

Abstract
Neurotensin receptor 1 (NTR1) is a G protein coupled receptor that is widely expressed throughout the central nervous system where it acts as a neuromodulator. Neurotensin receptors have been implicated in a wide variety of CNS disorders but despite extensive efforts to develop small molecule ligands there are few reports of such compounds. Herein we describe the optimization of a quinazoline based lead to give 18 (SBI-553), a potent and brain penetrant NTR1 allosteric modulator.
Graphical Abstract
INTRODUCTION
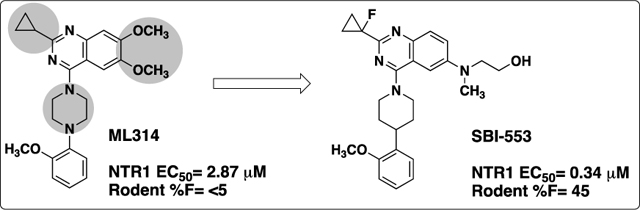
The tridecapeptide neurotransmitter neurotensin (NT)1 is widely distributed throughout the central and peripheral nervous systems2 where it acts as a neurotransmitter and neuromodulator, in particular for dopamine (DA) signaling.3 Neurotensin binds two G-protein-coupled receptors, NTR1 and NTR2,4,5 and neurotensin has been implicated in numerous CNS disorders such as schizophrenia,6 Parkinson’s disease7 and drug addiction.8 NTR1 is the most widely studied, mediates most of the known neurotensin effects, and holds the potential as an interesting therapeutic target. 9 In addition, two crystal structures of NTR1 have been recently reported.10,11 Despite the therapeutic promise of NTR1, it has proven to be difficult to develop ligands for the receptor. While there have been numerous reports of peptide agonists of NTR1,12,13 which in general suffer from poor oral bioavailability and CNS penetration, only a handful of small molecule antagonists and agonists (Figure 1) have been described. The most advanced compounds include the nM antagonists from Sanofi SR48692 (Meclinertant),14 which completed PhII clinical trials, and the related analog SR142948A.15 Positive modulators include sub-μM compounds from RTI,16 which was derived from the SR compounds, and the related imidazole ML301.17 In addition, a weakly active indole based partial agonist from Wyeth18 and an optimized full agonist analog from Scripps (SR-12062) have been reported.19 We have reported on a series of β-arrestin biased quinazoline positive modulators of NTR, exemplified by our probe compound ML314 (Figure 2).20 While ML314 was moderately potent, displayed good brain penetration after IP dosing and was active in vivo in a number of animal models of addiction,21 it displayed low oral bioavailability (<5%). We therefore embarked on an optimization campaign to improve both potency and oral bioavailability. Herein we disclose the discovery of SBI-553, an optimized derivative with a 10 fold potency improvement and significantly improved bioavailability that maintains good CNS penetration.
Figure 1:
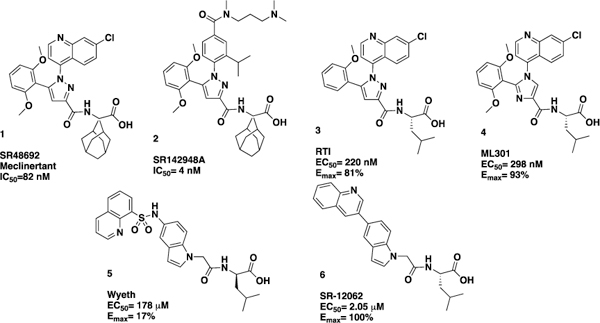
Selected previously reported NTR1 ligands
Figure 2:
Quinazoline NTR1 modulators
RESULTS AND DISCUSSION
Chemistry.
The compounds described in this paper consist of a quinazoline core structure with an N-linked piperdine or piperazine in the 4 position. The synthesis of most analogs followed our previously described routes20,22 (for full synthetic details see Supporting information). A representative synthesis is shown in Scheme 1. Starting from an appropriate substituted carboxylic acid (9), conversion to the acid chloride followed by reaction with a 2-cyanoaniline (11) gave intermediate 12. Cyclization under basic conditions gave quinazoline 13. The amino side chain 14 was introduced by a copper catalyzed coupling reaction to give intermediate 15, which was methylated using reductive alkylation to give 16. 16 was coupled with the substituted piperdine 17 to provide the final product 18.
Scheme 1.
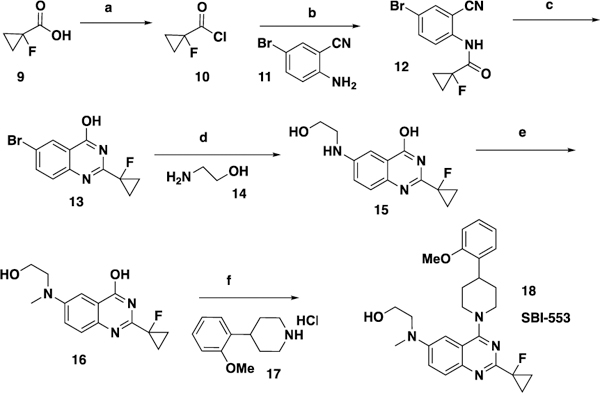
Representative synthesis of quinazoline compounds aReagents and conditions: (a) (COCl)2, 35 °C, 1.5h; (b) Pyridine, DCM, rt, 2h 86% over 2 steps; (c) NaOH, H2O2, EtOH, reflux, 12 h, 66%; (d) K3PO4, CuI, proline, DMSO, 100 °C, 12h; (e) NaBH(OAc)3, HCHO, MeOH, rt, 1h, 39% over 2 steps; (f) BOP, DBU, CH3CN, rt, 12h, 87%
SAR.
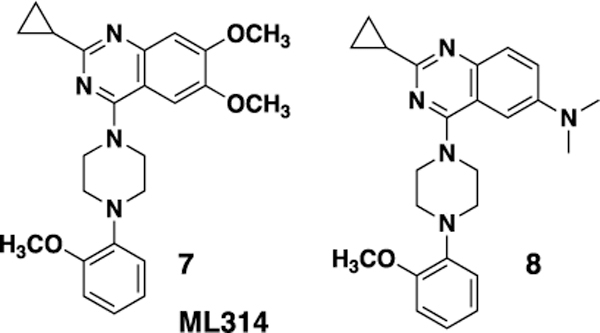
We have previously reported the preliminary SAR around the quinazoline scaffold to give the probe compound 7, ML314.20 However, ML314, while selective, displays only moderate potency (~2.8 μM) and modest pharmacokinetics, with low oral bioavailability (<5%). We therefore embarked on an SAR campaign to improve both of these parameters. For our primary SAR driving assay we used a high content assay measuring NT receptor mediated β-arrestin redistribution as previously reported.23 The primary counterscreen was an assay for NTR2 in the same format. Our initial SAR around the ML314 scaffold investigated a wide range of substituents around the quinazoline ring as well as around the pendant phenyl ring on the piperazine. Of the >100 analogs synthesized (data not shown) only one, the dimethylamine analog 8 displayed activity higher than the parent compound (NTR1 EC50= 0.71 μM). During the course of our preliminary investigation we also examined the piperazine linker; only 6 membered rings gave active compounds, and there was strong preference for a nitrogen-linkage to the quinazoline core. As shown in Table 1, piperdine 19b was approximately three fold more potent than the piperazine, while 19a, with the reverse linkage, was not active.
Table 1.
Structures and NTR1 activities of analogs 19
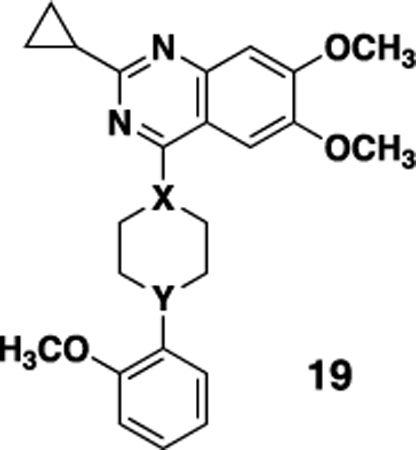 | ||||
|---|---|---|---|---|
| Cpd # | X | Y | EC50, μMa | Emax (%)b |
| 7 (ML314) | N | N | 2.87 ± 1.43 (171) | 100 |
| 19a | CH | N | >20 | -- |
| 19b | N | CH | 1.04 ± 0.14 | 89 |
β-Arrestin2-GFP NTR1 potency measured relative to the EC100 (100 nM) of the NT(8–13) peptide control average ± SEM (n=4 unless otherwise noted);
Emax was calculated as the % of the response obtained with NT(8–13) peptide. None of the compounds from this series showed activity in the NTR2 (>80 μM) counterscreen.
With this result in hand, we then re-examined the SAR around the core (Table 2). Similar to the results with the piperazine linked compound 8, a dimethylamine group in the 6 position (20a) was reasonably potent. Increasing the size to an ethyl group improved activity (20b), although there were no further gains in potency going to the propyl analog (20d). Similar to previous SAR, the 6 position was preferred, with substitution in the 5 (compound 20c) or 7 (compound 20e) giving substantially less potent analogs. Adding an additional methyl (compound 20f) or fluoro (compound 20g) in the 7 position gave compounds of lower potency. A range of analogs that were substituted with a methoxy (20h), hydroxy (20i) or morpholino (20j) group showed that all modifications were tolerated, with hydroxy compound 20i being the most potent. Piperdine (20k,20l) and pyrrolidine (20m-p) substitution also gave compounds that showed good activity.
Table 2.
Structures and NTR1 activities of analogs 20
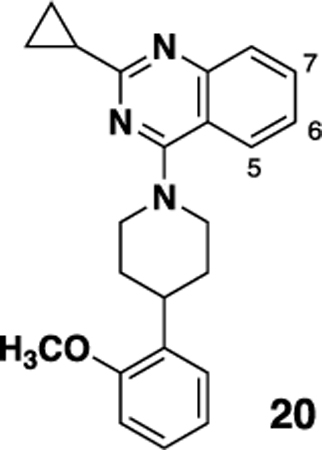 | |||||
|---|---|---|---|---|---|
| Cpd # |
5 | 6 | 7 | EC50, μMa | Emax (%)b |
| 19b | -H | -OCH3 | -OCH3 | 1.04 ± 0.14 | 89 |
| 20a | -H | -N(CH3)2 | -H | 0.888 ± 0.11 (3) |
89 |
| 20b | -H | 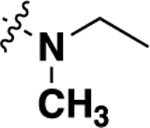 |
-H | 0.56 ± 0.13 | 80 |
| 20c | 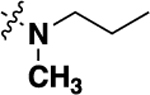 |
-H | -H | 6.71 ± 1.54 (3) |
60 |
| 20d | -H | 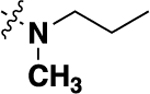 |
-H | 0.732 ± 0.12 | 68 |
| 20e | -H | -H | 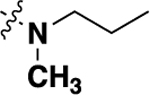 |
10.02 ± 3.95 | 86 |
| 20f | -H | 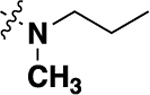 |
-CH3 | 0.99 ± 0.44 (8) |
86 |
| 20g | -H | 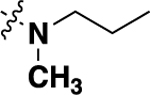 |
-F | 3.01 ± 0.18 (6) |
68 |
| 20h | -H | 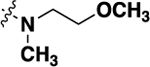 |
-H | 1.12 ± 0.13 | 79 |
| 20i | -H | 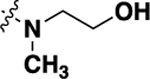 |
-H | 0.478 ± 0.08 | 92 |
| 20j | -H | 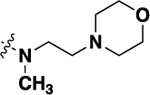 |
-H | 0.81 ± 0.10 | 92 |
| 20k | -H | 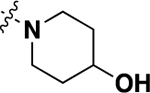 |
-H | 6.72 ± 1.59 | 82 |
| 20l | -H | 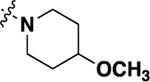 |
-H | 2.78 ± 0.57 | 79 |
| 20m | -H | 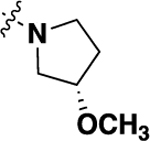 |
-H | 0.81 ± 0.25 (8) |
82 |
| 20n | -H | 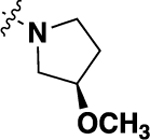 |
-H | 0.64 ± 0.29 (12) |
93 |
| 20o | -H | 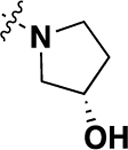 |
-H | 1.01 ± 0.23 (8) |
78 |
| 20p | -H | 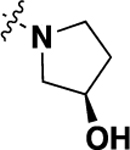 |
-H | 0.44 ± 0.16 (8) |
79 |
β-Arrestin2-GFP NTR1 potency measured relative to the EC100 (100 nM) of the NT(8–13) peptide control average ± SEM (n=4 unless otherwise noted);
Emax was calculated as the % of the response obtained with NT(8–13) peptide. None of the compounds from this series showed activity in the NTR2 (>80 μM) counterscreen.
We then turned to examining the effect of substitution on the cyclopropyl ring off the quinazoline core (Table 3). Substitution at the 2 and 3 positions of the cyclopropyl ring were generally poorly tolerated (data not shown); however substitution at the 1 position was tolerated and also improved the PK properties (vide infra). While in some cases potency was not improved (for example 21a compared to 20d or 21e to 20a), the combination of the hydroxyethyl group with either a fluorine (18, SBI-553) or methyl group (21e) gave up to a five fold increase in potency. Interestingly the corresponding analogs with either a methoxyethyl (21c, 21d) or morpholinoethyl (21g-i) did not show a similar potency boost.
Table 3.
Structures and NTR1 activities of analogs 21
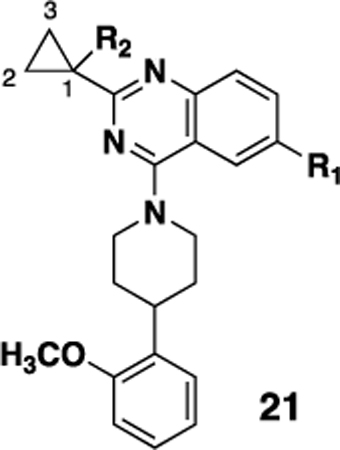 | ||||
|---|---|---|---|---|
| Cpd # | R1 | R2 | EC50, μMa | Emax (%)b |
| 21a | 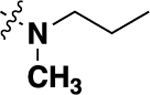 |
-F | 1.45 ± 0.31 (6) |
80 |
| 21b | -N(CH3)2 | -F | 0.92 ± 0.28 (6) |
84 |
| 21c | 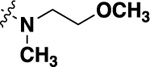 |
-CH3 | 2.23 ± 0.37 | 60 |
| 21d | 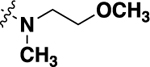 |
-F | 1.06 ± 0.08 | 86 |
|
18 (SBI-553) |
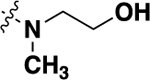 |
-F | 0.34 ± 0.21 (99) |
100 |
| 21e | 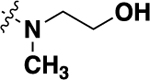 |
-CH3 | 0.098 ± 0.01 | 90 |
| 21f | 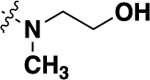 |
-CF3 | 0.34 ± 0.21 (8) |
94 |
| 21g | 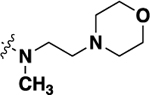 |
-F | 1.72 ± 0.13 | 97 |
| 21h | 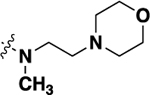 |
-CH3 | 3.36 ± 0.28 | 71 |
| 21i | 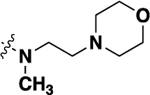 |
-CF3 | 3.81 ± 1.57 (8) |
78 |
β-Arrestin2-GFP NTR1 potency measured relative to the EC100 (100 nM) of the NT(8–13) peptide control average + SEM (n=4 unless otherwise noted);
Emax was calculated as the % of the response obtained with NT(8–13) peptide. None of the compounds from this series showed activity in the NTR2 (>80 μM) counterscreen.
We also investigated a range of other groups in place of the cyclopropyl group (Table 4). Cyclobutyl (22a) was well tolerated, but potency dropped off when the ring size was increased to cylcopentyl (22b). A phenyl substituent (22c) was also less active. As with the cyclopropyl compounds 18 and 21e, substitution of the cyclobutyl group in the 1 position with either F (22d) or methyl (22e) gave potent compounds, although activity was not substantially improved in the case of methyl analog 22e, which is in contrast with the corresponding analog 21e.
Table 4.
Structures and NTR1 activities of analogs 22
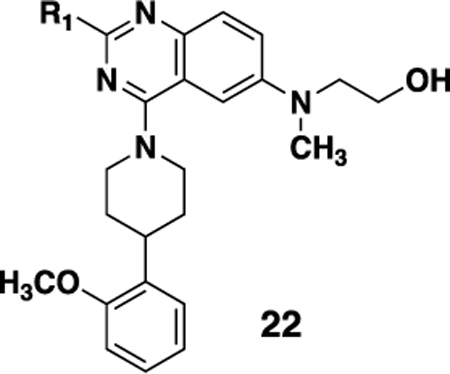 | |||
|---|---|---|---|
| Cpd # | R1 | EC50, μMa | Emax (%)b |
| 22a | 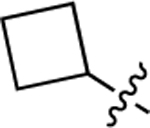 |
1.05 ± 0.32 | 80 |
| 22b | 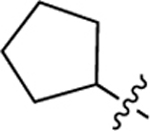 |
3.88 ± 1.12 (6) |
78 |
| 22c | Ph- | 4.23 ± 0.88 | 70 |
| 22d | 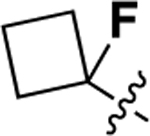 |
0.27 ± 0.05 | 96 |
| 22e | 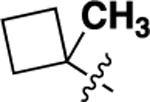 |
1.48 ± 0.38 | 75 |
β-Arrestin2-GFP NTR1 potency measured relative to the EC100 (100 nM) of the NT(8–13) peptide control average ± SEM (n=4 unless otherwise noted);
Emax was calculated as the % of the response obtained with NT(8–13) peptide. None of the compounds from this series showed activity in the NTR2 (>80 μM) counterscreen.
Lastly, we examined substitution of the quinoline core as outlined in Table 5. Compared with the unsubstituted analog (20i) we observed only modest variations in potency with F, Cl or methyl substitution, with the 8 position being preferred for substitution (compare 23b, 23e and 23h).
Table 5.
Structures and NTR1 activities of analogs 23
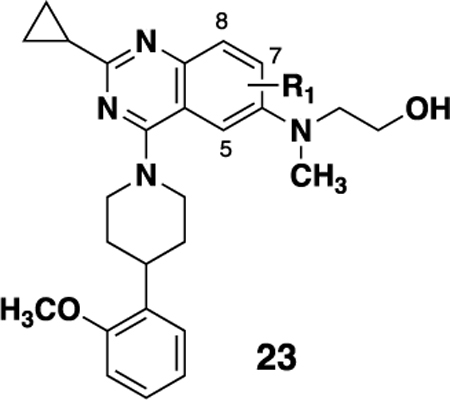 | |||
|---|---|---|---|
| Cpd # |
R1 | EC50, μMa | Emax (%)b |
| 23a | 5-F | 0.59 ± 0.08 (8) | 95 |
| 23b | 8-F | 0.16 ± 0.04 (8) | 100 |
| 23c | 5-Me | 1.26 ± 0.36 (8) | 93 |
| 23d | 7-Me | 0.56 ± 0.11 | 88 |
| 23e | 8-Me | 0.16 ± 0.04 (8) | 87 |
| 23f | 5-Cl | 0.50 ± 0.16 | 91 |
| 23g | 7-Cl | 0.60 ± 0.24 (23) | 98 |
| 23h | 8-Cl | 0.22 ± 0.14 | 95 |
β-Arrestin2-GFP NTR1 potency measured relative to the EC100 (100 nM) of the NT(8–13) peptide control average ± SEM (n=4 unless otherwise noted);
Emax was calculated as the % of the response obtained with NT(8–13) peptide. None of the compounds from this series showed activity in the NTR2 (>80 μM) counterscreen.
Pharmacokinetics.
We examined a range of analogs for their rodent PK properties, with the results for the fluoro-cyclopropyl (18, SBI-553) and methyl-cyclopropyl (21e) analogs shown in Table 6. Both analogs showed high clearance but also a high volume of distribution in both mouse and rat with good oral bioavailability (~50%) and half life in both species. In addition, the brain:plasma ratio 1 hour post dose was good for SBI-553 in both mouse (0.54) and rat (0.98). Similar brain:plasma ratios in rat were observed 4 and 8 hours post dose (PO, data not shown). Due to the combination of potency, bioavailability and brain penetration, 18 (SBI-553) was selected for further in vitro and in vivo pharmacological profiling.
Table 6.
Selected mouse or rat PK parameters for compounds 18 and 21e
| Compd sa |
Species | Clpb (mL/min/kg) |
Vdb (L/kg) |
Cmaxc (ng/mL) |
AUCc (ng.hr/mL) |
t1/2c (hr) |
%F | Brain:Plasma @1 hrc |
|---|---|---|---|---|---|---|---|---|
| 18 (SBI-553) | Mouse | 44.8 | 6.16 | 1460 | 4824 | 5.28 | 45 | 0.54 |
| 21e | Mouse | 79.4 | 7.68 | 627 | 3324 | 2.90 | 52 | 0.35 |
| 18 (SBI-553) | Rat | 81.0 | 7.02 | 3482 | 2693 | 2.23 | 48 | 0.98 |
Compounds dosed 5 mpk iv and 30 mpk po,
iv,
po
In vitro profiling.
This series of compounds display a unique profile in that they are allosteric modulators of NTR1 that selectively enhance the interaction with β-arrestin while blocking the classical Ca2+ signaling. Extensive in vitro and in vivo characterization, including the consequences of this biased signaling, will be reported elsewhere.24 Similar to ML314, 18 (SBI-553) alone showed no activity in a Ca2+ flux assay.20 Also, as observed with ML314, 18 (SBI-553) appears to act as a positive allosteric modulator of neurotensin peptide binding. 18 (SBI-553) was evaluated by Eurofins Panlabs Discovery Services25 in a radioligand binding assay that examined the binding of radiolabeled neurotensin (NT) to membranes prepared from cells overexpressing human NTR1 or NTR2, or mouse NTR. Unlike orthosteric ligands, which displace neurotensin, significant enhancement of neurotensin peptide binding to the human NTR1 and mouse NTR1, but not human NTR2, was observed in the presence of increasing concentrations of 18 (SBI-553) (Figure 3), indicating both the selectivity of the compound as well as confirming its unique mechanism of action.
Figure 3.
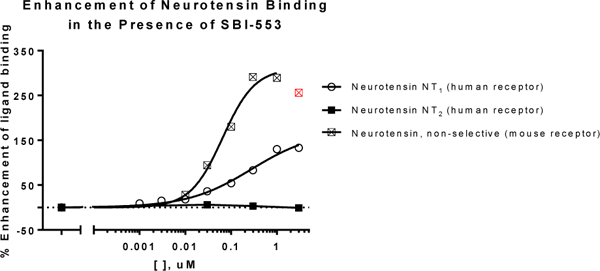
Augmentation of [125I]-NT binding (0.02 nM) to cell membranes in the presence of increasing doses of 18 (SBI-553). The EC50 for SBI-553 at the human receptor for this experiment was 0.14 μM, with a maximum efficacy of 140% (Data were plotted and analyzed using GraphPad Prism V.7, n=2). Efficacy is relative to the maximal displacement of orthosteric ligands (i.e. −100%). Data point excluded from curve fitting is shown in red. NT peptide in these assays give Kds of 0.082 nM (hNTR1), 0.59 nM (hNTR2) and 0.17 nM (mNTR) respectively.
The presence of 18 (SBI-553) had no effect on the potency of NT in the NTR1 high-content (β-arrestin2-GFP) assay (Figure 4A). EC50 values for NT in this assay ranged from 0.37 to 0.40 nM across the range of 18 (SBI-553) doses tested (0.5 −356 nM), while the EC50 for NT in the absence of 18 (SBI-553) was 0.40 nM. In contrast, the EC50 for NT in the Ca2+ flux assay (Figure 4B) was shifted to the right over the tested concentration range of 18 (SBI-553), 0.0088–2.15 μM, but did not quite reach saturation as indicated by the separation of the response curves at higher concentrations. In the absence of 18 (SBI-553), the EC50 for NT in this assay was 0.035 nM, while the EC50 for the NT was shifted >30-fold (1.18 nM) in the presence of 2.15 μM 18 (SBI-553). A Schild analysis of these results (Figure S1) supports the notion that 18 (SBI-553) acts functionally similar to a competitive antagonist over this range. Thus, from Figures 3,4, and S1, 18 (SBI-553) can be considered a β-arrestm-biased modulator and acts functionally like a “PAM antagonist” of the Ca2+ pathway with respect to its effects on NT/NTR1 signaling.
Figure 4.
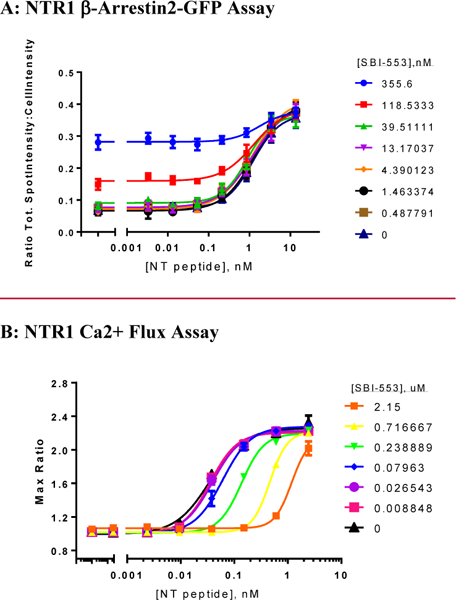
Dose response of NT peptide in the presence of 18 (SBI-553) in the β-Arrestin (A) and Ca2+ flux (B) assays. Data were plotted and analyzed using GraphPad Prism V.7 and are presented as an average ± SEM (n=4). The compound shows divergent behavior, with no effect on the EC50 in the β-Arrestin assay but a strong rightward shift in the Ca2+ flux assay.
As with ML314, 18 (SBI-553) showed no cross reactivity against a range of related and unrelated GPCRs using the same high-content assay format (NTR2, GPR35, GPR55, K-opioid). 18 (SBI-553) was also screened in a Eurofins/Panlabs panel of 80 targets and showed moderate promiscuity with binding (>30%) against 10 receptors at 10 μM concentration. Subsequent dose titrations indicated binding at the adrenergic α2a (55% at 5 μM), dopamine D1 (36% at 5 μM), histamine H1 (75% at 5 μM) and 5-HT2B (42% at 5 μM) receptors.
In vivo efficacy.
We then examined the effect of 18 (SBI-553) in vivo, after both IP (Figure 5, Panels A and B) and PO (Panels C and D, Figure 5) in the dopamine transporter knockout mouse model model of hyperdopaminergic activity.26 Unlike the peptide NTR1 agonist PD149163, which causes significant hypotension and hypothermia, animals dosed with 18 did not show any adverse effects. In addition, 18 attenuated hyperlocomotion after both IP and PO dosing. Full in vivo profiling of 18 will be reported elsewhere. 24
Figure 5: 18 (SBI-553) attenuates basal hyperlocomotion in dopamine transporter knockout (DAT−/−) mice.
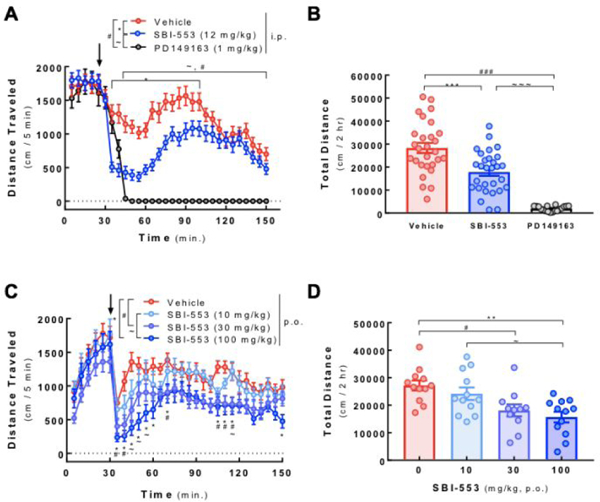
Locomotor activity was evaluated using open field automated activity monitors (AccuScan Instruments, Columbus, OH). Male and female DAT−/− mice were acclimated to the open field chamber for 30 min prior to administration of NTR1 ligands (indicated by arrow). After treatment, animals were immediately returned to chambers and horizontal locomotion was monitored over the next 2 hrs. (A) Animals received SBI-553 (12 mg/kg), PD149163 (1 mg/kg) or vehicle (4% DMSO in saline; 10 ml/kg) i.p. Beam breaks in 5 min intervals were recorded to generate horizontal distance traveled per time bin. *p<0.05-p<0.001 Vehicle vs. SBI-553; #p<0.05-p<0.001 Vehicle vs. PD149163; ~p<0.05-p<0.001 SBI-553 vs. PD149163, using a two-way, repeated measures ANOVA (FTime(23, 1679)=13.5, p<0.0001; FTreatment(2, 73)=45.1, p<0.0001; FInteraction(46, 1679)=10.4, p<0.0001) followed by the post hoc Tukey test. (B) Time course data in (A) was summed to determine the total distance traveled in the 2 hr post-treatment. ***,###,~~~p<0.001, as indicated, using one-way ANOVA (F(2, 72)=46.6, p<0.0001) followed by the post hoc Tukey test. Data are represented as mean ± SEM of 18–29 animals per group. (C) Animals received SBI-553 10, 30 or 100 mg/kg or vehicle (distilled H2O) p.o. Beam breaks in 5 min intervals were recorded to generate horizontal distance traveled per time bin. *p<0.05-p<0.001 Vehicle vs. 100 mg/kg SBI-553; #p<0.05 Vehicle vs. 30 mg/kg SBI-553; ~p<0.05 10 mg/kg SBI-553 vs. 100 mg/kg SBI-553, using a two-way, repeated measures ANOVA (FTime(23, 989)=8.6, p<0.0001; FTreatment(3, 43)=6.8, p<0.001; FInteraction(69, 989)=1.1, p=0.2726) followed by the post hoc Tukey test. (D) Time course data in (C) was summed to determine the total distance traveled in the 2 hr post-treatment. **p<0.01, #p<0.05,~p<0.05, as indicated, using one-way ANOVA (F(3, 43)=6.8, p<0.001) followed by the post hoc Tukey test. Data are represented as mean ± SEM of 11–12 animals per group.
CONCLUSION
In conclusion we have optimized a series of quinazoline based NTR1 modulators to give a potent and orally available compound. In the process we have maintained a unique mechanism of action, wherein the compound enables biased β-arrestin interactions and acts as an antagonist on the Ca2+ pathway for Gq mediated activation. An extensive account of the implications and benefits of this biased signaling in vitro and in vivo, including important differences in their toxicity profiles relative to standard neurotensin ligands,24 and a structural basis28 for their mechanism of action will be published elsewhere.
EXPERIMENTAL SECTION
All compounds were purified to >95% as determined by LC/MS as well as 1H (and 13C in select cases) NMR and HRMS. The analytical methods, general chemistry, experimental information, and syntheses of all other compounds are supplied in the Supporting Information.
Synthesis of 2-({2-(1-Fluoro-cyclopropyl)-4-[4-(2-methoxy-phenyl)-piperidin-1-yl]-quinazolin-6-yl}-methyl-amino)-ethanol (18):
Steps a,b: To a solution of 1-fluoro-cyclopropanecarboxylic acid (Oakwood Products, West Columbia, SC, 9, 50.8 g, 0.49 mol) in methylene chloride (500 mL), was added oxalyl dichloride (74.7 g, 0.59 mol) and DMF (0.5 mL). The mixture was stirred at 35 °C for 1.5 h. The resulting solution was added slowly to a solution of 2-amino-5-bromo-benzonitrile (Sigma Aldrich, St. Louis, MO, 11, 96 g, 0.49 mol) in methylene chloride (1 L) and pyridine (300 mL). The reaction was stirred at room temperature for 2 h, then was washed with water (1 L), 1N HCl (1 L) and saturated NaHCO3 solution (1 L), dried over Na2SO4 and filtered. The filtrate was concentrated and the residue was triturated with MeOH to give 1-fluoro-cyclopropanecarboxylic acid (4-bromo-2-cyanophenyl)-amide (12, 78 g).22 The mother liquid was concentrated and purified by silica gel column (petroleum ether/ethyl acetate = 10/1) to give additional 1-fluoro-cyclopropanecarboxylic acid (4-bromo-2-cyano-phenyl)-amide (12, 41 g, total yield: 86%) as a white solid. Step c: To a suspension of 1-fluoro-cyclopropanecarboxylic acid (4-bromo-2-cyano-phenyl)-amide (12, 78 g, 276 mmol) in EtOH (1.6 L) and H2O2 (30%, 300 mL), was added NaOH (13.23 g, 0.33 mol). The mixture was stirred at 85 °C overnight. The reaction was worked up together with another batch (using 1-fluoro-cyclopropanecarboxylic acid (4-bromo-2-cyano-phenyl)-amide, (12, 41 g, 145 mmol), EtOH, 850 mL, H2O2 (30%, 150 mL) and NaOH (7 g, 0.17 mol)). The combined reaction solution was concentrated to 800 mL and poured into cold water (1.2 L). The precipitate was collected by filtration to give 6-bromo-2-(1-fluoro-cyclopropyl)-quinazolin-4-ol (13, 39 g).22 The filtrate was treated with excess Na2SO4 and extracted with ethyl acetate (0.6 L x2). The organic layer was concentrated to 200 mL and filtered to remove Na2SO4. The filtrate was taken up with ethyl acetate (100 mL) and stored at room temperature for 2 days. The solid that was formed was collected by filtration, the cake was washed with H2O, and dried to give 6-bromo-2-(1-fluoro-cyclopropyl)-quinazolin-4-ol (13, 40 g of recrystallized product, total of 79 g, total yield: 66%)22 as a light yellow solid. 1H NMR (300 MHz, DMSO-d6): δ = 12.71 (brs, 1H), 8.17 (d, J = 2.4 Hz, 1H), 7.92 (dd, J = 8.7, 2.1 Hz, 1H), 7.52 (d, J = 8.7 Hz, 1H), 1.60–1.48 (m, 4H). Steps d,e: A mixture of 6-bromo-2-(1-fluoro-cyclopropyl)-quinazolin-4-ol (13, 67 g, 237 mmol), 2-amino-ethanol (14, 21.6 g, 355 mmol), CuI (4.5 g, 23.7 mmol), L-proline (2.73 g, 23.7 mmol) and K3PO4 (100 g, 474 mmol) in DMSO was stirred at 100 °C under N2 overnight. The reaction was concentrated to give crude 2-(1-fluoro-cyclopropyl)-6-(2-hydroxy-ethylamino)-quinazolin-4-ol (15). The crude material was dissolved in MeOH (800 mL) and water (400 mL). To the mixture was added formaldehyde solution (37%, 45 g), NaBH(AcO)3 (59 g, 280 mmol) and NaBH3CN (27 g, 420 mmol). The reaction was stirred at room temperature for 1 h. The solution was poured into water (3 L) and extracted with ethyl acetate (1.5 L x3). The organic layers were combined, dried over Na2SO4, concentrated and purified by silica gel column (methylene chloride/MeOH = 20/1) to give 2-(1-fluoro-cyclopropyl)-6-[(2-hydroxy-ethyl)-methyl-amino]-quinazolin-4-ol (16, 32 g, yield: 39% over 2 steps)22 as a yellow solid. 1H NMR (400 MHz, DMSO-d6): δ = 12.25 (brs, 1H), 7.43 (d, J = 9.2 Hz, 1H), 7.28 (dd, J = 9.2, 3.2 Hz, 1H), 7.17 (d, J = 2.8 Hz, 1H), 4.75 (brs, 1H), 3.57–3.47 (m, 4H), 3.01 (s, 3H), 1.52–1.36 (m, 4H). Step f: To a suspension of 2-(1-fluoro-cyclopropyl)-6-[(2-hydroxy-ethyl)-methyl-amino]-quinazolin-4-ol (16, 14.2 g, 51.3 mmol) and BOP (34 g, 77 mmol) in MeCN (500 mL) was added DBU (31.2 g, 205 mmol) and the mixture was stirred for 5 min. To the reaction, 4-(2-methoxy-phenyl)-piperidine (17, HCl salt, 12.8 g, 56.4 mmol) was added. The reaction was stirred at room temperature overnight and filtered. The filter cake was washed with MeCN (100 mL) and dried to give 2-({2-(1-fluoro-cyclopropyl)-4-[4-(2-methoxy-phenyl)-piperidin-1-yl]-quinazolin-6-yl}-methyl-amino)-ethanol (18, 20 g, yield: 87%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 7.98 (d, J = 9.2 Hz, 1H), 7.41 (dd, J = 9.3, 2.6 Hz, 1H), 7.24 (t, J = 7.4 Hz, 2H), 6.99 (t, J = 7.9 Hz, 2H), 6.91 (d, J = 8.2 Hz, 1H), 4.42 (d, J = 13.2 Hz, 2H), 3.90 (t, J = 5.7 Hz, 2H), 3.87 (s, 3H), 3.60 (t, J = 5.6 Hz, 2H), 3.36 – 3.27 (m, 1H), 3.19 (t, J = 12.6 Hz, 2H), 3.08 (s, 3H), 2.30 (broad s, 1H), 2.00 (d, J = 12.8 Hz, 2H), 1.95 – 1.85 (m, 2H), 1.59 – 1.50 (m, 4H). 13C NMR (126 MHz, CDCl3) 8 164.33 (d, J C-F = 3.6 Hz), 159.17 (d, JC-F = 20.3 Hz), 156.80, 146.99, 144.97, 133.65, 129.07, 127.17, 126.48, 121.63, 120.65, 116.49, 110.41, 103.79, 79.01 (d, JC-F = 218.0 Hz), 60.08, 55.47, 55.30, 50.51, 38.95, 35.74, 31.81, 15.00 (d, JC-F = 10.8 Hz). HRMS: calculated for C26H31FN4O2: m/z = 451.2509; (M+H+) and observed m/z = 451.2528. HPLC Purity: 99%.
In Vivo Studies.
PD149163 was purchased from MilliporeSigma (St. Louis, MO). All compounds were freshly prepared in indicated vehicles on the day of testing. AnimalsExperiments employed adult male and female DAT KO mice27 maintained on a C57BL/6J background. Animals within studies were 12–24 week old, 18–30 g body weight and were age- and sex-matched. Animals were maintained on a 12/12 hour light/dark cycle and supplied tap water and standard laboratory chow ad libitum, except during testing. Mice were randomly assigned to treatment groups, and all experiments were carried out at the beginning of the light cycle. All mouse studies were conducted in accordance with the National Institutes of Health Guidelines for Animal Care and Use and with approved animal protocols from the Duke University Animal Care and Use Committee. Locomotor activity. Locomotor activity was evaluated by open field automated activity monitors (AccuScan Instruments, Columbus, OH) featured with eight photo-beams equally distributed along the border, as previously described.21 To assess the effect of NTR1 ligands on the basal hyperlocomotion exhibited by DAT KO animals, adult male and female DAT KO mice were acclimated to the open field for 30 min prior to treatment. For i.p. dosing studies, treatments included 12 mg/kg SBI-553, 1 mg/kg PD149163 or vehicle (4% DMSO in saline) delivered in a 10 ml/kg volume. For p.o. dosing studies treatments included 10, 30, or 100 mg/kg SBI-553 as the HCl salt or vehicle (distilled H2O) delivered in a 10 ml/kg volume. Prior to oral dosing studies animals were habituated to oral dosing via 5 consecutive daily sessions in which 10 ml/kg H2O was delivered p.o. After treatment on test days, animals were immediately returned to activity monitors and locomotor activity was recorded over the next 2 hr. Infrared beam breaks in 5 min intervals were recorded to generate horizontal distance traveled per time bin. Distance values were averaged across mice, and the means ± SEM were plotted. Total distance traveled in the 2 hr post-treatment was calculated by summing the time bins between 30 and 150 min. Data were plotted and analyzed using GraphPad Prism V.7.
Supplementary Material
ACKNOWLEDGEMENTS
Funding. This work was supported by National Institutes of Health grants R21/33 DA038019 to A.B.P. and 5P30DA029925 to M.G.C. and L.S.B. We acknowledge Peter Wilson at Almac, Craigavon, United Kingdom for scale up work on 18, WuXi, Shanghai, China, for measurements of the PK parameters, Eurofins, Taipei, Taiwan, for the binding studies in Figure 3, and Alexander Braddock and Emmanuel Theodorakis at the University of California, San Diego for assistance with measuring optical rotations.
ABBREVIATIONS USED
- NT
neurotensin
- NTR1
neurotensin receptor 1
- NTR2
neurotensin receptor 2
- DA
dopamine
- DAT
dopamine transporter
- GFP
green fluorescent protein
- PAM
positive allosteric modulator
- BOP
benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate
- DBU
1,8-Diazabicyclo[5.4.0]undec-7-ene
Footnotes
ASSOCIATED CONTENT
Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org. Experimental procedures and spectroscopic data for selected compounds, details of the PK studies, and assay protocols for the binding studies. (PDF).
The authors declare the following competing financial interest: Patent applications covering SBI-553 and related derivatives have been filed by SBP and Duke University.
REFERENCES
- 1.Carraway R, Leeman SE The isolation of a new hypotensive peptide, neurotensin, from bovine hypothalami. J. Biol. Chem, 1973, 248, 6854–6861. [PubMed] [Google Scholar]
- 2.Uhl GR Distribution of neurotensin and its receptor in the central nervous system. Ann. N. Y. Acad. Sci. 1982, 400, 132–149. [DOI] [PubMed] [Google Scholar]
- 3.Quirion R, Rowe WB, Lapchak PA, Araujo DM, and Beaudet A Distribution of neurotensin receptors in mammalian brain. What it is telling us about its interactions with other neurotransmitter systems. Ann. N. Y. Acad. Sci. 1992, 668, 109–119. [DOI] [PubMed] [Google Scholar]
- 4.Vincent J-P; Mazella J; Kitabgi P Neurotensin and neurotensin receptors. Trends Pharmacol. Sci, 1999, 20, 302–309. [DOI] [PubMed] [Google Scholar]
- 5.Hermans E; Maloteaux JM Mechanisms of Regulation of Neurotensin Receptors. Pharmacol. Ther, 1998, 79, 89–104. [DOI] [PubMed] [Google Scholar]
- 6.Nemeroff CB The interaction of neurotensin with dopaminergic pathways in the central nervous system: basic neurobiology and implications for the pathogenesis and treatment of schizophrenia. Psychoneuroendocrinology, 1986, 11, 15–37. [DOI] [PubMed] [Google Scholar]
- 7.Schimpff RM, Avard C, Fenelon G, Lhiaubet AM, Tenneze L, Vidailhet M Increased plasma neurotensin concentrations in patients with Parkinson’s disease. J. Neurol. Neurosurg. Psychiatr, 2001, 70, 784–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richelson E, Boules M, and Fredrickson P Neurotensin agonists: possible drugs for treatment of psychostimulant abuse. Life Sci., 2003, 73, 679–690. [DOI] [PubMed] [Google Scholar]
- 9.Boules M; Li Z; Smith K. Fredrickson P, Richelson E Diverse Roles of Neurotensin Agonists in the Central Nervous System Front. Endocrinol, 2013, 4, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White JF; Noinaj N; Shibata Y; Love J; Kloss B; Xu F; Gvozdenovic-Jeremic J; Shah P; Shiloach J; Tate CG; Grisshammer R Structure of the agonist-bound neurotensin receptor, Nature 2012, 490, 508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Egloff P; Hillenbrand M; Klenk C; Batyuk A; Heine P; Balada S; Schlinkmann KM; Scott DJ; Schütz M; Plückthun A Structure of signaling-competent neurotensin receptor 1 obtained by directed evolution in Escherichia coli., PNAS, 2014, 111 (6), E655–E662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Griebel G, Holsboer F Neuropeptide receptor ligands as drugs for psychiatric diseases: the end of the beginning? Nat Rev Drug Discov ., 2012, 6, 462–478. [DOI] [PubMed] [Google Scholar]
- 13.Hadden MK; Orwig KS; Kokko KP; Mazella J; Dix TA Design, synthesis, and evaluation of the antipsychotic potential of orally bioavailable neurotensin (8–13) analogues containing non-natural arginine and lysine residues. Neuropharmacology 2005, 49, 1149–1159. [DOI] [PubMed] [Google Scholar]
- 14.Gully D; Canton M; Boigegrain R; Jeanjean F; Molimard JC; Poncelet M; Gueudet C; Heaulme M; Leyris R; Brouard A Biochemical and pharmacological profile of a potent and selective nonpeptide antagonist of the neurotensin receptor. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 65–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gully D; Labeeuw B; Boigegrain R; Oury-Donat F; Bachy A; Poncelet M; Steinberg R; Suaud-Chagny MF; Santucci V; Vita N; Pecceu F; Labbe-Jullie C; Kitabgi P; Soubrie P; Le Fur G; Maffrand JP Biochemical and pharmacological activities of SR 142948A, a new potent neurotensin receptor antagonist. J. Pharmacol. Exp. Ther. 1997, 280, 802–812. [PubMed] [Google Scholar]
- 16.Thomas JB; Navarro H; Warner KR; Gilmour B The identification of nonpeptide neurotensin receptor partial agonists from the potent antagonist SR48692 using a calcium mobilization assay. Bioorg. Med. Chem. Lett. 2009, 19, 1438–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hershberger PM, Hedrick MP, Peddibhotla S, Mangravita-Novo A, Gosalia P, Li Y, Gray W, Vicchiarelli M, Smith LH, Chung TD, Thomas JB, Caron MG, Pinkerton AB, Barak LS, and Roth GP Imidazole-derived agonists for the neurotensin 1 receptor. Bioorg. Med. Chem. Lett. 2014, 24, 262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan Y; Lai MH; Sullivan K; Popiolek M; Andree TH; Dollings P; Pausch MH The identification of neurotensin NTS1 receptor partial agonists through a ligand-based virtual screening approach. Bioorg. Med. Chem. Lett. 2008, 18, 5789–5791. [DOI] [PubMed] [Google Scholar]
- 19.Fruscia PD; He Y; Koenig M; Tabrizifard S; Nieto A; McDonald PH; Kamenecka TM The discovery of indole full agonists of the neurotensin receptor 1 (NTSR1) Bioorg. Med. Chem. Lett. 2014, 24, 3974–3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peddibhotla S, Hedrick MP, Hershberger P, Maloney PR, Li Y, Milewski M, Gosalia P, Gray W, Mehta A, Sugarman E, Hood B, Suyama E, Nguyen K, Heynen-Genel S, Vasile S, Salaniwal S, Stonich D, Su Y, Mangravita-Novo A, Vicchiarelli M, Roth GP, Smith LH, Chung TD, Hanson GR, Thomas JB, Caron MG, Barak LS, and Pinkerton AB Discovery of ML314, a Brain Penetrant Non-Peptidic beta-Arrestin Biased Agonist of the Neurotensin NTR1 Receptor. ACS Med. Chem. Lett. 2013, 4, 846–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barak LS, Bai Y, Peterson S, Evron T, Urs NM, Peddibhotla S, Hedrick MP, Hershberger P, Maloney PR, Chung TDY, Rodriguiz RM, Wetsel WC, Thomas JB, Hanson GR, Pinkerton AB and Caron MG ML314: A Biased Neurotensin Receptor Ligand for Methamphetamine Abuse. ACS Chem. Biol. 2016, 11, 1880–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinkerton AB; Hershberger PM; Peddibhotla S; Maloney PR; Hedrick MP Preparation of substituted quinazolines as small molecule agonists of neurotensin receptor 1. PCT WO 2015200534, December 30, 2015.
- 23.Hershberger P, Hedrick M, Peddibhotla S, Maloney P, Li Y, Milewski M, Gosalia P, Gray W, Mehta A, Sugarman E, Hood B, Suyama E, Nguyen K, Heynen-Genel S, Vasile S, Salaniwal S, Stonich D, Su Y, Mangravita-Novo A, Vicchiarelli M, Smith LH, Roth G, Diwan J, Chung TDY, Caron MG, Thomas JB, Pinkerton AB, Barak LR Small Molecule Agonists for the Neurotensin 1 Receptor (NTR1 Agonists) Probe Reports from the NIH Molecular Libraries Program. National Center for Biotechnology Information; (US: ); 2010–2012 . https://pubchem.ncbi.nlm.nih.gov/bioassay/493036 [PubMed] [Google Scholar]
- 24.Slosky LM; Bai Y; Toth K; Rochelle LK; Ray C; Badea A; Pogorelov V; Chandrasekhar R; Abraham DM; Atluri N; Peddibhotla S; Hedrick MP; Hershberger P; Maloney P; Yuan H; Li Z; Wetsel W; Pinkerton AB; Barak L; Caron MG Allosteric Neurotensin Receptor 1 Modulator Confers p-arrestm Bias and Selectively Attenuates Addiction-Associated Behaviors. Unpublished results. [Google Scholar]
- 25.https://www.eurofinsdiscoveryservices.com/catalogmanagement/viewitem/NT1-Human-Neurotensin-GPCR-Binding-Agonist-Radioligand-Assay-Panlabs/258010 (accessed August 1, 2019)
- 26.Efimova EV; Gainetdinov RR; Budygin EA; Sotnikova TD Dopamine transporter mutant animals: a translational perspective, J Neurogenet, 2016, 30(1), 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giros B; Jaber M; Jones SR; Wightman RM; Caron MG Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter., Nature, 1996, 379(6566), 606–612. [DOI] [PubMed] [Google Scholar]
- 28.Krumm BE; DiBerto JF; Olsen RHJ; Kang HJ; Che T; Slocum ST; Strachan RT; Slosky LM; Pinkerton AB; Barak LS; Caron MG; Kenakin T; Roth BL Neurotensin receptor allosterism revealed in complex with a biased allosteric modulator. Unpublished results [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.