

Abstract
Active secretion of bile salts into the canalicular lumen drives bile formation and promotes biliary cholesterol and phospholipid output. Disrupting hepatic bile salt uptake, by inhibition of sodium‐taurocholate cotransporting polypetide (NTCP; Slc10a1) with Myrcludex B, is expected to limit bile salt flux through the liver and thereby to decrease biliary lipid excretion. Here, we show that Myrcludex B–mediated NTCP inhibition actually causes an increase in biliary cholesterol and phospholipid excretion whereas biliary bile salt output and bile salt composition remains unchanged. Increased lysosomal discharge into bile was excluded as a potential contributor to increased biliary lipid secretion. Induction of cholesterol secretion was not a consequence of increased ATP‐binding cassette subfamily G member 5/8 activity given that NTCP inhibition still promoted cholesterol excretion in Abcg8 −/− mice. Stimulatory effects of NTCP inhibition were maintained in Sr‐b1 −/− mice, eliminating the possibility that the increase in biliary lipids was derived from enhanced uptake of high‐density lipoprotein–derived lipids. NTCP inhibition shifts bile salt uptake, which is generally more periportally restricted, toward pericentral hepatocytes, as was visualized using a fluorescently labeled conjugated bile salt. As a consequence, exposure of the canalicular membrane to bile salts was increased, allowing for more cholesterol and phospholipid molecules to be excreted per bile salt. Conclusion: NTCP inhibition increases biliary lipid secretion, which is independent of alterations in bile salt output, biliary bile salt hydrophobicity, or increased activity of dedicated cholesterol and phospholipid transporters. Instead, NTCP inhibition shifts hepatic bile salt uptake from mainly periportal hepatocytes toward pericentral hepatocytes, thereby increasing exposure of the canalicular membrane to bile salts linking to increased biliary cholesterol secretion. This process provides an additional level of control to biliary cholesterol and phospholipid secretion.
Abbreviations
- ABCB4
ATP‐binding cassette subfamily B member 4
- ABCG5/8
ATP‐binding cassette subfamily G member 5/8
- ATP11C
ATPase phospholipid transporting 11C
- BW
body weight
- HBV
hepatitis B virus
- HDL
high‐density lipoprotein
- hOATP1B1
human OATP1B1
- NTCP
sodium‐taurocholate cotransporting polypeptide
- OATP
organic anion transporting protein
- SR‐B1
scavenger receptor class B type 1
- TC
taurocholate
- WT
wild type
Bile formation is driven by active secretion of bile salts from hepatocytes into the canalicular lumen by the bile salt export pump.1 Biliary bile salts can be derived from de novo synthesis in the liver or from reuptake of intestine‐derived bile salts from the portal circulation, a process that is mediated primarily by the sodium‐taurocholate cotransporting polypeptide (NTCP) and organic anion transporting proteins (OATPs).2 Hepatobiliary secretion of bile salts promotes biliary phospholipid and cholesterol output,3 and changes in bile salt secretion are therefore usually paralleled by similar changes in biliary phospholipid and cholesterol output.4 The latter provides an important elimination route for excess body cholesterol.3
Recently, it has been shown that NTCP is the entry receptor for the hepatitis B virus (HBV), which has led to the development of drugs targeting NTCP.5, 6 The drug Myrcludex B binds selectively to NTCP, thereby inhibiting HBV entry into hepatocytes, and it is currently being tested as treatment for chronic HBV/hepatitis delta virus infection in a phase 2 trial.6, 7 NTCP inhibition by Myrcludex B has also been demonstrated to inhibit the physiological function of NTCP, namely bile salt transport.8 As such, Myrcludex B is a potent tool to investigate the physiological consequences of interrupting enterohepatic cycling of bile salts by targeting hepatic bile salt (re)uptake. Previously, we have already shown that NTCP inhibition by Myrcludex B reduces cholestatic liver injury in mice by lowering the bile salt load on the liver and altering the phospholipid to bile salt ratio in bile, thereby reducing bile salt toxicity.9
In this study, we set out to evaluate whether NTCP inhibition by Myrcludex B specifically affects the bile formation process and hepatobiliary output of major bile components in healthy, noncholestatic mice. Surprisingly, NTCP inhibition increased biliary phospholipid and cholesterol output, whereas output of endogenous bile salts remained unaffected. Upon exclusion of known parameters that could explain this remarkable increase of phospholipid and cholesterol secretion relative to that of bile salts, we propose that NTCP inhibition leads to a shift in hepatic bile salt uptake from periportal to pericentral zones of the liver. This shift in zonated bile salt uptake enforces bile‐salt–induced biliary lipid secretion by prolonging exposure of the bile canalicular membrane domain to the stimulatory actions of bile salts.
Materials and Methods
Animal Experiments
Wild‐type (WT) and Abcg8 −/− mice (C57BL/6J background) were bred and housed in the University Medical Center Groningen, Groningen. Sr‐bI −/− mice (C57BL/6J background) were bred and housed in the Gorlaeus Laboratories of the Leiden Academic Center for Drug Research, Leiden. ATPase phospholipid transporting 11C (Atp11c)‐deficient mice (kindly provided by Drs. B. Beutler and O. Siggs)10 were bred and housed in the Amsterdam University Medical Centers, Amsterdam. The human OATP1B1 (hOATP1B1) Oatp1a/1b −/− mice were purchased from Taconic (Silkeborg, Denmark) and housed at the Amsterdam University Medical Centers. All mice used were male. Mice received single subcutaneous injections with Myrcludex B (2.5 µg/g body weight [BW]) or vehicle. One hour after Myrcludex B or vehicle administration, bile duct cannulation was performed using a PE‐10 catheter, as described in a previous work.11 Bile was collected in aliquots every 10 minutes for a total of 60 minutes (Sr‐bI −/− mice, WT C57BL/6J mice, and Abcg8 −/− mice). Taurocholate (TC) infusion and bile collection in Atp11c‐deficient mice was performed as described.12 Bile flow was determined gravimetrically assuming a density of 1 g/mL for bile. A heating pad maintained body temperature at 37°C. For visualization of bile salt uptake in vivo, WT C57BL/6J mice were purchased from Envigo (Venray, the Netherlands) and Ntcp −/− were bred and housed at the Amsterdam University Medical Centers. WT and Ntcp −/− mice (C57BL/6J) received 62.5 nmol of tauro‐nor‐hyocholic acid‐24‐DBD (GenoMembrane, Kanagawa, Japan) in a volume of 100 µL of NaCl 0.9% by the portal vein. WT mice were injected with vehicle or Myrcludex B (2.5 µg/g BW) before injection of the fluorescent bile salt. Five minutes after administration of tauro‐nor‐HCA‐24‐DBD, the liver was harvested. Mice were randomized to treatment using online randomization software, and investigators were blinded for treatments. Organs were snap‐frozen in liquid N2 and stored at –80°C for further analysis. The study design and all protocols for animal care and handling were approved by the Institutional Animal Care and Use Committee of the University of Amsterdam (WT C57BL/6J, hOATP1B1 Oatp1a/1b −/−, Atp11C −/−, and Ntcp −/−), Leiden University (Sr‐bI −/−), and the University of Groningen (WT C57BL/6J and Abcg8 −/−).
Microscopy and Imaging of Bile Salt Uptake
Livers of mice injected with tauro‐nor‐HCA‐24‐DBD were cut in sections of 6 μm on a Leica CM 1950 cryostat at −20°C. Sections were mounted on slides with Vectamount (H‐5000, Vector Laboratories, Burlingame, CA). Images were obtained with a Leica DM‐6000B microscope.
Western Blotting
Crude mouse liver membranes were isolated as described in a previous work.2 Proteins were transferred by wet blotting to polyvinylidene difluoride membrane and probed with antimouse scavenger receptor class B type 1 (SR‐b1; NB400‐104; Novus Biologicals, Centennial, CO), antimouse ABCG5 (generation described in a previous work13), or antimouse MDR1‐3 (C219; Invitrogen, Carlsbad, CA). Immune complexes were detected with a horseradish‐peroxidase–conjugated secondary antibody (Bio‐Rad, Hercules, CA), visualized using enhanced chemiluminescence detection reagent (Lumi‐light; Roche, Basel, Switzerland), and detected using ImageQuant LAS 4000 (GE Healthcare, Chicago, IL).
RNA Isolation and qPCR
Total RNA isolation and qPCR was performed as described.9
Quantification of Bile Salts by High‐Performance Liquid Chromatography
Concentrations of different bile salt species in plasma and bile were determined by reverse‐phase high‐performance liquid chromatography (HPLC) as described.2
Liver Lipid Isolation
Liver lipids were isolated using methanol/chloroform extraction as described.14
Assays
Bile salts, choline containing phospholipids, and cholesterol in bile were determined enzymatically as described.15 Free cholesterol and total cholesterol content of liver lipid isolations were determined using a cholesterol cholesterol oxidase/peroxidase aminophenazone assay according to the manufacturer’s instruction (catalogus nr. 80106 and 88656; BIOLABO, Maizy, France). Total β‐hexosaminidase activity in bile was determined using a 4‐methylumbelliferyl‐based substrate assay.16 Determination of changes in fluorescence was performed on a CLARIOstar analyzer (BMG‐labtech, Offenburg, Germany).
Statistical Analysis
Data are provided as the median and interquartile range or 95% confidence interval. Differences between groups were analyzed using the Mann‐Whitney U test, and statistical significance was considered when P < 0.05. To assess whether NTCP inhibition altered the relation between biliary bile salt output and biliary cholesterol or phospholipid output, linear regression analysis was performed on log‐transformed data; statistical significant differences were considered when either the slope or intercepts were determined to be nonequal with P < 0.05. Graph generation and statistical analysis were performed using GraphPad Prism (version 7.0; GraphPad Software Inc., La Jolla, CA).
Results
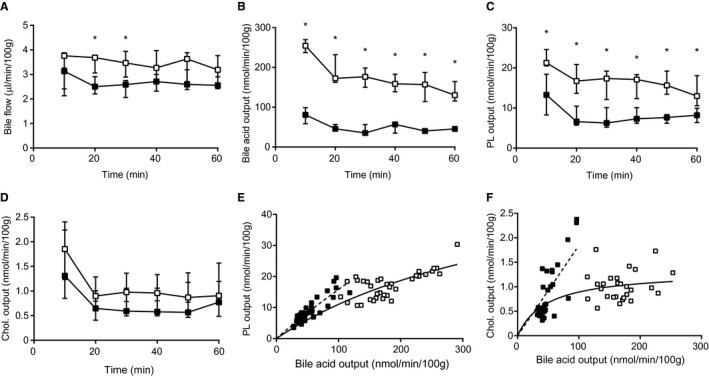
To investigate the effects of NTCP inhibition on bile formation and biliary bile salt, cholesterol, and phospholipid excretion, WT mice were injected with the NTCP inhibitor, Myrcludex B, or vehicle and bile was collected upon gallbladder cannulation. NTCP inhibition resulted in an increase in cholesterol output (Fig. 1A). The higher rate of biliary cholesterol excretion after NTCP inhibition was not accompanied by changes in biliary bile salt secretion (P < 0.0001; Fig. 1B,C), as tested by linear regression of log‐transformed data. Similar analysis of biliary phospholipid output indicated that NTCP inhibition also causes an increase in phospholipid output over bile salt output (P < 0.0001, Fig. 1D), even though absolute phospholipid output was not significantly increased (Supporting Fig. S1A). NTCP inhibition did not affect bile flow, plasma bile salt levels, or hepatic mRNA expression levels of small heterodimer partner and cytochrome P450 7a1 (Supporting Fig. S1B‐E). Altered bile composition after NTCP inhibition led to a higher molar ratio of cholesterol to bile salts, but did not increase cholesterol saturation of bile (calculated using the critical cholesterol saturation tables published previously17) given that NTCP inhibition also tended to increase the phospholipid to bile salt ratio (Supporting Fig. S1F‐H). The remarkable increase of biliary cholesterol and phospholipid output over biliary bile salt output prompted further investigation of the underlying mechanism.
Figure 1.
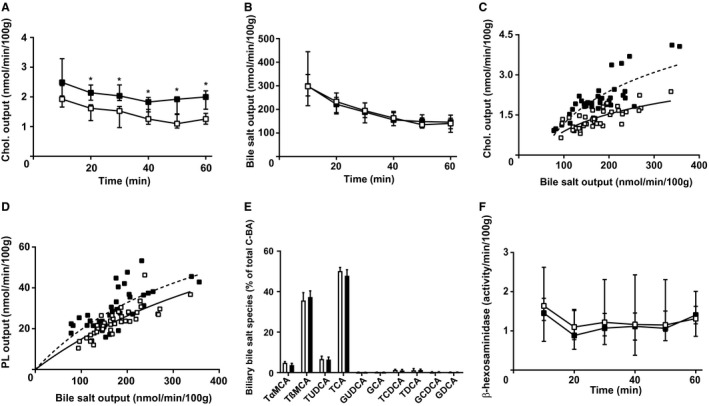
NTCP inhibition acutely increases biliary cholesterol and phospholipid secretion. Gallbladders of mice were cannulated 1 hour after treatment with vehicle or Myrcludex B; standardized 10‐minute bile samples were collected and data were plotted in time. (A) Total cholesterol and (B) bile salt output (in nmol/min/100 g BW). (C) Cholesterol output and (D) phospholipid output were plotted as a function of biliary bile salt output to assess whether this relation was changed by Myrcludex B administration (linear regression was performed on log‐transformed data and significance was assessed by comparing slopes or intercepts). (E) Biliary bile salt composition was quantified using HPLC, expressed as percentage of total conjugated bile acids in bile. (F) β‐hexosamindase activity in bile. Data are presented as median and interquartile range. White squares/bars indicate the vehicle group, and black squares/bars indicate the Myrcludex B group. Differences between groups were analyzed using the Mann‐Whitney U test. Asterisk (“*”) indicates P < 0.05; n = 8 mice/group. Abbreviations: C‐BA, conjugated bile acids; Chol., cholesterol; GCA, glycocholic acid; GCDCA, glycochenodeoxycholic acid; GDCA, glycodeoxycholic acid; GUDCA, glycoursodeoxycholic acid; PL, phospholipid; TCA, taurocholic acid; TCDCA, taurochenodeoxycholic acid; TDCA, taurodeoxycholic acid; TUDCA, tauroursodeoxycholic acid; TαMCA, tauro‐alpha‐muricholic acid; β‐MCA, beta‐muricholic acid; TβMCA, tauro‐beta‐muricholic acid.
A few processes have been described that either uncouple cholesterol and/or phospholipid output from biliary bile salt output or alter the relation between biliary bile salt and lipid output.4, 18, 19 A potential contributor is a change in biliary bile salt species. It has been described that hydrophobic bile salt species provoke more biliary lipid secretion than hydrophilic species.20 In the case of acute NTCP inhibition, however, no change in biliary bile salt composition was observed (Fig. 1E). A second potential contributor is an increased expulsion of lysosomal contents into bile, which is quite limited under normal physiological circumstances,21 upon Myrcludex B injection. It has been demonstrated that bile salts directly stimulate lysosomal discharge into bile.22 Yet, assessment of the lysosomal enzyme, β‐hexosaminodase, in bile indicated that NTCP inhibition did not affect lysosomal discharge into bile (Fig. 1F).
A third possible contributor to higher rates of cholesterol and phospholipid excretion is increased activity of dedicated cholesterol (ATP‐binding cassette subfamily G member 5/8; ABCG5/8) and phospholipid (ATP‐binding cassette subfamily B member 4; ABCB4) transporters.23, 24 NTCP inhibition did not increase mRNA or protein levels of the ABCG5/8 or ABCB4 transporter (Fig. 2A). However, the absence of changes in expression level does not exclude a change in activity of these transporters. To investigate whether ABCG5/8 serves as a mediator of the increase in cholesterol output upon NTCP inhibition, gallbladder cannulation experiments were performed in Abcg8‐deficient mice. Absence of either ABCG5 or ABCG8 turns the ABCG5/G8 complex fully dysfunctional and results in ~70% reduced cholesterol excretion.25 Administration of Myrcludex B to Abcg8 −/− mice did result in a clear increase in biliary cholesterol excretion (Fig. 2B). Therefore, NTCP inhibition mostly seems to affect ABCG5/8‐independent cholesterol secretion, although the absolute secretion rates were lower than those in WT mice (both in the absence and presence of Myrcludex B; Fig. 2B). Biliary phospholipid secretion was increased in Abcg8 −/− mice as well upon Myrcludex B injection (Fig. 2C) and the changes in phospholipid and cholesterol output occurred without any changes in biliary bile salt output (Fig. 2D‐F) or bile flow (Supporting Fig. S2A). Biliary phospholipid secretion relies completely on ABCB4 activity, and we previously showed that NTCP inhibition does not stimulate phospholipid or cholesterol secretion in Abcb4‐deficient mice, indicating that the induction of lipid secretion by Myrcludex B completely relies on ABCB4‐mediated phospholipid translocation.9 Hence, these experiments demonstrate that induction of phospholipid and cholesterol secretion by Myrcludex B requires activity of ABCB4, but increased activity of this transporter is unlikely to drive this effect.
Figure 2.
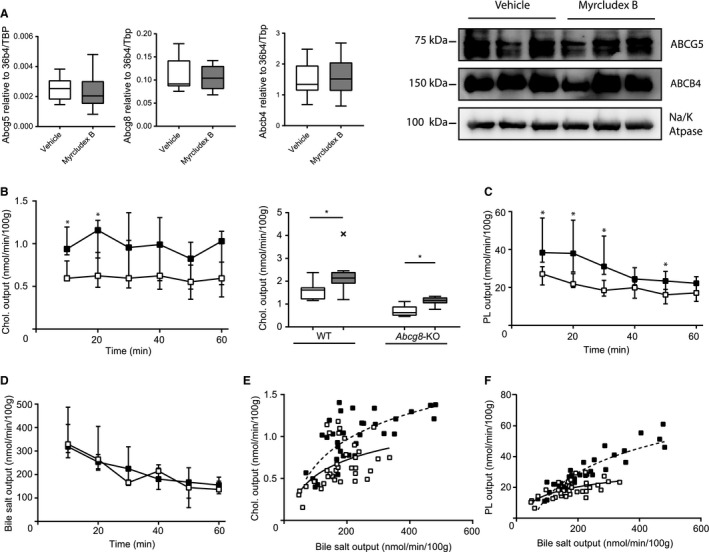
Increased biliary cholesterol secretion after NTCP inhibition is largely independent of ABCG5/8. (A) Hepatic mRNA and protein expression of Abcg5, Abcg8, and Abcb4 in WT mice. RNA expression is compared to the geometric mean of reference genes 36b4 and Tbp. Protein levels were compared to the sodium/potassium ATPase (n = 3/group). (B) Cholesterol output (in nmol/min/100 g BW) in Abcg8 −/− mice. One 10‐minute interval of biliary cholesterol output is compared between WT and Abcg8 −/− mice. (C) Phospholipid and (D) bile salt output into bile were determined in Abcg8 −/− mice. (E) Cholesterol output and (F) phospholipid output were plotted as a function of biliary bile salt output (linear regression was performed on log‐transformed data and significance was assessed by comparing slopes or intercepts). Data are presented as median and interquartile range. White squares/bars indicate the vehicle group, and black/gray squares/bars indicate the Myrcludex B group. Differences between groups were analyzed using the Mann‐Whitney U test. Asterisk (“*”) indicates P < 0.05; n = 8 mice/group. × Indicates statistical outlier in Tukey plot. Abbreviations: Chol., cholesterol; PL, phospholipid; Tbp, TATA‐box binding protein.
Next, we considered that an increased supply of cholesterol and phospholipid to the canalicular membrane may occur after NTCP inhibition, leading to increased biliary lipid output. It was previously postulated that biliary cholesterol excretion originates mainly from a (preexisting) hepatic pool of free cholesterol,26, 27 given that both microsomal acyl‐CoA:cholesterol acyltransferase (ACAT) activity and hepatic cholesterol ester concentration correlate in a reciprocal manner with biliary cholesterol output.28 The large increase in biliary cholesterol output observed upon treatment with diosgenin was suggested to be explained by this mechanism.28 However, it was later demonstrated that ACAT2 deficiency in mice does not lead to increased biliary cholesterol output.29 We analyzed hepatic esterified cholesterol content, but this was not affected by Myrcludex B treatment (Supporting Fig. S3A). Also, hepatic 3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A reductase mRNA levels were not affected by Myrcludex B treatment (Supporting Fig. S3B). Taken together, it does not seem likely that an increased supply of cholesterol to the canalicular membrane is derived from larger intracellular stores.
Enhanced uptake of cholesterol and phospholipids present in lipoproteins could theoretically stimulate both cholesterol and phospholipid excretion. It was demonstrated that especially high‐density lipoprotein (HDL)‐derived cholesterol ends up rapidly in bile in rodents.30 The uptake process is likely mediated by the scavenger receptor class B type 1 (SCARB1/SR‐B1), the main receptor for HDL. This receptor has also been shown to be able to mediate biliary cholesterol secretion independent of Abcg5/8.31, 32 In addition, liver‐specific overexpression of Sr‐bI in mice also stimulates biliary phospholipid output.31 To test the potential contribution of SR‐B1/HDL–derived lipids to the biliary phenotype of NTCP inhibition, Sr‐bI–deficient mice were injected with Myrcludex B or vehicle and gallbladder cannulation was performed. Myrcludex B induced an absolute increase in biliary cholesterol output of >50% in Sr‐bI −/− mice, but this did not reach statistical significance (Fig. 3A). The effects of NTCP inhibition on phospholipid secretion were clearly preserved in the Sr‐bI −/− mice (Fig. 3B) and bile salt output itself was not affected (Fig. 3C). Similar to WT and Abcg8 −/− mice, Myrcludex B treatment increased cholesterol and phospholipid output relative to bile salt output (P = 0.0002, Fig. 3D; P < 0.0001, Fig. 3D,E) and without changing bile flow (Supporting Fig. S4A). These experiments do not support the hypothesis that increased uptake of HDL‐derived cholesterol and phospholipids explains the increase in biliary cholesterol and phospholipid secretion upon NTCP inhibition. In accord with this conclusion is the absent change in expression of Sr‐bI in livers of Myrcludex B–treated WT mice (Fig. 3F).
Figure 3.
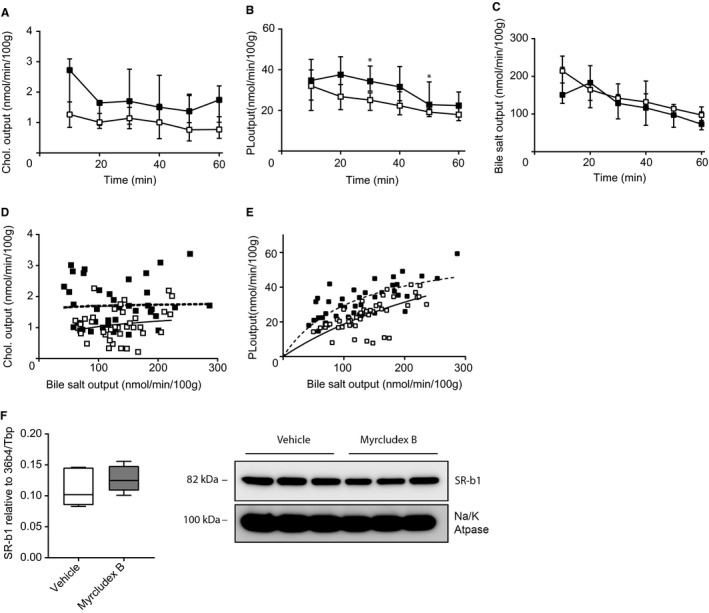
Increased biliary cholesterol and phospholipid secretion is not mediated by SR‐B1. (A) Cholesterol output, (B) phospholipid output, and (C) bile salt output into bile (in nmol/min/100 g BW) in Sr‐bI −/− mice. (D) Cholesterol output and (E) phospholipid output were plotted as a function of biliary bile salt (linear regression was performed on log‐transformed data and significance was assessed by comparing slopes or intercepts). (F) Hepatic mRNA and protein expression level of Sr‐bI in WT mice. RNA expression is compared to the geometric mean of reference genes 36b4 and Tbp. Protein levels were compared to the sodium/potassium ATPase (n = 3/group). Data are presented as median and interquartile range. White squares/bars indicate the vehicle group, and black/gray squares/bars indicate the Myrcludex B group. Differences between groups were analyzed using the Mann‐Whitney U test. Asterisk (“*”) indicates P < 0.05; n = 8 mice/group. Abbreviations: Chol., cholesterol; PL, phospholipid; Tbp, TATA‐box binding protein.
Given that neither of the investigated mechanisms provided an explanation for the findings of the current study, we propose a concept to explain why NTCP inhibition induces secretion of biliary lipids. We considered that blockade of NTCP induces a shift in bile salt uptake from mainly periportal to mainly pericentral hepatocytes. Bile salt uptake by hepatocytes displays a clear zonal distribution where periportal hepatocytes encounter the highest concentration of bile salts and contribute most to the clearance of intestine‐derived bile salts under physiological conditions33 (Fig. 4A,B). Administration of the fluorescent bile salt, tauro‐nor‐HCA‐24‐DBD, to vehicle‐treated WT mice demonstrates this zonal distribution, given that canalicularly located fluorescent bile salts are mainly restricted to the canaliculi around portal veins (Fig. 4B). Thus far, it has been described that only at high bile salt loads, for example, upon infusion with TC, pericentral hepatocytes also contribute to bile salt uptake.33 We hypothesized that upon NTCP inhibition, bile salts will reach pericentral hepatocytes also at low bile salt loads. During NTCP inhibition, hepatic bile salt uptake is maintained by OATP proteins and therefore biliary bile salt output is unaffected.2 OATP proteins are predominantly expressed in pericentral regions of the liver lobules,34 contributing to a shift in bile salt uptake from periportal to pericentral regions during NTCP inhibition. To visualize a shift in zonated bile salt uptake upon NTCP inhibition, we administered the fluorescent bile salt, tauro‐nor‐HCA‐24‐DBD, to Myrcludex B–treated WT mice and to Ntcp −/− mice. Of note, tauro‐nor‐HCA‐24‐DBD is transported by both NTCP and OATP proteins.35 In contrast to periportal distribution of the fluorescent bile salts in vehicle‐treated mice (Fig. 4B), cytoplasmic and canalicular distribution of bile salts in both Myrcludex B–treated WT mice and Ntcp −/− mice extended beyond the periportal zone toward the central zone (Fig. 4C,D). These experiments indicate that a significant portion of bile salt uptake during NTCP inhibition is mediated by more pericentral hepatocytes. We hypothesized that because of countercurrent bile flow, increased pericentral bile salt uptake will lead to a longer exposure, of a larger area of the canalicular membrane, to bile salts, allowing for higher biliary lipid secretion (Fig. 4C).
Figure 4.
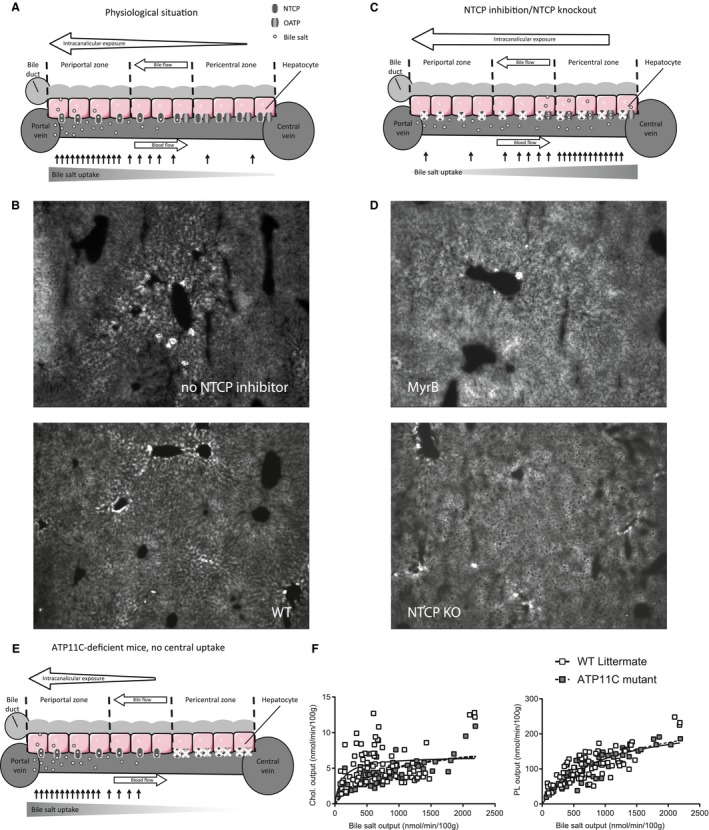
NTCP inhibition shifts hepatic bile salt uptake to pericentral hepatocytes, which links to higher biliary phospholipid and cholesterol secretion rates. (A) Graphic depicting exposure of the canalicular membrane to bile salts and zonated bile salt uptake in physiological situation. (B) Image of a liver of an WT mouse 5 minutes after injection with the fluorescent bile salt, tauro‐nor‐HCA‐24‐DBD. (C) Proposed mechanism of increased biliary lipid secretion after NTCP inhibition showing increased intracanalicular exposure to bile salts attributed increased pericentral bile salt uptake. (D) Images of livers of a Myrcludex B–treated WT mouse (top) and a Ntcp −/− mouse (bottom) 5 minutes after injection with the fluorescent bile salt, tauro‐nor‐HCA‐24‐DBD. (E) Graphic depicting zonated bile salt uptake and intracanalicular exposure in Atp11c‐deficient mice. (F) Cholesterol and phospholipid output as function of bile salt output in TC‐infused Atp11c‐deficient mice (linear regression was performed on log‐transformed data and significance was assessed by comparing slopes or intercepts). White squares indicate WT mice; gray squares indicate Atp11c‐deficient mice. Differences between groups were analyzed using the Mann‐Whitney U test. Asterisks (“*”) indicate P < 0.05; n = 5‐7 WT versus Atp11c. Abbreviations: Chol., cholesterol; KO, knockout; MyrB, Myrcludex B; PL, phospholipid; tauro‐nor‐HCA‐24‐DBD, N‐(24‐(7‐(4‐N,N‐dimethylaminosulfonyl‐2,1,3‐benzoxadiazole))amino‐3alpha,7alpha,12alpha‐trihydroxy‐27‐nor‐5beta‐cholestan‐26‐oyl)‐2′‐aminoethanesulfonate.
To assess whether altered zonated hepatic bile salt uptake can contribute to an increase in biliary lipid secretion, we analyzed the relation between biliary cholesterol and bile salt secretion in two models with zonation‐related phenotypes. First, we reassessed a TC‐infusion experiment, which was previously performed in WT and Atp11c‐deficient mice.12 Atp11c‐deficient mice are a model with a zonation difference in bile salt uptake given that basolateral bile salt uptake transporters are absent in the pericentral region restricting bile salt uptake to periportal regions, where NTCP expression is maintained in the periportal hepatocyte (Fig. 4E).12, 36 In this experiment, the relation between biliary cholesterol and bile salt levels was not significantly different between Atp11c‐deficient mice and WT littermates under conditions where the periportal region predominates uptake (before TC infusion). However, infusion of TC revealed an attenuated increase in cholesterol (P = 0.0023; but not phospholipid) secretion (Fig. 4F) in Atp11c‐deficient mice compared to control littermates, whereas bile salt output was similar. These results suggest that if pericentral hepatocytes become involved in hepatic bile salt uptake, more cholesterol may be secreted into bile, even at similar bile salt output rates.
Second, we assessed the consequences of Myrcludex B administration on biliary output of bile salts and lipids in a model where biliary bile salt excretion is largely limited to pericentral hepatocytes upon NTCP inhibition. For this purpose, hOATP1B1 Oatp1a/1b −/− mice were used. In these mice, the murine Oatp1a/1b‐class proteins are lacking and bile salt uptake is completely dependent upon NTCP.11 The human OATP1B1 protein was introduced into these mice and facilitates the uptake of Oatp1a/1b substrates such as bilirubin and steroid hormones.37 The human OATP1B1 transports bile salts, but only to a very limited extent.2 In the hOATP1B1 Oatp1a/1b −/− mice, bile flow, bile salt output, and phospholipid output were dramatically reduced after Myrcludex B administration (Fig. 5A‐C). Cholesterol output, on the other hand, was maintained after Myrcludex B administration (Fig. 5D). Myrcludex B treatment increased the proportional secretion of both cholesterol and phospholipid relative to bile salt output (P = 0.0154, Fig. 5E; P = 0.0039, Fig. 5F). When NTCP is inhibited and Oatp‐mediated uptake is virtually absent, hepatic bile salt uptake is essentially abolished and only newly synthesized bile salts are secreted into the biliary tree. Bile salt synthesis occurs primarily in central hepatocytes.38 Given that the bile salts are secreted by the central hepatocytes, the canalicular membrane of hepatocytes is exposed for a longer period of time to these bile salts, allowing for extraction of more cholesterol per amount of bile salt secreted. The results of these two models are in line with the proposed stimulatory role of shifting biliary bile salts from the periportal to the pericentral area on biliary cholesterol secretion.
Figure 5.
Total inhibition bile salt uptake reduces biliary bile salt and phospholipid output, but maintains biliary cholesterol output. (A) Bile flow, (B) biliary bile salt output, (C) biliary phospholipid output, and (D) biliary cholesterol output into bile (in nmol/min/100 g BW) in hOATP1B1 Oatp1a/1b −/− mice. (E) Cholesterol output and (F) phospholipid output were plotted as a function of biliary bile salt (linear regression was performed on log transformed data and significance was assessed by comparing slopes or intercepts). Data are presented as median and interquartile range. White squares/bars indicate the vehicle group, and black/gray squares/bars indicate the Myrcludex B group. Differences between groups were analyzed using the Mann‐Whitney U test. Asterisk (“*”) indicates P < 0.05; n = 8 mice/group. Abbreviations: Chol., cholesterol; PL, phospholipid.
Discussion
Taken together, we demonstrated that NTCP inhibition induces a greater contribution of pericentral hepatocytes to total hepatic bile salt uptake, which links to the observed increase in biliary cholesterol secretion. We postulate that the increase in pericentral bile salt processing, combined with the countercurrent bile flow, increases the exposure (time and area) of the canalicular membrane to intracanalicular bile salts (Fig. 4D). The potential importance of exposure time was already proposed by Verkade et al., who suggested that prolonged exposure of canalicular membranes to intracanalicular bile salts, attributed to reduced bile‐salt–independent bile flow, allows for more phospholipids and cholesterol molecules to be excreted per bile salt molecule.39 Our study suggests that increased pericentral secretion of bile salts plays a similar role in increasing exposure.
In conclusion, we discovered that NTCP inhibition in mice induces biliary cholesterol and phospholipid excretion, without affecting biliary bile salt output rates. Changes in bile salt hydrophobicity, lysosomal discharge, increased expression of transporter proteins, or stimulated uptake of plasma lipids were excluded as potential contributors to the increased biliary lipid output. Instead, a shift of bile salt uptake from periportal toward pericentral hepatocytes is the most likely the driving force for the increase in biliary cholesterol and phospholipid output. As such, the current study provides a view on the concept of bile‐salt–driven biliary lipid secretion, showing that location of hepatic bile salt uptake is an important contributing factor.
Supporting information
Acknowledgment
The authors thank Dr. Dirk Rudi de Waart for assistance with bile acid measurements in perfusion studies and Vincent W. Bloks for statistical advice.
S.F.J.vdG. is supported by the Netherlands Organization for Scientific Research (VIDI 91713319) and the European Research Council (Starting grant 337479).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Gerloff T, Stieger B, Hagenbuch B, Madon J, Landmann L, Roth J, et al. The sister of P‐glycoprotein represents the canalicular bile salt export pump of mammalian liver. J Biol Chem 1998;273:10046‐10050. [DOI] [PubMed] [Google Scholar]
- 2. Slijepcevic D, Roscam Abbing RLP, Katafuchi T, Blank A, Donkers JM, van Hoppe S, et al. Hepatic uptake of conjugated bile acids is mediated by both NTCP and OATPs and modulated by intestinal sensing of plasma bile acid levels in mice. Hepatology 2017;66:1631‐1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Northfield TC, Hofmann AF. Biliary lipid output during three meals and an overnight fast. I. Relationship to bile acid pool size and cholesterol saturation of bile in gallstone and control subjects. Gut 1975;16:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Coleman R, Rahman K. Lipid flow in bile formation. Biochim Biophys Acta 1992;1125:113‐133. [DOI] [PubMed] [Google Scholar]
- 5. Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012;1:e00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lempp FA, Urban S. Inhibitors of hepatitis B virus attachment and entry. Intervirology 2014;57:151‐157. [DOI] [PubMed] [Google Scholar]
- 7. Bogomolov P, Alexandrov A, Voronkova N, Macievich M, Kokina K, Petrachenkova M, et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: first results of a phase Ib/IIa study. J Hepatol 2016;65:490‐498. [DOI] [PubMed] [Google Scholar]
- 8. Yan H, Peng B, Liu Y, Xu G, He W, Ren B, et al. Viral entry of hepatitis B and D viruses and bile salts transportation share common molecular determinants on sodium taurocholate cotransporting polypeptide. J Virol 2014;88:3273‐3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Slijepcevic D, Roscam Abbing RLP, Fuchs CD, Haazen LCM, Beuers U, Trauner M, et al. Na(+) ‐taurocholate cotransporting polypeptide inhibition has hepatoprotective effects in cholestasis in mice. Hepatology 2018;68:1057‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Siggs OM, Arnold CN, Huber C, Pirie E, Xia Y, Lin P, et al. The P4‐type ATPase ATP11C is essential for B lymphopoiesis in adult bone marrow. Nat Immunol 2011;12:434‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Slijepcevic D, Kaufman C, Wichers CG, Gilglioni EH, Lempp FA, Duijst S, et al. Impaired uptake of conjugated bile acids and hepatitis b virus pres1‐binding in na(+) ‐taurocholate cotransporting polypeptide knockout mice. Hepatology 2015;62:207‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Waart DR, Naik J, Utsunomiya KS, Duijst S, Ho‐Mok K, Bolier AR, et al. ATP11C targets basolateral bile salt transporter proteins in mouse central hepatocytes. Hepatology 2016;64:161‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kosters A, Frijters RJ, Schaap FG, Vink E, Plosch T, Ottenhoff R, et al. Relation between hepatic expression of ATP‐binding cassette transporters G5 and G8 and biliary cholesterol secretion in mice. J Hepatol 2003;38:710‐716. [DOI] [PubMed] [Google Scholar]
- 14. Srivastava NK, Pradhan S, Mittal B, Kumar R, Gowda GAN. An improved, single step standardized method of lipid extraction from human skeletal muscle tissue. Analytical Letters 2006;39:297‐315. [Google Scholar]
- 15. Paulusma CC, Groen A, Kunne C, Ho‐Mok KS, Spijkerboer AL, Rudi de Waart D, et al. Atp8b1 deficiency in mice reduces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile salt transport. Hepatology 2006;44:195‐204. [DOI] [PubMed] [Google Scholar]
- 16. Tropak MB, Reid SP, Guiral M, Withers SG, Mahuran D. Pharmacological enhancement of beta‐hexosaminidase activity in fibroblasts from adult Tay‐Sachs and Sandhoff Patients. J Biol Chem 2004;279:13478‐13487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Carey MC. Critical tables for calculating the cholesterol saturation of native bile. J Lipid Res 1978;19:945‐955. [PubMed] [Google Scholar]
- 18. Verkade HJ, Havinga R, Gerding A, Vonk RJ, Kuipers F. Mechanism of bile acid‐induced biliary lipid secretion in the rat: effect of conjugated bilirubin. Am J Physiol 1993;264:G462‐G469. [DOI] [PubMed] [Google Scholar]
- 19. Verkade HJ, Wolbers MJ, Havinga R, Uges DR, Vonk RJ, Kuipers F. The uncoupling of biliary lipid from bile acid secretion by organic anions in the rat. Gastroenterology 1990;99:1485‐1492. [DOI] [PubMed] [Google Scholar]
- 20. Bilhartz LE, Dietschy JM. Bile salt hydrophobicity influences cholesterol recruitment from rat liver in vivo when cholesterol synthesis and lipoprotein uptake are constant. Gastroenterology 1988;95:771‐779. [DOI] [PubMed] [Google Scholar]
- 21. Rahman K, Coleman R. Effect of chloroquine on biliary lipid and lysosomal enzyme output in the isolated perfused rat liver at low bile salt output rates. Biochim Biophys Acta 1987;922:395‐397. [DOI] [PubMed] [Google Scholar]
- 22. LeSage GD, Robertson WE, Baumgart MA. Bile acid‐dependent vesicular transport of lysosomal enzymes into bile in the rat. Gastroenterology 1993;105:889‐900. [DOI] [PubMed] [Google Scholar]
- 23. Dikkers A, de Boer JF, Groen AK, Tietge UJ. Hepatic ABCG5/G8 overexpression substantially increases biliary cholesterol secretion but does not impact in vivo macrophage‐to‐feces RCT. Atherosclerosis 2015;243:402‐406. [DOI] [PubMed] [Google Scholar]
- 24. Smith AJ, de Vree JM, Ottenhoff R, Oude Elferink RP, Schinkel AH, Borst P. Hepatocyte‐specific expression of the human MDR3 P‐glycoprotein gene restores the biliary phosphatidylcholine excretion absent in Mdr2 (−/−) mice. Hepatology 1998;28:530‐536. [DOI] [PubMed] [Google Scholar]
- 25. Kosters A, Kunne C, Looije N, Patel SB, Oude Elferink RP, Groen AK. The mechanism of ABCG5/ABCG8 in biliary cholesterol secretion in mice. J Lipid Res 2006;47:1959‐1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Robins SJ, Brunengraber H. Origin of biliary cholesterol and lecithin in the rat: contribution of new synthesis and preformed hepatic stores. J Lipid Res 1982;23:604‐608. [PubMed] [Google Scholar]
- 27. Turley SD, Dietschy JM. Regulation of biliary cholesterol output in the rat: dissociation from the rate of hepatic cholesterol synthesis, the size of the hepatic cholesteryl ester pool, and the hepatic uptake of chylomicron cholesterol. J Lipid Res 1979;20:923‐934. [PubMed] [Google Scholar]
- 28. Nervi F, Bronfman M, Allalon W, Depiereux E, Del Pozo R. Regulation of biliary cholesterol secretion in the rat. Role of hepatic cholesterol esterification. J Clin Invest 1984;74:2226‐2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brown JM, Bell TA III, Alger HM, Sawyer JK, Smith TL, Kelley K, et al. Targeted depletion of hepatic ACAT2‐driven cholesterol esterification reveals a non‐biliary route for fecal neutral sterol loss. J Biol Chem 2008;283:10522‐10534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schwartz CC, Halloran LG, Vlahcevic ZR, Gregory DH, Swell L. Preferential utilization of free cholesterol from high‐density lipoproteins for biliary cholesterol secretion in man. Science 1978;200:62‐64. [DOI] [PubMed] [Google Scholar]
- 31. Wiersma H, Gatti A, Nijstad N, Oude Elferink RP, Kuipers F, Tietge UJ. Scavenger receptor class B type I mediates biliary cholesterol secretion independent of ATP‐binding cassette transporter g5/g8 in mice. Hepatology 2009;50:1263‐1272. [DOI] [PubMed] [Google Scholar]
- 32. Dikkers A, Freak de Boer J, Annema W, Groen AK, Tietge UJ. Scavenger receptor BI and ABCG5/G8 differentially impact biliary sterol secretion and reverse cholesterol transport in mice. Hepatology 2013;58:293‐303. [DOI] [PubMed] [Google Scholar]
- 33. Groothuis GM, Hardonk MJ, Keulemans KP, Nieuwenhuis P, Meijer DK. Autoradiographic and kinetic demonstration of acinar heterogeneity of taurocholate transport. Am J Physiol 1982;243:G455‐G462. [DOI] [PubMed] [Google Scholar]
- 34. Tachikawa M, Sumiyoshiya Y, Saigusa D, Sasaki K, Watanabe M, Uchida Y, Terasaki T. Liver zonation index of drug transporter and metabolizing enzyme protein expressions in mouse liver acinus. Drug Metab Dispos 2018;46:610‐618. [DOI] [PubMed] [Google Scholar]
- 35. De Bruyn T, Sempels W, Snoeys J, Holmstock N, Chatterjee S, Stieger B, et al. Confocal imaging with a fluorescent bile acid analogue closely mimicking hepatic taurocholate disposition. J Pharm Sci 2014;103:1872‐1881. [DOI] [PubMed] [Google Scholar]
- 36. Matsuzaka Y, Hayashi H, Kusuhara H. Impaired hepatic uptake by organic anion‐transporting polypeptides is associated with hyperbilirubinemia and hypercholanemia in Atp11c mutant mice. Mol Pharmacol 2015;88:1085‐1092. [DOI] [PubMed] [Google Scholar]
- 37. van de Steeg E, Wagenaar E, van der Kruijssen CM, Burggraaff JE, de Waart DR, Elferink RP, et al. Organic anion transporting polypeptide 1a/1b‐knockout mice provide insights into hepatic handling of bilirubin, bile acids, and drugs. J Clin Invest 2010;120:2942‐2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang J, Olin M, Rozell B, Bjorkhem I, Einarsson C, Eggertsen G, Gafvels M. Differential hepatocellular zonation pattern of cholesterol 7alpha‐hydroxylase (Cyp7a1) and sterol 12alpha‐hydroxylase (Cyp8b1) in the mouse. Histochem Cell Biol 2007;127:253‐261. [DOI] [PubMed] [Google Scholar]
- 39. Verkade HJ, Wolters H, Gerding A, Havinga R, Fidler V, Vonk RJ, Kuipers F. Mechanism of biliary lipid secretion in the rat: a role for bile acid‐independent bile flow? Hepatology 1993;17:1074‐1080. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials