to be interpreted with the caveat that the study did not reach its originally selected sample size. ANN NEUROL 2020;87:313–323
Seizures are a common manifestation of autoimmune encephalitis and paraneoplastic disorders.1 A considerable minority of patients with focal epilepsy of unknown etiology have neural‐specific antibodies,2, 3 with voltage‐gated potassium channel complex (VGKC) antibodies as a common serological biomarker among these reported cases.4, 5, 6
VGKC‐IgG was initially reported in association with neurological autoimmunity in 2004.7 However, discovery of autoantibodies against the extracellular domains of leucine‐rich, glioma‐inactivated 1 (LGI1)8 and contactin‐associated protein‐like 2 (CASPR2)9 facilitated a change in our understanding of clinical implications of VGKC antibodies.10, 11 LGI1‐IgG is typically associated with seizures and/or memory deficits among older adults, whereas CASPR2‐IgG seropositive cases predominantly have peripheral nervous system involvement (neuromyotonia, myokymia, or dysautonomia).12, 13 However, some CASPR2‐IgG seropositive patients present with epilepsy and/or encephalopathy as the primary neurological manifestation.9, 12 Both conditions are male predominant and affect patients in later life. Currently, the presence of VGKC‐IgG in the absence of LGI1‐ and/or CASPR2‐IgG seropositivity is not considered to be a specific biomarker of neurological autoimmunity, and such patients are typically not responsive to immunotherapy and do not carry the strikingly robust human leukocyte antigen (HLA) associations seen in patients with LGI1‐ or CASPR2‐IgG.10, 11, 12, 14, 15
Management of autoimmune epilepsy currently centers on immunotherapies.16 There are clear data supporting a variety of immunotherapies over antiepileptic drugs (AEDs).16, 17, 18 However, the current immunotherapy evidence base is limited to retrospective case series with largely retrospective data collection and to expert opinions.7, 13, 18, 19, 20, 21, 22, 23 To date, there have been no randomized control trials evaluating the efficacy of immunotherapy in autoimmune epilepsy.
Designing a clinical trial in autoimmune epilepsy is fraught with many specific and generic challenges. First, the heterogeneity of clinical presentation makes a unifying outcome measure difficult. Second, outcome measures such as brain magnetic resonance imaging (MRI),19, 24, 25 brain positron emission tomography, formal neuropsychological assessment, and seizure diaries have not been validated. Third, there remain difficulties in establishing the diagnosis due to limited clinician recognition, difficult logistics, costs, and limited scalability of serologic testing. Fourth, study size is limited by the rarity of the condition23, 26 and the potential rates of dropout in the event of complete recovery or suspected adverse events. Furthermore, due to increased recognition of the importance of early immunotherapy among neurologists,7, 16 most physicians will treat the patients acutely rather than delaying treatment to allow enrollment in a randomized controlled clinical trial.
Despite these limitations, there is a clear need for a randomized clinical trial evaluating the efficacy of immunotherapy in the setting of LGI1‐ or CASPR2‐IgG–associated autoimmune epilepsy. The existing data are biased due to the lack of a comparator arm and of reported placebo‐treated outcomes. In the absence of class I or class II evidence, most patients or physicians have difficulty in getting approval for insurance coverage of immunotherapy costs, especially intravenous immunoglobulin (IVIG). Third, some studies have suggested a self‐limiting nature, albeit with more limited recovery, of some of the LGI1‐IgG–associated epilepsies, further necessitating a placebo‐controlled study assessing efficacy of immunotherapy in the clinical outcomes of these patients.27
We designed a randomized placebo‐controlled clinical trial evaluating the efficacy of IVIG in LGI1‐ and/or CASPR2‐IgG associated autoimmune epilepsy. We hypothesized that IVIG would be more efficacious than placebo in achieving 50% reduction in seizure frequency.
Patients and Methods
Participants
Placebo was the chosen comparator, and a maximum possible duration of 5 weeks on placebo was determined acceptable following ethical consideration and recognition that no approved therapies or controlled trials had been performed to establish the risk/benefit profile of off‐label medications in LGI1/CASPR2‐IgG associated autoimmune epilepsy. The trial protocol (http://clinicaltrials.gov registration number NCT02697292) and supporting documentation were approved by the Mayo Clinic Institutional Review Board (IRB# 15‐005649).
Between February 19, 2016 and December 6, 2018, patients were identified through the Mayo Clinic Neuroimmunology Laboratory service line testing and directly recruited into a randomized double‐blind placebo‐controlled trial at Mayo Clinics in Rochester, Minnesota, following consent of the patient and managing physician. Inclusion and exclusion criteria are summarized in Supplementary Table 1. Patients provided written informed consent at enrollment.
Antibody Testing
Radioimmunoprecipitation assays were performed to detect VGKC‐IgG. All positive sera and cerebrospinal fluid (≥0.03nmol/l)13 were tested for LGI1‐IgG and CASPR2‐IgG specificities using a transfected cell‐based immunofluorescence assay (CBA; EUROIMMUN, Lübeck, Germany). Cerebrospinal fluid (CSF) was tested undiluted and serum at 1:10 dilution. Other neural‐specific antibodies that are a part of the Mayo Neuroimmunology Laboratory's comprehensive neural autoantibody evaluation were also tested as previously described.28
LGI1‐ and CASPR2‐IgG Subclass Quantificantion
Human embryonic kidney (HEK) 293 cells were transiently transfected with membrane‐tethered LGI1 or CASPR2 C‐terminally fused to enhanced green fluorescent protein.7, 12 After 24 to 48 hours, cells were detached and mixed with untransfected HEK293T cells at a ~1:1 ratio. Next, cells were incubated with serum/CSF dilutions for 30 minutes at room temperature. After 2 washing steps, a single mouse antihuman secondary antibody (IgG1–3: AF647, IgG4: PE; SouthernBiotech, Birmingham, AL [IgG1 Hinge–Alexa Fluor 647 (9052‐31), IgG2 Fc–Alexa Fluor 647 (9070‐31), IgG3 Hinge‐Alexa Fluor 647 (9210‐31), IgG4 Fc‐PE (9200‐09]) was incubated for 30 minutes at room temperature. Subsequently, cells were washed once with buffer containing 4′,6‐diamidino‐2‐phenylindole (DAPI) and once without DAPI, and analyzed on an Attune NxT flow cytometer. The serum dilution used for LGI1‐IgG1–3 was 1:50 and for LGI1‐IgG4 was 1:200 (normalized to 1:50). The serum dilution used for CASPR2‐IgG1–3 was 1:100 and for CASPR2‐IgG4 was 1:400 to 1:8,000 (normalized to 1:100). The CSF dilution used for all samples was 1:2.
The antibody levels were then calculated by subtracting the median IgG fluorescence of the green fluorescent protein (GFP)‐negative from GFP‐positive cells (to generate delta medians of the fluorescence intensity HEK/single cells/viable/GFP positive–HEK/single cells/viable/GFP negative) and normalized to calibration beads (Quantum Simply Cellular microspheres; Bangs Laboratories, Fishers, IN), resulting in antibody binding capacities of the cells. The cutoff was calculated for each subclass individually using 9 to 10 healthy control serum samples (mean + 3 standard deviations).
HLA Analysis
All patients (except Patient 6) were genotyped for HLA‐DRB, using polymerase chain reaction sequence‐specific primers.14
Procedures
Once consent was obtained and it was determined that the participant met all inclusion criteria, the participants were enrolled into the study. Patients were randomized into 2 groups (A vs B) in a 1 to 1 ratio using a simple randomization method by utilizing JMP Pro 13 software (SAS Institute, Cary, NC). The details of the randomization table were unknown to the researcher who enrolled the participants (J.S.) and the clinicians who evaluated the patients. The randomization code (A was the active arm and B was the placebo arm) was kept sealed until the completion of the blinded phase of the study. Patients in the active arm received IVIG and patients in control arm received placebo for 5 weeks. During the period between enrollment and unblinding, no changes were made to their antiseizure medication regimen. All patients were instructed to keep a seizure diary documenting seizure frequency and seizure semiologies; spouses and family members were also asked to assist. Following completion of 5 weeks of treatment with either IVIG or placebo, participants returned to Mayo Clinic for evaluation.
Sample size of 30 (ie, 15 per group) was calculated based on preliminary observations of 70% efficacy in the experimental group versus 10% anticipated as the placebo effect (power = 89% and significance level of alpha = 5% [Nquery advisor software version 7.0]). We proceeded with the target sample of 30 due to budgetary restrictions, limited IVIG availability for the research study, and feasibility of patient enrollment for a rare disease within 2 years (target study duration at the time of initiation).
Blinded Phase of the Trial (All Infusions Given at Mayo Clinic Infusion Center)
Active Comparator: IVIG (Gamunex‐C ) Group
The IVIG dose was determined based on ideal weight, with a dose not to exceed 80g. Patients received IVIG (0.5g/kg) on day 1 (week 1 day 1), then received IVIG (1g/kg not exceeding 80g) on day 2 (week 1 day 2). Patients also received 500ml normal saline before and after the higher‐dose infusion of 1g/kg. Then, once every 2 weeks, patients received 0.6g/kg IVIG (week 3 and week 5) for 2 more infusions. After completion of all 4 infusions, all patients were again evaluated at Mayo Clinic and unblinded.
Placebo Comparator: Placebo/Normal Saline Group
Patients received volume‐matched placebo on day 1 (week 1 day 1), then they received placebo on day 2 (week 1 day 2). Then, once every 2 weeks, patients received placebo (week 3 and week 5) for 2 more infusions. After completion of all 4 infusions, all patients were again evaluated at Mayo Clinic and unblinded.
Open Label of Arm of the Trial
The patients in the placebo group with persistent symptoms received IVIG in an open label fashion in their homes through Option Care home infusion services. Patients received IVIG (0.5g/kg) on day 1, and then they received IVIG (1g/kg not exceeding 80g) on day 2. Patients also received 500ml normal saline before and after the higher‐dose infusion of 1g/kg. Then, once every 2 weeks, patients received 0.6g/kg IVIG for 2 more infusions in their homes through Option Care. Patients were evaluated at Mayo Clinic following completion of all 4 infusions.
Evaluations
During the clinic visit (baseline and 5 weeks), complete neurological examination and formal cognitive assessment were performed for each patient. Electroencephalographic data were obtained during the initial visit (Supplementary Table 2). Estimation of seizure frequency prior to enrollment was based on history provided by the patient and family at the initial visit. All patients and family members were instructed to maintain seizure dairies. Data in the seizure diaries were corroborated with history taken at the time of the clinical visits. Where a discrepancy was identified, the frequency reported at the time of clinical visits was used. Maximum seizure frequency over the past 3 days prior to clinic evaluation was established utilizing patient report and/or documentation in seizure dairy. Cognitive status was measured using the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS).29 The RBANS is a well‐standardized battery that includes 12 subtests with widely used task formats tapping 5 major cognitive domains. It includes alternate forms to minimize practice effect. For patients who entered into the open label IVIG arm, the above testing was performed following completion of all 4 infusions. The study coordinators completed adverse event questionnaires as appropriate (J.S. and K.D).
Clinical Outcome
The primary clinical outcome was reduction in seizure frequency by ≥50% during the follow‐up visit following completion of the blinded phase at 5 weeks. Secondary outcome measures included RBANS.29 The proportion of patients with improvement in seizure frequency or seizure freedom was also calculated. Withdrawal from the blinded phase of the study due to worsening of seizure frequency and cognitive status was considered an intervention failure.
Statistical Analysis
Descriptive summaries are reported as median (range) for continuous variables and frequency (%) for categorical variables. Comparisons of categorical and continuous outcomes of interest were done with Fisher exact test and Mann–Whitney U test, respectively. A one‐sided statistical test was utilized to demonstrate improved efficacy of IVIG compared to placebo. Graphical analysis illustrated comparison of responder rate and seizure freedom between IVIG and placebo arms. All analyses were done in SPSS (v23; IBM, Armonk, NY) and JMP Pro 13. Odds ratio estimates were not reported when cells with 0 are observed.
Role of the Funding Source
The sponsors of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report.
Results
Patient Disposition
Between February 19, 2016 and December 6, 2018, the physicians of 594 LGI1/CASPR2‐IgG–positive patients were contacted by telephone regarding possible enrollment. Among these, the majority of patients (570) either did not meet the criteria (see Supplementary Table 1) or were unable to travel to Mayo Clinic, Rochester, Minnesota for evaluation (Fig 1). Twenty‐four patients were evaluated at Mayo Clinic for possible enrollment, but 7 failed the screening process. Because the planned duration of the study was 2 years and a decline in patient enrollment occurred in 2018 (none from July to December 2018), the investigators terminated the study before the statistically based sample size target of 30 was reached.
Figure 1.
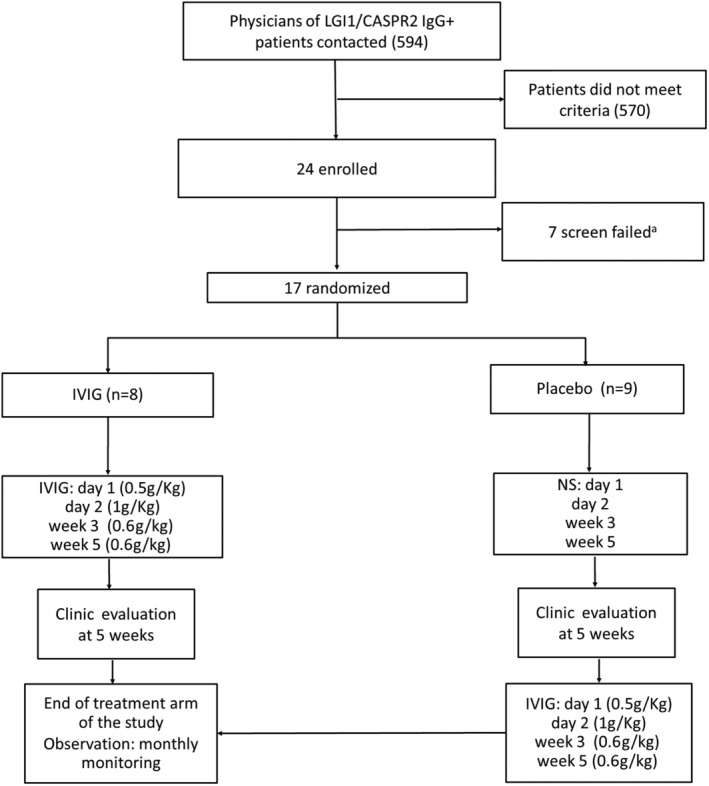
Patient enrollment summary and trial design. IVIG = intravenous immunoglobulin; NS = normal saline. aReasons for failed screening: patient refused due to concern of receiving placebo, n = 2; patient refused to participate in the study due to inability to return for required follow‐up visits, n = 1; change in regimen of antiepileptic drugs <1 week prior to enrollment, n = 1; seizure frequency < 2 per week, n = 1; patient refused to keep a seizure dairy, n = 1; IgA deficiency, n = 1).
Baseline Demographic and Clinical Characteristics
At the time of study termination, 17 patients (12 men, 5 women) had undergone randomization to receive either placebo or IVIG. None of the patients enrolled in the study were on any immunosuppressive medication at the time of enrollment, and only 1 patient had received any immunotherapy prior to enrollment: a 3‐day course of IVIG >8 months previously. As expected, at baseline 11 of 17 reported >10 seizures daily and 6 of 17 experienced >60 seizures per day. The majority (n = 14, 82%) were seropositive for LGI1‐IgG, and 3 patients had CASPR2‐IgG. None were positive for both antibodies. The demographic and clinical characteristics of all 17 patients are outlined in Supplementary Table 2. Baseline characteristics are summarized separately for each treatment arm in Table 1. There was no significant difference in demographic and clinical characteristics, including duration from symptom onset to time of enrollment between the 2 study arms. One LGI1‐IgG–seropositive patient (Patient 9) required hospitalization due to significant deterioration of her neurological conditions with increasing seizures (≥3 seizures per hour) and worsening cognitive status. She was unblinded at 3 weeks and found to be in the placebo arm.
Table 1.
Clinical and Demographic Characteristics of the 2 Groups
| Characteristic | IVIG, n = 8 | Placebo, n = 9 | p |
|---|---|---|---|
| Median age, yr (range) | 70 (66–77) | 70 (59–77) | 0.773 |
| Male, n (%) | 6 (75) | 6 (67) | 0.707 |
| LGI1‐IgG, n (%) | 8 (57) | 6 (43) | 0.206 |
| CASPR2‐IgG, n (%) | 0 | 3 (100) | 0.206 |
| Median duration from symptom onset to enrollment, mo (range) | 5 (1–12) | 8 (1–13) | 0.359 |
| ≥5 seizures every day, n (%) | 4 (50) | 5 (55) | 1.000 |
| Faciobrachial dystonic seizures, n (%) | 4 (50) | 5 (66) | 1.000 |
| Secondarily generalized seizures, n (%) | 1 (13) | 3 (33) | 0.576 |
| Cognitive dysfunction, n (%) | 8 (100) | 8 (89) | 0.331 |
| Mesial temporal hyperintensity on MRI, n (%) | 1 (13) | 4 (44) | 0.294 |
| ASMs, n (%) | 3 (38) | 5 (56) | 0.457 |
| Levetiracetam, n (%) | 0 | 4 (44) | 0.637 |
| Sodium channel blocking ASMs: ZNS, OXC, LMT, LCM, n (%) | 3 (38) | 2 (22) | 0.620 |
ASM = antiseizure medication; CASPR2 = contactin‐associated protein‐like 2; IVIG = intravenous immunoglobulin; LCM = lacosamide; LGI1 = leucine‐rich, glioma‐inactivated 1; LMT = Lamictal; MRI = magnetic resonance imaging; OXC = oxcarbazepine; ZNS = zonisamide.
All LGI1‐IgG–seropositive patients tested carried 1 copy of HLA‐DRB1*07:01, and all CASPR2‐IgG–seropositive patients carried 1 copy of HLA‐DRB1*11:01. In addition, 1 patient with LGI1 antibodies also carried DRB1*11:01 (Patient 4).
Efficacy
Primary Outcome
A higher proportion of patients in the IVIG arm experienced ≥50% reduction in seizure frequency compared to the placebo arm at the completion of the blinded phase (75% [6 of 8 patients] vs 22% [2 of 9 patients], p = 0.044, odds ratio = 10.5, 95% confidence interval [CI] = 1.1–98.9; Table 2, Fig 2). Among the LGI1‐IgG seropositive subgroup who received IVIG, the 50% responder rate was 75% (6 of 8 patients), compared to 0 (0 of 6 patients) among those in the placebo arm. Two LGI1‐IgG–seropositive patients receiving IVIG, but none in the placebo arm, were seizure‐free at the end of the blinded phase. All CASPR2‐IgG patients (n = 3) were randomized to receive placebo; 1 reported ≥50% reduction in seizure frequency and another reported seizure freedom at the 5‐week visit.
Table 2.
Comparison of Clinical Outcomes and Adverse Effects between IVIG and Placebo Arms
| LGI1‐ and CASPR2‐IgG | |||
|---|---|---|---|
| IVIG | Placebo | p (OR, 95% CI) | |
| 50% seizure rate reduction, week 5, n (%) | 6/8 (75) | 2/9 (22) | 0.044 (10.5, 1.1–98.9) |
| Improvement in seizure frequency, week 5, n (%) | 6/8 (75) | 2/9 (22) | 0.044 (10.5, 1.1–98.9) |
| Seizure freedom, week 5, n (%) | 2/8 (25) | 1/9 (13) | 0.453 (2.7,0.11–176.6) |
| Improvement in FBDS frequency, week 5, n (%) | 2/4 (50) | 0/5 | 0.167 (NR, 0.3–∞) |
| Change in seizure semiology, n (%) | 1/7 (14) | 1/6 (17) | 0.731 (0.8, 0.01–78.4) |
| Stable or improved RBANS percentile, week 5, n (%) | 8/8 (100) | 5/8 (62) | 0.100 (NR, 0.45–∞) |
| Adverse effects, n (%) | 1/8 (13) | 2/9 (22) | 0.547 (0.5, 0.01–12.3) |
| Open label IVIG phase, n (%) | |||
| 50% seizure rate reduction, week 11 | — | 4/6 (67) | — |
| Improvement in seizure frequency, week 11 | — | 5/6 (83) | — |
| Seizure freedom, week 11 | — | 0/6 | — |
| Stable or improved RBANS percentile, week 11 | — | 4/7 (57) | — |
CASPR2 = contactin‐associated protein‐like 2; CI = confidence interval; FBDS = faciobrachial dystonic seizure; IVIG = intravenous immunoglobulin; LGI1 = leucine‐rich, glioma‐inactivated 1; NR = not reported (OR estimates are not reported when cells with 0 are observed); OR = odds ratio; RBANS = Repeatable Battery for the Assessment of Neuropsychological Status.
Figure 2.
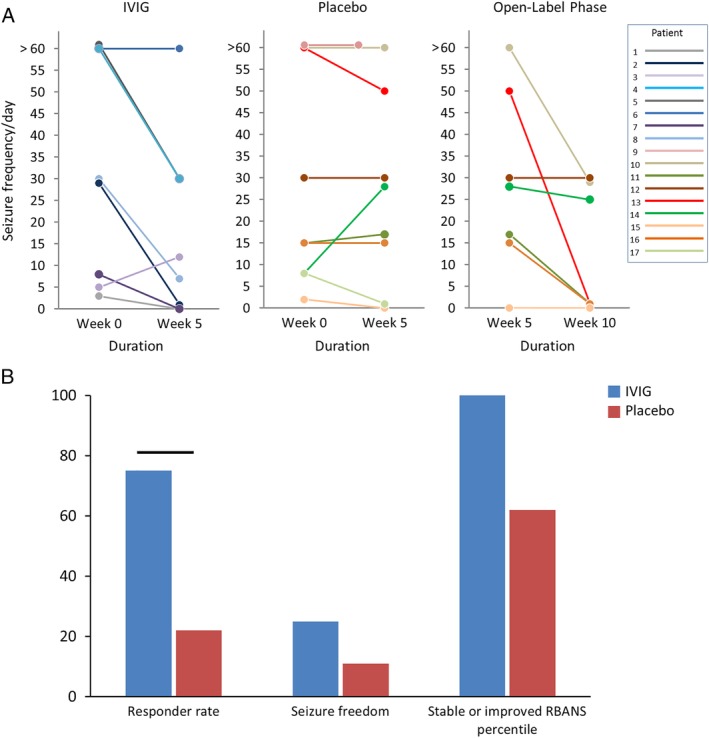
Clinical outcomes. A, Change in seizure frequency following administration of placebo or intravenous immunoglobulin (IVIG) in the blinded and open label phase of the study. B, Proportion of the patients achieving ≥50% reduction in seizure frequency, seizure frequency improvement, seizure freedom, and stabilization or improvement in a cognitive measure (Repeatable Battery for the Assessment of Neuropsychological Status [RBANS]) in the 2 study arms during the blinded phase of the study. Bars represent significant difference (p < 0.05).
Secondary Outcome
The median change in RBANS total score among LGI1‐IgG seropositive patients showed a trend toward improvement for the IVIG group compared to placebo (3 [0 to 13] vs −1 [−12 to 6], p = 0.077, 95% CI = 0.84–∞; Table 3). Eight of 8 LGI1‐IgG seropositive patients in the IVIG group showed zero change or increase in RBANS total‐score percentiles, compared with 2 of 5 in the placebo group (Fig 3). There were no significant differences in the RBANS index scores for immediate memory, visuospatial/constructional, language, attention, or delayed memory (see Table 3). Although there was no statistical difference between IVIG and placebo in the combined cohort, a trend toward a favorable cognitive outcome was observed in the IVIG arm (100% [8 of 8] vs 40% [2 of 5], p = 0.100, 95% CI = 0.45–∞).
Table 3.
Comparison of Clinical Outcomes and Adverse Effects between Intravenous Immune Globulin and Placebo Arms of the Study in the LGI1‐IgG Subgroup
| LGI1‐IgG | |||
|---|---|---|---|
| IVIG | Placebo | p (OR, 95% CI) | |
| 50% seizure rate reduction, week 5, n (%) | 6/8 (75) | 0/6 | 0.009 (NR, 1.51–∞) |
| Improvement in seizure frequency, week 5, n (%) | 6/8 (75) | 0/6 | 0.009 (NR, 1.51–∞) |
| Seizure freedom, week 5, n (%) | 2/8 (25) | 0/6 | 0.308 (NR, 0.14–∞) |
| Improvement in FBDS frequency, week 5, n (%) | 2/4 (50) | 0/5 | 0.167 (NR, 0.254–∞) |
| Change in seizure semiology, n (%) | 1/7 (14) | 0/5 | 0.583 (NR, 0.02–∞) |
| Stable or improved RBANS percentile, week 5, n (%) | 8/8 (100) | 2/5 (40) | 0.035 (NR, 0.84–∞) |
| Median change in immediate memory score (range) | 6.5 (−4 to 14) | −4 (−4 to 9) | 0.174 (−13.4 to 2.2) |
| Median change in visuospatial memory score (range) | −1.5 (−9 to −11) | −9 (−27 to 19) | 0.378 (−26.7 to 15.6) |
| Median change in language score (range) | −1 (−7 to 7) | 0 (−3 to 11) | 0.505 (−4.5 to 9.7) |
| Median change in attention score (range) | 0 (−11 to 9) | 0 (−12 to 6) | 0.412 (−12.2 to 5.6) |
| Median change in delayed memory score (range) | 2.5 (−9 to 35) | −8 (−47 to 13) | 0.188 (−14.3 to −0.01) |
| Median change in total RBANS score (range) | 3 (0–13) | −1 (−12 to 6) | 0.077 (−15.9 to 1.6) |
| Adverse effects, n (%) | 1/8 (13) | 1/6 (17) | 0.692 (0.75, 0.03–14.56 |
| Open label IVIG phase, n (%) | |||
| 50% seizure rate reduction, week 12 | — | 3/5 (60) | — |
| Improved cognitive score, week 12 | — | 3/5 (60) | — |
CI = confidence interval; FBDS = faciobrachial dystonic seizure; IVIG = intravenous immunoglobulin; LGI1 = leucine‐rich, glioma‐inactivated 1; NR = NR not reported (OR estimates are not reported when cells with 0 are observed); OR = odds ratio; RBANS = Repeatable Battery for the Assessment of Neuropsychological Status.
Figure 3.
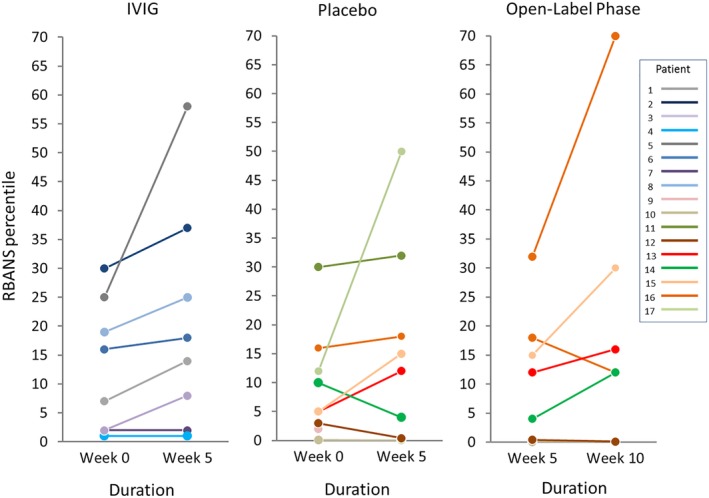
Cognitive outcomes among leucine‐rich, glioma‐inactivated 1 (LGI1)‐IgG–seropositive cases. Change in Repeatable Battery for the Assessment of Neuropsychological Status (RBANS) percentiles among LGI1‐IgG–seropositive cases following administration of placebo or intravenous immunoglobulin (IVIG) in the blinded and open label phase of the study is shown.
Open Label Arm
Seven of the 9 patients who initially received placebo were subsequently adminstered open label IVIG.
Two patients did not enter the open label arm of the study: 1 LGI1‐IgG patient who was unblinded prior to completion of the 5‐week regimen and another CASPR2‐IgG patient who had nearly complete resolution of symptoms on placebo. One CASPR2‐IgG patient, who was seizure‐free at week 6 follow‐up, received IVIG for management of ataxia attributed to his condition.
Six patients with persistent seizures who were nonresponders at week 5 on placebo during the blinded phase of the study entered the open label IVIG arm of the study. Among these, 4 patients reported ≥50% reduction in seizure frequency during the week 11 evaluation. However, none achieved seizure freedom. Furthermore, 5 of the 7 patients (including the CASPR2‐IgG seropositive patient with ataxia but no seizures; see Supplementary Table 2) who received IVIG in the open label part of the study showed stabilization or improvement of RBANS percentiles.
LGI1‐ and CASPR2‐IgG Subclasses
All LGI1 and CASPR2 autoimmune epilepsy cases had IgG4 as the predominant subclass (serum). Both the LGI1‐IgG seropositive patients who achieved seizure freedom during the blinded phase of the study had undetectable LGI1‐IgG1 antibodies. However, there was no significant difference in percentage IgG1–4 subclasses among the 2 LGI1 patients who did not have a favorable response to IVIG compared to others (see Supplementary Table 2). Proportion of LGI1‐IgG1–4 subclass did not differ significantly based on seizure semiology. Among 8 patients with faciobrachial dystonic seizures (FBDSs), 4 had undetectable or barely detectable (0.2%) LGI1‐IgG1 antibody subclass.
Treatment Tolerance and Toxicity
One LGI1‐IgG–seropositive patient (Patient 9) was withdrawn from the study due to progressive decline in her neurological status. Three weeks into the study period, she became stuporous and nonresponsive to questions or commands with frequent witnessed seizures. She was taken to the local emergency department. MRI of the brain demonstrated bilateral limbic encephalitis. Due to significant clinical deterioration, she was unblinded and found to be in the placebo arm.
Two falls without serious injuries were reported during the blinded phase of the study; one in a CASPR2‐IgG–seropositive patient receiving placebo and another in a LGI1‐IgG–seropositive patient receiving IVIG, likely secondary to an FBDS. No other drug‐related adverse effects were reported during the blinded phase of the study.
During the open label phase, 1 patient developed mild to moderate headaches attributed to IVIG infusion, and another patient developed generalized body rash. The skin rash persisted even after completion of the IVIG regimen, and was later attributed to levetiracetam.
Immunotherapy after Blinded/Open Label IVIG
All patients were recommended to continue immunotherapy following completion of the trial period (see Supplementary Table 2). Neurology assessment follow‐up details were available on 15 cases. Among these, 3 patients had achieved seizure freedom prior to clinical trial completion. Nine of the remaining 12 patients received high‐dose intravenous corticosteroids along with additional immunotherapies (mycophenolate mofetil [n = 2], plasmapheresis [n = 1], oral prednisone [n = 5], IVIG every 2 weeks [n = 2]) and AEDs. Seizure freedom was achieved in 56% of cases (n = 5/9; see Supplementary Table 2). The median duration from intravenous corticosteroids infusion to seizure freedom was 1 month (range = 0.5–4 months). One patient who received intravenous methylprednisolone (IVMP; 1g daily for 5 days) followed by oral prednisone 60mg daily developed acute psychosis and hyperglycemia (measured serum glucose = 600mg/dl). He was admitted to a hospital, and corticosteroids were held. Subsequently, he was treated with rituximab (1,000mg, 2 infusions 2 weeks apart), IVIG (0.4g/kg every week for 12 weeks), plasmapheresis (5 sessions), and azathioprine (2mg/kg/day), leading to seizure freedom. Two patients achieved seizure freedom following treatment with IVIG (1g/kg every 2 weeks for 12 weeks) and mycophenolate mofetil (1,000mg twice daily), and did not receive corticosteroids. The patient in the placebo arm of the study who was unblinded at 3 weeks due to clinical deterioration received rituximab (1,000mg) and IVIG (2g/kg over 5 days) with partial clinical improvement (more conscious and communicative).
Discussion
This study represents the first double‐blind randomized placebo‐controlled clinical trial of any immunotherapy in antibody‐mediated autoimmune epilepsy. The data support the use of IVIG and present the first placebo data in this condition. Due to limited enrollment and early termination, the study did not reach its statistically based sample size and was underpowered. Nevertheless, among the randomized patients, administration of IVIG was associated with favorable responder rate, especially among patients with LGI1‐IgG, compared to placebo. Furthermore, LGI1‐IgG seropositive patients who received IVIG also had a significant association with stabilization/improvement of RBANS cognitive scores. The efficacy of IVIG was further supported by the open label arm of the study, in which the majority of cases demonstrated ≥50% reduction in seizure frequency following 6 weeks of IVIG. Additionally, in the setting of autoimmune epilepsy, IVIG administration showed a promising safety profile compared with the frequent side effects seen with AEDs in these patients.14, 17
This study also highlights the morbidity associated with autoimmune epilepsies, with the majority of patients (11/17) reporting >10 seizures daily. High seizure frequency and coexisting cognitive deficits limited precise seizure counting in some patients.30, 31 However, corroborative history during clinical visits by the nonresponders and their family members denoting similar or increased seizure frequency estimates during follow‐up clinic evaluation helped with determination of efficacy.
A potential limitation of the study is that the median duration from symptom onset to enrollment was 3 months longer for the placebo group compared to the IVIG group (see Table 1); however, this time the difference was not found to be statistically significant.
Despite the significant improvement in seizure frequency associated with IVIG, only 2 patients achieved seizure freedom at the end of the blinded phase of the study. Additionally, 63% (5/8) of the patients continued to have frequent (≥5) daily seizures. High‐dose intravenous corticosteroid infusion was initiated for 9 of 12 patients following completion of blinded or open label IVIG infusion, with the majority attaining seizure freedom (see Supplementary Table 2). This modest effect appears to contrast with the more dramatic effect in patients treated with steroids as a consistent medication, often alongside plasma exchange/IVIG.7, 17
Prior studies have reported limited efficacy of IVIG in MuSK IgG myasthenia32 and neurofascin 155 IgG–associated chronic inflammatory demyelinating polyradiculoneuropathy, both IgG4‐dominant diseases.33 Patients with LGI1‐ and CASPR2‐IgG both show an IgG4 predominance,7, 34 and the IgG1:4 ratio has been reported to correlate with clinical outcomes in LGI1‐antibody–associated diseases.7, 13 Therefore, we evaluated the proportion of IgG1–4 subclasses (see Supplementary Table 2). All the LGI1‐ or CASPR2‐IgG–seropositive cases were IgG4 subclass predominant, including the 2 LGI1‐IgG–seropositive patients who did not have a favorable response to IVIG during the blinded phase. Interestingly, 2 LGI1‐IgG–seropositive patients receiving IVIG who achieved seizure freedom during the blinded phase of the study had undetectable LGI1‐IgG1 antibodies. Therefore, at least among patients with LGI1/CASPR2 autoimmune epilepsy, the IgG4 predominance does not appear to limit IVIG efficacy.
Consistent with their autoantibody profiles and phenotypes,14 patients carried the established HLA associations with LGI1‐IgG and CASPR2‐IgG seropositivity. LGI1‐IgG or CASPR2‐IgG patients with variable responses to IVIG and different seizure semiologies did not have variability in HLA‐DRB geneotypes, suggesting that HLA association does not distinguish between outcomes or subphenotypes.
This study is especially important for patients in whom corticosteroids are relatively contraindicated, or for those refractory to corticosteroid therapy. Additionally, for management of some cases with high neurological morbidity, coadministration of both IVIG and IVMP, or addition of plasma exchange, may be considered.
In this randomized control trial, we limited our assessment to patients with epilepsy associated with LGI1/CASPR2 autoimmunity. Patients with other clinical presentations of LGI1/CASPR2‐IgG associated autoimmunity such as cognitive impairment and/or peripheral hyperexcitability syndromes without coexisting seizures were excluded. Therefore, efficacy of IVIG among LGI1‐ or CASPR2‐IgG seropositive patients with inclusion of a broader clinical spectrum may need to be analyzed in the future. Nearly all patients in our study had not received any other immunotherapy prior to enrollment in the clinical trial. Future studies should also compare the safety profile and efficacy of IVIG to other first‐line immunotherapies such as high‐dose corticosteroids or plasma exchange. Its efficacy as an add‐on therapy to corticosteroids or second‐line agents should also be evaluated. Furthermore, utility of scheduled or monthly IVIG to prevent disease relapse should be assessed in a long‐term study.
These data support consideration of IVIG as a therapeutic option in the acute management of autoimmune epilepsy due to LGI1‐IgG, and provide preliminary data supportive of the development of larger, multicenter randomized controlled studies to evaluate this rare treatable condition.
Author Contributions
S.J.P., J.B., and A.M. contributed to concept and design of the study. All authors contributed to acquisition and analysis of the data. D.D., M.D., and S.J.P. contributed to drafting the text and preparing the figures. All authors contributed to critical revision of the manuscript.
Potential Conflicts of Interest
D.D. and J.B. have received research support from Grifols (the company manufactures a drug used in the study). A.M. has consulted for Grifols. S.R.I. is a coapplicant and receives royalties on patent application WO/2010/046716 (UK patent PCT/GB2009/051441) entitled "Neurological Autoimmune Disorders"; the patent has been licensed to EUROIMMUN for the development of assays for LGI1 and other VGKC‐complex antibodies. S.J.P. reports grants from Grifols. The other authors have nothing to report.
Supporting information
Supplementary Table 1: Inclusion and exclusion criteria
Supplementary Table 2: Seizure semiology, frequencies and immunotherapies utilized.
Acknowledgment
This study was supported by Grifols Shared Services North America, Option Care, and the Mayo Clinic Center for Multiple Sclerosis and Autoimmune Neurology. Patient travel and accommodation expenses were supported by a grant from the Autoimmune Encephalitis Alliance. M.R. is supported by the Austrian Science Fund (FWF J4157‐B30). S.R.I. is supported by the Wellcome Trust (104079/Z/14/Z), the British Medical Association research grants Vera Down grant (2013) and Margaret Temple grant (2017), Epilepsy Research UK (P1201), and the Encephalitis Society. The research was funded/supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (the views expressed are those of the authors and not necessarily those of the National Health Service, the NIHR, or the Department of Health). The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication.
We thank G. Ayer for methodological guidance and continued support for the study; E. Polley, S. Bryant, and M. Masoud for statistical support; all the physicians who kindly referred their patients for inclusion in the study; and the patients and their families for participation in this study.
References
- 1. Bien CG, Elger CE. Limbic encephalitis: a cause of temporal lobe epilepsy with onset in adult life. Epilepsy Behav 2007;10:529–538. [DOI] [PubMed] [Google Scholar]
- 2. Dubey D, Alqallaf A, Hays R, et al. Neurological autoantibody prevalence in epilepsy of unknown etiology. JAMA Neurol 2017;74:397–402. [DOI] [PubMed] [Google Scholar]
- 3. Dubey D, Singh J, Britton JW, et al. Predictive models in the diagnosis and treatment of autoimmune epilepsy. Epilepsia 2017;58:1181–1189. [DOI] [PubMed] [Google Scholar]
- 4. Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody‐associated encephalopathy: a potentially immunotherapy‐responsive form of limbic encephalitis. Brain 2004;127:701–712. [DOI] [PubMed] [Google Scholar]
- 5. Thieben MJ, Lennon VA, Boeve BF, et al. Potentially reversible autoimmune limbic encephalitis with neuronal potassium channel antibody. Neurology 2004;62:1177–1182. [DOI] [PubMed] [Google Scholar]
- 6. Barber PA, Anderson NE, Vincent A. Morvan's syndrome associated with voltage‐gated K+ channel antibodies. Neurology 2000;54:771–772. [DOI] [PubMed] [Google Scholar]
- 7. Thompson J, Bi M, Murchison AG, et al. The importance of early immunotherapy in patients with faciobrachial dystonic seizures. Brain 2018;141:348–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lai M, Huijbers MG, Lancaster E, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 2010;9:776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lancaster E, Huijbers MG, Bar V, et al. Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol 2011;69:303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van Sonderen A, Petit‐Pedrol M, Dalmau J, Titulaer MJ. The value of LGI1, Caspr2 and voltage‐gated potassium channel antibodies in encephalitis. Nat Rev Neurol 2017;13:290–301. [DOI] [PubMed] [Google Scholar]
- 11. Lang B, Makuch M, Moloney T, et al. Intracellular and non‐neuronal targets of voltage‐gated potassium channel complex antibodies. J Neurol Neurosurg Psychiatry 2017;88:353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel‐complex proteins leucine‐rich, glioma inactivated 1 protein and contactin‐associated protein‐2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain 2010;133:2734–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gadoth A, Pittock SJ, Dubey D, et al. Expanded phenotypes and outcomes among 256 LGI1/CASPR2‐IgG‐positive patients. Ann Neurol 2017;82:79–92. [DOI] [PubMed] [Google Scholar]
- 14. Binks S, Varley J, Lee W, et al. Distinct HLA associations of LGI1 and CASPR2‐antibody diseases. Brain 2018;141:2263–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Binks SNM, Klein CJ, Waters P, et al. LGI1, CASPR2 and related antibodies: a molecular evolution of the phenotypes. J Neurol Neurosurg Psychiatry 2018;89:526–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Toledano M, Britton JW, McKeon A, et al. Utility of an immunotherapy trial in evaluating patients with presumed autoimmune epilepsy. Neurology 2014;82:1578–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011;69:892–900. [DOI] [PubMed] [Google Scholar]
- 18. de Bruijn M, van Sonderen A, van Coevorden‐Hameete MH, et al. Evaluation of seizure treatment in anti‐LGI1, anti‐NMDAR, and anti‐GABABR encephalitis. Neurology 2019;92:e2185–e2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Irani SR, Stagg CJ, Schott JM, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain 2013;136:3151–3162. [DOI] [PubMed] [Google Scholar]
- 20. Dubey D, Kothapalli N, McKeon A, et al. Predictors of neural‐specific autoantibodies and immunotherapy response in patients with cognitive dysfunction. J Neuroimmunol 2018;323:62–72. [DOI] [PubMed] [Google Scholar]
- 21. Gadoth A, Zekeridou A, Klein CJ, et al. Elevated LGI1‐IgG CSF index predicts worse neurological outcome. Ann Clin Transl Neurol 2018;5:646–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shin YW, Lee ST, Shin JW, et al. VGKC‐complex/LGI1‐antibody encephalitis: clinical manifestations and response to immunotherapy. J Neuroimmunol 2013;265:75–81. [DOI] [PubMed] [Google Scholar]
- 23. van Sonderen A, Thijs RD, Coenders EC, et al. Anti‐LGI1 encephalitis: clinical syndrome and long‐term follow‐up. Neurology 2016;87:1449–1456. [DOI] [PubMed] [Google Scholar]
- 24. Finke C, Pruss H, Heine J, et al. Evaluation of cognitive deficits and structural hippocampal damage in encephalitis with leucine‐rich, glioma‐inactivated 1 antibodies. JAMA Neurol 2017;74:50–59. [DOI] [PubMed] [Google Scholar]
- 25. Nantes JC, Thomas AG, Voets NL, et al. Hippocampal functional dynamics are clinically implicated in autoimmune encephalitis with faciobrachial dystonic seizures. Front Neurol 2018;9:736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dubey D, Pittock SJ, Kelly CR, et al. Autoimmune encephalitis epidemiology and a comparison to infectious encephalitis. Ann Neurol 2018;83:166–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Szots M, Marton A, Kover F, et al. Natural course of LGI1 encephalitis: 3‐5 years of follow‐up without immunotherapy. J Neurol Sci 2014;343:198–202. [DOI] [PubMed] [Google Scholar]
- 28. Klein CJ, Lennon VA, Aston PA, et al. Insights from LGI1 and CASPR2 potassium channel complex autoantibody subtyping. JAMA Neurol 2013;70:229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Randolph C, Tierney MC, Mohr E, Chase TN. The Repeatable Battery for the Assessment of Neuropsychological Status (RBANS): preliminary clinical validity. J Clin Exp Neuropsychol 1998;20:310–319. [DOI] [PubMed] [Google Scholar]
- 30. Hoppe C, Poepel A, Elger CE. Epilepsy: accuracy of patient seizure counts. Arch Neurol 2007;64:1595–1599. [DOI] [PubMed] [Google Scholar]
- 31. Fisher RS, Blum DE, DiVentura B, et al. Seizure diaries for clinical research and practice: limitations and future prospects. Epilepsy Behav 2012;24:304–310. [DOI] [PubMed] [Google Scholar]
- 32. Pasnoor M, Wolfe GI, Nations S, et al. Clinical findings in MuSK‐antibody positive myasthenia gravis: a U.S. experience. Muscle Nerve 2010;41:370–374. [DOI] [PubMed] [Google Scholar]
- 33. Querol L, Nogales‐Gadea G, Rojas‐Garcia R, et al. Neurofascin IgG4 antibodies in CIDP associate with disabling tremor and poor response to IVIg. Neurology 2014;82:879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. van Sonderen A, Arino H, Petit‐Pedrol M, et al. The clinical spectrum of Caspr2 antibody‐associated disease. Neurology 2016;87:521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: Inclusion and exclusion criteria
Supplementary Table 2: Seizure semiology, frequencies and immunotherapies utilized.