

Abstract
A synthetic protocol for the preparation of 5‐(aryl)dibenzothiophenium salts starting from inexpensive dibenzothiophene S‐oxide and simple arenes is reported. The scope of the method regarding the nature of the arene is evaluated, intermediates along the reaction sequence have been trapped, and side‐reactions identified. In addition, the X‐ray structures of a complete set of these salts are reported and their reactivities studied. Specifically, chemoselective Suzuki coupling is observed at the dibenzothiophenium in the presence of iodides.
Keywords: cross-coupling, palladium, structure elucidation, sulfur, synthetic methods
Easy on, easy off: 5‐(aryl)dibenzothiophenium triflates were prepared by highly selective metal‐free C−H sulfenylation of arenes. Interestingly, these salts undergo site‐selective Suzuki–Miyaura coupling in the presence of C−I bonds, enabling the iterative synthesis of polyaromatics.
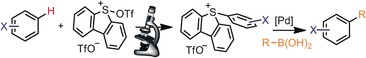
Introduction
Although the use of sulfonium moieties as linchpins in cross‐coupling chemistry,1 and as source of organic radicals, has been known for more than two decades,2 it has not been until very recently that the actual synthetic potential of these reagents has been broadly recognized. Variants of the Pd‐catalyzed Suzuki–Miyaura,3 Mizoroki–Heck,4 Sonogashira,5 Negishi,6 Stille,7 and related reactions, such as Buchwald–Hartwig aminations,8, 9 are now available by employing aryl sulfonium salts as electrophilic partners. Moreover, either by direct reaction with nucleophiles or through photoredox catalyzed processes, the sulfonium moiety can be selectively exchanged with alkyl groups, halogens, or amines under mild reaction conditions.10, 11 Aside from this wide range of applications, additional beneficial attributes are associated with the use of sulfonium salts. Namely, their easy and regioselective preparation from commercially available sulfides12 or sulfoxides,13 nonvolatility, and comparatively higher thermal stability than diazonium salts.
Very recently, our group reported on the synthesis of related 5‐(cyano)‐ and 5‐(alkynyl) dibenzothiophenium salts by triflic acid anhydride activation of the parent dibenzothiophene S‐oxide.14 Given the synthetic potential of S‐(aryl)sulfonium salts and the lack of a detailed study on the reaction sequence leading to their formation from the dibenzothiophene S‐oxide 1 (see Figure 1), we decided to carefully study this synthetic route, evaluate its scope regarding the nature of the arene, and further explore the reactivity of the sulfonium salts obtained.15
Figure 1.
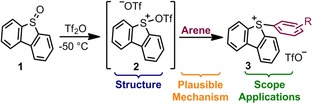
Schematic synthesis of 5‐(aryl)dibenzothiophenium triflates.
Results and Discussion
Trapping of 2 and Characterization of Its Thermally Induced Decomposition Products
We started our investigation by attempting the isolation of compound 2, the species expected from the activation of 1 with Tf2O (Figure 1). Bistriflates of that general structure are usually proposed as the products of activation of sulfoxides when treated with Tf2O. However, no structural characterization of salts with such connectivity has been reported to date. Thus, we slowly added Tf2O to a cooled (−50 °C) dichloromethane solution of 1, and obtained a red suspension.14 All our attempts to obtain monocrystals of 2 under these reaction conditions failed. Warming the reaction mixture to −20 °C resulted in the disappearance of the red color and precipitation of a beige solid. Throughout this transformation the sample remained EPR silent. Chromatographic analysis of the reaction mixture allowed the isolation of the salts 4 and 5 in moderate yields, together with a highly insoluble material (Scheme 1 a). The formation of 4 and 5 suggests a disproportionation of 2 into the corresponding sulfide and sulfone, followed by reaction of both species with either one or two equivalents of the remaining 2. The X‐ray structure of 4, in which the sulfonium substituent is meta to the SO2 group, evidences that this compound was not formed by oxidation of 6. In contrast, the necessary intermediate for the formation of 5, the sulfonium salt 6, was not observed in this experiment, but it can be obtained in nearly quantitative yield by reaction of 2 with one equivalent of dibenzothiophene. Moreover, isolated 6 can further react with a second equivalent of 2 to afford 5 (Scheme 1 a). Hence, we believe that the disproportionation of 2 is a slow process compared with the electrophilic aromatic substitution steps leading to 4 and 5. Finally, MS analysis of the remaining insoluble fraction indicated the formation of highly charged oligomeric species containing four and five dibenzothiophene units, which can be explained by further reaction of 5 with additional 2 (see the Supporting Information). Attempts to inhibit these side reactions by blocking positions 2 and 8 of the dibenzothiophene core with fluorine substituents did not improve the reaction outcome, and instead the sulfonium salt 8 was the only product isolated from the complex mixture (Scheme 1 c). Further analysis is still necessary to explain the formation of 8.
Scheme 1.
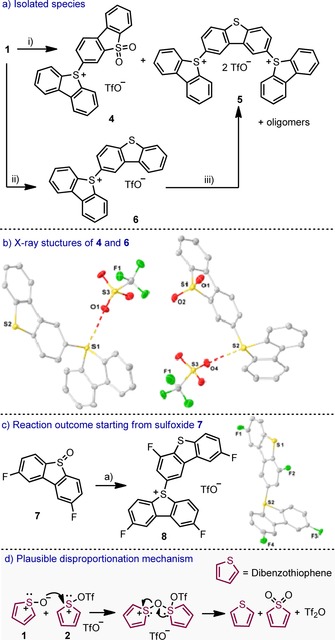
Thermally induced decomposition products of 2. Reagents and conditions: i) Tf2O (1.1 equiv), −50°→−20 °C, 4 h; 4, 11 %; 5, 19 %; ii) Tf2O (1.1 equiv), −50° and then dibenzothiophene (1.1 equiv), 6, 95 %; iii) Tf2O (1.1 equiv), −50° and then 6; 5, 62 %. Molecular structures of 4, 6, and 8 in the solid state. Anisotropic displacement shown at 50 % probability level. Hydrogen atoms and triflate anions omitted for clarity.16.
Finally, a plausible reaction pathway for the formation of sulfone derivatives is depicted in Scheme 1 d. We hypothesize an equilibrium for the formation of 2 from 1 and Tf2O. The attack of 1 onto the sulfur atom in 2, leads to the formation of an oxygen‐bridged disulfonium salt, [17] which presumably rearranges to dibenzothiophene and dibenzothiophene‐5,5‐dioxide by asymmetric cleavage of the bridge.
Being aware of the difficulty that the isolation of 2 entails, we decided to chemically trap it. It was hypothesized that the use of a monodentate neutral ligand, able to engage in strong σ‐donation to the SIV center of 2, might provide sufficient stabilization to allow the isolation of the corresponding adduct before side‐reactions take place. Hence, one equivalent of DMAP was added to a freshly prepared suspension of 2 at −50 °C. This addition causes the slow formation of an orange/yellow solid (9), which was obtained in analytically pure form after washing with dry dichloromethane (Scheme 2 a). X‐ray diffraction analysis of the monocrystals obtained by cooling a saturated dichloromethane solution confirmed the formation of the expected Lewis adduct 9. In the solid state, 9 adopts a dimeric structure in which the geometry around each dibenzothiophene S‐atom can be defined as distorted octahedral. Two vertices are occupied by the aryl rests of the dibenzothiophene moiety, while the pyridine ligand is positioned at a third vertex, adopting a cis relationship with both arene substituents. The other three vertices of the octahedron are each occupied by one triflate anion, which additionally act as a bridge to a second S center. Five from the six S–O contacts [S1⋅⋅⋅O1, 2.991(1); S1⋅⋅⋅O4, 3.821(1); S1⋅⋅⋅O7, 2.769(1); S2⋅⋅⋅O3, 2.986(1); S2⋅⋅⋅O6, 2.963(1); S2⋅⋅⋅O8, 2.534(1)] are significantly shorter than the sum of the van der Waals radii of the corresponding elements (3.32 Å), evidence of a significant Lewis acid character at the S atom. The isolation of 9 unequivocally demonstrates that the primary product of activation of 1 with Tf2O is a dicationic SIV species. Moreover, 9 is still electrophilic and upon treatment with one equivalent of a stronger σ‐donor, such as 1,3‐diisopropyl‐4,5‐dimethyl‐imidazol‐2‐ylidene, ligand exchange takes place to afford the carbene adduct 10 (Scheme 2 b).18
Scheme 2.
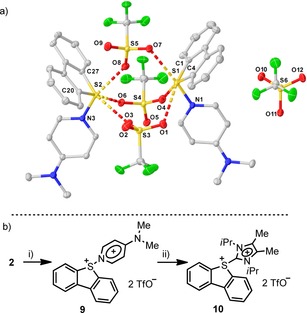
Coordination of Lewis bases to 2. Reagents and conditions: i) DMAP (1 equiv), −50 °C→RT, DCM, 9, 37 %; ii) 1,3‐Diisopropyl‐4,5‐dimethyl‐imidazol‐2‐ylidene (1.0 equiv), THF 10, 20 %. Molecular structure of compound 9. Anisotropic displacement shown at 50 % probability level. Hydrogen atoms, non‐coordinating triflate anion and solvent omitted for clarity. Selected bond lengths [Å]: C1–S1 1.773(2), C4–S1 1.777(2), S1–N1 1.734(1), S1⋅⋅⋅O1 2.991(1), S1⋅⋅⋅O4 3.821(1), S1⋅⋅⋅O7 2.769(1), S1⋅⋅⋅O8 3.232(1), S2–C20 1.778(2), S2–C27 1.781(1), S2–N3 1.735(1), S2⋅⋅⋅O3 2.986(1), S2⋅⋅⋅O6 2.963(1), S2⋅⋅⋅O8 2.534(1).16
Synthesis of 5‐(aryl)Dibezothiophenium Triflates: Scope and Limitations
The next step in our investigation was the study of the reaction between 2 and aromatic substrates. Hence, a series of functionalized arenes were carefully added to freshly prepared suspensions of 2 at −50 °C, and the mixture was warmed to −20 °C and stirred for a 12 hours. Under these reaction conditions, deleterious side‐reactions were minimized and a remarkable scope of sulfonium salts (3 a–r) was obtained (Scheme 3). This synthetic route allows access to the desired compounds without the use of IIII reagents.19 Ideal substrates for this transformation are moderate electron‐rich arenes and heteroarenes such as anisole derivatives, alkyl‐ or aryl‐substituted benzenes, haloarenes, and thiophenes. In most of the cases the reaction proceeded in good to excellent yields and satisfactory para/ortho ratios. In addition, it is tolerant to halogens, ethers, esters, and heteroaromatic skeletons. The reactivity of 2 and 9 is similar in terms of conversion into 3 a when benzene is used as substrate. For this range of aromatic compounds we have no evidence against the formation of sulfonium salts 3 by electrophilic aromatic substitution, even the cyclopropane ring of 3 h survives. However, single‐electron oxidation of the substrate by 2 followed by immediate recombination of the two radicals cannot be excluded (Scheme 4).20
Scheme 3.
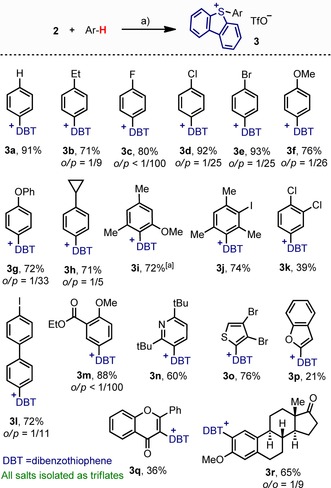
Synthesis of 5‐(aryl)dibenzothiophenium triflates. If not indicated, only one regioisomer was detected. Reagents and conditions: a) Arene (1.1 equiv) −50°→‐20 °C, 15 h. [a] Isolated as a 1:2 mixture of rotamers.
Scheme 4.
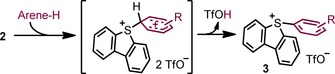
Proposed mechanism for the sulfonium salt formation when using substrates of moderate electron richness.
Figure 2 depicts the molecular diagrams of the salts 3 m and 3 o in the solid state (see the Supporting Information for the crystallographic structures of 3 b–l). It worth noting that interactions between the S atom and the triflate counteranion, reminiscent of the ones observed in 9, are again detected along the complete series. The S1⋅⋅⋅O1 distances [2.990(1) Å for 3 m and 3.057(2) Å for 3 o] are shorter than the sum of the van der Waals radii, revealing the still remarkable electrophilic character at that position.
Figure 2.
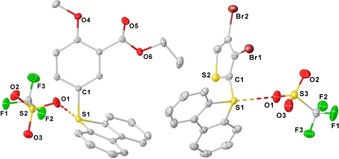
Molecular structures of 3 m (left) and 3 o (right) in the solid state. Anisotropic displacement shown at 50 % probability level. Hydrogen atoms omitted for clarity. Selected bond lengths [Å] and angles [deg]: 3 m, C1–S1 1.776(1), S1⋅⋅⋅O1 2.990(1); C1‐S1‐O1 80.8(1); 3 o, C1–S1 1.763 (3), S1⋅⋅⋅O1 3.057(2); C1‐S1‐O1 77.1(1).16
Electron‐poor rings such as 1,2‐dichlorobenzene are still able to render the corresponding sulfonium salt (3 k) by reaction with 2, albeit in moderate to low yields. Careful column chromatography is necessary to separate 3 k from the side‐products 4 and 5, which are formed in considerable amounts. Electron poorer substrates such as 1,4‐difluorobenzene or trifluoromethylbenzene are inert to the attack of 2. In these essays only decomposition products 4 and 5 together with polycationic dibenzothiophene oligomers are observed.
Electron‐rich (hetero)aromatic rings are problematic as well, but for different reasons. For example, reaction of 2 with N‐methylindole (11) afforded a complex mixture of products from which dibenzothiophene is the main product. The sulfonium salts 6 and 3 s, and bis(indole) 12 were isolated as well (Scheme 5 a). Additionally, tri‐ and tetraindolic structures were detected by MS analysis. This outcome suggests, as reported by Kita et al. for the treatment of pyrroles with hypervalent IIII reagents,21 that single‐electron oxidation of methylindole by 2 takes place, yielding an indole radical cation, which oxidatively couples with more indole producing 12 and indole oligomers. Simultaneously, the dibenzothiophene being formed partially reacts with 2 to afford 6. Additional evidence of the feasibility of a radical mechanism when easy to oxidize substrates are employed derives from the reaction of 2 with stoichiometric amounts of TEMPO. This essay affords a clean mixture of 6 (75 % yield of isolated product) and the TEMPO oxoamonium triflate 13 (Scheme 5 b).
Scheme 5.
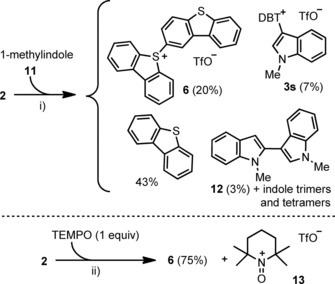
a) Reaction of 2 with 1‐methylindole (11). b) Reaction of 2 with TEMPO. Reagents and conditions: i) 1‐methylindole (1.1 equiv), CH2Cl2, −50→−20 °C. ii) CH2Cl2, −50→−20 °C.
Post‐Synthetic Modification of Sulfonium Salts by Nucleophilic Aromatic Substitution and Synthetic Applications
Given their electron richness, aniline substrates are not well‐tolerated by the protocol just described. However, sulfonium salts containing these substituents might be synthetically useful compounds because haloanilines are not always easy to obtain. In fact, classical Friedel–Crafts aniline halogenation is difficult to control, with polyhalogenation taking place readily.22 We rationalized at this stage that aniline‐substituted sulfonium salts might be obtained in a two‐step sequence starting from the fluorobenzene derivative 3 c. In this salt the fluoride is expected to be activated towards nucleophilic aromatic substitution because of the strong electron‐withdrawing nature of the sulfonium moiety at the para position. Should that substitution be feasible, reaction of 3 c with amines may deliver the desired aniline‐derived sulfonium salts. Actually, it was found that a variety of secondary amines were suitable for the reaction, leading to a set of sulfonium salts (14 a–e) containing structurally differentiated aniline substituents (Scheme 6). The chlorobenzene derivative 3 d is unable to deliver the products 14 a–e under identical reaction conditions.
Scheme 6.
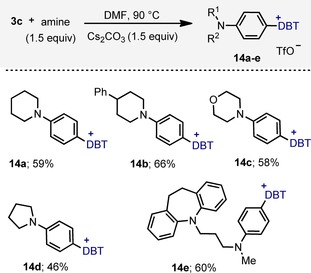
Synthesis of aniline‐substituted dibenzothiophenium triflates by nucleophilic aromatic substitution.
Finally, it was recently reported that thianthrenium cations participate in site‐selective coupling reactions over typical linchpins such as C−Br and C−OTf bonds.11b We rationalized that with the DBT unit being more electron‐poor than the thianthrenium one, the insertion of Pd along the C−DBT bond should be even more favored. Hence, we decided to challenge the well‐established C−I site selectivity in the Suzuki coupling versus the C−DBT.23 Specifically, the salt 3 l was chosen as a substrate because the iodo and dibenzothiophenium units are in identical stereoelectronic environments but on independent rings. One equivalent of 4‐(trifluoromethyl)phenyl boronic acid was used for this experiment (Scheme 7). From this assay, carried out at 35 °C and employing only 0.5 mol % of Pd(AcO)2 as catalyst, the product of selective arylation at the C−DBT bond (15) was isolated in 90 % yield. By MS we detected traces of the thioether 16, which derives from initial insertion of Pd into one of the internal C−S bonds of the dibenzothiophene moiety, and traces of the product resulting from coupling at the C−I site. This result demonstrates intrinsic chemoselectivity for C−DBT over C−I in the Suzuki reaction. Given the mild reaction conditions and simple catalyst employed to promote the coupling, as well as the exquisite site selectivity observed, we envision the use of DBT units as a promising linchpin to enable rapid access to densely functionalized polyarenes through coupling reactions.
Scheme 7.
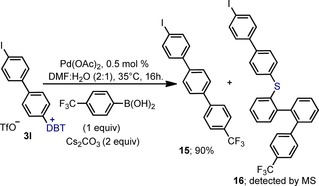
Chemoselective Suzuki coupling at C−DBT over C−I bonds.
Conclusion
In summary, we report herein a straightforward method for the synthesis 5‐(aryl)dibenzothiophenium triflates by metal‐free sulfenylation of C−H bonds. Preliminary mechanistic studies based on trapping intermediates, and the careful isolation of decomposition products, suggests that an electrophilic aromatic substitution is operative for the sulfenylation of electron‐neutral or moderately‐rich arenes. The generation of radicals by single‐electron transfer between 2 and the substrate is more conceivable for more electron‐rich aromatics, leading to complex reaction mixtures. Comparison of the reactivity of the DBT linchpin with that of C−I bonds in a model Suzuki reaction reveals full selectivity for the DBT moiety over the competing C−I coupling site. Hence, we anticipate a widespread range of applications for 5‐(aryl)dibenzothiophenium triflates in Pd chemistry, in particular for the execution of sequential biaryl couplings leading to the construction of polyarenes.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Support from the European Research Council (ERC CoG 771295), the Deutsche Forschungsgemeinschaft (INST 186/1237‐1), and the University of Göttingen is gratefully acknowledged.
K. Kafuta, A. Korzun, M. Böhm, C. Golz, M. Alcarazo, Angew. Chem. Int. Ed. 2020, 59, 1950.
References
- 1. Srogl J., Allred G. D., Liebeskind L. S., J. Am. Chem. Soc. 1997, 119, 12376–12377. [Google Scholar]
- 2.
- 2a. Chung S.-K., Sasamoto K., Chem. Commun. 1981, 346–347; [Google Scholar]
- 2b. Beak P., Sullivan T. A., J. Am. Chem. Soc. 1982, 104, 4450–4457; [Google Scholar]
- 2c. Saeva F. D., Tetrahedron 1986, 42, 6123–6129; [Google Scholar]
- 2d. Dektar L., Hacker N. P., J. Am. Chem. Soc. 1990, 112, 6004–6015; [Google Scholar]
- 2e. Saeva F. D., Breslin D. T., Luss H. R., J. Am. Chem. Soc. 1991, 113, 5333–5337; [Google Scholar]
- 2f. Kampmeier J., Nalli T. W., J. Org. Chem. 1993, 58, 943–949; [Google Scholar]
- 2g. Wang X., Saeva F. D., Kampmeier J. A., J. Am. Chem. Soc. 1999, 121, 4364–4368. [Google Scholar]
- 3.
- 3a. Vasu D., Yorimitsu H., Osuka A., Synthesis 2015, 47, 3286–3291; [Google Scholar]
- 3b. Wang S. M., Wang X. Y., Qin H. L., Zhang C. P., Chem. Eur. J. 2016, 22, 6542–6546; [DOI] [PubMed] [Google Scholar]
- 3c. Wang X.-Y., Song H.-X., Wang S.-M., Yang J., Qin H.-L., Jiang X., Zhang C.-P., Tetrahedron 2016, 72, 7606–7612; [Google Scholar]
- 3d. Cowper P., Jin Y., Turton M. D., Kociok-Kohn G., Lewis S. E., Angew. Chem. Int. Ed. 2016, 55, 2564–2568; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2610–2614. [Google Scholar]
- 4.
- 4a. Wang S.-M., Song H.-X., Wang X.-Y., Liu N., Qin H.-L., Zhang C.-P., Chem. Commun. 2016, 52, 11893–11896. [DOI] [PubMed] [Google Scholar]
- 5. Tian Z.-Y., Wang S.-M., Jia S.-J., Song H.-X., Zhang C.-P., Org. Lett. 2017, 19, 5454–5457. [DOI] [PubMed] [Google Scholar]
- 6. Aukland M. H., Talbot F. J. T., Fernández-Salas J. A., Ball M., Pulis A. P., Procter D. J., Angew. Chem. Int. Ed. 2018, 57, 9785–9789; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9933–9937. [Google Scholar]
- 7. Zhang S., Marshall D., Liebeskind L. S., J. Org. Chem. 1999, 64, 2796–2804. [DOI] [PubMed] [Google Scholar]
- 8. Vasu D., Yorimitsu H., Osuka A., Angew. Chem. Int. Ed. 2015, 54, 7162–7166; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7268–7272. [Google Scholar]
- 9. Engl P. S., Häring A. P., Berger F., Berger G., Pérez-Bitrián A., Ritter T., J. Am. Chem. Soc. 2019, 141, 13346–13351. [DOI] [PubMed] [Google Scholar]
- 10. Gendron T., Sander K., Cybulska K., Benhamou L., Sin P. K. B., Khan A., Wood M., Porter M. J., Årstad E., J. Am. Chem. Soc. 2018, 140, 11125–11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Donck S., Baroudi A., Fensterbank L., Goddard J.-P., Ollivier C., Adv. Synth. Catal. 2013, 355, 1477–1482; [Google Scholar]
- 11b. Berger F., Plutschack M. B., Riegger J., Yu W., Spiecher S., Ho M., Frank N., Ritter T., Nature 2019, 567, 223–228; [DOI] [PubMed] [Google Scholar]
- 11c.M. H. Aukland, M. Šiaučiulis, A. West, G. J. P. Perry, D. J. Procter, ChemRxiv Preprint 10.26434/chemrxiv.9158564.v1 and ref. [9]. [DOI]
- 12.
- 12a. Kim K., Hull V. J., Shine H. J., J. Org. Chem. 1974, 39, 2534–2537; [Google Scholar]
- 12b. Miyatake K., Yamamoto K., Endo K., Tsuchida E., J. Org. Chem. 1998, 63, 7522–7524; [DOI] [PubMed] [Google Scholar]
- 12c. Racicot L., Kasahara T., Ciufolini M. A., Org. Lett. 2014, 16, 6382–6385; [DOI] [PubMed] [Google Scholar]
- 12d. Zhang L., Li X., Sun Y., Zhao W., Luo F., Huang X., Lin L., Yang Y., Peng B., Org. Biomol. Chem. 2017, 15, 7181–7189. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Hartke K., Teuber D., Gerber H. D., Tetrahedron 1988, 44, 3261–3270; [Google Scholar]
- 13b. Miller R. D., Renaldo A. F., Ito H., J. Org. Chem. 1988, 53, 5571–5573; [Google Scholar]
- 13c.A. Fürstner, M. Alcarazo, K. Radkowski, C. W. Lehmann, Angew. Chem. Int. Ed 2008, 47, 8302–8306; [DOI] [PubMed]
- 13d.J. Wu, X.-Y. Chen, Y. Wu, D. Wang, P. Wang, ChemRxiv Preprint 10.26434/chemrxiv.8148725.v2. [DOI]
- 14.
- 14a. Waldecker B., Kraft F., Golz C., Alcarazo M., Angew. Chem. Int. Ed. 2018, 57, 12538–12542; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12718–12722; [Google Scholar]
- 14b. Li X., Golz C., Alcarazo M., Angew. Chem. Int. Ed. 2019, 58, 9496–9500; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9596–9600; [Google Scholar]
- 14c. Waldecker B., Kafuta K., Alcarazo M., Org. Synth. 2019, 96, 258–276. [Google Scholar]
- 15.5-(aryl)dibenzothiophenium salts have been very recently prepared in situ, and directly used as substrates in photoredox catalyzed C−H/C−H coupling reactions; their systematic characterization is not reported in that article (Ref. [11c]).
- 16.CCDC https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/anie.201912383 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/.
- 17. Fascione M. A., Adshead S. J., Mandal P. K., Kilner C. A., Leach A. G., Turnbull W. B., Chem. Eur. J. 2012, 18, 2987–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Compound 10 is isolobal to α-cationic phosphines. See:
- 18a. Alcarazo M., Acc. Chem. Res. 2016, 49, 1797–1805; [DOI] [PubMed] [Google Scholar]
- 18b. Nicholls L. D. M., Alcarazo M., Chem. Lett. 2019, 48, 1–13. [Google Scholar]
- 19.
- 19a. Zhang B., Li T., Kang Y., Res. Chem. Intermed. 2017, 43, 6617–6625. [Google Scholar]
- 20.No EPR signal was detected while monitoring these reactions.
- 21.
- 21a. Dohi T., Morimoto K., Maruyama A., Kita Y., Org. Lett. 2006, 8, 2007–2010; [DOI] [PubMed] [Google Scholar]
- 21b. Dohi T., Ito M., Morimoto K., Iwata M., Kita Y., Angew. Chem. Int. Ed. 2008, 47, 1301–1304; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 1321–1324. [Google Scholar]
- 22. Smith M. B., March J., Advanced Organic Chemistry, 5th ed., Wiley, New York, 2001, chap. 11. [Google Scholar]
- 23. Rossi R., Bellina F., Lessi M., Adv. Synth. Catal. 2012, 354, 1181–1255. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary