

Abstract
Background & Aims
The loss‐of‐function rs72613567 T > TA‐variant in the 17β‐hydroxysteroid dehydrogenase 13 (HSD17B13) gene might protect from alcoholic and non‐alcoholic fatty liver disease (ALD/NAFLD) and associated fibrosis/cirrhosis. We investigated the impact of the T > TA‐variant on hepatic decompensation and mortality and investigated its implications on retinol and sex steroid metabolism in patients who had already developed advanced chronic liver disease (ACLD).
Methods
Retrospective analysis in prospectively characterized patients with viral hepatitis‐ and ALD/NAFLD‐induced portal hypertension (hepatic venous pressure gradient (HVPG) ≥ 6 mmHg) diagnosed at the Medical University of Vienna.
Results
Among 487 patients who were followed longitudinally, 166 (34%) were heterozygous and 24 (5%) were homozygous for the ‘protective’ TA‐allele. Patients harbouring at least one TA‐allele had a lower MELD (9 (8‐12) vs 10 (8‐13) points; P = .003) and showed a trend towards lower HVPG (16 ± 6 vs 17 ± 7 mmHg; P = .067). Interestingly, in competing risk analyses adjusted for age, HVPG and MELD, harbouring the TA‐allele was associated with numerically increased risks for mortality (adjusted subdistribution hazard ratio (aSHR): 1.3 (95% confidence interval (95% CI): 0.888‐1.91); P = .18), liver‐related death (aSHR: 1.34 (95% CI: 0.9‐1.98); P = .15) and hepatic decompensation (aSHR: 1.29 (95% CI: 0.945‐1.77); P = .11). This might be explained by trends towards worse outcomes (eg liver‐related death: aSHR: 1.64 (95% CI: 0.95‐2.84); P = .076) in patients with viral hepatitis‐induced ACLD. In a cross‐sectional analysis of 211 additional patients, serum retinol levels were comparable between HSD17B13 genotypes, but in males, serum testosterone levels numerically decreased with an increasing number of TA‐alleles.
Conclusion
In patients with viral hepatitis‐ and ALD‐induced portal hypertension, the T > TA‐variant was not protective of hepatic decompensation and mortality. Further studies should investigate the pathophysiological mechanisms underlying the effects of HSD17B13 genotype at different stages of liver disease.
Keywords: alcoholic liver disease, cirrhosis, non‐alcoholic fatty liver disease, viral hepatitis
Abbreviations
- 95% CI
95% confidence interval
- ACLD
advanced chronic liver disease
- ALD
alcohol‐related liver disease
- aSHR
adjusted subdistribution hazard ratio
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HSD17B13
17β‐hydroxysteroid dehydrogenase 13
- HVPG
hepatic venous pressure gradient
- INR
international normalized ratio
- IVD
in vitro diagnostics
- MELD
model for end‐stage liver disease
- NAFLD
non‐alcoholic fatty liver disease
- PNPLA3
patatin‐like phospholipase domain containing 3
- SERPINA1
serpin family A member 1
- TM6SF2
transmembrane 6 superfamily 2
Keypoints.
The 17β‐hydroxysteroid dehydrogenase 13 (HSD17B13) rs72613567 T > TA variant might protect from alcoholic and non‐alcoholic fatty liver disease (ALD/NAFLD) and associated fibrosis/cirrhosis.
Data in patients who had already developed advanced chronic liver disease (ACLD) are limited.
Patients with ACLD exhibiting at least one TA allele had a lower model for end‐stage liver disease score and showed a trend towards a lower hepatic venous pressure gradient.
The T > TA variant was not protective of hepatic decompensation and mortality during follow‐up and even tended to increase the risks in patients with viral hepatitis.
Serum retinol levels were comparable between HSD17B13 genotypes, but in males, serum testosterone levels numerically decreased with an increasing number of TA‐alleles.
1. INTRODUCTION
Advanced chronic liver disease (ACLD), which subsumes advanced liver fibrosis, cirrhosis and portal hypertension, is a major cause of morbidity and mortality worldwide. In Europe, it accounts for about 150 000 deaths per year, with alcohol‐related liver disease (ALD), chronic hepatitis B and C (ie viral hepatitis) and non‐alcoholic fatty liver disease (NAFLD) being the most common aetiologies.1
Importantly, liver disease progression shows substantial interindividual variability, and thus, research has focused on the identification of genetic factors which accelerate the progression to ACLD/cirrhosis and predispose for the development of liver‐related events.2, 3
The patatin‐like phospholipase domain containing 3 (PNPLA3) I148M variant has been linked to NAFLD and ALD, as well as hepatic steatosis and liver fibrosis in patients with viral hepatitis.4 In addition, a series of common (eg transmembrane 6 superfamily 2 (TM6FS2) E167K) and rare (eg serpin family A member 1 (SERPINA1) E342) genetic variants have repeatedly been associated with NAFLD and ALD as well as its severity,5, 6, 7, 8 and less consistently, hepatic steatosis and liver fibrosis in patients with viral hepatitis.9
We have recently demonstrated that homozygosity for the PNPLA3 I148M variant doubles the risks of hepatic decompensation and (liver‐related) mortality in patients who had already developed portal hypertension owing to ALD and NAFLD.10 Similarly, heterozygosity for SERPINA1 E342 increased the risks of liver‐related events in patients with cirrhosis,11 whereas longitudinal data on the impact of other genetic variants in patients with ACLD are still limited.12
In contrast to all of these detrimental variants, a recent study comprising data from the DiscovEHR cohort 13 revealed, that the common loss‐of‐function rs72613567 T > TA variant in the 17β‐hydroxysteroid dehydrogenase 13 (HSD17B13) gene protects from NAFLD and ALD, as well as associated liver fibrosis/cirrhosis.14 The 17β‐hydroxysteroid dehydrogenases (17β‐HSD) are a family of 14 enzymes playing key roles in sex steroid metabolism, cholesterol biosynthesis, as well as elongation and oxidation of fatty acids.15, 16, 17, 18 Similar to 17β‐HSD11, 17β‐HSD13 has been localized to both lipid droplets and the endoplasmic reticulum.19 Lipid droplets are cytoplasmic organelles dedicated to lipid storage and formed by budding from the membrane of the endoplasmic reticulum. The active site of 17β‐HSD13 protrudes into the cytosol 16 and both wild‐type 17β‐HSD13 and the truncated protein derived from the T > TA variant were found to be expressed on lipid droplets,14 with an upregulation in NAFLD, as compared to healthy controls.20 The immunohistochemistry performed in the study by Pirola et al21 suggests, that the levels of 17β‐HSD13, which was only found in hepatocytes, decreased according to the number of TA alleles. In contrast to other members of the 17β‐HSD family, little is known about the functions of 17β‐HSD13 in health and disease. Upregulation of this lipid droplet‐associated protein has previously been observed in NAFLD patients and also increased the number and size of lipid droplets in cultured hepatocytes.20, 22 Despite the limited knowledge on its role in the pathophysiology of chronic liver diseases, 17β‐HSD13 has already been proposed as a therapeutic target.21, 23
Based on its protective effect on NAFLD and ALD progression, we hypothesized that the T > TA variant decreases the risks of liver‐related events in patients who had already developed ACLD owing to these aetiologies. Moreover, we also included patients with viral hepatitis, since this common aetiology was not assessed in the initial study by Abul‑Husn et al.14 Thus, we aimed to investigate the impact of the T > TA variant on hepatic decompensation and (liver‐related) mortality in patients with viral hepatitis as well as ALD/NAFLD‐induced portal hypertension.
2. PATIENTS AND METHODS
2.1. Study population
This retrospective analysis included 487 (cohort A) and 211 (cohort B) prospectively characterized patients undergoing hepatic venous pressure gradient (HVPG)‐measurement at the Vienna Hepatic Hemodynamic Lab of the Medical University of Vienna from 01/2004‐06/2017 (cohort A) or after this time period (cohort B). Only patients with portal hypertension as defined by a HVPG ≥ 6 mmHg were included. As the impact of HSD17B13 genotype may vary between different aetiologies, our analysis was restricted to patients with viral hepatitis or ALD/NAFLD‐induced ACLD. The aetiology of ACLD was determined after detailed chart review by the hepatologist performing the HVPG‐measurement. The subgroup of patients with viral hepatitis comprised patients with chronic hepatitis B (HBV) or C virus (HCV) infection and also included patients who had achieved viral suppression or sustained virological response. Of note, in a considerable proportion of patients who were diagnosed with ALD/NAFLD, the diagnostic work up did not include a liver biopsy. Additional relevant clinical (ie alcohol consumption) and laboratory information was collected from patients’ medical records.
2.2. 17β‐hydroxysteroid dehydrogenase 13 genotyping (cohort A and B)
DNA was extracted using the QIAamp DNA Blood Mini Kit (QIAGEN) and stored at −20°C until HSD17B13 rs72613567 genotyping was performed by StepOnePlus Real‐Time PCR System and a TaqMan SNP Genotyping Assay (Applied Biosystems).
2.3. Hepatic venous pressure gradient measurement (cohort A and B)
HVPG was measured in accordance with a standardized operating procedure24 using a balloon catheter.25 Potential treatment with non‐selective β‐blockers and/or nitrates was interrupted prior to the assessment of HVPG. Subclinical portal hypertension was defined by a HVPG ≥ 6 mmHg and HVPG ≥ 10 mmHg denoted clinically significant portal hypertension (CSPH).26, 27
2.4. Assessment of alcohol consumption and clinical events (cohort A)
Quantitative information on alcohol consumption was documented in patient discharge letters as part of the anamnesis as well as during visits in the cirrhosis outpatient ward. Alcohol consumption was classified as follows: abstinent, moderate (ie below the cut‐off for differentiating between NAFLD and ALD ‐ ≤30 g/day and ≤20 g/day for males and females, respectively28), or heavy drinking.
Clinical events were retrospectively assessed by reviewing patients’ digital medical records at Medical University of Vienna, which is the only transplant centre in the east of Austria. Moreover, we also assessed patients’ digital medical records from other hospitals within the Vienna Hospital Association, as well as the Austrian register of deaths. The following events defined (further) hepatic decompensation: requirement of paracentesis, hospital admission for grade 3/4 hepatic encephalopathy, variceal bleeding and liver‐related death.10, 29, 30, 31, 32 Deaths owing to complications of ACLD33 or hepatocellular carcinoma (HCC) were classified as liver‐related death.
We abstained from analysing clinical events in cohort B owing to the short duration of follow‐up.
2.5. Assessment of serum retinol and testosterone levels (cohort B)
In cohort B, serum retinol levels were measured by high performance liquid chromatography with ultraviolet detection (Shimadzu) using an in‐vitro diagnostics (IVD) CE‐certified assay (ChromSystems), whereas serum testosterone levels were assessed by quantitative electrochemiluminescence (cobas e 602 module, Roche Diagnostics) using IVD CE‐certified assays.
2.6. Statistical analyses
Statistical analyses were performed using IBM SPSS Statistics 25 (IBM), GraphPad Prism 7 (GraphPad Software) and R 3.4.1 (R Core Team, R Foundation for Statistical Computing).
Continuous variables are reported as mean ± standard deviation or median (interquartile range), and categorical variables are shown as numbers (n) and proportions (%) of patients. Comparisons of continuous variables were performed using Student's t test/one‐way analysis of variance or Mann‐Whitney U test/Kruskal‐Wallis one‐way analysis of variance, as applicable.
Patients entered the survival analyses at the time of HVPG‐measurement. Variables showing a statistically significant difference between genotypes in univariate analyses, or which we considered highly relevant for the prediction of the endpoints (ie age, HVPG and model for end‐stage liver disease (MELD) score) were included as covariates. Cox regression models were calculated to investigate the impact of HSD17B13 rs72613567 genotype on the risk of liver transplantation or (liver‐related) death. Moreover, its effect on overall mortality, liver‐related death and hepatic decompensation was assessed using competing risk analysis considering liver transplantation, and, if applicable (non‐liver‐related) death as competing risks. Therefore, Fine and Gray competing risks regression models (cmprsk: subdistribution analysis of competing risks, https://CRAN.R-project.org/package=cmprsk)34 were calculated.
P ≤ .05 was considered statistically significant.
2.7. Ethics
This study was conducted in accordance with the Declaration of Helsinki and approved by the ethics committee of the Medical University of Vienna (EK 1526/2017) and (EK 1262/2017). All subjects were consented for genetic testing.
3. RESULTS
3.1. Study population (cohort A and B)
In total, 887 (cohort A) and 421 (cohort B) patients underwent HVPG‐measurement within the study period (Figure 1). According to in‐ and exclusion criteria, 190 (cohort A) and 107 (cohort B) patients had to be excluded because of missing information on HSD17B13 rs72613567 genotype, 167 (cohort A) and 96 (cohort B) were not included owing to earlier stages of liver disease (HVPG < 6 mmHg). Additionally, 43 (cohort A) and 1 (cohort B) were excluded since they had other aetiologies of liver disease than viral hepatitis or ALD/NAFLD and in six patients of cohort B no information on retinol and steroid hormone levels were available.
Figure 1.
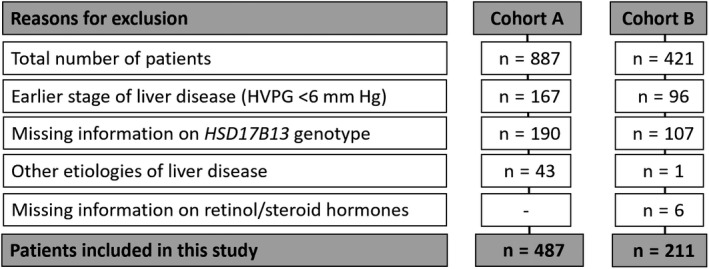
Study flow chart. Abbreviations: HVPG hepatic venous pressure gradient; HSD17B13, 17β‐hydroxysteroid dehydrogenase 13
Finally, 487 thoroughly characterized patients were included in cohort A to investigate the impact of HSD17B13 genotype on liver‐related events and 211 patients were included in cohort B to study potential underlying pathophysiological mechanisms.
3.2. Patient characteristics according to HSD17B13 rs72613567 genotype (cohort A and B)
The majority of patients included in cohort A were male (75%) with a mean age of 54 ± 11 years. Fifty‐nine percent had viral hepatitis, whereas 41% had ALD/NAFLD. Of the 202 patients with alcoholic (ALD)/non‐alcoholic fatty liver disease (NAFLD) included in our study, 146 had ALD, whereas only 56 had NAFLD. Overall, 297 patients harboured the wild‐type (T/T), whereas 166 patients had one and 24 had two TA alleles. Comparisons of patient characteristics between the three genotypes are depicted in Table 1. Because of the low number of patients homozygous for the TA allele (only 5% of the study population), we decided to merge patients with at least one TA allele for all further analyses (Table 1). Thirty‐nine percent harboured at least one TA allele, whereas 61% of patients presented with wild‐type (T/T). In accordance with the differences observed when comparing the three genotypes separately, comparisons of patients with at least one T/A allele, or without, revealed that T/T patients had higher INR (1.3 ± 0.2 vs 1.2 ± 0.2; P < .001) and bilirubin levels (1.2 (0.8‐2) vs 1 (0.7‐1.6) mg × dL−1; P = .011) as well as MELD scores (10 (8‐13) vs 9 (8‐12) points; P = .003). Furthermore, these patients tended to have a higher HVPG (17 ± 7 vs 16 ± 6 mmHg; P = .067) and lower serum albumin levels (36 ± 6 vs 37 ± 6 g × L−1; P = .077).
Table 1.
Comparison of patient characteristics according to the 17β‐hydroxysteroid dehydrogenase 13 (HSD17B13) rs72613567 genotype
| Patient characteristics | HSD17B13 | HSD17B13 | |||||
|---|---|---|---|---|---|---|---|
|
TA/TA, n = 24 |
T/TA, n = 166 |
T/T, n = 297 |
P value |
T/TA or TA/TA, n = 190 |
T/T, n = 297 |
P value | |
| Age, y | 57 ± 12 | 55 ± 10 | 54 ± 11 | .417 | 55 ± 10 | 54 ± 11 | .313 |
| Sex | |||||||
| Male | 18 (75%) | 121 (73%) | 226 (76%) | .748 | 139 (73%) | 226 (76%) | .466 |
| Female | 6 (25%) | 45 (27%) | 71 (24%) | 51 (27%) | 71 (24%) | ||
| Etiology | |||||||
| ALD/NAFLD | 12 (50%) | 65 (39%) | 125 (42%) | .568 | 77 (41%) | 125 (42%) | .733 |
| Viral | 12 (50%) | 101 (61%) | 172 (58%) | 113 (59%) | 172 (58%) | ||
| Alcohol consumption | |||||||
| Abstinent | 18 (75%) | 117 (70%) | 210 (71%) | .16 | 135 (71%) | 210 (71%) | .052 |
| Moderatea | 6 (25%) | 28 (17%) | 42 (14%) | 34 (18%) | 42 (14%) | ||
| Severeb | 0 (0%) | 3 (2%) | 17 (6%) | 3 (2%) | 17 (6%) | ||
| Unknown | 0 (0%) | 18 (11%) | 28 (9%) | 18 (9%) | 28 (9%) | ||
| Intravenous drug use | 0 (0%) | 2 (1%) | 1 (0%) | .513 | 2 (1%) | 0 (0%) | .566 |
| HCC | 1 (4%) | 4 (2%) | 18 (6%) | .205 | 5 (3%) | 18 (6%) | .082 |
| HVPG, mmHg | 16 ± 6 | 16 ± 6 | 17 ± 7 | .172 | 16 ± 6 | 17 ± 7 | .067 |
| MELD, points | 10 (8‐12) | 9 (7‐11) | 10 (8‐13) | .009 | 9 (8‐12) | 10 (8‐13) | .003 |
| Albumin, g × L−1 | 37 ± 5 | 37 ± 6 | 36 ± 6 | .257 | 37 ± 6 | 36 ± 6 | .077 |
| Bilirubin, mg × dL−1 | 1 (0.8‐1.8) | 1 (0.7‐1.5) | 1.2 (0.8‐2) | .032 | 1 (0.7‐1.6) | 1.2 (0.8‐2) | .011 |
| INR | 1.2 ± 0.2 | 1.2 ± 0.2 | 1.3 ± 0.2 | .002 | 1.2 ± 0.2 | 1.3 ± 0.2 | <.001 |
| Creatinine, mg × dL−1 | 0.8 (0.7‐1) | 0.8 (0.7‐0.9) | 0.8 (0.7‐0.9) | .909 | 0.8 (0.7‐0.9) | 0.8 (0.7‐0.9) | .671 |
| Sodium, mmol × L−1 | 137 ± 3 | 138 ± 4 | 138 ± 4 | .672 | 138 ± 4 | 138 ± 4 | .746 |
Abbreviations: ALD, alcoholic liver disease; HCC, hepatocellular carcinoma; HVPG, hepatic venous pressure gradient; INR, international normalized ratio; MELD, model for end‐stage liver disease; NAFLD, non‐alcoholic fatty liver disease.
≤30 g/d and ≤20 g/d for males and females respectively.
>30 g/d and >20 g/d for males and females respectively.
Cohort B showed similar patient characteristics, as compared to cohort A (Table S1).
3.3. Impact of HSD17B13 rs72613567 genotype on overall mortality, liver‐related death and (further) hepatic decompensation, as well as HCC in the whole cohort (cohort A)
Patients were followed for a median of 26 (15.4‐41.7) months. In total, 119 patients (24%) died during follow‐up and 111 of these deaths were considered liver‐related. Furthermore, 27 patients (6%) underwent liver transplantation.
Cox regression models adjusted for age, HVPG and MELD score, indicated that carriers of the TA allele had numerically increased risks of liver transplantation or death (adjusted hazard ratio (aHR): 1.29 (95% confidence interval (95% CI): 0.912‐1.82); P = .152) as well as liver transplantation or liver‐related death (aHR: 1.27 (95% CI: 0.759‐2.13); P = .362) (Figure 2, Table 2). To investigate whether sex modifies the effect of the TA allele on liver‐related outcomes, sex and the interaction term of sex and HSD17B13 genotype (HSD17B13 * sex) were added to the above‐mentioned Cox regression models. HSD17B13 * sex did not modify the risks liver transplantation or death (aHR: 0.76 (95% CI: 0.33‐1.76); P = .520) as well as liver transplantation or liver‐related death (aHR: 0.86 (95% CI: 0.36‐2.03); P = .724).
Figure 2.
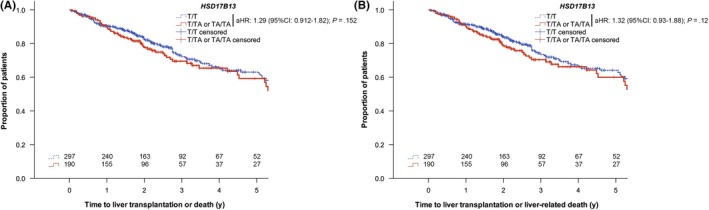
Risks of (A) liver transplantation or death and (B) liver transplantation or liver‐related death according to 17β‐hydroxysteroid dehydrogenase 13 (HSD17B13) rs72613567 genotype. Abbreviations: 95% CI, 95% confidence interval; aHR adjusted hazard ratio
Table 2.
Cox regression analyses on the influence of the 17β‐hydroxysteroid dehydrogenase 13 (HSD17B13) rs72613567 T > TA variant on A liver transplantation or death and B liver transplantation or liver‐related death. Competing risk regression analyses on the influence of the TA allele on C mortality, D liver‐related death and E (further) hepatic decompensation in the overall cohort
| Patient characteristics |
A Liver transplantation or deatha |
B Liver transplantation or liver‐related deathb |
C Mortalitya |
D Liver‐related deathb |
E (Further) hepatic decompensationb |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aHR | 95% CI | P value | aHR | 95% CI | P value | aSHR | 95% CI | P value | aSHR | 95% CI | P value | aSHR | 95% CI | P value | |
| Age, per 10 y | 1.28 | 1.09‐1.51 | .003 | 1.24 | 1.05‐1.46 | .013 | 1.41 | 1.15‐1.73 | .001 | 1.31 | 1.07‐1.6 | .01 | 1.18 | 1.02‐1.37 | .027 |
| HVPG, per mmHg | 1.06 | 1.03‐1.09 | <.001 | 1.06 | 1.03‐1.12 | <.001 | 1.05 | 1.01‐1.09 | .009 | 1.05 | 1.01‐1.09 | .012 | 1.09 | 1.05‐1.12 | <.001 |
| MELD, per point | 1.08 | 1.03‐1.12 | <.001 | 1.07 | 1.03‐1.12 | .001 | 1.04 | 0.98‐1.1 | .21 | 1.02 | 0.96‐1.09 | .44 | 1.05 | 1.0‐1.09 | .034 |
| T/TA or TA/TA, vs T/T (reference) | 1.29 | 0.912‐1.82 | .152 | 1.32 | 0.93‐1.88 | .12 | 1.3 | 0.89‐1.91 | .18 | 1.34 | 0.90‐1.98 | .150 | 1.29 | 0.95‐1.77 | .11 |
Abbreviations: 95% CI, 95% confidence interval; aHR, hazard ratio; aSHR, adjusted subdistribution hazard ratio; HVPG, hepatic venous pressure gradient; MELD, model for end‐stage liver disease.
Considering liver transplantation as a competing risk.
Considering liver transplantation and non‐liver‐related death as competing risks.
To assess the impact of HSD17B13 genotype on overall mortality, non‐liver‐related death and hepatic decompensation in the presence of competing risks, Fine and Gray competing risks regression models were used for all further analyses. When considering liver transplantation (for overall mortality) and liver transplantation as well as non‐liver‐related death (for liver‐related death) as competing risks and adjusting for age, HVPG and MELD score, harbouring at least one TA allele was associated with numerically increased risks for overall mortality (adjusted subdistribution hazard ratio (aSHR): 1.3 (95% CI: 0.89‐1.91); P = .18; 2‐year cumulative incidence: 21% vs 13%; 5‐year: 36% vs 31%) and liver‐related death (aSHR: 1.34 (95% CI: 0.9‐1.98); P = .15; 2‐year cumulative incidence: 17% vs 13%; 5‐year: 33% vs 29%) (Figure 3). Furthermore, competing risks regression analysis indicated that age (per 10 years: aSHR: 1.41 (95% CI: 1.15‐1.73); P = .001) and HVPG (per mmHg: aSHR: 1.05 (95% CI: 1.01‐1.09); P = .009) were independently predictive of mortality (Table 2). The same factors were associated with liver‐related death: age (per 10 years: aSHR: 1.31 (95% CI: 1.07‐1.60); P = .010) and HVPG (per mmHg: aSHR: 1.05 (95% CI: 1.01‐1.09); P = .012).
Figure 3.
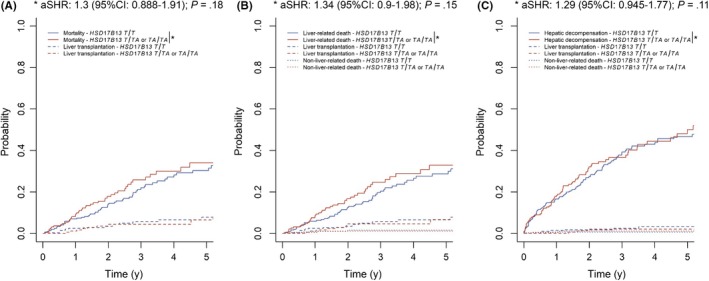
(A) Mortality (considering liver transplantation as a competing risk), as well as (B) liver‐related death and (C) (further) hepatic decompensation (considering liver transplantation and non‐liver‐related death as competing risks) according to 17β‐hydroxysteroid dehydrogenase 13 (HSD17B13) rs72613567 genotype. Abbreviations: 95% CI, 95% confidence interval; aSHR, adjusted subdistribution hazard ratio
Hepatic decompensation occurred in 185 patients (38%). Harbouring at least one TA allele was not protective of (further) hepatic decompensation (aSHR: 1.29 (95% CI: 0.95‐1.77); P = .11; 2‐year cumulative incidence: 32% vs 27%; 5‐year cumulative incidence: 50% vs 47%), if liver transplantation and non‐liver‐related death were considered as competing risks (Figure 3). Age (per 10 years: aSHR: 1.18 (95% CI: 1.02‐1.37); P = .027), HVPG (per mmHg: aSHR: 1.09 (95% CI: 1.05‐1.12); P < .001) and MELD score (per point: aSHR: 1.05 (95% CI: 1‐1.09); P = .034) were independently predictive of (further) hepatic decompensation (Table 2).
Finally, the proportion of patients developing HCC during follow‐up did not differ between carriers of the TA allele (4%) and wild‐type patients (6%; P = .246).
3.4. Patient characteristics according to the aetiology of liver disease and HSD17B13 rs72613567 genotype (cohort A)
As the effects of HSD17B13 genotype may depend on the aetiology of ACLD, cohort A was stratified by underlying aetiology (viral hepatitis vs ALD/NAFLD). Detailed patient characteristics at the time of HVPG‐measurement are shown in Table 3.
Table 3.
Comparison of patient characteristics according to the 17β‐hydroxysteroid dehydrogenase 13 (HSD17B13) rs72613567 genotype in patients with A viral hepatitis and B alcoholic (ALD)/non‐alcoholic fatty liver disease (NAFLD)
| Patient characteristics |
A Viral hepatitis, n = 285 |
B ALD/NAFLD, n = 202 |
||||
|---|---|---|---|---|---|---|
|
T/TA or TA/TA, n = 113 |
T/T, n = 172 |
P value |
T/TA or TA/TA, n = 77 |
T/T, n = 125 |
P value | |
| Age, years | 52 ± 9 | 52 ± 10 | .811 | 59 ± 10 | 56 ± 12 | .056 |
| Sex | ||||||
| Male | 80 (71%) | 132 (77%) | .26 | 59 (77%) | 94 (75%) | .819 |
| Female | 33 (29%) | 40 (23%) | 18 (23%) | 31 (25%) | ||
| Etiology | ||||||
| HBV | 6 (5%) | 15 (9%) | .281 | — | — | — |
| HCV | 107 (95%) | 157 (91%) | — | — | ||
| Viraemiaa | 102 (95%) | 146 (95%) | .849 | — | — | — |
| GT 3b | 16 (19%) | 17 (14%) | .378 | — | — | — |
| HCC | 3 (3%) | 12 (7%) | .11 | 2 (3%) | 6 (5%) | .493 |
| HVPG, mmHg | 14 ± 6 | 15 ± 6 | .221 | 18 ± 6 | 20 ± 6 | .159 |
| MELD, points | 8 (7‐10) | 9 (7‐11) | .121 | 11 (9‐13) | 12 (10‐15) | .003 |
| Albumin, g × L−1 | 37 ± 6 | 37 ± 5 | .679 | 36 ± 6 | 34 ± 7 | .057 |
| Bilirubin, mg × dL−1 | 1 (0.6‐1.4) | 1.1 (0.7‐1.5) | .263 | 1.2 (0.8‐1.9) | 1.6 (0.9‐2.9) | .005 |
| INR | 1.17 ± 0.17 | 1.21 ± 0.21 | .043 | 1.3 ± 0.3 | 1.4 ± 0.2 | .002 |
| Creatinine, mg × dL−1 | 0.8 (0.7‐0.9) | 0.8 (0.7‐0.9) | .193 | 0.8 (0.7‐1) | 0.8 (0.7‐0.9) | .478 |
| Sodium, mmol × L−1 | 138 ± 3 | 138 ± 3 | .874 | 136 ± 4 | 137 ± 4 | .475 |
Abbreviations: GT, genotype; HBV, hepatitis virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; HVPG, hepatic venous pressure gradient; INR, international normalized ratio; MELD, model for end‐stage liver disease.
Of HCV patients.
Of 207 viraemic HCV patients with information on GT.
Among 285 patients with viral hepatitis‐induced portal hypertension, 113 (40%) had at least one TA allele and presented with higher INR (1.21 ± 0.21), when compared with TT patients (1.17 ± 0.17; P = .043). Importantly, HBV and HCV were equally distributed across HSD17B13 genotypes and there were no differences in the proportion of viraemic HCV patients or patients with genotype 3. Moreover, the proportions of HCV patients who were viraemic at the time of HVPG‐measurement and achieved sustained virological response during follow‐up was comparable between HSD17B13 genotypes (T/TA or TA/TA: 70% vs T/T: 74%; P = .559).
In 202 patients with ALD/NAFLD‐induced portal hypertension, 38% were either heterozygous or homozygous for the TA allele. When compared with the overall cohort, similar findings regarding differences in liver disease severity at baseline were observed. T/T patients had higher bilirubin levels (1.6 (0.9‐2.9) vs 1.2 (0.8‐1.9) mg × dL−1; P = .005), INR (1.4 ± 0.2 vs 1.3 ± 0.3; P = .002), as well as MELD scores (12 (10‐15) vs 11 (9‐13) points; P = .003) and tended to have lower serum albumin levels (34 ± 7 vs 36 ± 6 g × L−1; P = .057) than carriers of the T/A allele. Notably, even though presenting with less pronounced liver disease, carriers of the TA allele tended to be older at the time of HVPG measurement (59 ± 10 vs 56 ± 12 years; P = .056).
Separate analyses in patients with ALD‐ and NAFLD‐induced portal hypertension are shown in Table S4.
3.5. Impact of HSD17B13 genotype on overall mortality, liver‐related death and (further) hepatic decompensation, as well as HCC in the subgroups of patients with viral hepatitis and ALD/NAFLD (cohort A)
The findings obtained in the whole cohort seemed to be more pronounced in patients with viral hepatitis (Table S2; Figure 4). Again, harbouring at least one TA allele tended to increase the risks for overall mortality (aSHR: 1.42 (95% CI: 0.83‐2.42); P = .2) and liver‐related death (aSHR: 1.64 (95% CI: 0.95‐2.84); P = .076). High HVPG increased the risks for overall mortality (per mmHg: aSHR: 1.07 (95% CI: 1.02‐1.13); P = .006) as well as liver‐related mortality (per mmHg: aSHR: 1.07 (95% CI: 1.01‐1.13); P = .015), independently of the other covariates. Finally, TA allele carriers had a numerically increased risk for (further) hepatic decompensation (aSHR: 1.44 (95% CI: 0.9‐2.3); P = .12). Again, HVPG was independently related to the outcome of interest (per mmHg: aSHR: 1.11 (95% CI: 1.06‐1.16); P < .001).
Figure 4.
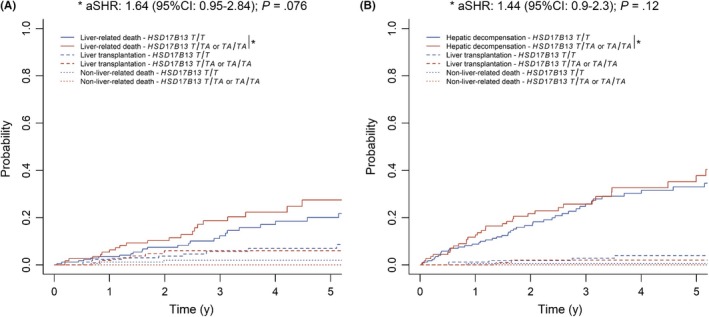
(A) Liver‐related death and (B) (further) hepatic decompensation according to 17β‐hydroxysteroid dehydrogenase 13 (HSD17B13) rs72613567 genotype in viral hepatitis patients, considering liver transplantation and non‐liver‐related death as competing risks. Abbreviations: 95% CI, 95% confidence interval; aSHR, adjusted subdistribution hazard ratio
There was not difference in the proportion of viral hepatitis patients developing HCC when comparing patients with (4%) or without (5%; P = .374) the TA allele.
The TA allele did not reduce the risks for overall mortality (aSHR: 1.18 (95% CI: 0.68‐2.04); P = .55), liver‐related death (aSHR: 1.06 (95% CI: 0.6‐1.9); P = .83), or (further) hepatic decompensation (aSHR: 1.18 (95% CI: 0.77‐1.82); P = .45) in patients with ALD/NAFLD‐induced portal hypertension (Table S3; Figure 5). With aSHR of 1.14 ((95% CI: 0.605‐2.13); P = .69) and 0.996 ((95% CI: 0.309‐3.22); P = 1) for mortality, HSD17B13 genotype seemed to have similar effects in patients with ALD and NAFLD respectively. Moreover, HSD17B13 genotype did not impact liver‐related death in ALD (aSHR: 1.25 (95% CI: 0.654‐2.4); P = .5) and NALFD (aSHR: 0.571 (95% CI: 0.153‐2.13); P = .41) patients.
Figure 5.
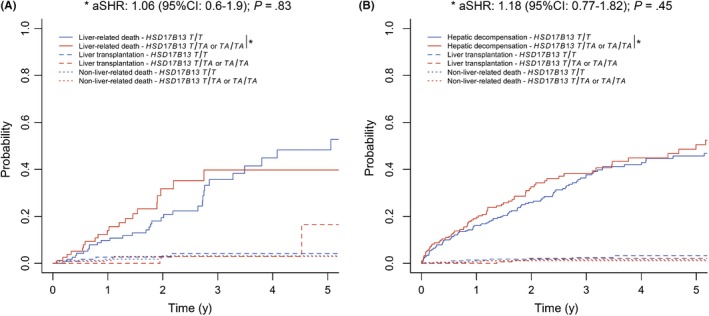
(A) Liver‐related death and (B) (further) hepatic decompensation according to 17β‐hydroxysteroid dehydrogenase 13 (HSD17B13) rs72613567 genotype in patients with alcoholic (ALD)/non‐alcoholic fatty liver disease (NAFLD), considering liver transplantation and non‐liver‐related death as competing risks. Abbreviations: 95% CI, 95% confidence interval; aSHR, adjusted subdistribution hazard ratio
Finally, the proportion of ALD/NAFLD patients developing HCC during follow‐up did not differ between carriers of the TA allele (3%) and wild‐type patients (5%; P = .493).
3.6. Impact of HSD17B13 genotype on retinol and testosterone serum levels (cohort B)
Serum retinol levels were comparable between cohort B patients with different HSD17B13 (TA/TA: 0.76 ± 0.54 vs T/TA: 0.75 ± 0.42 vs T/T: 0.8 ± 0.44; P = .817; Figure S2) as well as PNPLA3 genotypes (G/G: 0.77 ± 0.40 vs G/C: 0.77 ± 0.45 vs C/C: 0.8 ± 0.45; P = .904; Figure S3).
Among males, there was a numerical trend towards lower serum testosterone levels in patients harbouring a TA allele, which did not attain statistical significance (TA/TA: 3.75 ± 2.03 vs T/TA: 4.95 ± 3.62 vs T/T: 5.65 ± 3.02; P = .339; Figure S4).
4. DISCUSSION
Recently, a study linking exome sequence data from the DiscovEHR cohort13 to electronic health records reported, that the HSD17B13 rs72613567 T > TA variant is associated with reduced risks of ALD/NAFLD as well as alcoholic and non‐alcoholic cirrhosis. Moreover, the study reported associations with non‐alcoholic steatohepatits and liver fibrosis in patients undergoing bariatric surgery. Therefore, the authors concluded that T > TA variant protects against the progression to ACLD in fatty liver disease.14 The association between the T > TA variant and NASH as well as liver fibrosis has subsequently been confirmed by independent studies comprising subjects from Argentina21 and the USA as well as the UK.20 In contrast, we aimed to evaluate the impact of TA allele on the clinical course of patients who had already ACLD, as indicated by an HVPG ≥ 6 mmHg.26
In cohort A of this study, patients harbouring at least one TA allele presented with changes typically associated with a less pronounced liver disease. When compared with T/T patients, TA allele carriers had lower INR, bilirubin and MELD score and also showed a trend towards lower HVPG and higher albumin levels at the time of HVPG‐measurement. Of note, these associations were not reproduced in the considerably smaller cohort B. Importantly, the T > TA variant was not protective of hepatic decompensation and (liver‐related) mortality during follow‐up and even tended to increase the risks in the subgroup of patients with patients with viral hepatitis. Accordingly, once ACLD is established, its protective effect may vanish.
In search of potential pathophysiological mechanisms explaining the different findings in our longitudinal study in ACLD patients, as compared to previous cross‐sectional studies which primarily included non‐ACLD patients, we investigated the effect of HSD17B13 genotype on serum levels of retinol as well as testosterone (as an example for its potential impact on sex hormone metabolism). This analysis was performed in cohort B, which had similar patient characteristics, as compared to cohort A.
Interestingly, the PNPLA3 I148M variant has been shown to affect retinol metabolism because of the function of PNPLA3 as a retinyl‐palmitate lipase,35 with lower circulating levels of retinol in subjects with NAFLD or obesity harbouring the I148M variant.36 In cohort B, neither PNPLA3 nor HSD17B13 variants had an effect on circulating retinol levels measured by the same method as in the previous study,36 which may suggest, that the effects of PNPLA3 (and possibly, also HSD17B13) on retinol metabolism are less pronounced in patients with ACLD, as compared to earlier stages of liver disease. In line with these findings, PNPLA3 I148M only affected liver disease progression in patients with ALD/NAFLD‐induced ACLD when analysed in a recessive model (ie G/G vs other genotypes) and did not affect liver disease progression in patients with ACLD owing to viral hepatitis included in our previous study.10 Accordingly, also for PNPLA3 I148M, its effects in ACLD seemed to be less consistent than in earlier stages of liver disease, which could possibly be related to a decreased impact on retinol metabolism.
The 17β‐HSD13 isoform derived from the TA allele has been found to be catalytically inactive against oestradiol in a previous study.14 Although sex did not appear to modify the effect of HSD17B13 genotype on liver‐related events, we investigated serum testosterone levels in cohort B. Interestingly, we observed numerically lower serum levels of testosterone (Figure S4) in male patients harbouring the loss‐of‐function TA allele. As low testosterone levels have repeatedly been linked to liver‐related events by our group37 and others, a potential detrimental impact of the HSD17B13 TA allele in patients with ACLD via decreased testosterone biosynthesis warrants further study in a larger series of patients.
Of note, the impact of T > TA variant appeared to be aetiology‐dependent. While harbouring the T > TA variant was associated with numerically increased risks for adverse clinical outcomes in viral hepatitis patients, it did not seem to affect follow‐up events in ALD/NAFLD patients. Although there are differences regarding predisposing factors for NAFLD/ALD and pathophysiological mechanisms driving disease progression, variants in the PNPLA3, TM6SF2, as well as the SERPINA1 genes have been shown to increase the susceptibility for both diseases and have also been associated with adverse outcomes,5, 6, 8 whereas variants in HSD17B13 have been found to be protective.14 Therefore, we decided to merge these two entities in order to maximise the statistical power of our main analyses. Analysing both aetiologies separately, HSD17B13 genotype seemed to have similar effects on mortality in patients with NAFLD and ALD. However, considering the small sample size and broad 95% CI in the NAFLD subgroup, no firm conclusions can be drawn regarding the impact of the HSD17B13 genotype in NAFLD patients in particular. Importantly, this is the first study providing information on the impact of the TA allele in viral hepatitis, and thus, there is no information on its impact on the progression to ACLD in this aetiology. However, while showing similar associations ALD/NAFLD, another HSD17B13 variant (rs6834314) which is in strong linkage disequilibrium with the HSD17B13 rs72613567 T > TA variant had no impact on hepatic inflammation or liver fibrosis in patients with hepatitis C virus (HCV) infection.20 The HCV lifecycle is closely linked to lipid metabolism.38 Recently, the TM6FS2 E167K variant has been shown to be required for maturation, lipidation and secretion of infectious lipoviroparticles.39 Therefore, HCV upregulates the expression of TM6SF2 to facilitate productive infection. Accordingly, a potential interaction between the lipid droplet‐associated protein 17β‐HSD13 and HCV infection requires further study.
The prevalence of HCC at the time of HVPG‐measurement tended to be lower in carriers of the TA allele, which is in line with a recent report in patients with ALD.40 However, there were no differences in the proportion of patients developing HCC during follow‐up, which may be explained by limited sample size.
We would like to point out, that considering the very limited knowledge on the physiological function of 17β‐HSD13 and the lack of understanding of its role in the pathogenesis of (advanced) chronic liver disease and portal hypertension, all of these points are highly speculative and should be seen as suggestions for further research, rather than a definitive interpretation of our findings. Our results indicate liver disease severity‐ and possibly aetiology‐dependent differences in the clinical impact of the T > TA variant. Thus, it clearly requires further experimental and clinical studies before 17β‐HSD13 can be considered as a potential therapeutic target for chronic liver disease.21, 23
The main limitation of our study is its retrospective design; however, patients were thoroughly characterized at the time of HVPG‐measurement. The extensive characterization, which even included the measurement of HVPG,24 is an important strength of our study. The only other longitudinal study investigating the impact of the T > TA variant also included a subgroup of patient with a diagnosis of cirrhosis, however, it was population‐based, and thus, unadjusted for liver disease severity.12 Accordingly, the significance of its findings is substantially limited by the inability to adjust for the between‐genotype differences in liver disease severity at inclusion, which were observed and adjusted for in our study. The limited number of patients homozygous for the TA allele included in our study hindered the assessment of a dose‐dependent effect of the T > TA variant. Of note, when combining cohort 1 and 2, HSD17B13 genotype frequencies were in Hardy‐Weinberg equilibrium (χ 2 = 0.805)41 and the TA allele frequency (22.3%) was comparable to the frequency reported in the literature.14 Accordingly, it would require even larger study populations to investigate dose‐dependent effects of the TA allele. In comparison to the study by Abul‐Husn et al,14 our study has considerably smaller sample size, however, we would like to point out that our study is restricted to a specific stage of liver disease and provides additional longitudinal information. Since we have recently demonstrated that the PNPLA3 I148M variant impacts the risks of hepatic decompensation in a smaller, partially overlapping cohort,10 our study appears to have adequate statistical power to detect clinically meaningful effects. Lastly, our study did not include a validation cohort, as we are not aware of another cohort linking information on HVPG and genetic data.
In conclusion, the T > TA variant was not protective of hepatic decompensation and (liver‐related) mortality during follow‐up. Future studies should investigate the pathophysiological mechanisms underlying the effects of HSD17B13 genotype within different stages of liver disease. Importantly, further experimental and clinical studies with a special focus on ACLD are required before 17β‐HSD13 can be considered as a potential therapeutic target.
CONFLICTS OF INTEREST
The authors have nothing to disclose regarding the work under consideration for publication. However, the authors disclose the following financial activities outside the submitted work: B.Sc. has nothing to disclose. AFS has served as a speaker and/or consultant and/or advisory board member for Boehringer Ingelheim, Gilead, Janssen, MSD and Roche. PS has served as a speaker for Boehringer Ingelheim. TB has served as a speaker and/or consultant and/or advisory board member for Bristol‐Myers Squibb. RP has nothing to disclose. DB has nothing to disclose. B.Si. has nothing to disclose. RS has nothing to disclose. RM has nothing to disclose. AF has served as a speaker and/or consultant and/or advisory board member for AbbVie, Gilead and Intercept. AF owns a patent on a catheter for the measurement of hepatic venous pressure gradient. MP‐R. has served as a speaker and/or consultant and/or advisory board member for Abbott, AbbVie, Bayer, Boehringer Ingelheim, Bristol‐Myers Squibb, Gilead, Janssen, Lilly and MSD, and received research funding from AbbVie, ArQule, Bayer, Daiichi Sankyo, Gilead and MSD. MP has served as a speaker and/or consultant and/or advisory board member for Bayer, Bristol‐Myers Squibb, Eisai, Lilly and Ipsen. MT has served as a speaker and/or consultant and/or advisory board member for Albireo, Bristol‐Myers Squibb, Dr Falk Pharma, Gilead, Intercept, MSD, Novartis, Phenex Pharmaceuticals and Regulus, and has received research funding from Albireo, Dr Falk Pharma, Gilead, Intercept, MSD and Takeda. MT is listed as a co‐inventor on patents on the medical use of nor‐ursodeoxycholic acid. TR has served as a speaker and/or consultant and/or advisory board member for AbbVie, Bayer, Boehringer Ingelheim, Gilead, W. L. Gore & Associates and MSD and has received research funding from AbbVie, Boehringer Ingelheim, Gilead, Phenex Pharmaceuticals and Philips. PF has served as a speaker and/or consultant and/or advisory board member for AbbVie, Bristol Myer‐Squibb, Gilead, MSD and Roche and has received research funding from Gilead and Roche. MM has served as a speaker and/or consultant and/or advisory board member for AbbVie, Bristol‐Myers Squibb, Gilead, W. L. Gore & Associates and Janssen.
Supporting information
Scheiner B, Stättermayer AF, Schwabl P, et al. Impact of HSD17B13 rs72613567 genotype on hepatic decompensation and mortality in patients with portal hypertension. Liver Int. 2020;40:393–404. 10.1111/liv.14304
REFERENCES
- 1. Blachier M, Leleu H, Peck‐Radosavljevic M, Valla DC, Roudot‐Thoraval F. The burden of liver disease in Europe: a review of available epidemiological data. J Hepatol. 2013;58(3):593‐608. [DOI] [PubMed] [Google Scholar]
- 2. Karlsen TH, Lammert F, Thompson RJ. Genetics of liver disease: from pathophysiology to clinical practice. J Hepatol. 2015;62(1 Suppl):S6‐S14. [DOI] [PubMed] [Google Scholar]
- 3. Scheiner B, Mandorfer M, Schwabl P, et al. The impact of PNPLA3 rs738409 SNP on liver fibrosis progression, portal hypertension and hepatic steatosis in HIV/HCV coinfection. PLoS ONE. 2015;10(11):e0143429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Trepo E, Romeo S, Zucman‐Rossi J, Nahon P. PNPLA3 gene in liver diseases. J Hepatol. 2016;65(2):399‐412. [DOI] [PubMed] [Google Scholar]
- 5. Eslam M, Valenti L, Romeo S. Genetics and epigenetics of NAFLD and NASH: clinical impact. J Hepatol. 2018;68(2):268‐279. [DOI] [PubMed] [Google Scholar]
- 6. Stickel F, Moreno C, Hampe J, Morgan MY. The genetics of alcohol dependence and alcohol‐related liver disease. J Hepatol. 2017;66(1):195‐211. [DOI] [PubMed] [Google Scholar]
- 7. Mandorfer M, Bucsics T, Hutya V, et al. Liver disease in adults with alpha1‐antitrypsin deficiency. United European Gastroenterol J. 2018;6(5):710‐718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Strnad P, Buch S, Hamesch K, et al. Heterozygous carriage of the alpha1‐antitrypsin Pi*Z variant increases the risk to develop liver cirrhosis. Gut. 2019;68(6):1099‐1107. [DOI] [PubMed] [Google Scholar]
- 9. Liu Z, Que S, Zhou L, et al. The effect of the TM6SF2 E167K variant on liver steatosis and fibrosis in patients with chronic hepatitis C: a meta‐analysis. Sci Rep. 2017;7(1):9273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mandorfer M, Scheiner B, Stattermayer AF, et al. Impact of patatin‐like phospholipase domain containing 3 rs738409 G/G genotype on hepatic decompensation and mortality in patients with portal hypertension. Aliment Pharmacol Ther. 2018;48(4):451‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schaefer B, Mandorfer M, Viveiros A, et al. Heterozygosity for the alpha‐1‐antitrypsin Z allele in cirrhosis is associated with more advanced disease. Liver Transpl. 2018;24(6):744‐751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gellert‐Kristensen H, Nordestgaard BG, Tybjaerg‐Hansen A, Stender S. High risk of fatty liver disease amplifies the alanine transaminase‐lowering effect of a HSD17B13 variant. Hepatol. 2019;in press. [DOI] [PubMed] [Google Scholar]
- 13. Dewey FE, Murray MF, Overton JD, et al. Distribution and clinical impact of functional variants in 50,726 whole‐exome sequences from the DiscovEHR study. Science. 2016;354(6319). [DOI] [PubMed] [Google Scholar]
- 14. Abul‐Husn NS, Cheng X, Li AH, et al. A protein‐truncating HSD17B13 variant and protection from chronic liver disease. New Engl J Med. 2018;378(12):1096‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moeller G, Adamski J. Integrated view on 17beta‐hydroxysteroid dehydrogenases. Mol Cell Endocrinol. 2009;301(1–2):7‐19. [DOI] [PubMed] [Google Scholar]
- 16. Tsachaki M, Odermatt A. Subcellular localization and membrane topology of 17β‐hydroxysteroid dehydrogenases. Mol Cell Endocrinol. 2019;489:98‐106. [DOI] [PubMed] [Google Scholar]
- 17. Saloniemi T, Jokela H, Strauss L, Pakarinen P, Poutanen M. The diversity of sex steroid action: novel functions of hydroxysteroid (17beta) dehydrogenases as revealed by genetically modified mouse models. J Endocrinol. 2012;212(1):27‐40. [DOI] [PubMed] [Google Scholar]
- 18. Rotinen M, Villar J, Celay J, Encio I. Type 10 17beta‐hydroxysteroid dehydrogenase expression is regulated by C/EBPbeta in HepG2 cells. J Steroid Biochem Mol Biol. 2010;122(4):164‐171. [DOI] [PubMed] [Google Scholar]
- 19. Tsachaki M, Birk J, Egert A, Odermatt A. Determination of the topology of endoplasmic reticulum membrane proteins using redox‐sensitive green‐fluorescence protein fusions. Biochim Biophys Acta. 2015;1853(7):1672‐1682. [DOI] [PubMed] [Google Scholar]
- 20. Ma Y, Belyaeva OV, Brown PM, et al. HSD17B13 is a Hepatic retinol dehydrogenase associated with histological features of non‐alcoholic fatty liver disease. Hepatology. 2019;69(4):1504‐1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pirola CJ, Garaycoechea M, Flichman D, et al. Splice variant rs72613567 prevents worst histologic outcomes in patients with nonalcoholic fatty liver disease. J Lipid Res. 2019;60(1):176‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Su W, Wang Y, Jia X, et al. Comparative proteomic study reveals 17beta‐HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc Natl Acad Sci USA. 2014;111(31):11437‐11442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sookoian S, Arrese M, Pirola CJ. Genetics meets therapy? Exome‐wide association study reveals a loss‐of‐function variant in HSD17B13 (17‐beta‐hydroxysteroid dehydrogenase 13) that protects patients from liver damage and NAFLD‐progression. Hepatology. 2019;69(2):907‐910. [DOI] [PubMed] [Google Scholar]
- 24. Reiberger T, Schwabl P, Trauner M, Peck‐Radosavljevic M, Mandorfer M. Measurement of the hepatic venous pressure gradient and transjugular liver biopsy. J Vis Exp. 2019;in press. [DOI] [PubMed] [Google Scholar]
- 25. Ferlitsch A, Bota S, Paternostro R, et al. Evaluation of a new balloon occlusion catheter specifically designed for measurement of hepatic venous pressure gradient. Liver Int. 2015;35(9):2115‐2120. [DOI] [PubMed] [Google Scholar]
- 26. de Franchis R, Faculty BVI. Expanding consensus in portal hypertension: Report of the Baveno VI Consensus Workshop: Stratifying risk and individualizing care for portal hypertension. J Hepatol. 2015;63(3):743‐752. [DOI] [PubMed] [Google Scholar]
- 27. Reiberger T, Puspok A, Schoder M, et al. Austrian consensus guidelines on the management and treatment of portal hypertension (Billroth III). Wien Klin Wochenschr. 2017;129(Suppl 3):135‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. European Association for the Study of the L, European Association for the Study of D, European Association for the Study of O . EASL‐EASD‐EASO Clinical Practice Guidelines for the management of non‐alcoholic fatty liver disease. J Hepatol. 2016;64(6):1388‐1402. [DOI] [PubMed] [Google Scholar]
- 29. Ripoll C, Groszmann R, Garcia–Tsao G, et al. Hepatic venous pressure gradient predicts clinical decompensation in patients with compensated cirrhosis. Gastroenterology. 2007;133(2):481‐488. [DOI] [PubMed] [Google Scholar]
- 30. Berzigotti A, Albillos A, Villanueva C, et al. Effects of an intensive lifestyle intervention program on portal hypertension in patients with cirrhosis and obesity: The SportDiet study. Hepatology. 2017;65(4):1293‐1305. [DOI] [PubMed] [Google Scholar]
- 31. Scheiner B, Steininger L, Semmler G, et al. Controlled attenuation parameter does not predict hepatic decompensation in patients with advanced chronic liver disease. Liver Int. 2019;39(1):127‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mandorfer M, Kozbial K, Schwabl P, Chromy C, Semmler G, Stättermayer AF, et al. Changes in HVPG predict hepatic decompensation in patients who achieved SVR to IFN‐free therapy. Hepatology. 2019;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. European Association for the Study of the Liver . EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J Hepatol. 2018;69(2):406‐460. [DOI] [PubMed] [Google Scholar]
- 34. Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. Journal of the American Statistical Association. 1999;94(446):496‐509. [Google Scholar]
- 35. Pirazzi C, Valenti L, Motta BM, et al. PNPLA3 has retinyl‐palmitate lipase activity in human hepatic stellate cells. Hum Mol Genet. 2014;23(15):4077‐4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mondul A, Mancina RM, Merlo A, et al. PNPLA3 I148M variant influences circulating retinol in adults with nonalcoholic fatty liver disease or obesity. J Nutr. 2015;145(8):1687‐1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Paternostro R, Heinisch BB, Reiberger T, et al. Dysbalanced sex hormone status is an independent predictor of decompensation and mortality in patients with liver cirrhosis. Hepatol Res. 2019;49(2):201‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lavie M, Dubuisson J. Interplay between hepatitis C virus and lipid metabolism during virus entry and assembly. Biochimie. 2017;141:62‐69. [DOI] [PubMed] [Google Scholar]
- 39. Boyer A, Park SB, de Boer Y, Li Q, Liang TJ. TM6SF2 promotes lipidation and secretion of hepatitis C virus in infected hepatocytes. Gastroenterology. 2018;155(6):1923‐1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang J, Trépo E, Nahon P, et al. A 17‐beta‐hydroxysteroid dehydrogenase 13 variant protects from hepatocellular carcinoma development in alcoholic liver disease. Hepatology. 2019;70(1):231‐240. [DOI] [PubMed] [Google Scholar]
- 41. Rodriguez S, Gaunt TR, Day IN. Hardy‐Weinberg equilibrium testing of biological ascertainment for Mendelian randomization studies. Am J Epidemiol. 2009;169(4):505‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials