

Abstract
Background
Congenital anomalies are a major cause of co‐morbidity in children. Diagnostic code lists are increasingly used to identify congenital anomalies in administrative health records. Evidence is lacking on comparability of these code lists.
Objectives
To compare prevalence of congenital anomalies and prognostic outcomes for children with congenital anomalies identified in administrative health records using three different code lists.
Methods
We developed national cohorts of singleton livebirths in England (n = 7 354 363, 2003‐2014) and Scotland (n = 493 556, 2003‐2011). Children with congenital anomalies were identified if congenital anomaly diagnosis was recorded at birth, during subsequent hospital admission or as cause of death before 2 years old. We used three code lists: the EUROCAT list for congenital anomaly surveillance in Europe; the Hardelid list developed to identify children with chronic conditions (including congenital anomalies) admitted to hospital in England; and the Feudtner list developed to indicate children with complex chronic conditions (including congenital anomalies) admitted to hospitals in the United States. We compared prevalence, and risks of postnatal hospital readmission and death according to each code list in England and Scotland.
Results
Prevalence of congenital anomalies was highest using the EUROCAT list (4.1% of livebirths in England, 3.7% in Scotland), followed by Hardelid (3.1% and 3.0% of livebirths, respectively) and Feudtner (1.8% and 1.5% of livebirths, respectively). 67.2%‐73.3% of children with congenital anomalies in England and 65.2%‐77.0% in Scotland had at least one postnatal hospital admission across the three code lists; mortality ranged between 42.6‐75.4 and 41.5‐88.7 deaths per 1000 births in England Scotland, respectively. The risk of these adverse outcomes was highest using Feudtner and lowest using EUROCAT code lists.
Conclusions
The prevalence of congenital anomalies varied by congenital anomaly code list, over time and between countries, reflecting in part differences in hospital coding practices and admission thresholds. As a minimum, researchers using administrative health data to study congenital anomalies should report sensitivity analyses using different code lists.
Keywords: administrative data, congenital anomalies, international comparison, phenotyping
Synopsis
Study question
How can we measure the prevalence of congenital anomalies in administrative health data?
What is already known
Diagnostic code lists are increasingly used to identify congenital anomalies in administrative health records. Evidence is lacking on comparability of these code lists.
What this study adds
We compared prevalence of congenital anomalies, and risk of postnatal hospital admission and death in children aged less than two years old with a congenital anomaly using three code lists. Feudtner code list identified the least prevalent but most severe congenital anomalies. The EUROCAT code list identified the largest and least severely affected group. Prevalence of congenital anomalies in administrative health records was influenced by thresholds for coding and for hospital admission; therefore, sensitivity analyses using alternative code lists identifying congenital anomalies of different severities are recommended.
1. BACKGROUND
Congenital anomalies are one of the leading causes of childhood mortality and morbidity, accounting for nearly 30% of all deaths in children aged under five years old in high‐income countries.1 Congenital anomalies can be caused by environmental factors (such as exposure to toxic agents), use of certain medications during pregnancy, factors associated with maternal health (eg, infections or chronic illness) and adverse health behaviours during pregnancy (eg, smoking or alcohol consumption), or by genetic effects, although causes of most congenital anomalies remain unexplained.2 Monitoring congenital anomalies is important to detect emergence of hazards, but requires large, population‐based datasets because congenital anomalies are relatively rare. Historically, congenital anomaly registries such those participating in the European Surveillance of Congenital Anomalies Network (EUROCAT) have been used for surveillance,3, 4, 5 but they are resource‐intensive to maintain and therefore often restricted to regional rather than national coverage. Consequently, individual congenital anomaly registries may lack power to detect very rare anomalies, or to examine regional variation in birth prevalence.
In contrast, whole‐country birth cohorts created using administrative health datasets can be used to classify children with congenital anomalies based on clinical coding. These cohorts offer national coverage and sufficient power to compare prevalence of congenital anomalies across population subgroups at a relatively low cost.6, 7 In settings with universal health care and limited private sector provision such as in the UK, national administrative data capture complete hospitalisation trajectories, including all hospital contacts across the nation. A further advantage of administrative health data is the availability of information on birth characteristics (eg gestational age and birthweight) and longitudinal follow‐up (including outcomes such as mortality, surgical procedures, or hospital admission) among children with and without congenital anomalies. Birth cohorts from administrative health data can therefore be used to identify children with isolated or syndromic anomalies and those with multiple morbidities, explore risk factors for congenital anomalies, and assess risks of adverse outcomes and health care use in children with congenital anomalies compared with a population norm. Indicators of congenital anomalies based on clinical coding are also increasingly used to adjust for potential confounding effects of congenital anomalies.8 However, there is a lack of evidence on the comparability of codes used to identify congenital anomalies in administrative databases and how outcomes differ.
We compared three different International Classification of Disease version 10 (ICD‐10) code lists developed to identify congenital anomalies in health care or surveillance records: (a) the EUROCAT code list, developed for surveillance of congenital anomalies in Europe9; (b) the Hardelid code list developed to identify children with chronic conditions (including a subgroup of congenital anomalies) admitted to hospitals in the National Health Service (NHS) in England,10, 20 and (c) the Feudtner code list which aims to identify children with chronic complex conditions (with congenital anomalies as a subgroup) hospitalised in the United States (US).11, 12 We used birth cohorts from linked administrative health databases in England and Scotland to evaluate the overlap between the code lists and compare prognostic risks for key outcomes.
2. METHODS
2.1. Data sources
We developed whole‐country birth cohorts using de‐identified administrative health datasets in England and Scotland. We included singleton in‐hospital livebirths to resident mothers. Follow‐up information was obtained via linkage to birth records, hospital admission, and mortality records, and covered the period from birth until a child's second birthday or death, whichever occurred first.
In England, the birth cohort was based on birth admissions between 1 January 2003 and 31 December 2014 recorded in Hospital Episode Statistics (HES).8, 13 HES is an administrative hospital admission database covering 97% of all births in England.14 Birth records were linked to all subsequent admissions and to Office for National Statistics death registration data using a study‐specific identifier (HES‐ID) generated by NHS Digital, the data provider. NHD Digital routinely links the data based on child's NHS number, date of birth, postcode, sex, and local hospital identifier.15 The basic observation unit in HES is an episode of care under one consultant, and a hospital admission can consist of multiple episodes if multiple consultants are seen during an admission. Each episode can cover up to 14 diagnoses before 2007, and 20 thereafter. Up to 15 causes of death were available in the linked mortality data.10
For Scotland, we developed a birth cohort using National Records for Scotland birth certificates, which covered all registered births in Scotland from 1 January 2003 to 31 December 2011. Hospitalisation history was obtained through linkage to neonatal inpatient records (Scottish Birth Records, SBR) and subsequent hospital admissions (Scottish Morbidity Records for inpatients, SMR01). To assess deaths, we used Scottish Mortality Records (SMR99). The datasets were linked using study‐specific identifiers generated by the data provider—the electronic Data Research and Innovation Service—who routinely links these datasets using the CHI‐number, a unique identifier in the Scottish NHS.16 Hospital admission records in SMR01 contained up to 6 diagnostic fields, and up to 12 diagnoses in SBR. Up to 11 causes of death were available in SMR99.17
In both countries, diagnostic information and causes of death were recorded using ICD‐10 codes during the study period.10 Clinical coders in each hospital translate medical notes into diagnostic codes once patient care has finished. All coders are required to complete national accreditation training to ensure standardisation of recorded information between hospitals. Only information explicitly stated in the notes can be recorded; therefore, coding sensitivity can vary according to the quality and details recorded in medical notes.14, 18 In both countries, causes of death are coded at death registration.
2.2. Code lists
We evaluated three congenital anomaly code lists based on ICD‐10 codes. The EUROCAT code list was developed by the EUROCAT network to classify unstandardized text in registries for congenital anomaly surveillance in Europe. The EUROCAT list focuses on conditions diagnosed during pregnancy or in infancy, covering most codes from Chapter 17 of ICD‐10 (“Congenital malformations, deformations, and chromosomal abnormalities”19) and five additional codes from other ICD‐10 chapters. Minor or unspecified anomalies are excluded from the list.9
The second code list was developed by Hardelid et al 10 to identify children with chronic conditions, including congenital anomalies, who were admitted to NHS hospitals. The code list was derived using previous reports (including EUROCAT and Feudtner) and iterative review by a clinical panel to identify chronic conditions expected to require medical follow‐up for more than 12 months in more than 50% of cases.20 We used a subgroup of Hardelid codes from Chapter 17 of ICD‐10 (that is, codes starting with “Q”) to indicate congenital anomalies.
Third, we used the code list of paediatric complex chronic conditions developed by Feudtner et al to indicate conditions likely to last at least 12 months and involve multiple organ systems or require specialist paediatric care/hospitalisation in a tertiary care centre.21 The list was originally based on ICD version 9 and was later updated to ICD‐10.11 We indicated congenital anomalies using a subgroup of codes from Chapter 17 of ICD‐10 included in the Feudtner code list. For Hardelid and Feudtner code lists, we did not include congenital infections (recorded in chapter 16 of ICD‐10), which were included in the EUROCAT code list. All three code lists are provided in Table S1 in Appendix S1, together with additional information about code selection criteria.
2.3. Outcomes
The presence of congenital anomalies was indicated if at least one appropriate ICD‐10 code in the relevant code list was recorded as primary or subsidiary diagnosis at birth, during any hospital admission, or as any cause of death on the death record, before two years of age. This length of follow‐up enabled us to capture anomalies which may not be obvious at birth and diagnosed at later age.
2.4. Statistical analyses
We calculated the prevalence of congenital anomalies according to each code list (with 95% confidence intervals, 95% CI), defined as the number of children with at least one congenital anomaly divided by all livebirths in the cohorts. To assess trends in prevalence of congenital anomalies over time in each country, we estimated risk ratios (RR) for congenital anomalies by year of birth using log‐binomial regression models.22 We determined the overlap in the number of children identified between the code lists, and we compared the number of overlapping ICD‐10 codes included in the three code lists. We determined the number of children identified using each code list by ICD‐10 subchapter, and we listed five most commonly recorded ICD‐10 codes for congenital anomalies included in each code list per country.
We compared prognostic outcomes between the code lists in each country to assess differences in severity of congenital anomalies identified by each list. We estimated the proportion of children with a readmission in the first two years of life after birth (defined as the number of children with at least one hospital readmission after postnatal discharge divided by all births captured by a given congenital anomaly code list) and under‐2 mortality rate (defined as the number of all deaths in children aged less than 2 years old per 1000 births captured by a given congenital anomaly code list). We defined hospital admission as a continuous period in hospital; therefore, admissions and transfers within one day were treated as part of the same hospital stay.10 We counted all hospital admissions after birth including those before a congenital anomaly diagnosis was recorded. We estimated RR for postnatal readmission and mortality for each congenital anomaly code list over time and relative to children with no recorded anomalies using log‐binomial regression models. We report proportions of children in the two countries by number of postnatal readmissions (classified as 0, 1, 2‐3, 4+) and congenital anomaly code list. We also report number and proportion of child deaths by age at death (0‐27 days, 28‐364 days, and 1‐2 years). All analyses were performed using Stata SE version 15.0.
2.5. Sensitivity analyses
We compared the distribution of age at first recorded diagnosis of a congenital anomaly to examine whether anomalies captured by the code lists differed in timing of detection in hospital. Age at first diagnosis was grouped as 0‐27 days, 28‐89 days, 90‐364 days, and 1‐2 years. English data enabled further breakdown into diagnosis around the time of birth, in the first week (1‐6 days) and at 7‐27 days.
Since 2004, hospitals in England charge tariffs based on recorded diagnoses, thereby creating financial incentives for hospitals to improve coding of more complex patients.14 The same financial incentives are not present in Scotland. To assess whether changes in coding depth could have affected recording of congenital anomalies in HES, we examined changes in the mean and median number of unique diagnoses recorded during hospital admission per child by admission year.
3. RESULTS
3.1. Prevalence
We included 7 354 363 children born in England in 2003‐2014, and 493 556 children born in Scotland in 2003‐2011. The overall prevalence of congenital anomalies in England ranged from 4.1% (EUROCAT) to 3.1% (Hardelid) and 1.8% (Feudtner). Equivalent figures for Scotland were 3.7%, 3.0%, and 1.5% (Table 1). In England, prevalence of congenital anomalies increased by 2.7% per year using the Hardelid code list (RR = 1.03, 95% CI: 1.03, 1.03), by 3.0% using EUROCAT (RR = 1.03, 95% CI: 1.03, 1.03); the increase was slowest for congenital anomalies identified using Feudtner code list (2.1%, RR = 1.02, 95% CI: 1.02, 1.02, Table 1 and Figure 1A). In Scotland, annual increases in the prevalence of congenital anomalies were observed only for Hardelid and EUROCAT code lists, but were lower than in England (1.6%, RR = 1.02, 95% CI: 1.01, 1.02 and 1.7%, RR = 1.02, 95% CI: 1.01, 1.02, respectively).
Table 1.
Comparison of prevalence of congenital anomalies, risk of hospital admission after birth, and mortality rates according to each congenital anomaly code list in England and Scotland
| England (2003‐2014) | Scotland (2003‐2011) | |||||||
|---|---|---|---|---|---|---|---|---|
| EUROCAT | Hardelid | Feudtner | No congenital anomalies | EUROCAT | Hardelid | Feudtner | No congenital anomalies | |
| Prevalence of congenital anomalies | ||||||||
| Total number of children with congenital anomalies | 298 471 | 230 538 | 134 602 | 7 043 986 | 18 099 | 14 826 | 7294 | 474 481 |
| Prevalence of congenital anomalies | 4.06% | 3.13% | 1.83% | 95.78% | 3.67% | 3.00% | 1.48% | 96.14% |
| (4.04%, 4.07%) | (3.12%, 3.15%) | (1.82%, 1.84%) | (95.77%, 95.79%) | (3.61%, 3.72%) | (2.96%, 3.05%) | (1.44%, 1.51%) | (96.08%, 96.19%) | |
| Annual rate of change in prevalence | 1.03 | 1.03 | 1.02 | — | 1.02 | 1.02 | 1.00 | — |
| (1.03, 1.03) | (1.03, 1.03) | (1.02, 1.02) | (1.01, 1.02) | (1.01, 1.02) | (0.99, 1.01) | |||
| Hospital readmission after birth | ||||||||
| Number of children with at least 1 readmission | 200 426 | 165 955 | 98 680 | 1 963 043 | 11 794 | 10 335 | 5589 | 121 303 |
| % of children with at least 1 readmission | 67.2% | 72.0% | 73.3% | 27.9% | 65.2% | 69.7% | 77.0% | 25.6% |
| (67.0%, 67.3%) | (71.8%, 72.2%) | (73.1%, 73.5%) | (27.8%, 27.9%) | (64.5%, 65.9%) | (69.0%, 70.4%) | (76.0%, 78.0%) | (25.4%, 25.7%) | |
| Annual rate of change in risk of readmission | 1.00 | 1.01 | 1.01 | 1.02 | 0.99 | 0.99 | 1.00 | 1.01 |
| (1.00, 1.01) | (1.01, 1.01) | (1.01, 1.01) | (1.02, 1.02) | (0.99, 1.00) | (0.99, 1.00) | (1.00, 1.01) | (1.00, 1.01) | |
| Risk ratios for readmission in children with & without congenital anomalies | 2.41 | 2.58 | 2.63 | 2.55 | 2.73 | 3.01 | ||
| (2.40, 2.42) | (2.58, 2.59) | (2.62, 2.64) | (2.52, 2.58) | (2.70, 2.76) | (2.97, 3.05) | |||
| Under‐2 mortality rate per 1000 livebirths | ||||||||
| Number of deaths | 13 606 | 13 418 | 10 919 | 20 833 | 751 | 748 | 647 | 1358 |
| Under‐2 mortality rate | 42.6 | 53.4 | 75.4 | 2.7 | 41.5 | 50.5 | 88.7 | 2.9 |
| (41.8, 43.3) | (52.5, 54.3) | (74.0, 76.8) | (2.7, 2.8) | (38.6, 44.4) | (46.9, 54.0) | (82.2, 95.2) | (2.7, 3.0) | |
| Annual rate of change in mortality | 0.97 | 0.98 | 0.97 | 0.96 | 0.95 | 0.95 | 0.98 | 0.97 |
| (0.96, 0.97) | (0.97, 0.98) | (0.96, 0.97) | (0.95, 0.96) | (0.92, 0.97) | (0.92, 0.98) | (0.95, 1.00) | (0.95, 0.99) | |
| Risk ratios for death in children with & without congenital anomalies | 15.5 | 19.5 | 27.5 | 14.5 | 17.6 | 31.0 | ||
| (15.2, 15.9) | (19.0, 19.9) | (26.9, 28.1) | (13.3, 15.8) | (16.2, 19.3) | (28.3, 34.0) | |||
Annual rate of change was derived from log‐binomial regression models with year of birth included as a continuous covariate. Separate model was run for each country and each code list. All numbers in brackets are 95% confidence intervals.
Figure 1.
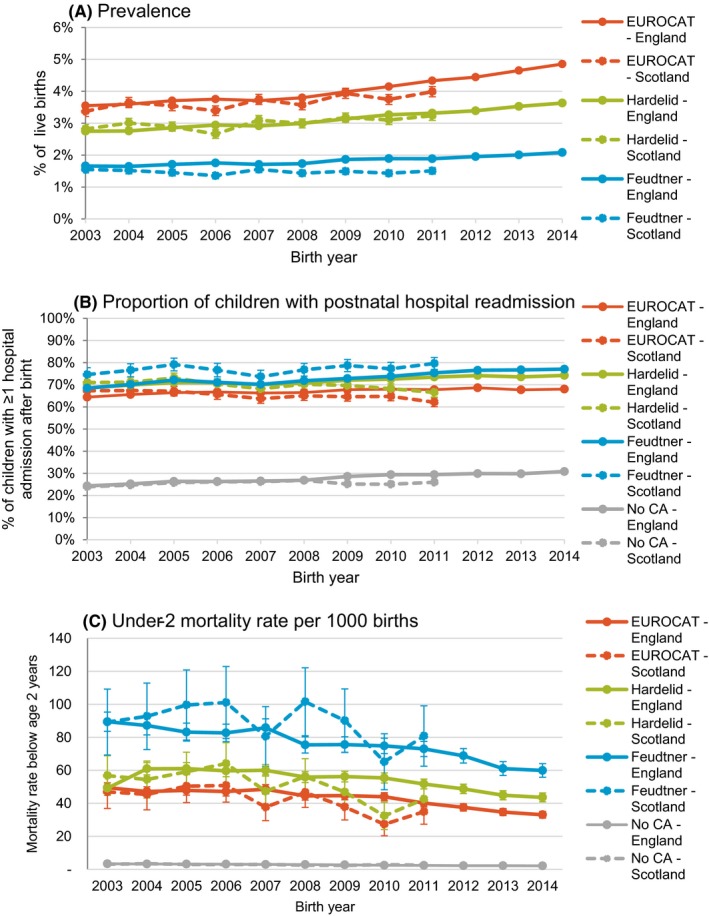
Time trends (with 95% confidence intervals) in A) prevalence of congenital anomalies in all livebirths according to each code list, B) proportion of children with a congenital anomaly according to each code list and with no anomalies who had at least one readmission in the two years after birth according and C) under‐2 mortality per 1000 livebirths with a congenital anomaly according to each code list and with no congenital anomalies who in England (solid lines) and Scotland (dotted lines)
3.2. Overlap between the code lists
Across all three code lists, 4.2% (310 377) children in England and 3.9% (19 075) children in Scotland had a record of any congenital anomaly. There was considerable overlap in children identified using each code list—40.9% (126 884) of children with any congenital anomaly in the English cohort and 34.0% (6483) in the Scottish cohort were captured using all three code lists, 30.1% (93 329) and 39.2% (7479), respectively, were captured by both Hardelid and EUROCAT codes (Table 2). The EUROCAT code list alone captured the highest proportion of children—24.2% (75 114) in England and 20.8% (3977) in Scotland. These results mirror the number of overlapping ICD‐10 codes. Overall, there were 561 unique four‐character ICD‐10 codes used (478 in the Hardelid list, 298 in the Feudtner list, and 536 in the EUROCAT list out of 628 4‐character codes in Chapter 17 of ICD‐10). 47.4% of all unique codes were included in all three code lists, 34.6% were captured by EUROCAT and Hardelid lists, and 10.3% were captured only by EUROCAT (Table 2).
Table 2.
Overlap in number of children identified and ICD‐10 codes included in each congenital anomaly code list in England and Scotland
| England (2003‐2014) | Scotland (2003‐2011) | ||
|---|---|---|---|
| Total number of births | 7 354 363 | 493 556 | |
| Number (%) of births with any congenital anomalies | 310 377 (4.2%) | 19 075 (3.9%) |
| Overlap between code lists | N | % of children with any congenital anomalies | N | % of children with any congenital anomalies | N | % of unique codes |
|---|---|---|---|---|---|---|
| Hardelid only | 7332 | 2.4 | 361 | 1.9 | 11 | 2.0 |
| Feudtner only | 1581 | 0.5 | 112 | 0.6 | 7 | 1.2 |
| EUROCAT only | 75 114 | 24.2 | 3977 | 20.8 | 58 | 10.3 |
| Hardelid + Feudtner | 2993 | 1.0 | 503 | 2.6 | 7 | 1.2 |
| Hardelid + EUROCAT | 93 329 | 30.1 | 7479 | 39.2 | 194 | 34.6 |
| Feudtner + EUROCAT | 3144 | 1.0 | 160 | 0.8 | 18 | 3.2 |
| all 3 codelists | 126 884 | 40.9 | 6483 | 34.0 | 266 | 47.4 |
ICD‐10 = International Classification of Disease version 10. Table shows number and % of all children with any congenital anomalies identified using each congenital anomaly code list, and overlap in ICD‐10 codes included by each code list.
All three code lists covered all congenital anomalies of the nervous system included in ICD‐10, and largely overlapping codes for congenital anomalies of the respiratory and urinary systems and chromosomal anomalies (Table S1), leading to comparable absolute numbers of children identified with these conditions between the code lists (Table S2). Congenital anomalies of circulatory system covered the highest number of children identified using each code list (Table S2); patient ductus arteriosus, and atrial and ventricular septal defects were included in the five commonly recorded congenital anomalies for Hardelid and EUROCAT code lists (Table S3). In both countries, congenital malformations of the skin were the most common condition diagnosed using the EUROCAT code list, and patent ductus arteriosus was the most frequently recorded condition based on Hardelid code list, while congenital hydronephrosis (England) and congenital laryngeal stridor (Scotland) were the most frequently recorded conditions using the Feudtner codes (Table S3). Unlike EUROCAT and Hardelid, Feudtner code list did not include any codes for congenital anomalies of the eye, ear, face, neck, cleft lip or palates, or of genital organs. Fifty‐eight codes unique to the EUROCAT list indicated primarily physical anomalies (eg, webbing of neck and anomalies of fingers and toes) and malformations of male genital organs.
3.3. Postnatal readmissions
The proportion of children with any hospital readmission after postnatal discharge and before age 2 years was similar between the two countries and was consistent with a gradient of severity (Table 1). The proportion was lowest for the EUROCAT code list (67.2% in England and 65.2% in Scotland), followed by the Hardelid code list (72.0% and 69.7%, respectively), and highest for the Feudtner code list (73.3% and 77.0%, respectively). The risk of hospitalisation relative to children with no congenital anomalies ranged from 2.4 times higher risk in England and 2.6 times higher risk in Scotland using EUROCAT codes, to 2.6‐ and 3.0‐ fold higher risks, respectively, for Feudtner codes (RR with 95% CIs are presented in Table 1). Depending on the congenital anomaly list used, 19.8%‐25.4% of children had 1 hospital readmission after birth, 21.3%‐25.9% had 2‐3 readmissions. The number of children with 4 + readmissions was the most variable between congenital anomaly groups, ranging from 1.6% for children with no congenital anomalies in England and 1.3% in Scotland, to a high of 29.0% (England) and 30.9% (Scotland) for children with congenital anomalies identified using the Feudtner code list (Figure 2).
Figure 2.
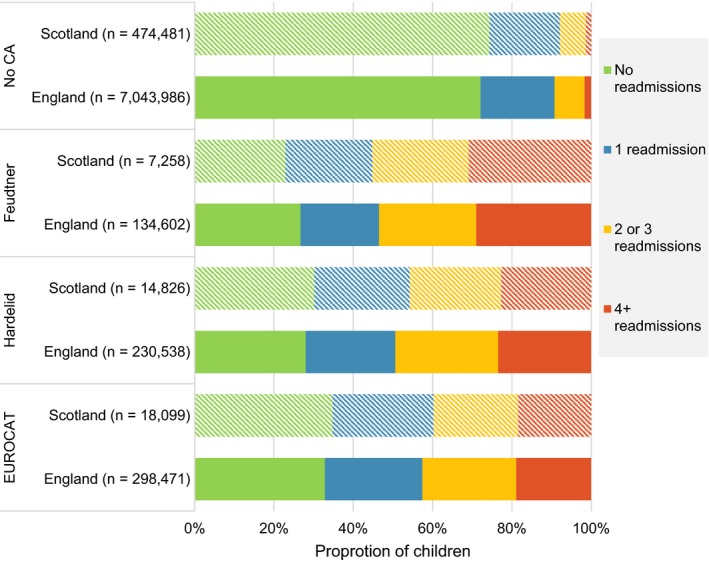
Proportion of children by congenital anomaly code list and number of hospital readmissions after birth in the first two years of life. Each bar represents the proportion of children who had 0, 1, 2‐3, and 4 + hospital readmissions after birth for England (solid bars) and Scotland (patterned bars). No CA, no congenital anomalies
3.4. Mortality
The mortality rates before 2 years old were highest for children identified using the Feudtner code list (75.4 deaths/1000 children with a congenital anomaly in England and 88.7/1000 in Scotland). Rates were lower for those identified by the Hardelid code list (53.4/1000 and 50.5/1000, respectively) and lowest using the EUROCAT code list (42.6/1000 and 41.5/1000, respectively, Table 1 and Figure 1C). Depending on the code list, mortality in children with congenital anomalies was 15‐32 times higher than for children with no congenital anomalies (2.7/1000 in England and 2.9/1000 in Scotland, RR and 95% CIs are presented in Table 1). For all three code lists, 51.7%‐52.4% of deaths in children with congenital anomalies in England and 56.5%‐57.3% in Scotland were in children aged 0‐27 days, and 32.7%‐35.2% were in children aged 28‐364 days (Table S4).
3.5. Sensitivity analyses
The majority of children with congenital anomalies across all code lists were identified in the first month of life; in England, 56.4%‐67.4% of congenital anomalies were first recorded at birth. The EUROCAT codes were most likely to be recorded during the first month of life (accounting for 76.1% and 75.1% of children diagnosed using EUROCAT code list in England and Scotland, respectively), whereas Feudtner codes were least likely to be recorded in that time (69.0% and 67.8%, Table 3).
Table 3.
Distribution of children in England and Scotland according to age at first diagnosis of a congenital anomaly by code list
| Timing of diagnosis | England | Scotland | ||||
|---|---|---|---|---|---|---|
| Eurocat | Hardelid | Feudtner | Eurocat | Hardelid | Feudtner | |
| First month (age 0‐27 d) | 226 996 (76.1%) | 166 130 (72.1%) | 92 887 (69.0%) | 14 222 (75.1%) | 10 898 (70.0%) | 5238 (67.8%) |
| Birth | 201 062 (67.4%) | 141 704 (61.5%) | 75 923 (56.4%) | |||
| 0‐6 d | 14 580 (4.9%) | 13 524 (5.9%) | 9085 (6.7%) | |||
| 7‐27 d | 11 354 (3.8%) | 10 902 (4.7%) | 7879 (5.9%) | |||
| First 3 mo (age 28‐89 d) | 17 168 (5.8%) | 15 152 (6.6%) | 11 521 (8.6%) | 1351 (7.1%) | 1373 (29.3%) | 937 (37.6%) |
| First year (age 90‐365 d) | 29 915 (10.0%) | 27 402 (11.9%) | 19 363 (14.4%) | 1960 (10.4%) | 1960 (41.9%) | 1102 (44.2%) |
| Second year (age 1‐2 y) | 24 387 (8.2%) | 21 849 (9.5%) | 10 827 (8.0%) | 1404 (7.4%) | 1347 (28.8%) | 453 (18.2%) |
In England, the mean number of recorded ICD‐10 codes per hospital admission increased from 2.0 diagnostic codes per hospital admission in 2003 to 2.5 in 2013. The median number of diagnostic codes remained at 2 throughout the study period in England (Table S5).
4. COMMENT
4.1. Principal findings
The prevalence of congenital anomalies ranged between 1.8% and 4.1% in England and between 1.5% and 3.7% in Scotland, depending on the code list used. In both countries, the prevalence was highest using EUROCAT code list and lowest using Feudtner code list. Approximately seven out of ten children with a congenital anomaly had at least one postnatal hospital readmission before 2 years old and between four and nine per 100 children with a congenital anomaly died before two years old. Feudtner codes identified the smallest but most severely affected group, and the EUROCAT code list identified the largest and least severely affected group.
4.2. Strengths of the study
A strength of our study is our use of national birth cohorts based on administrative hospital databases for two UK countries, with similar NHS health systems. As 97% of deliveries occur in NHS hospitals, our data enabled us to calculate nationally representative prevalence of congenital anomalies, overcoming limitations of regional congenital anomaly registers. Whole‐country coverage allowed us to compare long‐term outcomes in children with congenital anomalies relative to children with no anomalies. National birth cohorts from administrative databases in England and Scotland offer an excellent, low cost resource for studying the health of children with congenital anomalies and associated risk factors, due to the availability of additional birth details (such as birthweight and gestational age), longitudinal follow‐up through linkage to hospital and mortality records,14 and possibility of linking babies to mother's records (eg to obtain information about maternal infections during pregnancy).13
4.3. Limitations of the data
One limitation is that we could not validate the accuracy of hospital records indicating congenital anomalies. We did not have permissions or capacity to validate hospital records directly by accessing case notes. Nor could we determine record accuracy by a comparison with a reference standard, for example through record linkage between high‐quality congenital anomaly registry data and hospital administrative data. However, coding is likely to be specific, given requirements to record only definitive diagnoses, and routine auditing within hospitals. In England, historical data from five regional congenital anomaly registers collected until April 2015 as part of the British and Isles Network of Congenital Anomaly Research Database (BINOCAR) is undergoing retrospective linkage to hospital admission records, birth and death registration, and educational databases. Once complete, de‐identified data will be available for research purposes.23 These data, however, cover only 36% of births in England excluding for example London, East and South East of England.24 Since April 2015, congenital anomaly surveillance is carried out by Public Health England as part of the National Congenital Anomaly and Rare Disease Registration Service (NCARDRS). NCARDRS collects data from BINOCAR registers and established additional regional reporting to ensure whole‐country coverage since April 2017.25 In Scotland, a national Congenital Anomalies and Rare Diseases (CARDRISS) register is under development since 2018.26 Therefore, cross‐validation of congenital anomaly coding in hospital against those resources will be possible in both countries in the future.
Additional linkage between administrative data sources and congenital anomaly registers would also likely improve case ascertainment in congenital anomaly registers. For example, according to BINOCAR data for England reported to the EUROCAT, the prevalence of congenital anomalies in livebirths in 2005‐2012 was 1.8%27 compared with 4.1% estimated using the EUROCAT code list in the English administrative data birth cohort in 2003‐2014. Figures for Scotland were 3.5% in 2003‐2011. These differences could be explained by differences in population coverage and in the data collection process. Specifically, BINOCAR/NCARDRS‐participating registers collect data reported by midwives, GPs and paediatricians, and reported congenital anomalies are mainly diagnosed at birth or during newborn examinations.24 This may miss internal organ anomalies, which are numerous and could be recorded in hospital data days or weeks after birth during a hospital admission for a related procedure. Linkage to primary care records is needed to capture congenital anomalies which might be diagnosed or confirmed after birth, and do not require hospitalisation. A cohort study from Bradford showed that linking congenital anomaly register data with primary care records with follow‐up until the fifth birthday improved the case ascertainment, increasing prevalence of congenital anomalies from 4.3% in infants to 6.2% in before 5 years old.28 Similarly, the prevalence of congenital heart disease was twice as high when using primary care records for England compared with the UK prevalence calculated using congenital anomaly registers participating in EUROCAT network and the National Congenital Anomaly System.29 Therefore, congenital anomaly registers need to be linked not only to hospital admissions, but also to primary care records to ensure complete case ascertainment.
Lastly, our study was limited to singleton livebirths only. In English data, same sex siblings from a multiple births are more likely to be falsely allocated the same study‐specific identifier. To minimise potential bias from linkage error, we excluded multiple births from the analyses.15 Multiple births, however, have a higher risk of most congenital anomalies, including more severe congenital anomalies of central nervous system, cardiovascular and musculo‐skeletal systems.30 The national prevalence of congenital anomalies is therefore likely to be slightly higher than that based on singleton births only.
4.4. Interpretation
The prevalence of congenital anomalies varied with the type of code list, between England and Scotland, and over time. The EUROCAT list identified the highest proportion of children affected by congenital anomalies, earliest in life, and with the lowest rate of readmission and mortality. The Feudtner code list identified the fewest children, with the highest risk of adverse outcomes such as mortality or postnatal hospital readmission. The Hardelid code list repeatedly identified an intermediary group. These differences are consistent with the purposes for which the lists were developed: surveillance (EUROCAT), identifying children in hospitalisation data who have chronic conditions (Hardelid), and those with complex chronic conditions (Feudtner).
We have shown that administrative health data can be used to derive nationally representative figures for prevalence of congenital anomalies which require hospital care or are likely to be diagnosed at birth. The estimated prevalence rates need to be interpreted with caution, however, as they may be affected by coding practices and thresholds for hospital admissions in a given country. For example, we showed differing prevalence rates of congenital anomalies in England and Scotland consistent with the number of diagnostic codes available for each admission (ie higher prevalence in England, where 20 diagnostic codes could be recorded, compared with six in Scotland). The risk of postnatal readmission to hospital was higher in Scotland, while prevalence of congenital anomaly was lower there. This suggests that children with congenital anomalies identified in Scotland might have been more severely affected, or that minor anomalies were less often recorded due to the limited number of diagnosis fields available in Scottish data. We observed increases in the prevalence of congenital anomalies in England over time, where financial incentives for increased coding depth are present. We did not observe comparable increase in Scotland. Increases over time in England could also partly reflect declining thresholds for hospital admissions, suggested by increases in the risk of hospital readmission after birth both for children with and without congenital anomalies over time. These findings illustrate that trends and inter‐country comparisons of congenital anomaly prevalence based on hospital admission data need to be interpreted with caution and sensitivity analyses using multiple code lists are recommended.
5. CONCLUSIONS
Multiple code lists are available to monitor the prevalence and type of congenital anomalies and identify children with different severities. Furthermore, different thresholds for coding and hospital admissions can influence prevalence rates and measured risk of adverse outcomes. Researchers need to bear this in mind when attempting to identify children with congenital anomalies as part of their studies. Future research should investigate whether linkage of primary records and hospital records for mild cases of congenital anomalies and hospital records alone for the detection of severe congenital anomalies would suffice. Monitoring of fetal losses, stillbirths, and terminations of pregnancy due to congenital anomalies would likely require linkage of hospital admission, death registration, maternity, prenatal ultrasound, cytogenetic laboratory, termination registrations, and primary care records. Such far‐reaching linkage of data about sensitive and potentially disclosive events is unlikely to be developed for real‐time monitoring. An enhanced congenital anomaly register, linking register data with hospital, primary care and civil registration records, including termination registrations, is needed for identification of cases and valuable follow‐up. In the absence of such wider linkage between health care data sources, we recommend that researchers who use administrative hospital data to measure congenital anomalies explore the recording of congenital anomalies in their data and repeat their analyses using alternative code lists to examine the sensitivity of results to the choice of code list.
CONFLICT OF INTEREST
We declare no competing interests.
Supporting information
Zylbersztejn A, Verfürden M, Hardelid P, Gilbert R, Wijlaars L. Phenotyping congenital anomalies in administrative hospital records. Paediatr Perinat Epidemiol. 2019;34:21–28. 10.1111/ppe.12627
Zylbersztejn and Verfürden shared first authors.
Funding information
This work was supported by NIHR funding through a Senior Investigator award (Gilbert), the GOSH BRC, Health Data Research UK, and the Children and Families Policy Research Unit. This research was funded by the Economic and Social Research Council (grant ES/L007517/1) establishing the Administrative Data Research Centre for England (ADRC‐E). MV was supported by the Economic and Social Research Council in form of a UBEL DTP studentship. RG, PH, LW, and AZ were (in part) supported by the National Institute for Health Research (NIHR) Children and Families Policy Research Unit. The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care. Research at UCL Great Ormond Street Institute of Child Health is supported by the NIHR Great Ormond Street Hospital Biomedical Research Centre. This work uses data provided by patients and collected by the NHS as part of their care and support.
DATA AVAILABILITY STATEMENT
Source data for England can be accessed by researchers applying to the NHS Digital. Copyright © 2017. Re‐used with the permission of NHS Digital. All rights reserved.
Source data for Scotland can be accessed by researchers applying to the electronic Data Research and Innovation Service (eDRIS). The data for this study were accessed under agreements PAC Reference 09/11 and 01/12 and PBPP 1516‐0405.
REFERENCES
- 1. Wang H, Naghavi M, Allen C, et al. Global, regional, and national life expectancy, all‐cause mortality, and cause‐specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015. The Lancet. 2016;388(10053):1459‐1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. EUROCAT . Special Report: Primary Prevention of Congenital Anomalies in European Countries. http://www.eurocat-network.eu/content/Special-Report-Primary-Preventions-of-CA.pdf. Accessed March 25, 2019. [Google Scholar]
- 3. Lechat MF, Dolk H. Registries of congenital anomalies: EUROCAT. Environ Health Perspect. 1993;101(Suppl 2):153‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dolk H, Loane M, Garne E. The prevalence of congenital anomalies in Europe. Adv Exp Med Biol 2010;686:349‐364. [DOI] [PubMed] [Google Scholar]
- 5. Dolk H, Loane M, Garne E. Group a ES of CA (EUROCAT) W. congenital heart defects in Europe. Circulation. 2011;123:841‐849. [DOI] [PubMed] [Google Scholar]
- 6. Wijlaars LPMM, Gilbert R, Hardelid P. Chronic conditions in children and young people: learning from administrative data. Arch Dis Child. 2016;101:881‐885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thygesen LC, Ersbøll AK. When the entire population is the sample: strengths and limitations in register‐based epidemiology. Eur J Epidemiol. 2014;29:551‐558. [DOI] [PubMed] [Google Scholar]
- 8. Zylbersztejn A, Gilbert R, Hjern A, Wijlaars L, Hardelid P. Child mortality in England compared with Sweden: a birth cohort study. The Lancet. 2018;391:2008‐2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. EUROCAT . EUROCAT Guide 1. 4 and Reference Documents (Last update version 28/12/2018). 2013.
- 10. Hardelid P, Dattani N, Davey J, Pribramska I, Gilbert R. Overview of child deaths in the four UK countries. 2013; 2013. [DOI] [PMC free article] [PubMed]
- 11. Feudtner C, Feinstein JA, Zhong W, Hall M, Dai D. Pediatric complex chronic conditions classification system version 2: Updated for ICD‐10 and complex medical technology dependence and transplantation. BMC Pediatr. 2014;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feudtner C, Christakis DA, Connell FA. Pediatric deaths attributable to complex chronic conditions: a population‐based study of Washington State, 1980–1997. Pediatrics. 2000;106:205‐209. [PubMed] [Google Scholar]
- 13. Harron K, Gilbert R, Cromwell D, Van Der Meulen J. Linking data for mothers and babies in de‐identified electronic health data. PLoS ONE. 2016;11:1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Herbert A, Wijlaars L, Zylbersztejn A, Cromwell D, Hardelid P. Data Resource profile: hospital episode statistics admitted patient care (HES APC). Int J Epidemiol. 2017;46:1093‐1093i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. The Health and Social Care Information Centre (HSCIC) . Methodology for creation of the HES Patient ID (HESID). 2014. [Google Scholar]
- 16. Verfurden M, Gilbert R, Sebire N, Hardelid P. Avoidable mortality from respiratory tract infection and sudden unexplained death in children with chronic conditions: A data linkage study. Arch Dis Child. 2018;103(12):1125‐1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hardelid P, Dattani N, Davey J, Pribramska I, Gilbert R. Overview of child deaths in the four UK countries. London; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. ISD Services . Terminology Services and Clinical Coding | Coding & Terminology Systems. https://www.isdscotland.org/Products-and-Services/Terminology-Services/Coding-and-Terminology-Systems/. Accessed May 1, 2019. [Google Scholar]
- 19. World Health Organization . International statistical classification of diseases and related health problems, 10th revision. Volume 2: instruction manual. 2010th ed Geneva, Switzerland: WHO Library Cataloguing‐in‐Publication Data International; 2011. [Google Scholar]
- 20. Hardelid P, Dattani N, Gilbert R, et al. Estimating the prevalence of chronic conditions in children who die in England, Scotland and Wales: a data linkage cohort study. BMJ Open. 2014;4:e005331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Feudtner C, Christakis DA, Connell FA. Pediatric deaths attributable to complex chronic conditions: a population‐based study of Washington state, 1980–1997. Pediatrics 2000;106:205‐209. [PubMed] [Google Scholar]
- 22. Spiegelman D, Hertzmark E. Easy SAS calculations for risk or prevalence ratios and differences. Am J Epidemiol. 2005;162:199‐200. [DOI] [PubMed] [Google Scholar]
- 23. British and Irish Network of Congenital Anomaly Researchers (BINOCAR) . BINOCARD: British and Irish Network of Congenital Anomaly Research Database. http://www.binocar.org/researchdatabasebinocard. Accessed May 1, 2019. [Google Scholar]
- 24. British and Irish Network of Congenital Anomaly Researchers (BINOCAR) . Congenital Anomaly Statistics 2012; 2014. [Google Scholar]
- 25. Public Health England . National Congenital Anomaly and Rare Disease Registration Service statistics 2016 summary report. https://www.gov.uk/government/publications/ncardrs-congenital-anomaly-annual-data/national-congenital-anomaly-and-rare-disease-registration-service-statistics-2016-summary-report. Accessed May 1, 2019. [Google Scholar]
- 26. ISD Scotland . CARDRISS: Congenital Anomalies and Rare Diseases. https://www.isdscotland.org/Health-Topics/Maternity-and-Births/CARDRISS/. Accessed May 1, 2019. [Google Scholar]
- 27. European Platform on Rare Diseases Registration . EUROCAT Prevalence charts and tables. https://eu-rd-platform.jrc.ec.europa.eu/eurocat/eurocat-data/prevalence/export/. Accessed October 25, 2019. [Google Scholar]
- 28. Bishop C, Small N, Mason D, et al. Improving case ascertainment of congenital anomalies: findings from a prospective birth cohort with detailed primary care record linkage. BMJ Paediatr Open. 2017;1:e000171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wurst KE, Ephross SA, Loehr J, Clark DW, Guess HA. Evaluation of the general practice research database congenital heart defects prevalence: Comparison to United Kingdom national systems. Birth Defects Res A. 2007;79:309‐316. [DOI] [PubMed] [Google Scholar]
- 30. Dawson AL, Tinker SC, Jamieson DJ, et al. Twinning and major birth defects, National Birth Defects Prevention Study, 1997–2007. J Epidemiol Community Health. 2016;70:1114‐1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data for England can be accessed by researchers applying to the NHS Digital. Copyright © 2017. Re‐used with the permission of NHS Digital. All rights reserved.
Source data for Scotland can be accessed by researchers applying to the electronic Data Research and Innovation Service (eDRIS). The data for this study were accessed under agreements PAC Reference 09/11 and 01/12 and PBPP 1516‐0405.