

Abstract
Protozoal infections are still a global health problem, threatening the lives of millions of people around the world, mainly in impoverished tropical and sub‐tropical regions. Thus, in view of the lack of efficient therapies and increasing resistances against existing drugs, this study describes the antiprotozoal potential of synthetic cinnamate ester analogues and their structure‐activity relationships. In general, Leishmania donovani and Trypanosoma brucei were quite susceptible to the compounds in a structure‐dependent manner. Detailed analysis revealed a key role of the substitution pattern on the aromatic ring and a marked effect of the side chain on the activity against these two parasites. The high antileishmanial potency and remarkable selectivity of the nitro‐aromatic derivatives suggested them as promising candidates for further studies. On the other hand, the high in vitro potency of catechol‐type compounds against T. brucei could not be extrapolated to an in vivo mouse model.
Keywords: antiprotozoal agents, cinnamate esters, trypanosomiasis, leishmaniasis, structure-activity relationships
Fighting parasitic diseases: A classical phenotypic approach combined with a comprehensive SAR study led to establishment of potent and selective cinnamate ester analogues acting in vitro against Leishmania donovani and Trypanosoma brucei rhodesiense. Key structural requirements for activity were determined resulting in interesting candidates for further development.
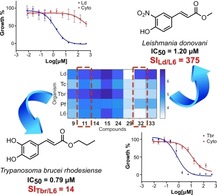
Introduction
Infectious diseases continuously represent a major worldwide human health problem, especially those occurring in tropical and sub‐tropical regions. Protozoan parasites are the causative agents of a diverse group of infectious diseases responsible for thousands of annual deaths. These diseases include malaria, sleeping sickness, Chagas disease and Leishmaniasis.1, 2 All of them are vector‐borne diseases with highest prevalence in low‐income countries. While malaria, caused by parasites of the genus Plasmodium and transmitted from host to host by mosquitos (Anopheles sp.), has received more attention in recent years resulting in a high number of relevant scientific publications, for the other three diseases bigger research efforts and investment in the development of new drugs are still due. Trypanosomiasis and Leishmaniasis hence remain on the list of the so‐called Neglected Tropical Diseases (NTDs) defined by the World Health Organization (WHO).3 Sleeping sickness (human African trypanosomiasis) and Chagas disease (American trypanosomiasis) are caused by Trypanosoma brucei and T. cruzi, respectively. The former is transmitted by tsetse flies, the latter by reduviid bugs. On the other hand, Leishmaniasis, caused by parasites of the genus Leishmania and transmitted by sand flies, has been reported as responsible for twice as many deaths as both Trypanosomiases together.1, 2, 4 Malaria infections caused around eighteen times more deaths than Leishmaniasis in the same period of time,1, 2 and thus, even though not on the list of NTDs, make it an urgent need, also in this case, to search for new active compounds for the development of efficient, safe and affordable new treatments.
Not only the complex biology of these organisms, but resistance to existing drugs and associated treatment failures are serious issues, making the search for new adequate cures a fundamental urge. The drugs currently used for the treatment of these diseases are not satisfactory due to poor/limited efficacy, increasing resistance, complex administration and major side effects. All of this makes the search for new adequate cures a challenge.
In this regard, cinnamate ester derivatives and analogues, according to various reports, have demonstrated attractive antiparasitic potential. In particular, caffeic acid bornyl esters have shown activity against L. major,5 whereas long chain esters of cinnamic acid possess activity against L. panamensis.6 Similarly, some cinnamic acid derivatives have demonstrated activity against L. donovani.7, 8 Isopentyl caffeic acid ester has been described as active against L. amazonensis,9 while caffeic acid phenethyl esters encapsulated into poly‐lactic‐co‐glycolic acid nanoparticles have demonstrated a positive effect against L. tropica.10 Additionally, antitrypanosomal,11, 12 antiplasmodial13, 14 and antischistosomal15 activity have also been described for related compounds. Therefore, a set of thirty‐four cinnamic acid ester analogues with variable aromatic substituents and ester groups was synthesized and tested in vitro against T. cruzi, T. brucei rhodesiense, L. donovani, and P. falciparum, in order to define their potential as antiprotozoal agents and to study the underlying structure‐activity relationships.
Results and Discussion
Chemistry
The set of cinnamate ester analogues in the present research included structural variations on the side chain and the aromatic ring. Based on these structural requirements, three different approaches were employed to obtain the esters through well‐known synthetic procedures previously described for similar compounds16, 17, 18, 19, 20, 21, 22 and outlined in Scheme 1. Fischer esterification of carboxylic acids I with primary and secondary alcohols16, 17, 18 was used to prepare a total of sixteen esters II (Scheme 1A; compounds 1–16, Figure 1). A second pathway consisted of Heck reaction of conveniently substituted bromo‐derivatives III with tert‐butyl acrylate (IV)19 to afford a set of seven tert‐butyl esters V (Scheme 1B; compounds 17–23, Figure 1). Activation of Meldrum's acid (VI) with a desired alcohol and subsequent Knoevenagel condensation with an aromatic aldehyde VII 21, 22 gave eight esters VIII (Scheme 1C; compounds 24–31, Figure 1). Finally, direct nitration of three esters IX with zirconyl nitrate23 afforded the corresponding nitro derivatives X (Scheme 1D; compounds 32–34, Figure 1). Only compounds 1, 5, 9, 17, and 24 have been previously obtained by the above‐mentioned methods. For the rest, the procedures were properly adapted affording yields in a wide range (Figure 1) but mostly comparable to those originally reported for such kind of reactions.
Scheme 1.
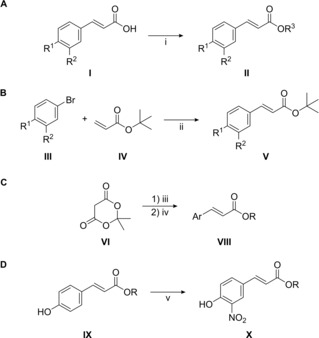
Synthesis of cinnamate ester analogues. Reaction conditions: i) R3OH, Dowex 50WX8 H+ resin or conc. H2SO4, reflux; ii) PdCl2, PPh3, K2CO3, DMF, 100 °C; iii) ROH, toluene, reflux; iv) ArCHO (VII), pyridine, piperidine, 60°C; v) ZrO(NO3)2•xH2O, acetone, RT.
Figure 1.
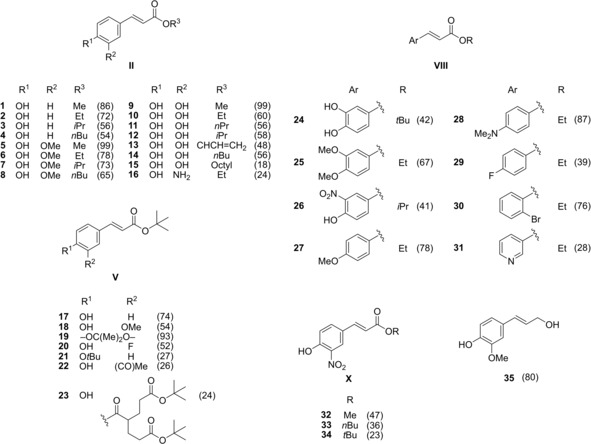
Chemical structures of the synthesized compounds. Yields are given in parentheses.
Interestingly, compound 22 appeared sensitive to the Heck reaction conditions and as a result, it was partially transformed into the unexpected side product 23 (24 %) during its preparation. This translated in an exceptionally low yield for compound 22 (26 %), compared to the other compounds obtained by this pathway. Apparently, the acidity of the protons in α‐position to the carbonyl moiety in 22 was high enough to afford a Michael addition reaction on another molecule of IV (Figure S1). The acidity of the α‐proton of the intermediate 23 a allowed addition of a further molecule of IV leading to 23. Studies on the mechanism of reaction were out of the scope of the present research, and therefore no further experimental information at this regard is presented here. However, the feasibility of this process seems to be attributable to the formation of enolates of 22 and 23 a, probably favored by Pd species, acting as bidentate ligands by their 3‐acetyl‐4‐hydroxyphenyl moieties. Pd catalysts have been used for different Michael addition reactions24 and involvement of Pd‐enolate intermediates within several catalytic cycles has also been recognized in literature.25, 26, 27, 28 Moreover, the presence of K2CO3 may have aided to the formation of those enolates. It might be reasonable that greater excesses of IV could improve the yield of 23, while smaller amounts of IV could result in better selectivity toward 22.
Antiprotozoal Activity and Cytotoxicity
The whole set of compounds was tested in vitro against L. donovani (axenic amastigotes), T. cruzi (intracellular amastigotes), T. brucei rhodesiense (bloodstream trypomastigotes) and P. falciparum (intraerythrocytic forms). Cytotoxicity against rat skeletal myoblasts (L‐6 cell line) was also determined. All assays were performed using validated protocols at the Swiss TPH. Coniferyl alcohol (35) was included to evaluate the possible effect of reduction of the carboxylate moiety on the biological potential. The results of the activity tests are shown in graphical form in Figures 2, 5 and 6. They are reported in full numerical form in the supplementary material (Table S1). The boxplot in Figure 2A makes evident the limited or non‐existent activity of the compounds against P. falciparum and T. cruzi, except for a few outliers represented by cross markers. Similarly, the low cytotoxicity of most of the compounds becomes evident from the plot. In contrast, a relatively wide range of activity values was observed when the compounds were tested against L. donovani and T. brucei rhodesiense, suggesting that more detailed analysis of the underlying structure‐activity relationships would be of interest. Particularly, the median activity against L. donovani was better than against T. brucei rhodesiense.
Figure 2.
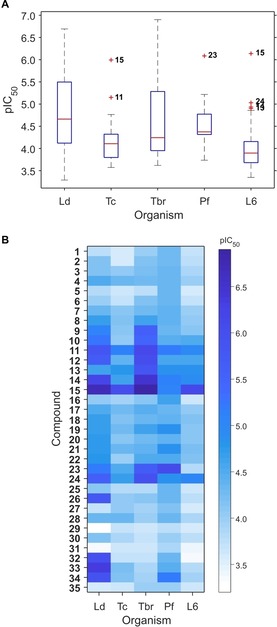
Boxplot (A) and heatmap (B) of the antiprotozoal activity and cytotoxicity data of compounds 1–35. Ld: L. donovani; Tc: T. cruzi; Tbr: T. brucei rhodesiense; Pf: P. falciparum; L6: rat skeletal myoblasts. Data are expressed as pIC50=−log(IC50 [M]) values.
A more detailed insight into the general trends of activity is shown in Figure 2B. The higher potential of the compounds on L. donovani and T. brucei rhodesiense becomes obvious from the darker shades of blue. Moreover, the wide range of blue shades for these two parasites indicates a marked structure dependence. Although some compounds (11, 12, 14, 15, and 24) were commonly active against both parasites, compounds 26 and 32–34 were selective toward L. donovani (being essentially inactive against T. brucei rhodesiense as observed from their very light blue color in the corresponding row). In general, 34 % and 26 % of the compounds could be defined as active against L. donovani and T. brucei rhodesiense, respectively (IC50<10 μM). Interestingly, compound 15 resulted to be very promiscuous acting not only against both L. donovani and T. brucei, but also being very toxic against the mammalian cell line (IC50=0.73 μM).
The diverse behavior of the synthesized esters towards the parasites and the L‐6 cells depicted in Figure 2 and already discussed, with high similarities in some cases but also with unparalleled differences in others, encouraged further data analysis looking for characteristic patterns. Firstly, in order to investigate direct relationships between the two more promising sets of antiparasitic activity and cytotoxicity against mammalian cells, plots of the respective pIC50 values vs. those for cytotoxic activity were prepared (Figure 3).
Figure 3.
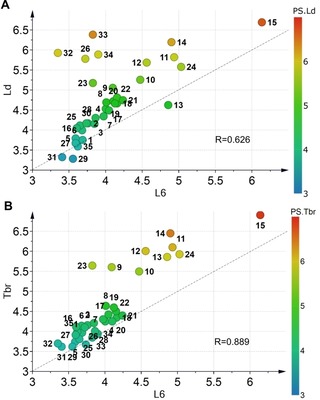
Scatter plots of (A) antileishmanial (L. donovani, Ld) and (B) antitrypanosomal (T. brucei rhodesiense, Tbr) activity vs. cytotoxicity against L‐6 cells (pIC50 values). Compounds are color‐coded according to their respective antiparasitic activity; R‐Pearson's correlation coefficient. The dashed grey lines are not trend lines but represent the diagonal for easier orientation.
It becomes clear that both desirable activities for most compounds are significantly correlated with cytotoxicity indicating that similar mechanisms are responsible for the antiprotozoal and cytotoxic activity. This is obviously even more prominent for the antitrypanosomal activity (Figure 3B). However, in both cases, deviations of particular compounds from the general trend become more prominent as antiparasitic activity increases. Here, especially in case of the antileishmanial activity, some compounds are much less cytotoxic than active (i. e. far more selective than would be expected from a linear trend line through the data). These compounds deviating from the general trend are the nitroaromatic derivatives 26 and 32–34 already mentioned above in the specific case of L. donovani and the catechol type compounds 11–14 as well as 24 in both cases. These compounds therefore obviously represent interesting hits that possibly act in a different manner than most of their congeners.
More insights about general trends were then obtained by means of a principal component analysis (PCA) on all five sets of bioactivity data in the form of pIC50 values. The resulting PC1‐PC2 score and loadings plots are shown in Figure 4. For an easy overview, the data in the scores plot were color‐coded according to a hierarchical clustering analysis (HCA) performed with the same data. The two resulting principal components explained a total variance of 91.6 %. The clustering obtained by HCA gave four main groups. In particular, the highly lipophilic compound 15 appeared as outlier, which correlates with its promiscuous behavior. Although compounds 32 and 33 also appeared as outliers, their relative location on the score space indicated definitely special properties (and different from those of 15; Figure 4A). In fact, they were clustered together with 26 and 34 (red marks), which is in agreement with the above‐mentioned special case of these nitro compounds. Additionally, compounds bearing a catechol moiety (o‐dihydroxyphenyl) were also clustered together demonstrating their distinguishable performance among the whole set of compounds (green marks). These compounds were characterized by comparable activities against L. donovani and T. brucei rhodesiense, which probably implies similar mechanisms of action on both parasites. More interestingly, selectivity over the other parasites as well as low toxicity to the L‐6 cells were always observed. The rest of the compounds were close in the PC1‐PC2 plane, but still discriminated into two groups by HCA. The group represented by yellow marks consisted exclusively of the tert‐butyl esters. Their appearance in the middle of the score plot suggested limited variation in the activity values regardless of their structural differences in the aromatic ring. Finally, compounds showing the poorest general effect on the protozoan parasites were also clustered separately (blue marks). Most of them corresponded to the methyl and ethyl esters.
Figure 4.
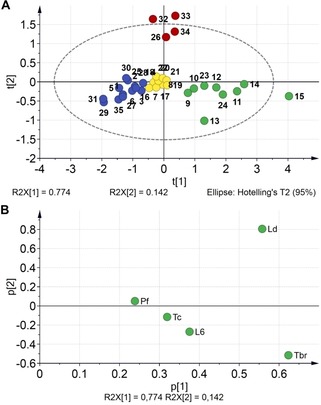
Score (A) and loadings (B) plots of a PCA on the pIC50 values. The scores are color‐coded according to clusters obtained by HCA. Ld: L. donovani; Tc: T. cruzi; Tbr: T. brucei rhodesiense; Pf: P. falciparum; L6: rat skeletal myoblasts.
Inspection of the loadings plot (Figure 4B) then shows that the overall differences among the compounds’ activities are indeed mainly due to the two activities already mentioned, i. e. antileishmanial activity and anti‐T. brucei activity. Both strongly contribute to the first and second PC. The differential behavior against these two parasites reflects in their opposite contributions to PC2 and it can safely be stated that this is mainly caused by the nitro‐compounds’ (26, 32–34) high activity against L. donovani and low activity against T. brucei.
Structure−Activity Relationships
The wide range of activity values found against L. donovani and T. brucei rhodesiense allowed more detailed analyses of the corresponding structure–activity relationships. In order to accomplish this in an easy way, the concept of SAR maps29 was adopted. The independent SAR maps for the two sets of antiparasitic activity data were constructed using an in‐house MATLAB algorithm and are shown in Figures 5 and 6.
Figure 5.
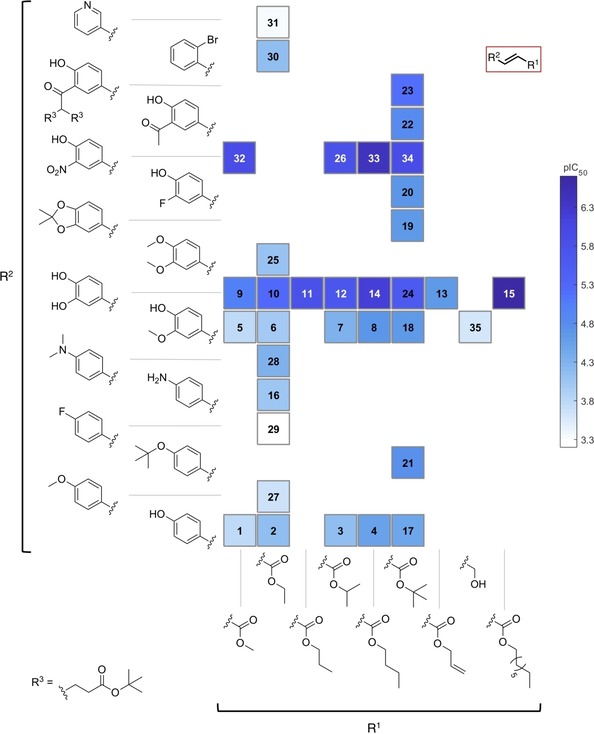
SAR map for anti‐leishmanial activity of compounds 1–35.
Figure 6.
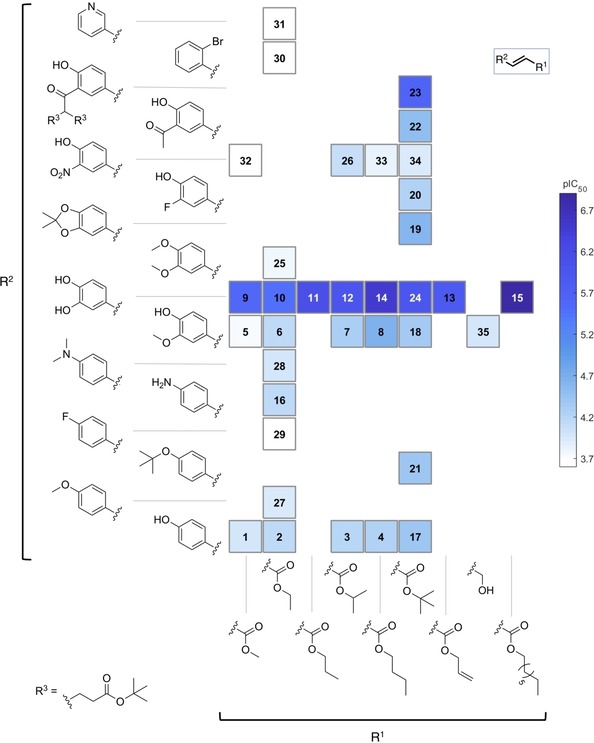
SAR map for anti‐trypanosomal activity of compounds 1–35.
The effect of the alkyl ester group (R1) on the activity against L. donovani was straightforward to observe (Figure 5). Within the series studied, a medium sized linear alkyl chain (nPr, nBu) provided the highest level of activity while smaller or branched alkyl groups gave somewhat lower activity. Thus compounds 4, 8, 14, and 33 were more potent compared to their corresponding analogues bearing methyl, ethyl, isopropyl and tert‐butyl groups. The only compound with a longer linear alkyl chain (15) was synthesized in order to find out whether this trend continues with even longer alkyl chains. Indeed, the activity of 15 was further increased but this was accompanied by a remarkable increase of toxicity against mammalian cells as mentioned earlier. The presence of an unsaturation in the lateral chain (compound 13) disfavored the activity. Moreover, reduction of the carboxylate to the corresponding alcohol (compound 35) led to decrease in activity, too. The changes in substitution of the aromatic ring (R2) also exhibited a strong impact on the antileishmanial activity (Figure 5). Monosubstituted rings displayed a deleterious effect on the activity. Such compounds were within an activity range considered not interesting for further development (IC50>40 μM). Only limited effects were achieved among these compounds by alkylation of the free amino and phenol groups (compounds 28 vs 16 and 27 vs 2). Particularly, 4‐fluorphenyl and 3‐pyridyl groups (compounds 29 and 31, respectively) led to total loss of activity. Introduction of a second substituent on the aromatic ring showed a more profound effect on the activity against L. donovani, although 3‐F (20), 3‐MeO (18) and 3‐acetyl (22) were not able to improve the activity of the monosubstituted compound (17). In contrast, the introduction of 3‐OH or 3‐NO2 groups significantly increased the leishmanicidal potential of the esters. Apparently, the alkylation of the OH groups was deleterious for the activity as inferred from the lowered potency of 6 and 25 compared to 10, and of 18 and 19 compared to 24. The series of 3‐nitro‐4‐hydroxy compounds displayed always stronger potency against the parasite than the corresponding catechol derivatives. The difference was highest in case of the methyl esters; for the rest, it increased from the isopropyl to the butyl esters (tert‐butyl higher difference than for n‐butyl). The most potent compounds were 15 (IC50=0.20 μM, which is less interesting, however, due to its high cytotoxicity), 14 and 33 (IC50 of 0.64 and 0.42 μM, respectively). Calculation of the corresponding selectivity indexes versus cytotoxicity (SI=IC50(L6)/IC50(Ld)) demonstrated that compound 33 was a far better candidate than 14, since the former possessed an SI of 355 whereas the latter reached only an SI of 20. As observed from Figure 2, compound 33 was exclusively selective towards Leishmania (against which it was 141, 335 and 90 times more active than against T. cruzi, T. brucei rhodesiense and P. falciparum, respectively). In contrast, compound 14 showed no discrimination between L. donovani and T. brucei rhodesiense. The other nitro compounds (26, 32 and 34) were somewhat less active than 33 and displayed a very similar activity level (IC50 of 1.7, 1.2, and 1.3 μM, respectively) but good selectivity; their SIs were quite different (SI of 113, 375 and 96, respectively), but promising. The outstanding selectivity and significant activity of the nitro compounds against L. donovani was however not totally surprising since the high potential of nitro compounds for treatment of Leishmaniasis has already been highlighted.30 Nitroaromatics like Nifurtimox, Benznidazole, and Fexinidazole have been commonly used for treatment of trypanosomatid diseases and continue inspiring studies on related compounds.31, 32, 33, 34, 35 The interesting antitrypanosomal activity of such compounds supported and encouraged the choice for the preparation of the nitro derivatives 26 and 32–34 in the present work. Although it has been suggested that nitroaromatics act on the parasite by alteration of the redox equilibrium within the parasite after enzymatic metabolization by a nitroreductase (NTR),30, 35 lack of correlation between antitrypanosomal activity and effect on NTR activity have sometimes been observed,34 implying that further studies are required in order to understand better the potential of nitro compounds as leishmanicidal agents, including 26 and 32–34. Involvement of alternative mechanisms of action may explain the significant difference in the potential of the nitro compounds against the two related parasites under study.
As already mentioned, the potential of the synthesized compounds as trypanocidal agents was significantly narrower than the leishmanicidal (higher number of compounds with lighter color in the SAR map; Figure 6). Fluoro and bromo derivatives 29 and 30 as well as the pyridyl analogue 31 were totally inactive. As observed for the leishmanicidal activity, replacement of OH by NH2 did not lead to any significant change in activity (compounds 2 vs 16). Methylated derivatives 27 and 28 did not show interesting activity either. Interestingly, the acetonide 19 exhibited a slightly better activity than the corresponding monomethylated analogue 18. This seems not to be related to the increased lipophilicity, since the dimethylated compound 25 was significantly less active than the monomethylated 6. However, apart from the unusual compound 23, which showed some activity (IC50 of 2.3 μM), the most active compounds were those bearing an o‐dihydroxyphenyl group. It is very noteworthy, that – in contrast to Leishmania – the 3‐nitro‐4‐hydroxy compounds were much less active than the catechols. Among the latter, compound 14 was the most potent with an IC50 value of 0.35 μM, followed by the compounds 11, 12, and 24 with values of 0.79, 1.0 and 1.2 μM, respectively. Regarding the effect of changes in the lateral chain of the ester, no clear trends could be established. While the n‐butyl group offered the best activity to compounds 14 and 8 within their respective series, the isopropyl and the tert‐butyl groups conferred the highest activity within the series of compounds 26 and 17. Concerning selectivity, compound 14 displayed an SI of 36, which was only outmatched by compound 23 (SI of 65). Overall, the potential of these compounds against T. brucei in terms of activity and selectivity appears less interesting than that against L. donovani. The potential of phenolic compounds as antitrypanosomatid agents have been already described in a number of scientific publications.36, 37, 38
In vivo Evaluation
While none of the target compounds showed an interesting potential against P. falciparum, the unexpected side product 23 displayed significant activity against this parasite (IC50=0.82 μM) and it was therefore considered for in vivo testing. Unfortunately, no reduction in parasitemia of mice infected with P. berghei was observed after four days of treatment at a regimen of 50 mg/kg/day i. p., despite its apparently high metabolic stability (in vitro determination using liver microsomes; data not shown). This negative outcome might be related to poor absorption/cell permeation of the compound as predicted by the SwissADME web tool.39
The seemingly promising in vitro activity and selectivity of compounds 11 and 14 also encouraged to move forward and test them against T. brucei brucei in an in vivo mouse model. Unfortunately, no significant reduction of the parasitemia was caused by either of them during four days of treatment at 30 mg/kg/day i. p. These compounds were predicted by the SwissADME as having suitable properties for oral bioavailability (including possibility of gastrointestinal absorption), but they were also predicted as likely reactive with some cytochrome enzymes (data not shown), which might explain the observed failure.
In vivo evaluation of the promising hits against L. donovani in suitable animal models, as well as studies on their activity against further Leishmania species will be subject to further investigations.
Conclusions
In summary, a total of thirty‐four cinnamate esters were prepared and tested for their antileishmanial, antitrypanosomal and antiplasmodial effects. Two groups of compounds demonstrated clearly superior performance over the rest being characterized by 4‐hydroxy‐3‐nitrophenyl and 3,4‐dihydroxyphenyl moieties. The presence of either of them resulted in high potency against L. donovani. In contrast, only the 3,4‐dihydroxyphenyl moiety afforded significant activity against T. brucei rhodesiense. Moreover, both types of compounds exhibited low cytotoxicity against the L‐6 cells (high SI values), making them even more relevant as possible candidates for further investigation. Detailed SAR analysis supported this idea and led to the identification of the n‐butyl ester group as the best option for improving the antileishmanial potency. This trend was less clear in case of the antitrypanosomal activity, although the most active compound against T. brucei rhodesiense was an n‐butyl ester as well. Surprisingly, enlargement of the lateral chain up to n‐octyl led to a highly promiscuous and cytotoxic compound (15). On the other hand, P. falciparum and T. cruzi were not sensitive to the esters, except for the unusual compound 23, an unintended side product from the synthesis of 22. Compound 23 showed a promising in vitro antiplasmodial activity. Efforts to evaluate the potential of the most potent antiplasmodial (compound 23) and antitrypanosomal compounds (11 and 14) in in vivo models lacked success, which highlighted the need of further structural optimization aiming at better pharmacokinetic properties. The most promising hits of this study were the 4‐hydroxy‐3‐nitrophenyl cinnamic acid esters against L. donovani. Further studies on this type of compounds are warranted.
Experimental Section
Chemistry
General: All the solvents and chemicals were used as obtained from Merck, Sigma‐Aldrich, Thermofisher Acros Organics, and Fluorochem. Routine 1H NMR experiments were recorded on an Agilent DD2 400 MHz spectrometer. 13C NMR and 2D experiments were recorded on an Agilent DD2 600 MHz equipped with a cryo‐probe. Exact mass determinations were accomplished by UHPLC/+ESI QqTOF MS as follows: Chromatographic separations were performed on a Dionex Ultimate 3000 RS Liquid Chromatography System (UHPLC) on a Dionex Acclaim RSLC 120, C18 column (2.1×100 mm, 2.2 μm) at 40 °C with a binary gradient (A: water with 0.1 % formic acid; B: acetonitrile with 0.1 % formic acid) at 0.8 mL/min: 0 to 0.4 min: isocratic at 5 % B; 0.4 to 9.9 min: linear from 5 % B to 100 %B; 9.9 to 15.0 min: isocratic at 100 % B; 15.0 to 15.1 min: linear from 100 % B to 5 % B; 15.1 to 20 min: isocratic at 5 % B. The injection volume was 2 μL. Eluted compounds were detected using a Dionex Ultimate DAD‐3000 RS over a wavelength range of 200–400 nm and a Bruker Daltonics micrOTOF‐QII time‐of‐flight mass spectrometer equipped with an Apolo electrospray ionization source in positive mode at 3 Hz over a mass range of m/z 50–1500 using the following instrument settings: nebulizer gas nitrogen, 4 bar; dry gas nitrogen, 9 L/min, 220 °C; capillary voltage 4500 V; end plate offset −500 V; transfer time 100 μs; collision gas nitrogen; collision energy and collision RF settings were combined to each single spectrum of 1250 summations as follows: 624 summations with 80 eV collision energy and 130 Vpp+313 summations with 16 eV collision energy and 200 Vpp+313 summations with 16 eV collision energy and 200 Vpp. Internal dataset calibration (HPC mode) was performed for each analysis using the mass spectrum of a 10 mM solution of sodium formiate in 50 % isopropanol that was infused during LC re‐equilibration using a divert valve equipped with a 20 μL sample loop.
Experimental details for all the compounds are reported in the Supporting Information. The general procedures for their synthesis are described as follows.
General Procedure A (Fischer esterification). The synthesis of compounds 1–16 was conducted according to literature.16, 18, 40 In brief, the corresponding cinnamic acid derivative (5 mmol) was dissolved in the corresponding alcohol (9 mL), and Dowex 50WX8 H+ resin (225 mg) or conc. H2SO4 (few drops) was added as catalyst. The resulting mixture was refluxed for 24–48 h. When the resin was used, the mixture was filtered, and the solvent removed under reduced pressure. In cases where sulfuric acid was used, the mixture was diluted with ethyl acetate (20 mL) and neutralized with 5 % NaHCO3 solution. Subsequently, the layers were separated, and the aqueous phase was extracted with ethyl acetate (3×10 mL). The combined organic layers were dried over anhydrous Na2SO4 and the solvent removed under reduced pressure. The residue was chromatographed to afford the corresponding pure products.
General Procedure B (Heck reaction). The procedure for the synthesis of compounds 17–23 was adapted from literature.19, 20 The corresponding aryl bromide (2.5 mmol) was dissolved in dry DMF (7.5 mL) under nitrogen. Tert‐butyl acrylate (5 mmol), anhydrous potassium carbonate (5 mmol), palladium dichloride (1 mol%) and triphenylphosphine (2 mol%) were added. The mixture was heated at 100 °C under nitrogen for 24–72 h. The mixture was then cooled down and filtered over a Celite® pad. The pad was washed several times with ethyl acetate and the layers separated. The organic one was washed with 1 M HCl (2×5 mL), 10 % NaCl (2×5 mL), and water (3×10 mL), and afterward dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue chromatographed to afford the corresponding pure products.
General Procedure C (Knoevenagel condensation). The procedure for synthesis of compounds 24–31 was adapted from literature.21, 22 Meldrum's acid (10 mmol) was mixed with toluene (20 mL) and the alcohol (10 mmol) was added. The mixture was refluxed for 5–6 h. The resulting mixture was cooled and the corresponding benzaldehyde derivative (4 mmol), pyridine (1 mL), and piperidine (0.1 mL) were added. The mixture was heated at 60 °C until complete transformation of the aldehyde as indicated by TLC. After cooling, it was diluted with ethyl acetate (20 mL) and washed with 7.5 % NaHCO3 (2×5 mL), 1 M HCl (2×5 mL), and water (3×10 mL). The organic layer was dried over anhydrous Na2SO4, the solvent removed under reduced pressure, and the residue chromatographed to afford the corresponding pure products.
General Procedure D (Nitration). The procedure for the nitration of 1, 4 and 17 to obtain compounds 32–34 was adapted from literature.23 The corresponding ester (0.35 mmol) was dissolved in acetone (3.9 mL) and zirconyl nitrate (0.35 mmol) was added. The mixture was stirred at RT for 1 week. The solvent was removed under reduced pressure and the residue re‐dissolved in ethyl acetate (5 mL) and washed with 1 M HCl (2×2 mL), 10 % NaCl (3 mL), water (2×5 mL), and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue chromatographed to afford the corresponding pure products.
Biological Assays
All the tests were performed applying validated methods of the Swiss TPH as described below. Each compound was tested once against each parasite and the determination repeated in case of active compounds, (IC50<5 μM) so that duplicate data were obtained in these cases. A full data table is found as Supporting Information.
In vitro activity against Trypanosoma brucei rhodesiense. The parasitic stock was isolated in 1982 from a human patient in Tanzania and after several mouse passages cloned and adapted to axenic culture conditions.41 Minimum Essential Medium (50 μL) supplemented with 25 mM HEPES, 1 g/L additional glucose, 1 % MEM non‐essential amino acids (100x), 0.2 mM 2‐mercaptoethanol, 1 mM Na‐pyruvate, and 15 % heat inactivated horse serum was disposed in 96‐well microtiter plates. Serial compound dilutions of eleven 3‐fold dilution steps covering a range from 100 to 0.002 μg/mL were prepared. Then, an aliquot containing 4x103 bloodstream forms (trypomastigotes) of T. b. rhodesiense STIB 900 in 50 μL were added to each well and the plate incubated at 37 °C under 5 % CO2 atmosphere for 70 h. After that, resazurin solution (12.5 mg in 100 mL water, 10 μL) was added to each well and the incubation continued for 2–4 h more.42 Then the plates were read using a Spectramax Gemini XS microplate fluorometer (Molecular Devices Cooperation, Sunnyvale, CA, USA) and excitation and emission wavelengths of 536 and 588 nm, respectively. Data were analyzed with the software Softmax Pro (Molecular Devices Cooperation, Sunnyvale, CA, USA), which calculated IC50 values by linear regression43 and 4‐parameter logistic regression from the sigmoidal dose inhibition curves. Melarsoprol was used as positive control.
In vitro activity against T. cruzi. Rat skeletal myoblasts (L‐6 cells) were seeded in 96‐well plates at 2000 cells/well in 100 μL RPMI 1640 medium with 10 % FBS and 2 mM L‐glutamine. After 24 h the medium was removed and replaced by 100 μL per well containing 5000 trypomastigote forms of T. cruzi Tulahuen strain C4 containing the β‐galactosidase (Lac Z) gene.44 After 48 h the medium was removed from the wells and replaced by 100 μL fresh medium with or without a serial compound dilution (as mentioned for T. brucei rhodesiense). After 96 h of incubation the plates were inspected under an inverted microscope to assure growth of the controls and sterility. Then substrate CPRG/Nonidet (50 μL) was added to each well. The color reaction was developed within 2–6 h and the plate read at 540 nm. Data were analyzed as mentioned in the previous section. Benznidazole was used as positive control.
In vitro activity against L. donovani axenic amastigotes. Amastigotes of L. donovani strain MHOM/ET/67/L82 were grown in axenic culture at 37 °C in SM medium45 at pH 5.4 supplemented with 10 % heat‐inactivated fetal bovine serum under a 5 % CO2 atmosphere. An aliquot containing 105 amastigotes from the axenic culture (100 μL) with or without a serial compound dilution (as specified before) were seeded in 96‐well plates. After 70 h of incubation the plates were inspected under an inverted microscope to assure growth of the controls and sterile conditions. Then, resazurin solution (12.5 mg in 100 mL water, 10 μL) was added to each well and the plates incubated for another 2 h. Finally, the plates were read using the same conditions mentioned for T. brucei rhodesiense. Data were analyzed in the same way. Miltefosine was used as positive control.
In vitro activity against P. falciparum. In vitro activity against erythrocytic stages of P. falciparum was determined using a 3H‐hypoxanthine incorporation assay,46, 47 employing the drug sensitive NF54 strain (Schipol Airport, The Netherlands48). Compounds were diluted with medium as indicated above before added to parasite cultures incubated in RPMI 1640 medium without hypoxanthine, supplemented with HEPES (5.94 g/L), NaHCO3 (2.1 g/L), neomycin (100 U/mL), AlbumaxR (5 g/L) and washed human red cells A+ at 2.5 % hematocrit (0.3 % parasitemia). Serial compound dilutions were prepared as before. The 96‐well plates were incubated in a humidified atmosphere at 37 °C; 4 % CO2, 3 % O2, 93 % N2. After 48 h, 50 μL of 3H‐hypoxanthine (0.5 μCi) was added to each well of the plate. The plates were incubated for a further 24 h under the same conditions. The plates were then harvested with a Betaplate™ cell harvester (Wallac, Zurich, Switzerland), and the red blood cells transferred onto a glass fiber filter then washed with distilled water. The dried filters were inserted into a plastic foil with 10 mL of scintillation fluid and counted in a Betaplate™ liquid scintillation counter (Wallac, Zurich, Switzerland). Data were treated as described above. Chloroquine and artemisinin were used as positive controls.
In vitro cytotoxicity with L‐6 cells. Assays were performed in 96‐well plates. Each well was seeded with 100 μL of RPMI 1640 medium supplemented with 1 % L‐glutamine (200 mM) and 10 % fetal bovine serum, and 4000 L‐6 cells.49, 50 Serial compound dilutions prepared as before were used. After 70 h of incubation the plates were inspected under an inverted microscope to assure growth of the controls and sterile conditions. Resazurin solution (12.5 mg in 100 mL water, 10 μL) was then added to each well and the plates incubated for another 2 h. Finally, the plates were read, and the data were treated as mentioned before. Podophyllotoxin was used as positive control.
In vivo antimalarial efficacy study. Groups of three female NMRI mice (20–22 g) intravenously infected with 2×107 parasitized erythrocytes on day 0 with GFP‐transfected P. berghei strain ANKA51 were used for the assessment of activity. Compounds were formulated in 100 % DMSO, diluted 10‐fold in distilled water and administered intraperitoneally or orally in a volume of 10 mL/kg on four consecutive days (4, 24, 48 and 72 h post infection). Parasitemia was determined on day 4 post‐infection (24 h after last treatment) by FACS analysis. Activity was calculated as the difference between the mean parasitemia percentage for the control (n=5 mice) and the treated groups expressed as a percentage relative to the control group. The survival time in days was also recorded up to 30 days after infection. The compound would be considered active if the animals survive to day 30 after infection with no detectable parasitemia. The in vivo studies in mice were conducted at the Swiss Tropical and Public Health Institute (Basel) (License number 1731) according to the rules and regulations for the protection of animal rights (“Tierschutzverordnung”) of the Swiss “Bundesamt für Veterinärwesen” and approved by the veterinary office of Canton Basel‐Stadt, Switzerland.
In vivo antitrypanosomal efficacy study. The T. brucei brucei STIB795_luc acute mouse model mimics the first stage of the disease. T. brucei brucei parasites were genetically modified with pTb‐AMluc construct52 kindly provided by JM Kelly, containing red‐shifted luciferase reporter gene (PpyRE9 h). Six female NMRI mice were used per experimental group. Each mouse was inoculated i. p. with 104 bloodstream forms of STIB795_luc, respectively. Heparinized blood from a donor mouse with approximately 5×106 parasites/mL was suspended in PSG to obtain a trypanosome suspension of 1×105 /mL. Each mouse was injected with 0.25 mL. Compounds were formulated in 100 % DMSO diluted 10‐fold in distilled water or 0.5 % Tween80 in 0.5 % methylcellulose. Compound treatment was initiated 3 days post‐infection on four consecutive days in a volume of 0.1 mL/10 g. Three mice served as infected‐untreated controls. They were not injected with the vehicle alone since we have established in our labs that these vehicles do not affect parasitemia nor the mice. Parasitemia was monitored by whole‐animal live imaging twice a week until day 31 post‐infection. For imaging, mice were inoculated i. p. with 200 μL D‐luciferin (15 mg/ml in PBS) (Perkin Elmer), and 10 min later anaesthetized with 2.5 % isofluorane. Light emission was recorded for 5 minutes using in vivo imaging systems (IVIS) (Perkin Elmer). Mice would be considered cured if the bioluminescence signal on day 31 post‐infection was not higher than the background level. The in vivo efficacy studies in mice were conducted at the Swiss Tropical and Public Health Institute (Basel) (License number 2813) according to the rules and regulations for the protection of animal rights (“Tierschutzverordnung”) of the Swiss “Bundesamt für Veterinärwesen”. They were approved by the veterinary office of Canton Basel‐Stadt, Switzerland.
Data Analysis
The pIC50 values (negative decadic logarithms of IC50 [M]) used in the analysis corresponded to geometric means of duplicate measurements or single value determination as indicated above. Statistically significant differences were evaluated by ANOVA when required. Assessment of general trends (Figure 2) by heatmap and boxplot were created in MATLAB R2015a (Mathworks, Inc.). The concept of SAR maps29 (Figures 5 and 6) was adopted herein using an in‐house MATLAB algorithm. PCA and HCA (Figure 4) were performed in SIMCA 14.1 (Umetrics) using centering as pretreatment. Pharmacokinetic properties and druglikeness of the most active compounds were predicted using the SwissADME web tool.39
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
F. Bernal acknowledges the Colombian governmental agency Colciencias for his doctoral fellowship in Münster, Germany (call No. 679 of 2014). The authors are grateful to J. Sendker and J. Köhler, University of Münster, for the support in performing LC–MS and NMR analyses. This study is part of the activities within the Research Network Natural Products against Neglected Diseases (ResNet NPND, http://www.resnetnpnd.org/).
F. A. Bernal, M. Kaiser, B. Wünsch, T. J. Schmidt, ChemMedChem 2020, 15, 68.
References
- 1. Shaw W. R., Catteruccia F., Nat. Microbiol. 2019, 4, 20–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. De Rycker M., Baragaña B., Duce S. L., Gilbert I. H., Nature 2018, 559, 498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization, “WHO | World Health Assembly,” 2018.
- 4. Mitra A. K., Mawson A. R., Trop. Med. Infect. Dis. 2017, 2, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Glaser J., Schultheis M., Hazra S., Hazra B., Moll H., Schurigt U., Holzgrabe U., Molecules 2014, 19, 1394–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Otero E., Robledo S. M., Díaz S., Carda M., Muñoz D., Paños J., Vélez I. D., Cardona W., Med. Chem. Res. 2014, 23, 1378–1386. [Google Scholar]
- 7. Tasdemir D., Kaiser M., Brun R., Yardley V., Schmidt T. J., Tosun F., Ru P., Antimicrob. Agents Chemother. 2006, 50, 1352–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Radtke O. A., Yeap Foo L., Lu Y., Kiderlen A. F., Kolodziej H., Z. Naturforsch. C 2003, 58, 395–400. [DOI] [PubMed] [Google Scholar]
- 9. Cabanillas B. J., Le Lamer A. C., Castillo D., Arevalo J., Rojas R., Odonne G., Bourdy G., Moukarzel B., Sauvain M., Fabre N., J. Nat. Prod. 2010, 73, 1884–1890. [DOI] [PubMed] [Google Scholar]
- 10. Abamor E. S., Asian Pac. J. Trop. Med. 2017, 10, 25–34. [DOI] [PubMed] [Google Scholar]
- 11. Steverding D., da Nóbrega F. R., Rushworth S. A., de Sousa D. P., Parasitol. Res. 2016, 115, 4397–4403. [DOI] [PubMed] [Google Scholar]
- 12. Otoguro K., Iwatsuki M., Ishiyama A., Namatame M., Nishihara-Tsukashima A., Kiyohara H., Hashimoto T., Asakawa Y., O'Mura S., Yamada H., J. Nat. Med. 2012, 66, 558–561. [DOI] [PubMed] [Google Scholar]
- 13. Alson S. G., Jansen O., Cieckiewicz E., Rakotoarimanana H., Rafatro H., Degotte G., Francotte P., Frederich M., J. Pharm. Pharmacol. 2018, 70, 1349–1356. [DOI] [PubMed] [Google Scholar]
- 14. Vongvanich N., Kittakoop P., Charoenchai P., Intamas S., Sriklung K., Thebtaranonth Y., Planta Med. 2006, 72, 1427–1430. [DOI] [PubMed] [Google Scholar]
- 15. Glaser J., Schurigt U., Suzuki B. M., Caffrey C. R., Holzgrabe U., Molecules 2015, 20, 10873–10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pieters L., Van Dyck S., Gao M., Bai R., Hamel E., Vlietinck a, Lemière G., J. Med. Chem. 1999, 42, 5475–5481. [DOI] [PubMed] [Google Scholar]
- 17. Ali S. H., Tarakmah A., Merchant S. Q., Al-Sahhaf T., Chem. Eng. Sci. 2007, 62, 3197–3217. [Google Scholar]
- 18. Allegretta G., Weidel E., Empting M., Hartmann R. W., Eur. J. Med. Chem. 2015, 90, 351–359. [DOI] [PubMed] [Google Scholar]
- 19. Estrada G. O. D., de Souza A. L. F., da Silva J. F. M., Antunes O. A. C., Catal. Commun. 2008, 9, 1734–1738. [Google Scholar]
- 20. Kiuchi H., Suzuki T., Suzuki T., Method for Producing Hydroxycinnamate, 2010, JP,2010-120894,A. [Google Scholar]
- 21. Hu W.-X., Xia C.-N., Wang G.-H., Zhou W., J. Chem. Res. 2006, 586–588. [Google Scholar]
- 22. Xia C., Li H., Liu F., Hu W., Bioorg. Med. Chem. Lett. 2008, 18, 6553–6557. [DOI] [PubMed] [Google Scholar]
- 23. Selvam J. J. P., Suresh V., Rajesh K., Reddy S. R., Venkateswarlu Y., Tetrahedron Lett. 2006, 47, 2507–2509. [Google Scholar]
- 24. Comelles J., Moreno-Mañas M., Vallribera A., Arkivoc 2005, 2005, 207–238. [Google Scholar]
- 25. Streuff J., White D. E., Virgil S. C., Stoltz B. M., Nat. Chem. 2010, 2, 192–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fox J. M., Huang X., Chieffi A., Buchwald S. L., J. Am. Chem. Soc. 2000, 122, 1360–1370. [Google Scholar]
- 27. Kawatsura M., Hartwig J. F., J. Am. Chem. Soc. 1999, 121, 1473–1478. [Google Scholar]
- 28. Hagiwara E., Fuji A., Sodeoka M., J. Am. Chem. Soc. 1998, 120, 2474–2475. [Google Scholar]
- 29. Agrafiotis D. K., Shemanarev M., Connolly P. J., Farnum M., Lobanov V. S., J. Med. Chem. 2007, 50, 5926–5937. [DOI] [PubMed] [Google Scholar]
- 30. Patterson S., Wyllie S., Trends Parasitol. 2014, 30, 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nocentini A., Osman S. M., Almeida I. A., Cardoso V., Alasmary F. A. S., AlOthman Z., Vermelho A. B., Gratteri P., Supuran C. T., J. Enzyme Inhib. Med. Chem. 2019, 34, 1164–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mendonça D. B. D., Silva R. E. C., Palace-Berl F., Takakura C. F., Soares S. R. C., Braz L. M. A., Tavares L. C., Lindoso J. A. L., J. Venomous Anim. Toxins Incl. Trop. Dis. 2019, 25, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Van den Kerkhof M., Mabille D., Chatelain E., Mowbray C. E., Braillard S., Hendrickx S., Maes L., Caljon G., Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Papadopoulou M. V., Bloomer W. D., Rosenzweig H. S., Wilkinson S. R., Szular J., Kaiser M., Eur. J. Med. Chem. 2016, 117, 179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. O'Shea I. P., Shahed M., Aguilera-Venegas B., Wilkinson S. R., Antimicrob. Agents Chemother. 2016, 60, 1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Amisigo C. M., Antwi C. A., Adjimani J. P., Gwira T. M., PLoS One 2019, 14, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Otero E., García E., Palacios G., Yepes L. M., Carda M., Agut R., Vélez I. D., Cardona W. I., Robledo S. M., Eur. J. Med. Chem. 2017, 141, 73–83. [DOI] [PubMed] [Google Scholar]
- 38. Lima T. C., Souza R. J., Santos A. D. C., Moraes M. H., Biondo N. E., Barison A., Steindel M., Biavatti M. W., Nat. Prod. Res. 2016, 30, 551–557. [DOI] [PubMed] [Google Scholar]
- 39. Daina A., Michielin O., Zoete V., Sci. Rep. 2017, 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ali S. H., Merchant S. Q., Ind. Eng. Chem. Res. 2009, 48, 2519–2532. [Google Scholar]
- 41. Baltz T., Baltz D., Giroud C., Crockett J., EMBO J. 1985, 4, 1273–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Räz B., Iten M., Grether-Bühler Y., Kaminsky R., Brun R., Acta Trop. 1997, 68, 139–147. [DOI] [PubMed] [Google Scholar]
- 43. Huber W., Koella J. C., Acta Trop. 1993, 55, 257–61. [DOI] [PubMed] [Google Scholar]
- 44. Buckner F. S., Verlinde C. L., La Flamme A. C., Van Voorhis W. C., Antimicrob. Agents Chemother. 1996, 40, 2592–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cunningham I., J. Protozool. 1977, 24, 325–9. [DOI] [PubMed] [Google Scholar]
- 46. Desjardins R. E., Canfield C. J., Haynes J. D., Chulay J. D., Antimicrob. Agents Chemother. 1979, 16, 710–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Matile H., Richard J., Pink L., Immunol. Methods 1990, 221–234. [Google Scholar]
- 48. Ponnudurai T., Leeuwenberg A. D., Meuwissen J. H., Trop. Geogr. Med. 1981, 33, 50–4. [PubMed] [Google Scholar]
- 49. Page B., Page M., Noel C., Int. J. Oncol. 1993, 3, 473–6. [PubMed] [Google Scholar]
- 50. Ahmed S. A., Gogal R. M., Walsh J. E., J. Immunol. Methods 1994, 170, 211–24. [DOI] [PubMed] [Google Scholar]
- 51. Franke-Fayard B., Trueman H., Ramesar J., Mendoza J., van der Keur M., van der Linden R., Sinden R. E., Waters A. P., Janse C. J., Mol. Biochem. Parasitol. 2004, 137, 23–33. [DOI] [PubMed] [Google Scholar]
- 52. McLatchie A. P., Burrell-Saward H., Myburgh E., Lewis M. D., Ward T. H., Mottram J. C., Croft S. L., Kelly J. M., Taylor M. C., PLoS Neglected Trop. Dis. 2013, 7, e2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary